
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of polyunsaturation – insights into the hydroformylation of oleochemicals†
Thomas Friedrich Hubertus
Roth
,
Maximilian Lukas
Spiekermann
 ,
David
Lütkenhaus
,
Fabian
Niefer
,
Dieter
Vogt
and
Thomas
Seidensticker
*
,
David
Lütkenhaus
,
Fabian
Niefer
,
Dieter
Vogt
and
Thomas
Seidensticker
*
Department for Biochemical and Chemical Engineering, Laboratory of Industrial Chemistry, TU Dortmund University, Emil-Figge-Straße 66, 44227 Dortmund, Germany. E-mail: thomas.seidensticker@tu-dortmund.de; Tel: +49 231 755 2310
First published on 12th September 2024
Abstract
Although the hydroformylation of vegetable oil-derived oleochemicals, such as methyl oleate, is a highly demanded reaction and has been intensively studied, little is known about the influence of naturally occurring polyunsaturated (PU) components and their effect on the homogeneous rhodium catalyst. This is now examined in detail in the presented work by conducting systematic perturbation experiments. For the first time, it can be verified that the isomerisation of double bonds in polyunsaturated oleochemicals generates conjugated double bond systems that form stable η3-Rh allyl species and thus temporarily inhibit the catalyst. However, based on these findings, hydroformylation activity can be significantly increased by selective hydrogenation of PU to monounsaturated components. In the case of sunflower methyl ester, the turnover frequency multiplied by a factor of 8 and reached 3201 h−1, the highest rate reported in the context of methyl oleate hydroformylation. These effects were shown for both phosphine and phosphite ligands under both mono- and biphasic conditions and for methyl esters with different PU content, stressing the magnitude of this effect. This work makes it possible to support long-observed phenomena with the underlying mechanism scientifically. This lays the scientific basis for efficiently converting oleochemicals into valuable intermediates through hydroformylation for an increased share of renewable carbon in the chemical value chain.
Introduction
In a time of increasing emphasis on sustainability, using renewable raw materials as molecular building blocks in the chemical industry is becoming increasingly important. Fats and oils, in particular, represent a promising resource with the potential for a wide range of chemical applications. Large quantities of these raw materials are already available as methyl esters from biodiesel production, especially in Europe and the US, allowing for swift market entry. Their use not only offers ecological benefits but also helps to reduce dependence on fossil fuels and their producers and minimise environmental impact.To date, the use of oleochemicals as a molecular building block has mainly focused on functionalizing carboxyl functionality, resulting in possible applications as surfactants and lubricants. However, functionalizing unsaturated alkyl chains would open up a wide range of new applications.1,2 One example might be using fat and oil derivatives to synthesize bifunctional molecules, e.g., for polymer precursors.3 Hydroformylation is a promising and atom-economic tool for this and many other purposes since the resulting aldehydes are valuable platform chemicals (Fig. 1).3–5
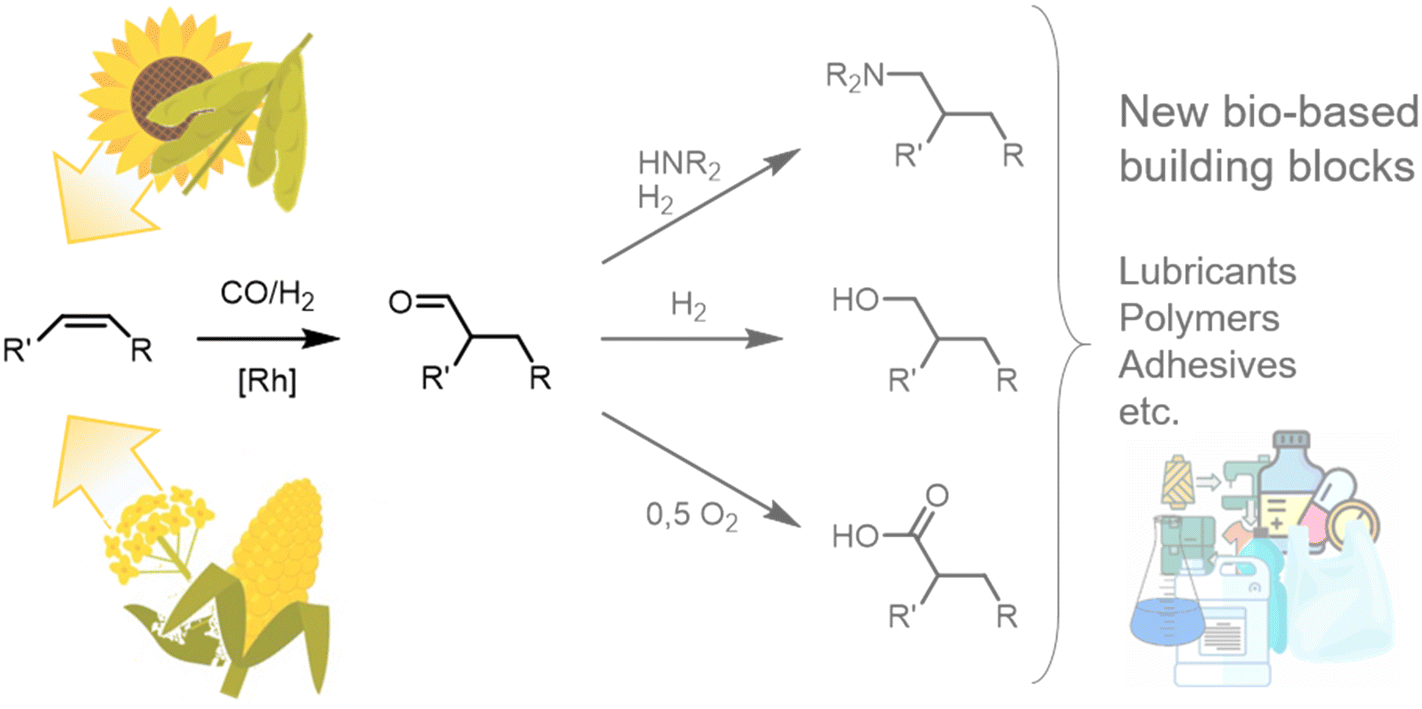 | ||
| Fig. 1 Hydroformylation on the internal alkene of a plant oil-derived oleochemical yielding an aldehyde as a bio-based platform chemical with a wide range of possible applications. | ||
