
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLaser-synthesized plasmonic HfN-based nanoparticles as a novel multifunctional agent for photothermal therapy†
A. I.
Pastukhov
a,
M. S.
Savinov
b,
I. V.
Zelepukin
cd,
J. S.
Babkova
 c,
G. V.
Tikhonowski
b,
A. A.
Popov
b,
S. M.
Klimentov
b,
A.
Devi
e,
A.
Patra
e,
I. N.
Zavestovskaya
bfg,
S. M.
Deyev
bcg and
A. V.
Kabashin
*ab
c,
G. V.
Tikhonowski
b,
A. A.
Popov
b,
S. M.
Klimentov
b,
A.
Devi
e,
A.
Patra
e,
I. N.
Zavestovskaya
bfg,
S. M.
Deyev
bcg and
A. V.
Kabashin
*ab
aAix-Marseille University, CNRS, LP3, 13288, Marseille, France. E-mail: andrei.kabashin@univ-amu.fr
bMEPhI, Institute of Engineering Physics for Biomedicine (PhysBio), 115409, Moscow, Russia
cShemyakin-Ovchinnikov Institute of Bioorganic Chemistry of Russian Academy of Sciences, 117997, Moscow, Russia
dUppsala University, Department of Medicinal Chemistry, 75310, Uppsala, Sweden
eInstitute of Nano Science and Technology, Mohali, 140306, India
fP. N. Lebedev Physical Institute of the Russian Academy of Sciences, 119991, Moscow, Russia
gNational Research Center “Kurchatov Institute”, 123182, Moscow, Russia
First published on 28th August 2024
Abstract
Hafnium nitride nanoparticles (HfN NPs) can offer appealing plasmonic properties at the nanoscale, but the fabrication of stable water-dispersible solutions of non-toxic HfN NPs exhibiting plasmonic features in the window of relative biological transparency presents a great challenge. Here, we demonstrate a solution to this problem by employing ultrashort (femtosecond) laser ablation from a HfN target in organic solutions, followed by a coating of the formed NPs with polyethylene glycol (PEG) and subsequent dispersion in water. We show that the fabricated NPs exhibit plasmonic absorption bands with maxima around 590 nm, 620 nm, and 650 nm, depending on the synthesis environment (ethanol, acetone, and acetonitrile, respectively), which are largely red-shifted compared to what is expected from pure HfN NPs. The observed shift is explained by including nitrogen-deficient hafnium nitride and hafnium oxynitride phases inside the core and oxynitride coating of NPs, as follows from a series of structural characterization studies. We then show that the NPs can provide a strong photothermal effect under 808 nm excitation with a photothermal conversion coefficient of about 62%, which is comparable to the best values reported for plasmonic NPs. MTT and clonogenic assays evidenced very low cytotoxicity of PEG-coated HfN NPs to cancer cells from different tissues up to 100 μg mL−1 concentrations. We finally report a strong photothermal therapeutic effect of HfN NPs, as shown by 100% cell death under 808 nm light irradiation at NP concentrations lower than 25 μg mL−1. Combined with additional X-ray theranostic functionalities (CT scan and photon capture therapy) profiting from the high atomic number (Z = 72) of Hf, plasmonic HfN NPs promise the development of synergetically enhanced modalities for cancer treatment.
Introduction
Owing to optical excitation of collective oscillations of free electrons (plasmons) in some metals (Au, Ag, Cu, etc.), plasmonic nanostructures are capable of providing a series of prominent properties, making them appealing candidates for a variety of applications in nanophotonics,1 nanoelectronics,2 photovoltaics,3 photocatalysis,4 optical biosensing,5,6etc. Some of these properties are beneficial for biomedical applications. In particular, plasmonic nanostructures can provide resonant absorption of light in a specific spectral range with the efficiency exceeding that of organic dyes by 3–4 orders of magnitude,7 which opens up avenues for their use as efficient sensitizers in photothermal therapy (PTT) to eliminate cancer cells via local overheating (hyperthermia)8–10 and as contrast agents in photoacoustic bioimaging.11 In addition, plasmonic nanostructures can exhibit near-field enhancement properties, which allows one to profit from the surface-enhanced Raman spectroscopy (SERS) channel for identification and imaging of molecules conjugated to their surface.12,13Applications of plasmonic nanomaterials in biological systems in vivo typically require their high chemical stability and good biological compatibility. Gold (Au) nanostructures mostly satisfy these requirements and have been considered the main plasmonic candidate for biomedical applications,14 although some acute toxicity effects were reported due to the presence of hazardous by-products (e.g., CTAB) on Au nanostructures fabricated by conventional chemical routes.15,16 However, since spherical Au nanoparticles (NPs) exhibit absorption peaks between 520 and 560 nm,7 which is outside the biological transparency window (650–900 nm), one has to use complex architectures such as Au core–shells,17,18 nanorods19 or nanocages20 to solve the mismatch problem. Meanwhile, the use of such structures in photothermal therapy applications is not free of problems, including the loss of shape of anisotropic structures under photo-induced heating21,22 or possible difficulty in the delivery of core-shells with sizes exceeding 120 nm to some organs.17 Therefore, alternative plasmonic nanomaterials providing absorption features in the biotransparency window with a relatively small size and low toxicity are still in high demand.
Transition metal nitrides (TMNs), including TiN, ZrN, and HfN, are now considered as possible plasmonic alternatives to conventional gold.23–26 Exhibiting a golden color similar to gold, these materials can also demonstrate prominent plasmonic properties at the nanoscale, which makes them highly valuable in a variety of applications, including photocatalysis,27,28 solar energy converters,29–31 and nanophotonics.32 Biomedical applications of TMN NPs have been less explored so far, but their prospects look promising if the NPs are properly designed and free of toxic contamination. Until now, titanium nitride (TiN) NPs are the most popular in these applications due to low cost, high abundance and the generation of plasmonic features in the region of relative tissue transparency (typically around 570–700 nm) with almost all light energy converted to absorption.26 Profiting from these properties, TiN NPs have been successfully tested in biomedical applications, including their use as efficient sensitizers in photothermal therapy33,34 and as contrast agents in photoacoustic imaging.33,35 All first data look very encouraging and promise the advancement of methods of diagnostics and therapy based on TMN plasmonic nanomaterials.
Such a success of TiN NPs stimulates further exploration of TMN NPs in biomedical applications. Of particular interest, we envision the use of nanomaterials, which could combine plasmonic properties and additional non-plasmonic functionalities to enhance the resulting theranostic outcome. In this sense, hafnium nitride (HfN) looks as a prominent example. In the pure state, HfN NPs exhibit a distinct plasmonic absorption feature outside the transparency window (around 480–510 nm), but their position can be red-shifted toward this window e.g. by coating HfN NPs with hafnium oxide (Fig. S1†), which provides opportunities for their use as sensitizers in photothermal therapy. Besides, being a high atomic number (Z = 72) element, hafnium is known to be one of the best candidates for X-ray radiation theranostics,36 which presents the combination of photon capture therapy37 and contrast-enhanced computed tomography (CT).38 Furthermore, NBTXR3, which is one of a few officially approved nanodrugs for cancer radiotherapy, is based on hafnium compounds, namely functionalized crystalline hafnium oxide nanoparticles with a size of 50 nm.39 Designed for injection directly into a malignant tumor, NBTXR3 can drastically enhance the radiotherapy outcome,39,40 while preclinical and clinical trials have proved the safety and efficacy of NBTXR3 for the treatment of various cancer types.41,42 We suppose that the use of HfN nanoformulations could also profit from the presence of Hf in their composition to enable X-ray diagnostics or therapy functionalities.
However, the synthesis of HfN NPs suitable for biological use faces the same problems as the preparation of other TMN NPs, while the literature on the fabrication of such NPs is scarce. Here, chemical routes typically involve hazardous products,43 which leads to the contamination of NP surfaces and, consequently, to possible toxic effects. On the other hand, the use of “dry” techniques such as the arc plasma process44 results in aggregated, hardly water-dispersible structures, which typically form very unstable colloids. As a solution to the above-stated problems in the synthesis of many nanomaterials for biological applications, we introduced a technique of femtosecond (fs) laser ablation in liquids,45,46 which relies on natural laser-ablative production of nanoclusters in a liquid environment, followed by their coalescence to form NPs. In contrast to chemical synthesis routes, laser ablation does not require fixed chemical products and particular synthesis conditions, while the ablation can be carried out in a clean environment (water, ethanol, acetone), which avoids any toxic contamination of formed NPs.47 In addition, in contrast to the dry synthesis routes, the ablated NPs are generated in a liquid medium, while their surface is charged in most cases due to the formation of a thin oxide shell arising as a result of NPs interacting with the environment, which affords exceptional stability to colloidal solutions.48 The technique of ultrashort (fs) laser ablation is especially efficient at controlling the size of NPs of virtually any material and we already used this technique for the fabrication of TiN NPs with a controlled position of the plasmonic peak depending on the oxidation state of NPs.34,49 Furthermore, after coating with polyethylene glycol, laser-synthesized NPs demonstrated high safety and favorable biodistribution in vivo,50 and were successfully used as sensitizers in photothermal therapy,34 contrast agents in photoacoustic imaging,51 and markers in point-of-care biosensing.52 We believe that the same strategy can be used to fabricate other TMN nanomaterials.
Here, we explore the use of fs laser ablation in different organic solutions (ethanol, acetone, acetonitrile) to fabricate HfN nanoparticles for biomedical use. We show that the laser synthesis leads to the formation of HfN-based NPs exhibiting plasmonic absorption with a peak between 590 and 650 nm, depending on the synthesis environment. The tuning of the peak position was explained by a variable inclusion of hafnium oxynitride and nitrogen-deficient hafnium nitride phases inside the core and oxynitride coating of NPs. After coating with PEG, HfN NPs demonstrated very low toxicity for different cancer cell lines, while their heating by radiation in the biological transparency window (808 nm) caused 100% cell death at low NP concentrations of 25 μg mL−1. Such plasmonic properties promise the advancement of methods of photothermal therapy and photoacoustic imaging based on HfN NPs, and in combination with X-ray theranostic functionalities due to the high Z number of Hf, providing access to possible synergetic modalities to improve the efficiency of cancer treatment.