Within fats and oils, oleic acid (C18:1) stands out, as it is formed in almost any oilseed, and its potential applications are numerous.6–8 However, fats and oils do not occur as pure substances; instead, a mixture of several different triglycerides with various fatty acid combinations is formed as a metabolic product of plants. This poses a particular challenge for chemical use in conventional processes, which usually work with almost pure compounds. It is noticeable that although many plant oils contain oleic acid, they always come with appreciable shares of polyunsaturated fatty acids (PU-FAs) like linoleic (C18:2) and linolenic (C18:3) acids (Table 1).
| ≤C16:0 | C18:0 | C18:1 | C18:2 | C18:3 | ≥C20:0 | ≥C20:1 | |
|---|---|---|---|---|---|---|---|
| Canola (old) | 2 | 1 | 15 | 16 | 10 | 2 | 54 |
| Canola (new) | 4 | 1 | 59 | 21 | 9 | 1 | 5 |
| Corn | 11 | 2 | 30 | 55 | 1 | 1 | 0 |
| Hemp | 6 | 2 | 11 | 60 | 21 | 0 | 0 |
| Linseed | 5 | 4 | 19 | 15 | 57 | 0 | 0 |
| Peanut | 11 | 3 | 55 | 29 | 1 | 1 | 0 |
| Soy | 9 | 4 | 24 | 54 | 8 | 0 | 1 |
| Sunflower (old) | 7 | 4 | 28 | 61 | 0 | 0 | 0 |
| Sunflower (high oleic) | 4 | 2 | 91 | 3 | 0 | 0 | 0 |
As we highlighted in an earlier study, the shares of polyunsaturated fatty acids, in particular, pose a problem in homogeneously catalyzed functionalization reactions of the double bonds.11 Several papers have already hinted at this phenomenon, especially in the case of hydroformylation of methyl oleate. The findings gathered in these studies now contribute to developing a deeper understanding of the underlying mechanisms through target-oriented experiments in this work.
In three consecutive studies with methyl esters of different origins (e.g., olive, safflower, soy) by Frankel et al. from 1971 onwards, it was demonstrated that more extreme reaction conditions are necessary to achieve similar conversions and yields with increasing polyunsaturated methyl ester (PU-FAME) shares.12–14 While oliveME (PU-FAME = 8 wt%) was converted at conditions of Rh = 0.1%, temperature of 110 °C and pressure of 138 bar with triphenylphosphine and heterogeneous supported Rh catalyst, for safflowerME (PU-FAME = 76 wt%) this was only achieved under significantly more extreme conditions (Rh = 1%, 120 °C, 241 bar). It is noteworthy that in multiple studies with complete conversion, in some cases, PUFA components remained in the form of conjugated dienes, which is why it was logically concluded that “these conjugated dienes were more difficult to hydroformylate […] than methyl linoleate”.14 In 1978, Friedrich demonstrated that pressures of only 14 bar at <120 °C are required for the hydroformylation of methyl oleate (monounsaturated methyl esters (MU-FAME) >98 wt%) using triphenyl phosphite.15 Similar observations were made in 1997 by the group of van Leeuwen during hydroformylation with rhodium and tris(2-tert-butyl-4-methylphenyl)phosphite ligands (20 bar, <100 °C).16 In the hydroformylation of pure methyl oleates, significantly higher reaction rates were observed than in the conversion of technical grade methyl oleates (PU-FAME = 14 wt%). They suggested that stable π-allylic intermediates are formed between polyunsaturated substrates and the catalyst, leading to a significantly slower hydroformylation reaction. These observations are by now known from the hydroformylation of butadiene17–20 and other 1,3-dienes,21–25 but there is no analytical validation for the conversion of PU-FAME yet. Similar observations were made in other studies in which hydroformylation of various vegetable oils (e.g. soy, sunflowers, safflower) was carried out to produce polymers and investigate their material properties subsequently.26–28 Additionally, in contrast to other studies, here 2–3 times higher activities were achieved with phosphines than with phosphites,28 although they are known as highly active ligands in hydroformylation29–33 due to their better pi-acceptor properties.34,35 It was assumed that these favour the conjugation of the double bonds and, thus, the formation of Rh-allyl intermediates, which is why phosphines were used.
Throughout the literature, it was repeatedly observed with various ligands that PUFAs have a disruptive effect on the hydroformylation of oleic acid-containing substrates. Although a convincing explanation exists, there is still a lack of quantification and analytical evidence to understand this phenomenon in detail. Hereby, it will be possible to identify specific measures to enable efficient hydroformylation with the help of active phosphites. With this work we would like to close this gap by first quantifying and proving the influence of methyl linoleate on the hydroformylation of methyl oleate. These findings will finally show that an additional harmonization step by selective hydrogenation enables much higher activities in the hydroformylation of oleochemical substrates of relevant and technical quality.
Result and discussion
In the hydroformylations of methyl oleate, a quite simple reaction network arises (Fig. 1): positional isomerization of the double bond can occur. For clarity, the resulting species are grouped as methyl oleates (MO) but can be identified individually. In addition, hydrogenation to methyl stearate (MS) and cis–trans-isomerisation may occur, resulting in various trans-configured isomers of methyl elaidate (iME). In the hydroformylation, different positional isomers of the formyl methyl stearates (FMS) can be formed. As soon as methyl linoleate is present, several other possible reactions arise. Firstly, it can be converted to various positional isomers with a multitude of cis–trans-configuration combinations; all summarised here to methyl linoleate (ML). Of particular importance is the conjugation of the double bonds to conjugated methyl linoleate (CML), which in turn can either be hydrogenated to MO or hydroformylated to formyl methyl oleate (FMO), which can then eventually react to form a diformyl methylstearate (dFMS).Ligand screening for hydroformylation of MO
For an initial ligand screening, the MO hydroformylation was tested under typical conditions.36–38 MO was obtained from high oleic (h.o.) sunflower ME (PU-FAME = 3.6 wt%) by our previously used partial hydrogenation method. By this a mixture consisting exclusively of MU-FAME and saturated species is obtained with a slightly increased proportion of C18:0 compared to the starting material (see ESI†). Only monodentate ligands were considered, as these generally offer higher activity than polydentate ligands, and their high regioselectivity is not required for the hydroformylation of methyl oleate. Two phosphines, triphenylphosphine (L1) and triazaadamantane (L2), as well as triphenylphosphite (L3) and tris(2,4-di-tert-butylphenyl)phosphite (L4) as phosphite ligands were investigated. The latter is a notably bulky ligand that has recently attracted particular attention. It was long assumed that L4 could not replace more than a single carbon monoxide in the complex because of its challenging steric.34 However, recent studies have shown that they can form significant amounts of a bis-ligand species even under classical hydroformylation conditions.39As expected, the phosphine ligands showed lower activities than the phosphite ligands used (Fig. 2). As a result, only the latter achieved quantitative conversion within four hours, while all ligands showed high aldehyde selectivity of >99%. Although no spectroscopic data are available to verify the character of the active species under reaction conditions, comparable reaction systems in the literature suggest that the species predominantly contributing to the reaction is the mono or bis phosphine/phosphite rhodium hydride.‡39,40 The reduced activity in the experiment without the addition of a ligand, except for L2 in toluene where the complex was not completely soluble, supports this.