Experimental
Synthesis of nanoparticles
To fabricate HfN-based NPs, a hot-pressed HfN target (99%, MatecK, Germany) was ablated by radiation from Yb:KGW lasers: (i) 490 fs pulse length, 1030 nm wavelength, 50 kHz repetition rate (Amplitude Systems, France); and (ii) 270 fs pulse length, 1030 nm wavelength, 50 kHz repetition rate (TETA-10, Avesta, Moscow, Russia) in ethanol (Merck, 96%), acetone (>99.5%, AcrosOrganics, Belgium), and acetonitrile (>99.5%, J.T. Baker, USA). Laser radiation was directed via a system of mirrors, the energy was attenuated to 40 μJ pulse−1 and then focused on the surface of a vertically fixed HfN target placed inside a glass cuvette filled with 50 mL of organic solvent (ethanol, acetone, or acetonitrile), as shown in Fig. 1a. The distance between the inner wall of the cuvette and the surface of the target was about 3 mm. The laser beam was moved across the target at 2.5–4 m s−1 velocity to avoid multiple ablation from the same target area, leading to the loss of NP production rate. Before characterization, colloidal solutions of HfN-based NPs were centrifuged for 1.5 min at 1000g to remove large nano- and microparticles produced due to the detachment of large fragments from the hot-pressed HfN target.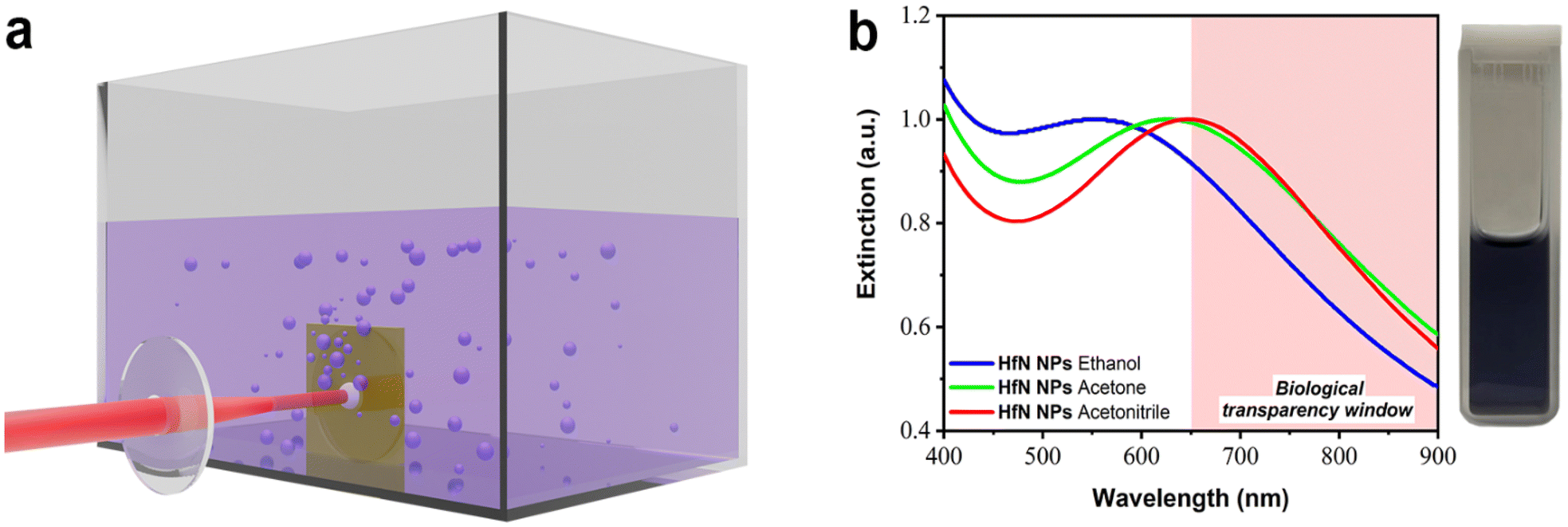 | ||
| Fig. 1 Laser-ablative synthesis of HfN-based NPs and their optical properties. (a) Schematic illustration of laser-ablative synthesis. A focused beam from an Yb:KGW fs laser is directed onto a HfN target placed inside a glass cuvette filled with an organic solvent (ethanol, acetone or acetonitrile). The laser ablation process leads to the formation of HfN-based NPs and their release into the surrounding liquid medium. (b) Extinction spectra from solutions of HfN-based NPs synthesized in ethanol, acetone and acetonitrile. The inset shows a photo of a typical solution of HfN-based NPs prepared in acetonitrile. | ||
Characterization of nanoparticles
Optical extinction spectra of the synthesized colloidal solutions were collected using 10 mm optical path length cuvettes using a UV 2600 spectrophotometer (Shimadzu, Japan) in the spectral range of 400–900 nm or an MC 122 spectrophotometer (SOL Instruments, Belarus) in the spectral range of 340–1000 nm. The morphology and size of HfN-based NPs synthesized in ethanol/acetone were characterized using a JEM-2100F high-resolution transmission electron microscope (TEM) (JEOL USA, Inc., USA) at an accelerating voltage of 200 keV. Samples for electron microscopy were prepared by dropping 1 μL of NPs solution onto a formvar-coated copper grid followed by drying at room temperature. The size distributions of NPs were obtained by analyzing the TEM images in the ImageJ software using a circle fit approximation. The chemical composition was characterized using an energy-dispersive X-ray spectroscopy module (EDX) X-Act (Oxford Instruments, UK) by dropping 1 μL of NP solution onto a cleaned monocrystalline silicon substrate, followed by drying at room temperature. EDX spectra were obtained at 20 keV accelerating voltage. The X-ray diffraction (XRD) measurements of HfN NP powder prepared in ethanol and acetone were performed in a transmission mode using a Rigaku Ultima IV X-Ray diffractometer (Rigaku, Japan) using Cu Kα radiation (λ = 1.5418 Å). The size of the beam was 0.5 × 0.5 mm2, and the maximum 2θ value was 65° (0.3° experimental resolution). The XRD measurements of HfN NPs synthesized in acetonitrile were performed on a Bruker D8 ADVANCE powder diffractometer with Co Kα radiation. The NP powder for XRD measurements was prepared by drying NP solutions under ambient conditions overnight in a glass Petri dish. Raman spectra were obtained using a compact confocal Raman microscope Confotec MR350 (SOL Instruments, Belarus) equipped with a 35 mW semiconductor laser at a wavelength of 633 nm. The samples were also characterized by X-ray photoelectron spectrometry (XPS) using a ThermoScientific Al K-alpha spectrometer equipped with a 180° double-focusing hemispherical analyzer, Al Kα X-ray source (1486.7 eV) with spot size 400 μm, and a 128-channel detector. For XPS analysis, the samples were drop-cast onto a clean glass slide surface.Study of photothermal properties
To evaluate the photothermal properties of bare (ligand-free) HfN-based NPs, they were irradiated in a glass cell with an optical path length of 10 mm by 808 nm continuous laser radiation. Temperature was measured using a FLIR C3 (FLIR Systems, USA) thermal imaging camera. Concentration of NPs was measured gravimetrically by weighing a mass of cover glass, then dropping 50 μl of NP solution onto the cover glass followed by drying at room temperature and reweighing. The NP photothermal conversion efficiency (η) was calculated by the previously developed method53,54 using η (%) = ΔTmaxmliquidCliquidB/(Iin − Iout) × 100%, where ΔTmax is the maximal difference between temperatures of the NP solution and the ambient medium, mliquid and Cliquid are mass and specific heat capacities of colloidal solutions, B is a temporal constant defined as the slope of the dependence of the cooling time on ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (ΔTmax/ΔT(t)), Iin is the initial radiation power, and Iout is the radiation power after passing the cell with a solution.
(ΔTmax/ΔT(t)), Iin is the initial radiation power, and Iout is the radiation power after passing the cell with a solution.
Coating of HfN nanoparticles with PEG
The HfN NPs were coated with PEG using the Stober reaction. 1 mg of HfN NPs was dispersed in 950 μL of 95% ethanol, and then, 65 μL of distilled water and 20 μL of 30% ammonia hydroxide were added. After thorough mixing and sonication, 100 μL of 1 g L−1 5 kDa mPEG-Silane solution in ethanol was quickly added to the HfN NP solution. Next, the reaction mixture was heated to 60 °C for 2 h and further incubated at room temperature overnight. The obtained HfN@PEG NPs were washed 3 times with 95% ethanol and with distilled water via centrifugation at 10000g for 15 min.
Characterization of HfN nanoparticles after coating
The hydrodynamic diameter and ζ-potential were measured by the dynamic light scattering technique using a Zetasizer Nano ZS device (Malvern Instruments, Malvern, UK). The mode values ± half-width of the peak of number-weighted size distributions were used for analysis. The measurements of ζ-potential were performed in a 10 mM NaCl water solution, and the Smoluchowski approximation was used for calculations. FTIR spectra were obtained from the polymer and nanoparticle powders using an FT-801 spectrometer (Simex, Russia) at room temperature with a wavenumber resolution of 4 cm−1 in the frequency range of 4000–500 cm−1 in a total internal reflection geometry.Cell culture
In vitro experiments were performed on BT474 (ATCC HTB-20™) and EMT6/P (ECACC catalog no. 96042344) cell lines. BT474 were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and Gibco antibiotic-antimycotic solution. EMT6/P cells were maintained in DMEM medium supplemented with 10% FBS, 2 mM L-glutamine, and Gibco antibiotic-antimycotic solution. Cells were incubated under a humidified atmosphere with 5% CO2 at 37 °C.MTT test
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test was used to determine the cytotoxicity of HfN@PEG NPs. Cells were seeded into 96-well plates at a density of 104 cells per well. HfN@PEG NPs were then added to the cells to final concentrations of 10, 25, 50, and 100 μg mL−1. Cells were incubated for 24 h or 72 h under a humidified atmosphere with 5% CO2 at 37 °C. After the incubation, the medium was removed, and 100 μL of MTT solution per well was added at the concentration of 0.5 mg mL−1 and cells were incubated for 1 h at 37 °C. Then, the MTT solution was removed and 100 μL of DMSO was added to each well. The measurement was carried out on the Infinite M1000Pro spectrophotometer (Tecan, Austria) at a wavelength of 570 nm with a reference wavelength of 630 nm. Cell viability was calculated relative to untreated cells without irradiation.To study photothermal efficiency after near-infrared irradiation, 105 cells were added to 1.5 mL tubes and incubated in 1 mL of colorless medium with particles at final concentrations of 10, 25, 50, and 100 μg mL−1 for 30 min at 37 °C. After the incubation, the samples were irradiated for 5 min by 808 nm laser radiation at 0.8 W under continuous shaking at 300 rpm. Then, the irradiated cells were transferred into a 96-well plate at a density of 104 cells per well. The cells were incubated for 24 h or 72 h under a humidified atmosphere with 5% CO2 at 37 °C. Then, cell viability was measured by the MTT test, as described above.