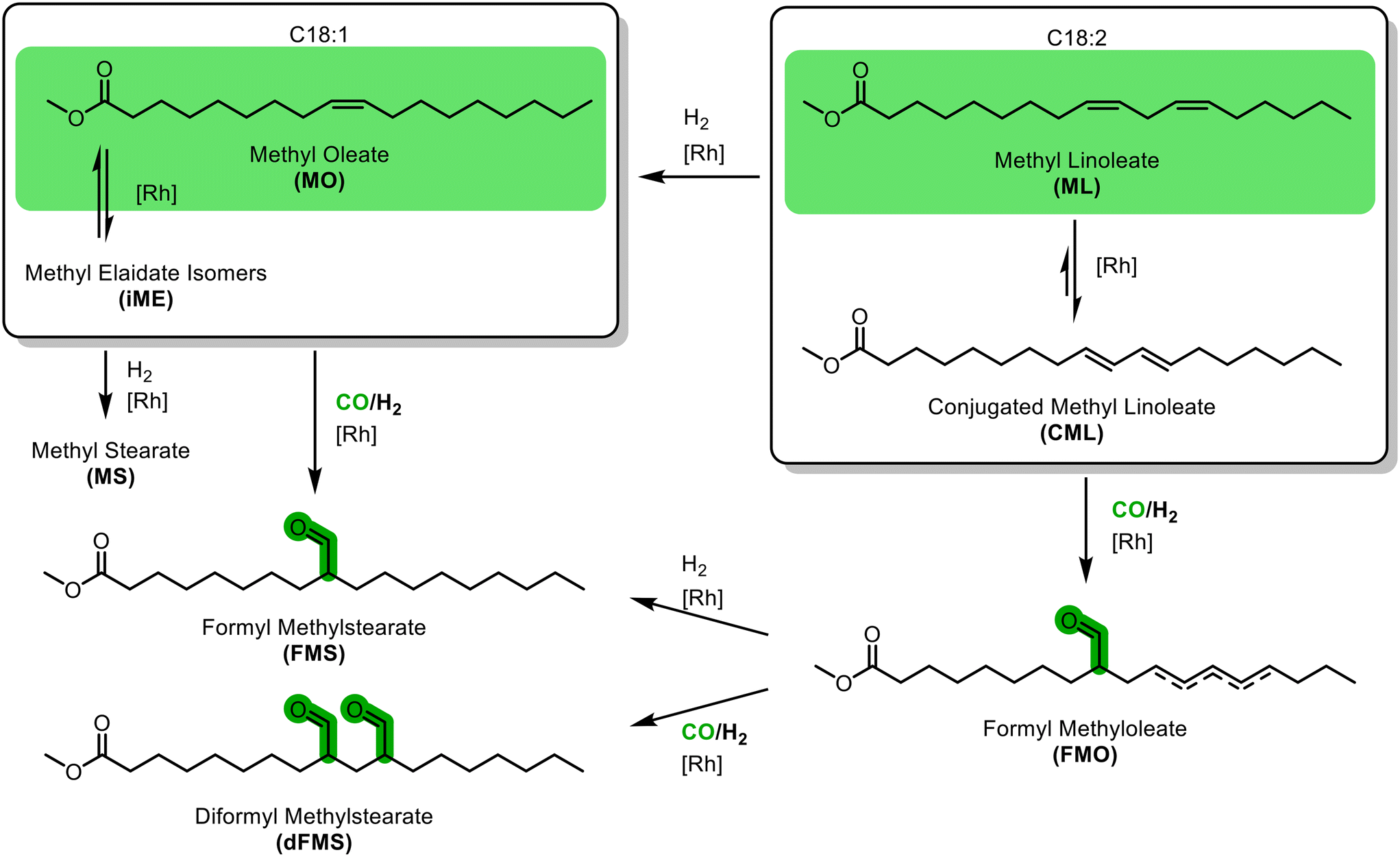 | ||
| Fig. 2 Most important species in the reaction network for the hydroformylation of methyl oleate with methyl linoleate present. Different positional isomers exist for all species except MS as well as different cis–trans-isomers for CML, which are not shown here for clarity. | ||
L3 also exhibited rapid positional isomerization, which led to a broader distribution of positional isomers of the product aldehyde MFS. L4 achieved the markedly highest turnover frequency at 20% conversion (TOF20) with 3201 h−1. To our knowledge, this is the highest reported activity for the hydroformylation of MO. It is to be considered that no prior optimisation of the reaction conditions was carried out.
Investigation of the ML influence
Subsequently, perturbation experiments were conducted by gradually replacing MO with ML using L4 (Fig. 4). Starting from an equimolar amount to the catalyst, the reaction is increasingly slowed down as the ML content increases. In addition, significantly lower aldehyde selectivities are observed at the beginning of the reaction, as positional and cis–trans-isomerization of the double bonds occurs. However, since these can still form aldehydes, all reactions achieve quantitative conversions with almost complete selectivity to mono or dialdehydes after a reaction time of 240 minutes. It is also noticeable that in all reactions, starting from a large cis-excess in the C18:1 species, a cis/trans ratio of 0.5 is formed within approximately thirty minutes (see ESI†). Since cis-double bonds make up a significantly higher proportion at the beginning, it is to be expected that these react much faster and the cis/trans ratio decreases. The fact that it stays at a level of 0.5 suggests that the hydroformylation of MO is about twice as fast as that of iME.Surprisingly, reaction profiles of the reactions with higher ML show an S-shape. This behaviour could already be observed under the influence of other accompanying substances than ML in the Rh-catalyzed hydroformylation.41 This indicates an initial inhibition that diminishes over time, resulting in an acceleration of the reaction rate before it decreases again due to a lack of reactant. The reason for the significantly lower reaction rate at the beginning of the experiments with a high ML content could be that ML has a higher affinity for coordination to the catalyst than MO. Still, reactions with ML proceed at a significantly slower rate. Only when ML is almost completely consumed is MO then converted in a faster reaction. This is supported by the observation that, among other minor species, conjugation of the double bonds occurs at the start of the reaction within the first 5 minutes (Fig. 5). With the help of commercially available chemicals, these could be identified as c,t 9,11 and t,c 9,11-CML (see ESI†). In the further course, the conjugated double bond system isomerise along the alkyl chain and accumulates in a particularly stable CML isomer, which elutes very close to the inert C20:0. As soon as this isomer is consumed, a sudden increase of the hydroformylation reaction rate is observed (chromatogram of 90 min sample in Fig. 4), which is in line with van Leeuwen's hypothesis that the rhodium catalyst forms a stable η3-π-allyl intermediate with CML.
Further experiments were carried out to verify this hypothesis. The precursor and ligand were first preformed in d8-toluene under syngas pressure, then the substrate was added, and the resulting solutions were analyzed using 1H- and 31P-NMR under syngas at ambient pressure. As expected, after preforming as well as after the addition of MO, rhodium species with one (31P-NMR: d, δ = 133 ppm, 134 ppm) and two (31P: d, δ = 117 ppm, 118 ppm) coordinated ligands and Rh-hydride (1H-NMR: s, δ = −9.5 ppm) could be detected, which is in accordance with the literature.39,42 Since the active Rh-hydride with two coordinated phosphite ligands is present, the hydroformylation of the added MO consequently occurs. In contrast, a rhodium species with only a single coordinated ligand and no Rh-hydride signals can be observed when the reaction is performed with both ML or CML (Fig. 6).
In addition, the formation of a signal (doublet of triplet, δ = 4.58 ppm, J = 2.1 Hz, J = 8.9 Hz) can be observed here, which can be assigned to the proton of the central allyl carbon.43 Similar results were found in the palladium-catalyzed methoxycarbonylation of ML, where an allylic species was also identified.44 In addition, significantly lower productivity was obtained with pure ML (TOF20 = 6) and with CML than with pure MO under the original reaction conditions (Fig. 3), requiring up to 7 days for full conversion of all C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. The even lower activity in the conversion of CML shows that hydroformylation and isomerization are competing reactions in the reaction of ML. Consequently, decelerating the overall reaction rate described here only occurs according to the mechanism described if CML is formed first. However, even small amounts of CML formation massively affect the reaction. The fact that even the presence of equimolar amounts (0.14 wt%) of the catalyst still causes a reduced activity further underlines this finding. It convincingly shows that the deactivation mechanism described here cannot be avoided by using high oleic substrates such as sunflowerME (PU-FAME = 3 wt%, Table 1).