Clonogenic assay
For the clonogenic assay, the same conditions were used as described for the MTT test. After the incubation of cells with HfN@PEG NPs and irradiation by an 808 nm laser, the cells were diluted, and 103 cells in 2 mL of culture medium with 10% FBS were added to each well of a 6-well plate and incubated for 14 days at 5% CO2 and 37 °C. Next, the medium was removed, and cells were washed with 1 mL of PBS (pH 7.4) and 1 mL of 50% ethanol solution in PBS. Then, 1 mL of 70% ethanol solution in PBS was added to the wells to fix the cells, followed by incubation at room temperature for 15 min. Then, 70% ethanol was removed, and 1 mL of 95% ethanol was added, repeating the incubation conditions. After the removal of 95% ethanol, the plates were washed with distilled water. The cells were stained with 0.1% crystal violet water solution for 30 min at room temperature. Then, the plates were washed 10 times with distilled water. Images of the colonies were obtained using an Epson Perfection 2400 photo scanner (Epson, USA). The number of colonies was counted manually under a microscope, considering colonies consisting of more than 50 cells. The surviving fraction was calculated relative to untreated control without irradiation.Results and discussion
Synthesis and characterization of HfN nanoparticles
For the formation of HfN-based NPs, we elaborated the technique of ultrashort (fs) laser ablation from a bulk hafnium nitride target in a liquid medium, using a methodology described in our previous works.34,49,55 The ablation was performed in three organic liquids (ethanol, acetone, and acetonitrile), as depicted in Fig. 1a. The laser ablation of material from a HfN target led to a visible coloration of solvents, suggesting the formation of colloidal solutions of NPs. When ablated in ethanol, colloidal solutions were dark purple, while similar ablation in acetone and acetonitrile led to a dark blue color. The maximal production rate was observed when the target was ablated in acetonitrile, as confirmed by solution coloration and concentration measurements.Extinction spectra of so-formed colloidal solutions revealed the appearance of resonance features, associated with the excitation of localized plasmons, as shown in Fig. 1b. One can see that each spectrum is broad, largely covering the window of relative biological transparency (650–900 nm), highlighted by a pink background. It is also seen that plasmonic absorption from NP samples prepared in ethanol, acetone, and acetonitrile had maxima at different parts of the spectrum, namely at 590 nm, 620 nm, and 650 nm, respectively. It should be noted that the extinction spectra of HfN-based NPs had narrower plasmon peaks compared to the spectra of TiN NPs obtained in our previous works,34,49 which can be explained by a lower oxidation rate of HfN NPs.
To better understand the mechanisms of the observed shift of plasmon peak positions, TEM measurements (Fig. 2a–d) were performed. As shown in Fig. 2a–c, the ablation process indeed led to the formation of spherical NPs. The samples prepared in ethanol and acetone had an average (mode) diameter of approximately 16 nm, while the mode diameter of NPs prepared in acetonitrile was slightly smaller (10 nm). To examine the structural properties of formed NPs, HR-TEM images of a single particle were analyzed. For samples synthesized in both acetonitrile (Fig. 2d) and ethanol/acetone (Fig. S2a and b†), the results of morphological analysis indicate the formation of polycrystalline phases, while the surface of NPs was coated with a shell, which could be attributed to oxidation phenomena. As shown in Fig. 2d, the interplanar spacing of the crystalline structure was approximately 0.26 nm, which corresponds to the table reference value for the (111) plane of crystalline HfN. Based on the analysis of size distributions, one may conclude that the size-dependent model of resonance peak position fails to explain the observations, as the mean size of the NPs remains nearly the same. Furthermore, samples of NPs prepared in acetonitrile had a red-shifted peak compared to the samples prepared in ethanol/acetone, while the mode size of these NPs was smaller (10 nm compared to 16 nm, respectively).
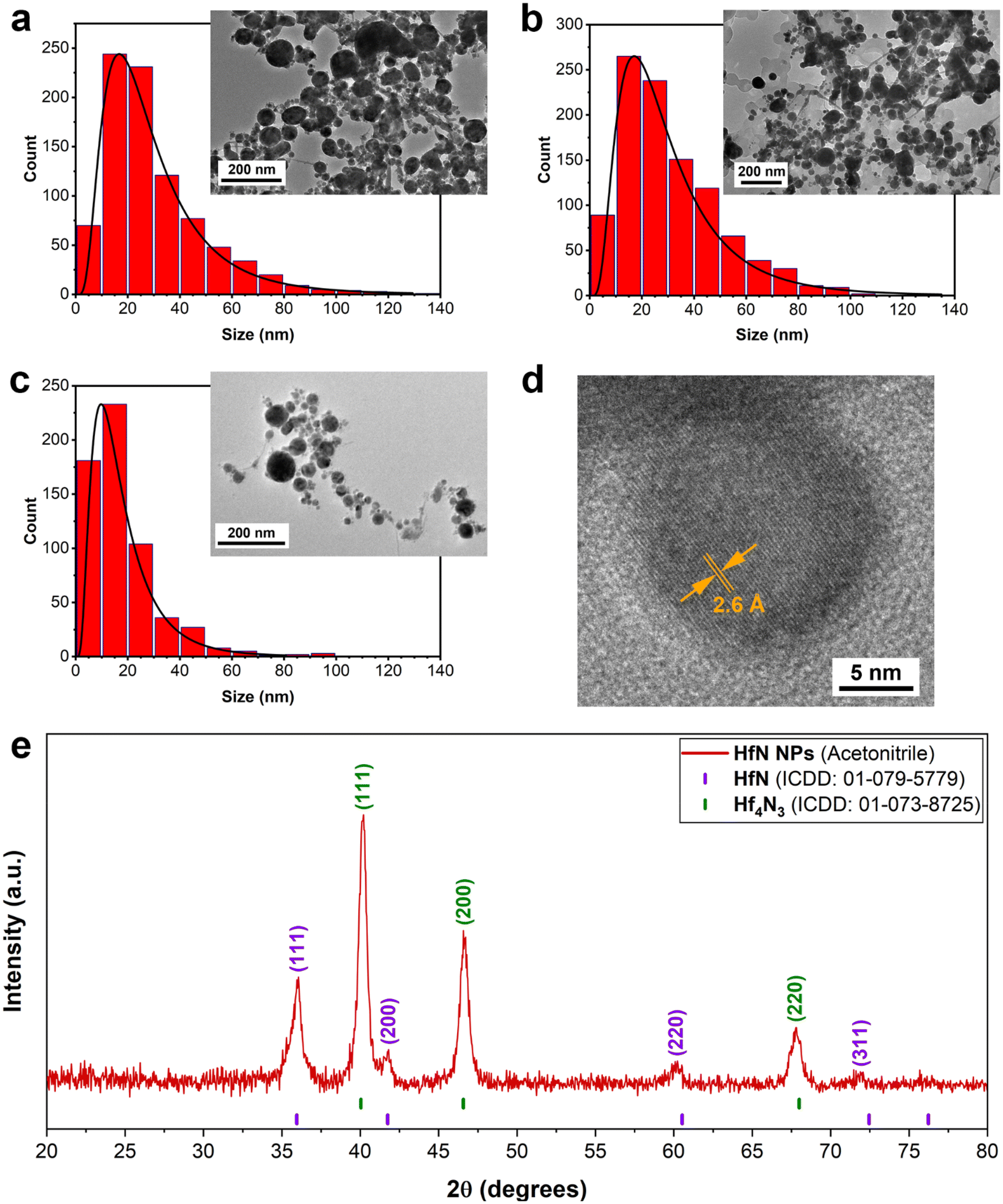 | ||
| Fig. 2 Characterization of HfN-based NPs. (a–c) Typical transmission electron microscopy images and corresponding size distributions of HfN-based NPs prepared in ethanol (a), acetone (b) and acetonitrile (c). (d) HR-TEM image of a nanoparticle synthesized in acetonitrile, showing clearly distinct crystalline structure of obtained NPs. The interplanar spacing of the laser-synthesized NPs is about 0.26 nm, which corresponds to the table value for the (111) plane of crystalline HfN. (e) XRD pattern of HfN-based NP powder. Table positions of crystalline HfN and Hf4N3 are given above the pattern. | ||
The elemental composition of HfN-based NPs was studied by energy-dispersive X-ray spectroscopy (EDX). A spectrum of HfN NPs prepared in acetonitrile is shown in Fig. S3.† Here, a strong signal associated with hafnium, as well as a weak nitrogen line, can be identified, indicating the presence of HfN-related structures in the composition. We also observed an oxygen signal, which is attributed to a partial oxidation of NPs.
To further investigate the NPs’ structural features and elucidate the mechanism responsible for plasmon resonance peak shift related to pure HfN NPs (Fig. S1†), we performed XRD measurements of HfN NP powders. A typical stoichiometric HfN signature was detected in all three samples prepared in acetonitrile, ethanol, and acetone (Fig. 2e and Fig. S4†). A detailed analysis of XRD patterns showed that HfN-based NPs synthesized in acetonitrile possess a sphalerite structure (F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m space group, Fig. 2e). In contrast, the samples prepared in acetone and ethanol have a rock salt structure (Fm
3m space group, Fig. 2e). In contrast, the samples prepared in acetone and ethanol have a rock salt structure (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, Fig. S4†). In addition, a comparative study of XRD patterns revealed the formation of hafnium derivatives. In particular, peaks related to nitrogen-deficient hafnium nitride (Hf4N3) and hafnium oxynitride (Hf7O8N4, ICDD: 00-050-1173) were identified.
m space group, Fig. S4†). In addition, a comparative study of XRD patterns revealed the formation of hafnium derivatives. In particular, peaks related to nitrogen-deficient hafnium nitride (Hf4N3) and hafnium oxynitride (Hf7O8N4, ICDD: 00-050-1173) were identified.