C bonds. The even lower activity in the conversion of CML shows that hydroformylation and isomerization are competing reactions in the reaction of ML. Consequently, decelerating the overall reaction rate described here only occurs according to the mechanism described if CML is formed first. However, even small amounts of CML formation massively affect the reaction. The fact that even the presence of equimolar amounts (0.14 wt%) of the catalyst still causes a reduced activity further underlines this finding. It convincingly shows that the deactivation mechanism described here cannot be avoided by using high oleic substrates such as sunflowerME (PU-FAME = 3 wt%, Table 1).
 | ||
Fig. 3 Ligands screened for the hydroformylation of MO. Left: Yield (Y) of aldehyde in the hydroformylation of MO. Right: TOFs are calculated as the aldehyde produced per amount of catalyst used per time required for 20% conversion, TOF = nProductnCat−1tX=20%−1. Calculations based on GC-FID analysis with n-dodecane as internal standard. Connections between data points are for visualization purposes only. Conditions: T = 120 °C, p = 30 bar, t = 4 h; nCO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nH2 = 1:1, VSTR = 100 ml, U = 1000 rpm, cRh = 0.5 mmol L−1, nP:nRh = 10, nMO:nRh = 1000, solvent:toluene, *solvent:2-propanol as solvent with nMO:nRh = 100, preforming: T = 120 °C, p = 30 bar, nCO:nH2 = 1:1, U = 1000 rpm, t = 30 min. :nH2 = 1:1, VSTR = 100 ml, U = 1000 rpm, cRh = 0.5 mmol L−1, nP:nRh = 10, nMO:nRh = 1000, solvent:toluene, *solvent:2-propanol as solvent with nMO:nRh = 100, preforming: T = 120 °C, p = 30 bar, nCO:nH2 = 1:1, U = 1000 rpm, t = 30 min. | ||
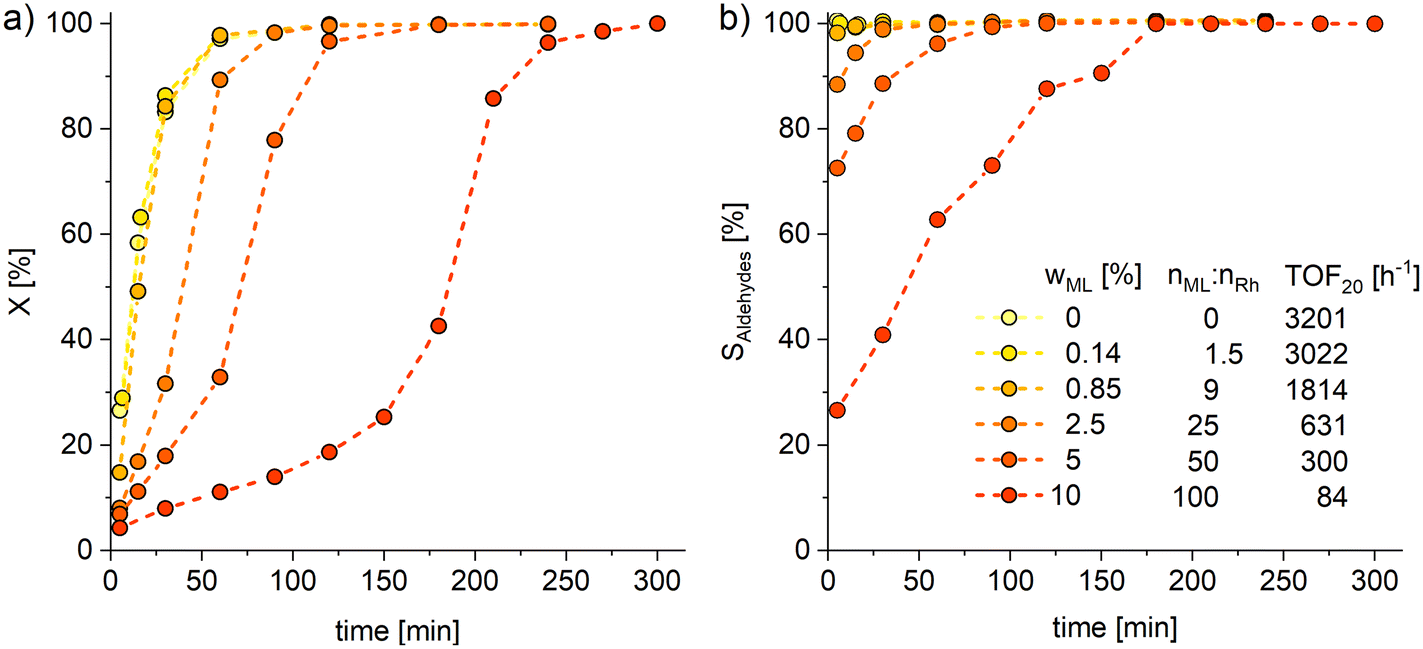 | ||
| Fig. 4 Conversions (X, a) of C18 species and combined selectivity (S, b) to mono and dialdehydes for the perturbation experiments of hydroformylation by gradually replacing MO with ML. X and S are calculated based on GC-FID analysis with n-dodecane as an internal standard. For more detailed diagram of the reaction start see ESI.† Connections between data points are for visualization purposes only. Conditions: T = 120 °C, p = 30 bar, nCO:nH2 = 1:1, VSTR = 100 ml, U = 1000 rpm, cRh = 0.5 mmol L−1, nP:nRh = 10, n(MO+ML):nRh = 1000, ligand: L4, preforming: T = 120 °C, p = 30 bar, nCO:nH2 = 1:1, U = 1000 rpm, t = 30 min. | ||
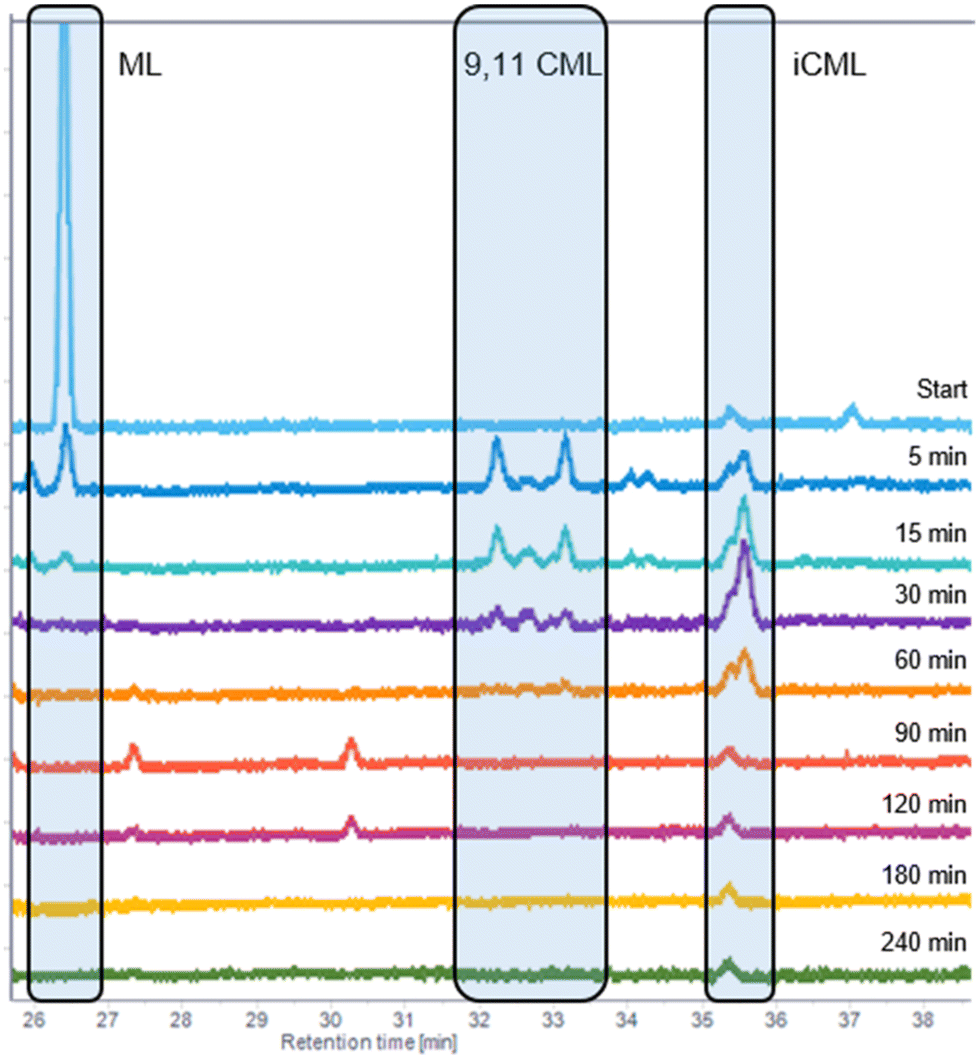 | ||
| Fig. 5 The C18:2 region of the GC-FID traces of the experiment with 5 wt% ML from Fig. 3. The peak at 35.6 is the inert C20:0. | ||
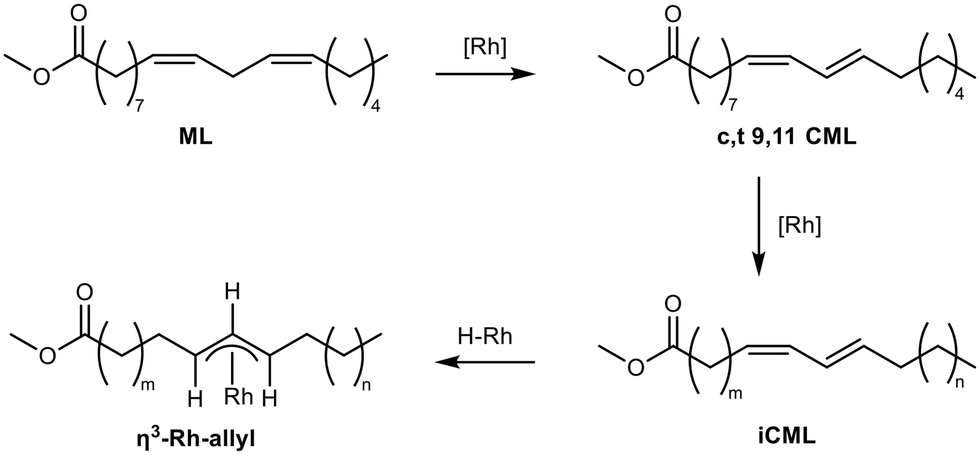 | ||
| Fig. 6 Observed reaction sequence of conjugation, isomerization and formation of Rh-allyl-complex. | ||