The surface composition of HfN-based NPs was examined using the X-ray photoelectron spectroscopy (XPS) technique. An XPS survey spectrum of the sample prepared in acetonitrile is shown in Fig. S5.† To elucidate the contribution of each component of HfN NPs’ composition, we deconvoluted the spectra in Hf 4f, N 1s and O 1s regions and these data are shown in Fig. 3 (panels (a), (b) and (c), respectively). It is worth noting that the deconvoluted spectrum in the Hf 4f region consists of 3 doublets corresponding to Hf 4f7/2 and 4 f5/2 core levels, which are typically explained by a spin–orbital splitting phenomenon. The first two doublets in Fig. 3a (orange and violet spectra) are attributed to hafnium oxynitride (16.56 eV and 18.2 eV) and hafnium nitride (16.05 eV and 17.72 eV) phases, respectively.56–62 The peaks of the third doublet (green spectra) centered at 14.52 eV and 16.1 eV can probably be attributed to a signal from HfNx (x < 1) with under-stoichiometric composition.63,64 In particular, the third doublet also indicates the formation of Hf4N3 structure,65 which is confirmed by our XRD measurements. In addition, we detected a weak signal in the C 1s region (spectrum is not shown) around 282 eV, which corresponds to minor hafnium carbide impurities. These results are in good agreement with XRD data, proving the absence of pure hafnium oxide structures in the NP composition. Nevertheless, we believe that the contribution of carbon-related peaks can be neglected when considering the physico-chemical properties of HfN NPs. Besides, additional peaks of ambiguous origin can be resolved in the N 1s and O 1s regions (Fig. 3b and c). While peaks located at 396.11 and 397.03 eV (N 1s) are related to hafnium nitride and oxynitride components, the three additional spectra may be attributed either to a satellite peak or N–O bonding (398.93 eV), surface N2 resulting from surface adsorption of nitrogen from the atmosphere58,66 (401.88 eV) in the NPs, or a shake-up feature67 (405.88 eV). Another ambiguity occurs in the deconvoluted spectrum in the O 1s region (Fig. 3c). The peak near 530.52 eV is nothing but Hf–O,56,58 whereas two additional components can be attributed to the surface oxygen56 (532.13 eV) and residual moisture68,69 (i.e. H2O; 535.93 eV). We believe that peaks associated with N2 in the N 1s region and O2/H2O peaks in the O 1s region appear as a result of NPs’ exposure to the ambient atmosphere due to sample preparation conditions before the XPS measurements. The XPS measurements also showed that the samples synthesized in ethanol and acetone possess very similar surface composition (Fig. S6a–c†) and despite the preparation conditions, the superficial layer remains strongly oxidized.
 | ||
| Fig. 3 X-ray photoelectron spectroscopy (XPS) characterization of laser-synthesized HfN-based NPs from different parts of the spectrum: Hf 4f (a), N 1s (b), O 1s (c). The data are presented for a sample prepared in acetonitrile. Shirley background correction is applied. (d) Raman spectra of HfN-based NPs. | ||
To unravel the composition features and to obtain a comprehensive insight into the structural properties of the formed HfN-based NPs, we carried out Raman scattering measurements (Fig. 3d). A Raman spectrum of the HfN target (Fig. 3d, blue line) recorded from a non-ablated area demonstrates the presence of two main peaks centered at around 150 cm−1 and 500 cm−1. The lines are typical of TMN (M = Ti, Zr, Hf) near-stoichiometric structures and attributed to first- and second-order optical and acoustic bands.70–73 In contrast, Raman spectra of the laser-ablated area of the target (red line), as well as that of produced HfN-based NPs (black line), contain three distinctive peaks including two already identified regions. The peak located near 600 cm−1 and the broad 2A band ranging from 200 to 400 cm−1 appear due to second-order Raman scattering induced by stoichiometry deviation.72,74 In general, such pronounced bands are typical of both slightly nitrogen-deficient and nitrogen-rich HfN structures.75,76 Nevertheless, according to our XRD, XPS, and EDX studies, the composition of HfN-based NPs includes not only nitrogen but also oxygen. This explains the coexistence of the hafnium oxynitride phase along with hafnium nitride. Therefore, the emergence of additional Raman bands can be attributed to a stoichiometry alteration of both oxygen and nitrogen components in the composition, rather than nitrogen alone.73 It is important to note that the scattering intensities in the acoustic and optical bands are determined by vibrations of hafnium and nitrogen/oxygen ions, respectively.72 Thus, when comparing the intensities of the O + A bands relative to the O band in both HfN-based NPs (black line) and target ablated area (red line) spectra, a slight decrease of intensity in the region of O + A of the latter spectrum is observed. In turn, this may be evidence that the formed NPs are nitrogen-/oxygen-rich compared to superficial Hf-based structures of the ablated area. The hypothesis is also supported by the fact that we observed a slight shift of the scattering peak position in the HfN ablated area spectrum, which agrees well with previously reported results for TMN samples.
Considering these results, we assume the main mechanism of the shift of the peak position of plasmon resonance for HfN-based NPs can be attributed to the formation of oxynitrides and nitrogen-deficient hafnium nitride. The clusters might be assembled during the growth stage into complex grain-like or monolith nanostructures consisting of hafnium nitride, and oxynitride within a single nanoformulation. It leads to the alteration of physical properties of the nanosystem such as permittivity, which in turn changes conditions of plasmon resonance excitation under an external electromagnetic field.
In general, HfN-based NPs prepared in different solvents (ethanol, acetone, and acetonitrile) had similar physicochemical properties, which was illustrated by TEM, EDX, XRD, XPS, and Raman spectroscopy measurements (under our experimental resolution). However, the samples prepared in acetonitrile had a slightly red-shifted position of the extinction peak compared to the counterparts prepared in ethanol and acetone. This feature can be explained by the difference of core compositions in prepared HfN-based NPs. In particular, according to the XRD and XPS measurements, the NPs synthesized in acetonitrile are less oxidized compared to samples prepared in ethanol and acetone, where the formation of oxynitrides is more favorable. Since the plasmonic peak of these NPs is closest to the biological transparency window, we selected the NPs prepared in acetonitrile for our subsequent biological tests.
Photothermal properties of HfN nanoparticles
Nanoparticles, which exhibit high light absorption efficiency in the visible and near-infrared spectra with subsequent light–heat conversion, are promising candidates for photothermal therapy. Therefore, the photothermal characteristics of HfN-based NPs were estimated (examined) under 808 nm laser irradiation, corresponding to the first window of biological transparency. Biological tissues and organs in this wavelength range have minimal absorption and scattering of light, which allows to increase the depth of light penetration.77 A colloidal HfN NP solution at different concentrations (0, 25, 50, 100 μg mL−1) was irradiated with a 1 W laser power, which led to the increase of temperature by 0.5 °C, 7.9 °C, 11.6 °C and 15.1 °C, respectively (Fig. 4a and b). However, such temperatures are excessive for effective destruction of cancer cells, and normally it is sufficient for NPs to be heated to a temperature of 42–43 °C, or 5–6 °C higher compared to the normal temperature of the human body. As can be seen from Fig. 4a, the solution of HfN-based NPs at 25 μg mL−1 concentration was able to reach these temperature values after 2.5–3.5 minutes of laser irradiation, while for the solution with 100 μg mL−1 particle concentration, such a temperature increase was observed within 1–1.5 minutes after the beginning of irradiation. | ||
| Fig. 4 Photothermal properties of bare and coated laser-synthesized HfN-based NPs. (a) Kinetics of temperature change of solutions with different concentrations of HfN NPs prepared in acetonitrile over a 15 min heating cycle (under irradiation at 1 W laser power) and a 20 min cooling cycle (at ambient conditions) and (b) the corresponding peak temperatures. (c) Photothermal stability cycling test with 10 min heating and 10 min cooling steps of a solution of 100 μg mL−1 particle concentration. (d) Kinetics of temperature change of 100 μg mL−1 HfN NP solution at different laser power values over a 15 min heating cycle and a 20 min cooling cycle and (e) the corresponding peak temperatures. (f) Kinetics of temperature change of 50 μg mL−1 HfN NP and 50 μg mL−1 HfN@PEG NP solutions over a 15 min heating cycle (under irradiation at 1 W laser power) and a 20-minute cooling cycle (under ambient conditions). | ||
A cooling time of 450 s (Fig. S7†) was used to calculate the photothermal conversion coefficient (η), whose value was estimated to be 62%. The calculated η of HfN-based NPs was found to be higher than the reported values for various NP types such as gold NPs (10.2%),78 Fe3O4 NPs (35.7%),79 Fe–Au core–shell nanocomposites (38%),80 TiN NPs (48%),33 and TiN nanoclusters (49%)81 and roughly agrees with the values for chemically synthesized ZrN (58%)81 and HfN nanocrystals (65%).81 Moreover, NPs demonstrated strong photothermal stability during on/off irradiation cycles, as can be seen from Fig. 4c.
The kinetics of temperature change of a 100 μg mL−1 HfN NP concentration solution depending on the illumination power was also studied (Fig. 4d and e). Peak temperature increments of 8.7 °C, 15.1 °C, 29 °C were recorded at power values of 0.5 W, 1 W, and 2 W, respectively (Fig. 4d). As shown in Fig. 4e, a linear relationship between the increase of temperature and laser power with a slope coefficient value of 15 °C W−1 was observed. So, one can say that HfN NPs have high photothermal efficiency over a wide range of irradiation powers, including safe low NIR light intensities commonly used for irradiation of healthy tissues. In addition, a comparison of the photothermal properties of uncoated and coated HfN NPs was carried out, and, as can be seen from Fig. 4e, the solutions of HfN and HfN@PEG heat up and cool down almost identically at the same concentration and irradiation power. Thus, the combination of η = 62%, high photostability, achieving the temperature difference required for the PTT procedure within 1–1.5 min (at a 100 μg mL−1 solution concentration), and no differences in photothermal properties between uncoated and coated particles, makes HfN-based NPs a promising candidate for photothermal therapy applications.
Coating of HfN nanoparticles with PEG
Biomedical applications of NPs require their colloidal stability in physiological environments. HfN NPs synthesized in acetonitrile could be transferred into distilled water, while maintaining a consistent hydrodynamic diameter of (80 ± 31) nm. However, these uncoated NPs tended to aggregate in salt buffers, such as PBS (pH 7.4) with a notable increase in the hydrodynamic diameter up to (299 ± 68) nm (Fig. 5a).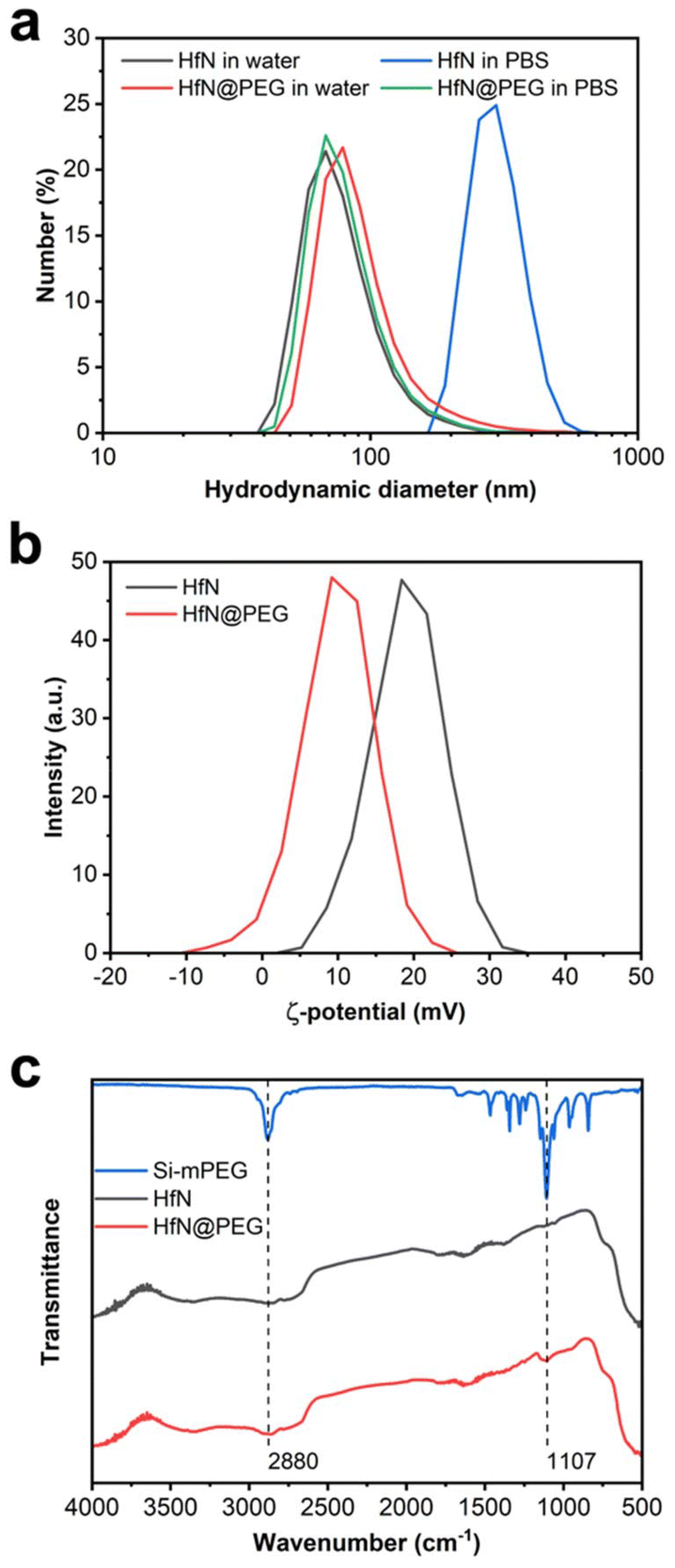 | ||
| Fig. 5 Coating of laser-synthesized HfN-based NPs with PEG. (a) Hydrodynamic diameter distribution for HfN and HfN@PEG NPs in water and PBS buffer. (b) ζ-potential distribution for HfN NPs before and after coating with mPEG-Silane polymer. (c) FTIR spectra of mPEG-Silane polymer, HfN and HfN@PEG NPs. | ||
To prevent the aggregation, we decided to coat HfN NPs with a polyethyleneglycol (PEG) polymer. For this aim we used the Stober reaction, condensing mPEG-Silane chains onto the hydrophilic surface of HfN NPs under alkaline conditions. PEGylation is a widely used method for improving the colloidal stability of nanoparticles by providing safe and biocompatible coating.82 Additionally, PEGylation generally improves the pharmacokinetic properties of nanoparticles in vivo.82
After the coating, HfN@PEG NPs exhibited a slight increase in hydrodynamic diameter in water to (96 ± 48) nm due to the attachment of the polymer layer. Moreover, HfN@PEG particles demonstrated excellent colloidal stability in the PBS buffer, maintaining the hydrodynamic diameter of 84 ± 32 nm 1 h after incubation. After the coating, the ζ-potential of nanoparticles changed from strongly positive +19 ± 4.7 mV to more neutral +9.8 ± 4.8 mV due to the incorporation of neutral PEG groups (Fig. 5b). To confirm the successful coating, FTIR spectra of the polymer and NPs before and after the coating were obtained (Fig. 5c). Both types of HfN NPs exhibited a main broad peak in the 500–800 cm−1 range, attributed to the presence of Hf–O bonds83 is due to a partial oxidation of the surface. After the coating, the HfN@PEG NPs exhibit two additional peaks at 1107 cm−1 and 2880 cm−1, corresponding to the symmetrical stretching vibration of C–O–C bonds and C–H stretching from PEG chains, respectively.84
Cytotoxicity and photothermal therapy
Next, we investigated the cytotoxicity of HfN@PEG NPs and their photothermal efficiency under irradiation by an 808 nm laser. In vitro tests were conducted on two cell lines: human ductal breast carcinoma BT474 and murine mammary carcinoma EMT6/P. The analysis of the metabolic activity of cells during incubation with nanoparticles, both before and after irradiation, was performed using the MTT test. This assay measures cell viability based on the activity of NAD(P)H-dependent oxidoreductases. To assess cytotoxicity, cells were incubated with HfN@PEG NPs at concentrations of 10, 25, 50, and 100 μg mL−1 for 24 and 72 hours. The relative cell viability remained above 88% and 74% after 72 h incubation at 100 μg mL−1 NP concentration for BT474 and EMT6/P cells, respectively. The cytotoxicity did not significantly differ between 24 and 72 h time-points, confirming the absence of long-term toxic effects of HfN@PEG NPs on cells (Fig. 6a). These cytotoxicity values are comparable to those observed with other biocompatible hafnium nanomaterials, such as HfO2 particles.85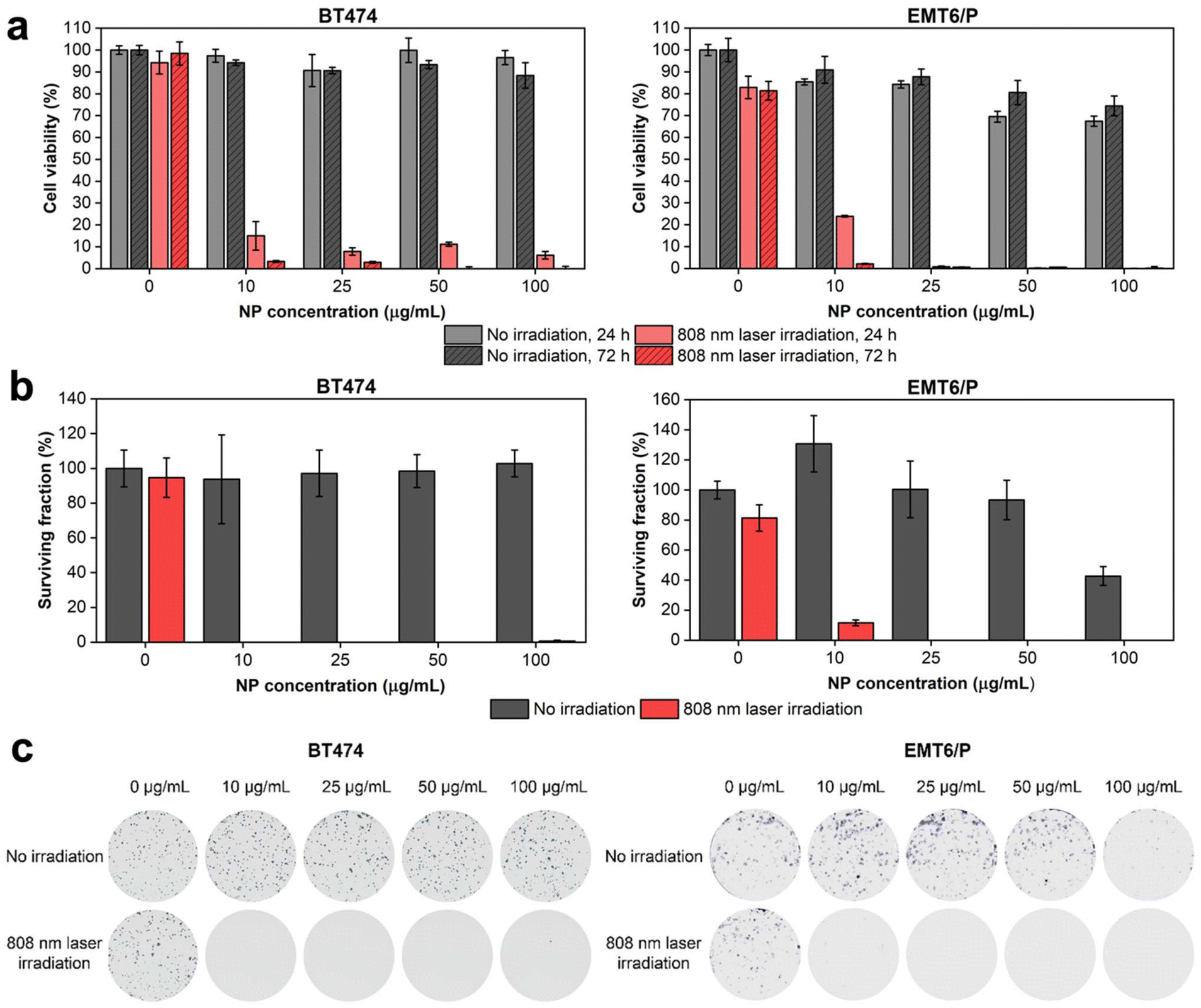 | ||
| Fig. 6 Analysis of cytotoxicity and photothermal efficiency of HfN@PEG NPs on BT474 and EMT6/P cell lines. (a) MTT test results after 24 h and 72 h of NP treatment and irradiation. (b) Clonogenic assay results after 14 days of NP treatment and irradiation. (c) Representative images of stained colonies. Irradiation was performed with an 808 nm laser for 5 min at 0.8 W power. Cell viability is shown as a percentage normalized to untreated cells without irradiation. In (a) and (b) data are presented as mean ± SD. | ||
To evaluate the impact of laser-induced hyperthermia, cells were incubated with NPs for 30 min and irradiated with an 808 nm laser for 5 min at a power of 0.8 W. After 24 hours post irradiation, the metabolic activity of cells was below 15% and 24% even at an NP concentration of 10 μg mL−1 for BT474 and EMT6/P cells, respectively. At 72 h timepoint, almost no cells with detectable metabolic activity were observed across all studied concentrations of HfN@PEG NPs (Fig. 6a). Notably, no photothermal damage was observed in cells that were not treated with NPs.