In summary, under hydroformylation conditions, the isolated alkene functions of ML partially isomerize to a conjugated system iCML before inserting into the rhodium hydride bond and forming an allylic-η3 complex, which slows down hydroformylation due to the slow conversion to the η1 species. If ML is present, this leads to a temporary binding of a part of the catalyst, which reduces the overall reaction rate. Only with increasing conversion of the ML is the entirety of the applied catalyst available, accelerating the overall reaction rate once again.
Hydroformylation of technical FAME and partially hydrogenated derivatives
To apply the findings made in the hydroformylation model mixtures, it is essential to transfer them to technically relevant substrates. Since ML significantly slows down the reaction rate, it can be assumed that methyl linolenate, which also occurs in natural raw materials, has a comparable, if not stronger, influence as the conjugation of double bonds is much more likely. Therefore, removing both components from the reaction mixture before hydroformylation is advisable to enable an efficient and fast reaction. Conventional separation processes such as distillation can only be achieved with considerable time and energy. As we have shown recently, the selective partial hydrogenation of PU-FAMEs to MU-FAME is an elegant way to remove the interfering components and simultaneously increase the efficiency of the overall process.11To demonstrate the influence of PU-FAME share on the hydroformylation of naturally derived ME with different compositions, a high oleic sunflowerME and a canolaME with a particularly high PU-FAME content were selected (Fig. 7).
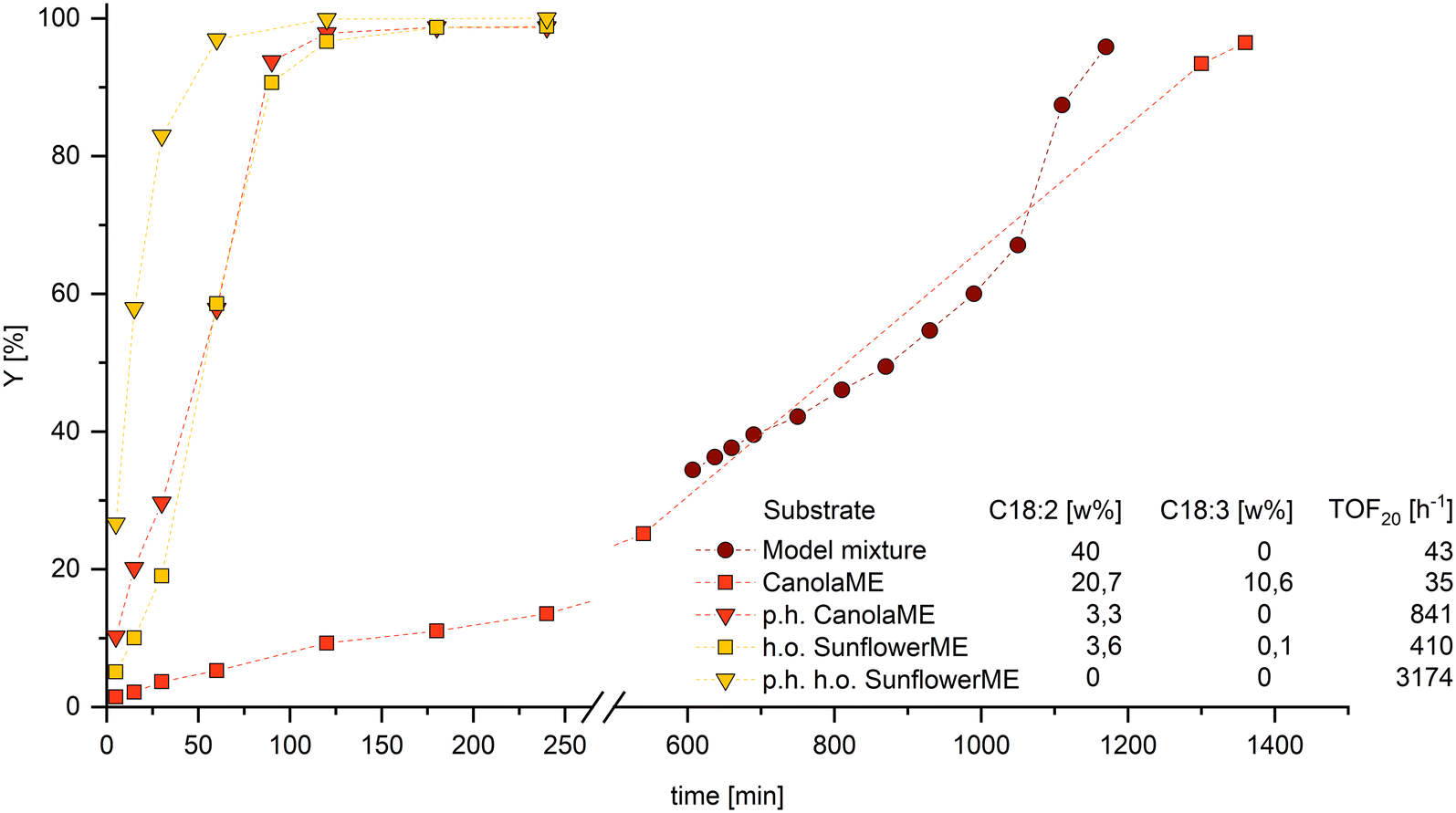 | ||
| Fig. 7 Results for the hydroformylation of a model mixture with 40 wt% ML, CanolaME (31 wt% PU-FAME), partially hydrogenated CanolaME (3.3 wt% PU-FAME), high oleic sunflowerME (3.7 wt% PU-FAME) and partially hydrogenated high oleic sunflowerME (0 wt% PU-FAME). Yields are calculated based on GC-FID analysis with n-dodecane as the internal standard. Connections between data points are for visualisation purposes only. Conditions: T = 120 °C, p = 30 bar, nCO:nH2 = 1:1, VSTR = 100 ml, U = 1000 rpm, cRh = 0.5 mmol L−1, nP:nRh = 10, nMO+PUFA:nRh = 1000, ligand: L4. Preforming: T = 120 °C, p = 30 bar, nCO:nH2 = 1:1, U = 1000 rpm, t = 30 min. | ||