A clonogenic assay indicating cell proliferative activity 14 days after the treatment was used to evaluate the long-term cytotoxicity of HfN@PEG NPs. Without laser irradiation, the surviving fraction of BT474 cells did not fall below the 94% level for 10–100 μg mL−1 HfN@PEG concentrations. EMT6/P cells had higher sensitivity at 100 μg mL−1 concentration, decreasing survival to 42% level. Nevertheless, at concentrations up to 50 μg mL−1, the percentage of surviving EMT6/P cells did not fall below 93%. Irradiation with an NIR laser significantly reduced clonogenic activity. No colony-forming cells remained at particle concentrations above 10 μg mL−1 and 25 μg mL−1 for BT474 and EMT6/P cells, respectively (Fig. 6b and c).
Discussion
Hafnium nitride nanoparticles present an appealing nanomaterial, which can be applied in a variety of biomedical and other applications. As pointed out above, HfN NPs are known to exhibit prominent plasmonic properties, although the plasmonic absorption/scattering feature for pure HfN NPs (centered around 480–510 nm) is outside the biological transparency window. In addition, due to the presence of Hf having a high Z number (Z = 72), HfN NPs look as promising candidates for photon capture therapy37 and contrast-enhanced computed tomography (CT)38 similar to NBTXR3 drug, based on functionalized crystalline hafnium oxide NPs, which has shown very high efficiency in the enhancement of radiotherapy outcome for a variety of cancers.41,42However, the fabrication of stable colloidal solutions of HfN NPs suitable for biological use still presents a great challenge. For instance, HfN NPs synthesized by conventional wet chemistry techniques are typically contaminated by toxic by-products and require multiple steps of purification to render their use in biological applications.43 Such a toxicity issue can be partially solved by the employment of dry synthesis techniques, namely plasma synthesis or magnetron sputtering,44 but TMN NPs synthesized by these techniques are typically aggregated and demonstrate poor water-dispersibility. In addition, the absorption band of NPs formed by chemical or dry synthesis usually lies outside of the biological transparency window, which complicates their further use in PTT.
In this study, we report a viable solution to problems of conventional pathways in the synthesis of HfN nanostructures by employing methods of ultrashort laser ablation in organic solutions (ethanol, acetone, acetonitrile). We show that such a technique can provide highly stable, water-dispersible, and non-toxic HfN-based nanoformulations with the manifestation of a tunable plasmonic feature in the biotransparency window. As follows from structural characterization, the formed HfN-based NPs are spherical and highly crystalline, while their average (mode) size depends on the used solvent (16 to 10 nm in ethanol/acetone and acetonitrile, respectively). We also show that all HfN-based NPs exhibit distinct plasmonic extinction peaks having their positions depending on the used solvent (590 nm, 620 nm and 650 nm for NPs prepared in ethanol, acetone and acetonitrile, respectively, Fig. 1). As follows from the results of our measurements and relevant analysis, the reported red-shift of the plasmonic peak position compared to pure near-stoichiometric HfN NPs can be attributed to a partial inclusion of nitrogen-deficient hafnium nitride (Hf4N3) and oxynitride (Hf7O8N4) phases into the core composition of NPs. It is worth noting that according to our XRD studies, crystalline oxynitride phases were only detected in NPs prepared in ethanol and acetone (Fig. S4†), which already contain oxygen in their molecular structure. In contrast, the synthesis of NPs in oxygen-free acetonitrile not only aids to suppress the formation of crystalline oxynitride derivatives, but also alters the formation mechanism of HfN, making possible the existence of a sphalerite structure (F3m) apart from the more common rock salt phase (Fmm). Nevertheless, according to our XPS measurements, the surface composition for all three types of HfN-based NPs remains almost unchanged and contains oxynitride derivatives in both amorphous and crystalline forms. Indeed, the shell composition also determines the red-shifted position of the resonance peak, rather than the core structure alone. This correlates well with our calculations (Fig. S1†) and previously reported models for different TMN (M = Ti, Zr) nanostructures.86,87 Thus, we suppose that a relative shift of plasmonic features for HfN-based NPs prepared in different solvents can be explained by the presence of different hafnium oxynitride and nitrogen-deficient nitride contents in the HfN core and the formation of a hafnium oxynitride shell.
It should be noted that the formation of HfN-based nanostructures during the laser-ablative process follows a slightly different scenario compared to the TiN-based counterparts. Indeed, while laser ablation of TiN in water or organic solvents typically led to oxidation phenomena with the inclusion of titanium oxide phases in the core and the shell,34,49 similar ablation of HfN resulted in the formation of oxynitride phases. We believe that such a difference can be explained by a higher catalytic activity of Hf compared to that of Ti. We also suppose that such a property can be used to enable catalytic applications of HfN NPs. Here, we should note that bare (ligand-free) nanomaterials are capable of providing much better catalytic activity compared to chemically synthesized counterparts. As an example, we showed that laser-synthesized bare Au NPs and PdAu nanoalloys can provide one order of magnitude higher activity toward electrooxidation of glucose compared to their chemically synthesized counterparts.88,89 We believe that the synthesized HfN NPs can be also highly valuable for catalytic applications.
In our tests, we also assessed the biological properties of laser-synthesized HfN-based NPs. Here, the NPs prepared in organic solutions were easily transferred to water without any loss of colloidal stability, which contrasts with data related to water-dispersion of TMN NPs prepared by dry methods. However, the transfer of NPs to a PBS buffer led to their destabilization in solution. To solve this problem, the NPs were coated with PEG using the Stober method. We then estimated cytotoxic and phototherapeutic effects in cancer cell models. As we demonstrated in the absence of laser irradiation, the cytotoxicity effect was very low (for example, the cell viability was more than 85% for the BT474 cell line), similar to what was previously reported for the officially registered hafnium oxide drug NBTXR3. We then demonstrated a high efficiency of HfN in the elimination of cancer cells. Even under very modest concentrations of NPs (25 μg mL−1), the excitation by 808 nm light with a moderate power of 0.8 W led to 100% cancer cell death, promising a very attractive application of the synthesized NPs in cancer photothermal therapy. It should be noted that the photothermal therapy modality can typically be completed by photoacoustic imaging functionality, as we recently demonstrated for laser-synthesized TiN NPs.35,51 We believe that the same imaging modality can be enabled for the HfN NPs and this topic will be the focus of our future tests.
Having a high photothermal conversion coefficient (η = 62%) and outstanding biocompatibility, the use of laser-synthesized HfN-based NPs looks very appealing for PTT applications promising higher photothermal efficacy compared to the conventional counterparts.9,90–92 Moreover, the broad absorption (Fig. 1b) makes possible the use of photoacoustic properties of HfN-based NPs, enabling the photoacoustic imaging modality similar to how it was earlier demonstrated for TiN NPs.35 We foresee that one of the main advantages of HfN NPs compared to other TMN-based counterparts consists in the possibility of enabling radiation theranostic modalities. Having a much higher Z number than that of Ti and Zr (72 compared to 22 and 40, respectively), one can enable efficient CT scan and radiotherapy modalities similar to how it was done using the hafnium oxide-based NBTXR3 drug. Such a radiotherapy modality can be employed in parallel with PTT and photoacoustic modality, promising a resulting synergetic theranostic effect.
Conclusions
Stable spherical water-dispersible solutions of HfN-based NPs were synthesized for the first time using the technique of femtosecond laser ablation in organic solutions. The fabricated NPs were crystalline and had a unimodal size distribution centered around 16–17 nm and 10 nm for the samples prepared in ethanol/acetone and acetonitrile, respectively. The NPs exhibited absorption peaks associated with the excitation of localized plasmons with the peak position between 590 to 650 nm, depending on the fabrication organic medium, and a long tail in the infrared region of relative biological tissue transparency. Such positions of the peaks were much red-shifted compared to what is expected from pure HfN, and such a mismatch of plasmonic peak positions was attributed to the presence of hafnium oxynitride and nitrogen-deficient hafnium nitride phases inside the core and oxynitride coating of NPs, as evidenced by X-ray photoelectron, Raman and energy-dispersive X-ray spectroscopy, transmission electron microscopy, and X-ray diffraction measurements. We showed that the uncoated HfN-based NPs can be heated under 808 nm laser irradiation up to 5–6 °C within 1–1.5 min at 100 μg mL−1 NP concentration. The NPs were stable under photoexcitation, while the photothermal conversion coefficient (about 62%) is comparable to the best values reported in the literature for plasmonic NPs. Moreover, it was found that HfN-based NPs do not cause any cytotoxic effect while having high photothermal efficiency in vitro, as illustrated by 100% cell death under 808 nm light irradiation at NP concentrations lower than 25 μg mL−1. We believe that due to their high NIR absorption efficiency, ultrapure surface, and low toxicity in vitro, laser-synthesized HfN-based NPs are promising for various applications in biomedicine and catalysis. We also suppose that such modalities can be combined with additional X-ray theranostic functionalities (CT scan and photon capture therapy) profiting from the high atomic number (Z = 72) of Hf, promising an enhanced synergetic outcome.Author contributions
Conceptualization: A.V.K.; methodology: A.V.K., A.A.P., S.M.D, I.V.Z., and A.P.; investigation: A.I.P., M.S.S., J.S.B., G.V.T., A.D., and I.V.Z.; validation: A.A.P., S.M.K., A.P., I.N.Z., S.M.D., and A.V.K.; formal analysis: A.I.P., M.S.S., I.V.Z., J.S.B., G.V.T., A.A.P., S.M.K., A.D., A.P., I.N.Z., S.M.D., and A.V.K.; writing – original draft preparation: A.I.P., I.V.Z., and A.V.K; writing – review and editing: A.V.K. after suggestions from all authors; supervision: A.V.K., A.A.P., S.M.K., I.N.Z., and S.M.D.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
A.I.P and A.V.K. acknowledge support from the French government under the France 2030 investment plan, as part of the Initiative d'Excellence d'Aix-Marseille Université – A*MIDEX AMX-22-RE-AB-107 and the French National Research Agency (project ANR-23-CE07-0051). The authors also acknowledge support from the Ministry of Science and Education of the Russian Federation under Agreement no. 075-15-2021-1347. The authors would like to thank Damien Chaudanson and Alexandre Altié from CINAM, Aix-Marseille University for their help in running TEM measurements and Vasile Heresanu from CINAM, Aix-Marseille University for his help in running XRD measurements.References
- S. A. Maier, M. L. Brongersma, P. G. Kik, S. Meltzer, A. A. G. Requicha and H. A. Atwater, Adv. Mater., 2001, 13, 1501–1505 CrossRef CAS.
- E. Ozbay, Science, 2006, 311, 189–193 CrossRef CAS PubMed.
- H. A. Atwater and A. Polman, Nat. Mater., 2010, 9, 205–213 CrossRef CAS.
- L. Zhou, J. M. P. Martirez, J. Finzel, C. Zhang, D. F. Swearer, S. Tian, H. Robatjazi, M. Lou, L. Dong, L. Henderson, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Nat. Energy, 2020, 5, 61–70 CrossRef CAS.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442–453 CrossRef CAS PubMed.
- A. V. Kabashin, V. G. Kravets and A. N. Grigorenko, Chem. Soc. Rev., 2023, 52, 6554–6585 RSC.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS PubMed.
- L. Zou, H. Wang, B. He, L. Zeng, T. Tan, H. Cao, X. He, Z. Zhang, S. Guo and Y. Li, Theranostics, 2016, 6, 762–772 CrossRef CAS.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Lasers Med. Sci., 2008, 23, 217–228 CrossRef PubMed.
- S. Lal, S. E. Clare and N. J. Halas, Acc. Chem. Res., 2008, 41, 1842–1851 CrossRef CAS PubMed.
- Y. Wang, X. Xie, X. Wang, G. Ku, K. L. Gill, D. P. O'Neal, G. Stoica and L. V. Wang, Nano Lett., 2004, 4, 1689–1692 CrossRef CAS.
- K. Kneipp, Phys. Today, 2007, 60, 40–46 CrossRef CAS.
- X. Qian, X.-H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. Nie, Nat. Biotechnol., 2008, 26, 83–90 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- Y.-P. Jia, B.-Y. Ma, X.-W. Wei and Z.-Y. Qian, Chin. Chem. Lett., 2017, 28, 691–702 CrossRef CAS.
- A. Sani, C. Cao and D. Cui, Biochem. Biophys. Rep., 2021, 26, 100991 CAS.
- L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, B. Rivera, R. E. Price, J. D. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549–13554 CrossRef CAS.
- C. Loo, A. Lowery, N. Halas, J. West and R. Drezek, Nano Lett., 2005, 5, 709–711 CrossRef CAS PubMed.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS PubMed.
- J. Chen, B. Wiley, Z.-Y. Li, D. Campbell, F. Saeki, H. Cang, L. Au, J. Lee, X. Li and Y. Xia, Adv. Mater., 2005, 17, 2255–2261 CrossRef CAS.
- Y. Liu, E. N. Mills and R. J. Composto, J. Mater. Chem., 2009, 19, 2704–2709 RSC.
- H. Petrova, J. Perez Juste, I. Pastoriza-Santos, G. V. Hartland, L. M. Liz-Marzán and P. Mulvaney, Phys. Chem. Chem. Phys., 2006, 8, 814–821 RSC.
- G. V. Naik, V. M. Shalaev and A. Boltasseva, Adv. Mater., 2013, 25, 3264–3294 CrossRef CAS PubMed.
- U. Guler, S. Suslov, A. V. Kildishev, A. Boltasseva and V. M. Shalaev, Nanophotonics, 2015, 4, 269–276 CAS.
- A. Lalisse, G. Tessier, J. Plain and G. Baffou, J. Phys. Chem. C, 2015, 119, 25518–25528 CrossRef CAS.
- A. Lalisse, G. Tessier, J. Plain and G. Baffou, Sci. Rep., 2016, 6, 38647 CrossRef CAS.
- A. Naldoni, U. Guler, Z. Wang, M. Marelli, F. Malara, X. Meng, L. V. Besteiro, A. O. Govorov, A. V. Kildishev, A. Boltasseva and V. M. Shalaev, Adv. Opt. Mater., 2017, 5, 1601031 CrossRef.
- Y. Liu, X. Zhang, L. Lu, J. Ye, J. Wang, X. Li, X. Bai and W. Wang, Chin. Chem. Lett., 2022, 33, 1271–1274 CrossRef CAS.
- S. Ishii, R. P. Sugavaneshwar and T. Nagao, J. Phys. Chem. C, 2016, 120, 2343–2348 CrossRef CAS.
- S. Farooq, C. V. P. Vital, G. Tikhonowski, A. A. Popov, S. M. Klimentov, L. A. G. Malagon, R. E. de Araujo, A. V. Kabashin and D. Rativa, Sol. Energy Mater. Sol. Cells, 2023, 252, 112203 CrossRef CAS.
- K. Biliak, M. Protsak, P. Pleskunov, D. Nikitin, J. Hanuš, S. Ali-Ogly, J. Šomvársky, M. Tosca, M. Cieslar, T. Košutová, M. Dopita, F. Lopes Ferreira and A. Choukourov, ACS Appl. Nano Mater., 2023, 6, 21642–21651 CrossRef CAS.
- S. H. C. Askes, N. J. Schilder, E. Zoethout, A. Polman and E. C. Garnett, Nanoscale, 2019, 11, 20252–20260 RSC.
- W. He, K. Ai, C. Jiang, Y. Li, X. Song and L. Lu, Biomaterials, 2017, 132, 37–47 CrossRef CAS PubMed.
- A. A. Popov, G. Tselikov, N. Dumas, C. Berard, K. Metwally, N. Jones, A. Al-Kattan, B. Larrat, D. Braguer, S. Mensah, A. Da Silva, M.-A. Estève and A. V. Kabashin, Sci. Rep., 2019, 9, 1194 CrossRef PubMed.
- A. Das, A. C. M. V. Pereira, A. A. Popov, A. Pastukhov, S. M. Klimentov, A. V. Kabashin and A. S. L. Gomes, Appl. Phys. Lett., 2022, 121, 083701 CrossRef CAS.
- X. Chen, J. Song, X. Chen and H. Yang, Chem. Soc. Rev., 2019, 48, 3073–3101 RSC.
- P. Retif, S. Pinel, M. Toussaint, C. Frochot, R. Chouikrat, T. Bastogne and M. Barberi-Heyob, Theranostics, 2015, 5, 1030–1044 CrossRef CAS.
- N. Lee, S. H. Choi and T. Hyeon, Adv. Mater., 2013, 25, 2641–2660 CrossRef CAS.
- L. Maggiorella, G. Barouch, C. Devaux, A. Pottier, E. Deutsch, J. Bourhis, E. Borghi and L. Levy, Future Oncol., 2012, 8, 1167–1181 CrossRef CAS.
- J. Marill, N. M. Anesary, P. Zhang, S. Vivet, E. Borghi, L. Levy and A. Pottier, Radiat. Oncol., 2014, 9, 150 CrossRef.
- S. Bonvalot, C. Le Pechoux, T. De Baere, G. Kantor, X. Buy, E. Stoeckle, P. Terrier, P. Sargos, J. M. Coindre, N. Lassau, R. Ait Sarkouh, M. Dimitriu, E. Borghi, L. Levy, E. Deutsch and J.-C. Soria, Clin. Cancer Res., 2017, 23, 908–917 CrossRef CAS PubMed.
- A. F. Bagley, E. B. Ludmir, A. Maitra, B. D. Minsky, G. Li Smith, P. Das, A. C. Koong, E. B. Holliday, C. M. Taniguchi, M. H. G. Katz, E. P. Tamm, R. A. Wolff, M. J. Overman, S. Patel, M. P. Kim, C.-W. D. Tzeng, N. Ikoma, M. S. Bhutani and E. J. Koay, Clin. Transl. Radiat. Oncol., 2022, 33, 66–69 Search PubMed.
- R. A. Karaballi, G. Humagain, B. R. A. Fleischman and M. Dasog, Angew. Chem., Int. Ed., 2019, 58, 3147–3150 CrossRef CAS PubMed.
- D. Zhang, K. Zhang, Z. Xie, B. Xu, M. Hou, Y. Lei, T. Watanabe, B. Yang and F. Liang, Materials, 2023, 16, 7469 CrossRef CAS PubMed.
- A. V. Kabashin and M. Meunier, J. Appl. Phys., 2003, 94, 7941–7943 CrossRef CAS.
- A. V. Kabashin and M. Meunier, J. Phys.: Conf. Ser., 2007, 59, 354 CrossRef CAS.
- A. V. Kabashin, P. Delaporte, A. Pereira, D. Grojo, R. Torres, T. Sarnet and M. Sentis, Nanoscale Res. Lett., 2010, 5, 454 CrossRef CAS.
- J.-P. Sylvestre, S. Poulin, A. V. Kabashin, E. Sacher, M. Meunier and J. H. T. Luong, J. Phys. Chem. B, 2004, 108, 16864–16869 CrossRef CAS.
- A. A. Popov, G. V. Tikhonowski, P. V. Shakhov, E. A. Popova-Kuznetsova, G. I. Tselikov, R. I. Romanov, A. M. Markeev, S. M. Klimentov and A. V. Kabashin, Nanomaterials, 2022, 12, 1672 CrossRef CAS.
- I. V. Zelepukin, A. A. Popov, V. O. Shipunova, G. V. Tikhonowski, A. B. Mirkasymov, E. A. Popova-Kuznetsova, S. M. Klimentov, A. V. Kabashin and S. M. Deyev, Mater. Sci. Eng., C, 2021, 120, 111717 CrossRef CAS PubMed.
- M. E. Maldonado, A. Das, A. S. L. Gomes, A. A. Popov, S. M. Klimentov and A. V. Kabashin, Opt. Lett., 2020, 45, 6695–6698 CrossRef.
- V. A. Bahamondes Lorca, O. Ávalos-Ovando, C. Sikeler, H. Ijäs, E. Y. Santiago, E. Skelton, Y. Wang, R. Yang, K. L. A. Cimatu, O. Baturina, Z. Wang, J. Liu, J. M. Slocik, S. Wu, D. Ma, A. Pastukhov, A. V. Kabashin, M. E. Kordesch and A. O. Govorov, Nano Lett., 2024, 24, 6069–6077 CrossRef CAS.
- D. K. Roper, W. Ahn and M. Hoepfner, J. Phys. Chem. C, 2007, 111, 3636–3641 CrossRef CAS.
- K. Jiang, D. A. Smith and A. Pinchuk, J. Phys. Chem. C, 2013, 117, 27073–27080 CrossRef CAS.
- A. I. Pastukhov, I. B. Belyaev, J. C. Bulmahn, I. V. Zelepukin, A. A. Popov, I. N. Zavestovskaya, S. M. Klimentov, S. M. Deyev, P. N. Prasad and A. V. Kabashin, Sci. Rep., 2022, 12, 9129 CrossRef CAS PubMed.
- Z. Xiao, K. Kisslinger, S. Chance and S. Banks, Crystals, 2020, 10, 136 CrossRef CAS.
- K. Mallem, S. V. Jagadeesh Chandra, M. Ju, S. Dutta, S. Phanchanan, S. Sanyal, D. P. Pham, S. Q. Hussain, Y. Kim, J. Park, Y.-H. Cho, E.-C. Cho and J. Yi, Thin Solid Films, 2019, 675, 16–22 CrossRef CAS.
- K. V. Egorov, Y. Y. Lebedinskii, A. A. Soloviev, A. A. Chouprik, A. Y. Azarov and A. M. Markeev, Appl. Surf. Sci., 2017, 419, 107–113 CrossRef CAS.
- F. Piallat, V. Beugin, R. Gassilloud, L. Dussault, B. Pelissier, C. Leroux, P. Caubet and C. Vallée, Appl. Surf. Sci., 2014, 303, 388–392 CrossRef CAS.
- S. Karwal, M. A. Verheijen, B. L. Williams, T. Faraz, W. M. M. Kessels and M. Creatore, J. Mater. Chem. C, 2018, 6, 3917–3926 RSC.
- P. Pleskunov, M. Protsak, Z. Krtouš, T. Košutová, M. Tosca, K. Biliak, V. Červenková, D. Nikitin, J. Hanuš, M. Cieslar, I. Gordeev, M. Dopita, M. Vorochta, J. Kousal, L. Martinu and A. Choukourov, Adv. Opt. Mater., 2024, 12, 2302715 CrossRef CAS.
- R. Zazpe, M. Ungureanu, F. Golmar, P. Stoliar, R. Llopis, F. Casanova, D. F. Pickup, C. Rogero and L. E. Hueso, J. Mater. Chem. C, 2014, 2, 3204–3211 RSC.
- I. Takano, S. Isobe, T. A. Sasaki and Y. Baba, Appl. Surf. Sci., 1989, 37, 25–32 CrossRef CAS.
- Y. Baba, T. A. Sasaki and I. Takano, J. Vac. Sci. Technol., A, 1988, 6, 2945–2948 CrossRef CAS.
- L.-C. Chang, B.-W. Liu and Y.-I. Chen, Coatings, 2018, 8, 354 CrossRef.
- E. O. Filatova, A. S. Konashuk, S. S. Sakhonenkov, A. A. Sokolov and V. V. Afanas'ev, Sci. Rep., 2017, 7, 4541 CrossRef CAS.
- A. Majumdar, S. C. Das, T. Shripathi and R. Hippler, Compos. Interfaces, 2012, 19, 161–170 CrossRef CAS.
- P. D. Schulze, S. L. Shaffer, R. L. Hance and D. L. Utley, J. Vac. Sci. Technol., A, 1983, 1, 97–99 CrossRef CAS.
- S. Yamamoto, H. Bluhm, K. Andersson, G. Ketteler, H. Ogasawara, M. Salmeron and A. Nilsson, J. Phys.: Condens. Matter, 2008, 20, 184025 CrossRef.
- W. Spengler and R. Kaiser, Solid State Commun., 1976, 18, 881–884 CrossRef CAS.
- X.-J. Chen, V. V. Struzhkin, S. Kung, H.-K. Mao, R. J. Hemley and A. N. Christensen, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 014501 CrossRef.
- W. Spengler, R. Kaiser, A. N. Christensen and G. Müller-Vogt, Phys. Rev. B: Solid State, 1978, 17, 1095–1101 CrossRef CAS.
- C. Moura, P. Carvalho, F. Vaz, L. Cunha and E. Alves, Thin Solid Films, 2006, 515, 1132–1137 CrossRef CAS.
- M. Stoehr, C.-S. Shin, I. Petrov and J. E. Greene, J. Appl. Phys., 2011, 110, 083503 CrossRef.
- A. P. Zavjalov, P. A. Nikiforov, D. Y. Kosyanov, A. M. Zakharenko, V. O. Trukhin, K. Y. Talskikh, O. O. Shichalin and E. K. Papynov, Adv. Eng. Mater., 2020, 22, 2000482 CrossRef CAS.
- M. Stoehr, H.-S. Seo, I. Petrov and J. E. Greene, J. Appl. Phys., 2008, 104, 033507 CrossRef.
- E. Hemmer, A. Benayas, F. Légaré and F. Vetrone, Nanoscale Horiz., 2016, 1, 168–184 RSC.
- P. Zhang, J. Wang, H. Huang, B. Yu, K. Qiu, J. Huang, S. Wang, L. Jiang, G. Gasser, L. Ji and H. Chao, Biomaterials, 2015, 63, 102–114 CrossRef CAS.
- T.-W. Chang, H. Ko, W.-S. Huang, Y.-C. Chiu, L.-X. Yang, Z.-C. Chia, Y.-C. Chin, Y.-J. Chen, Y.-T. Tsai, C.-W. Hsu, C.-C. Chang, P.-J. Tsai and C.-C. Huang, Chem. Eng. J., 2022, 428, 131237 CrossRef CAS.
- O. Y. Griaznova, I. B. Belyaev, A. S. Sogomonyan, I. V. Zelepukin, G. V. Tikhonowski, A. A. Popov, A. S. Komlev, P. I. Nikitin, D. A. Gorin, A. V. Kabashin and S. M. Deyev, Pharmaceutics, 2022, 14, 994 CrossRef CAS.
- R. A. Karaballi, Y. Esfahani Monfared and M. Dasog, Langmuir, 2020, 36, 5058–5064 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- A. Ramadoss, K. Krishnamoorthy and S. J. Kim, Mater. Res. Bull., 2012, 47, 2680–2684 CrossRef CAS.
- Y. Yang, W. Li, Y. Ren and X. Cai, Polym. Bull., 2019, 76, 2711–2724 CrossRef CAS.
- Y. Li, Y. Qi, H. Zhang, Z. Xia, T. Xie, W. Li, D. Zhong, H. Zhu and M. Zhou, Biomaterials, 2020, 226, 119538 CrossRef CAS PubMed.
- A. I. Pastukhov, V. O. Shipunova, J. S. Babkova, I. V. Zelepukin, M. Raab, R. Schmitt, A. Al-Kattan, A. Pliss, A. Kuzmin, A. A. Popov, S. M. Klimentov, P. N. Prasad, S. M. Deyev and A. V. Kabashin, ACS Appl. Nano Mater., 2024, 7, 18737–18754 CrossRef CAS.
- A. Alvarez Barragan, N. V. Ilawe, L. Zhong, B. M. Wong and L. Mangolini, J. Phys. Chem. C, 2017, 121, 2316–2322 CrossRef CAS.
- S. Hebie, Y. Holade, K. Maximova, M. Sentis, P. Delaporte, K. B. Kokoh, T. W. Napporn and A. V. Kabashin, ACS Catal., 2015, 5, 6489–6496 CrossRef CAS.
- Y. Holade, S. Hebié, K. Maximova, M. Sentis, P. Delaporte, K. B. Kokoh, T. W. Napporn and A. V. Kabashin, Catal. Sci. Technol., 2020, 10, 7955–7964 RSC.
- R. Lv, M. Raab, Y. Wang, J. Tian, J. Lin and P. N. Prasad, Coord. Chem. Rev., 2022, 460, 214486 CrossRef CAS.
- B. Li, D. Xu, Y. Chen, W. Li, H. Liu, A. A. Ansari and R. Lv, ACS Appl. Nano Mater., 2024, 7, 9428–9440 CrossRef CAS.
- D. Jaque, L. Martínez Maestro, B. del Rosal, P. Haro-Gonzalez, A. Benayas, J. L. Plaza, E. Martín Rodríguez and J. García Solé, Nanoscale, 2014, 6, 9494–9530 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02311k |
| This journal is © The Royal Society of Chemistry 2024 |