The significant influence of PU-FAME on the reaction can also be observed here. Since the technical canolaME has a 10 wt% share of the triple unsaturated component methyl linolenate, the probability of isomerization to a conjugated derivative increases compared to the model mixture with ML. It is, therefore, not surprising that the conversion of the technical canolaME has an even more reduced reaction rate at the start of the reaction and a longer time to reach complete conversion. The fact that by far the highest activity can be observed in p.h. sunflowerME (PU-FAME = 0 wt%) clearly shows that even the most minor fractions are detrimental to the hydroformylation of MO. On an industrial scale, hydroformylation is also carried out in aqueous biphasic systems using polar phosphines. The effects observed in this work could also be reproduced in this catalyst system (see ESI†). Consequently, a selective reduction of the PU-FAME content led to an increase in yield of up to 30%.
Conclusion
In this work, the influence of naturally occurring polyunsaturated methyl esters (PU-FAME) on the hydroformylation of methyl oleate was investigated. It has been observed for the first time that the isomerisation of the double bonds in the polyunsaturated components leads to the formation of conjugated double bond systems which subsequently form stable η3-Rh-allyl species. As a result, a large amount of catalyst is inhibited at the beginning of the reaction, which is gradually released during the slow reaction of the conjugated species, resulting in a characteristic S-shaped reaction profile. As little as 5 wt% PU-FAME results in a 90% activity loss when a bulky phosphite ligand is used under monophasic conditions. By selective hydrogenation of the PU-FAME to monounsaturated-FAME content in a canola methyl ester, the activity in subsequent hydroformylation could be increased 24-fold. In the case of a sunflowerME, the TOF was increased by a factor of 7.7 to a value of 3172 h−1, which, to our knowledge, is the highest reported rate for the hydroformylation of a methyl oleate. Overall, this work provides a deeper insight into the use of methyl esters as a molecular building block, paving the way for more sustainable chemistry.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the networking program ‘Sustainable Chemical Synthesis 2.0’ (SusChemSys 2.0) for the support and fruitful discussions across disciplines. The authors thank Dako AG for donating methyl oleate MB 9001. Finally, we are very thankful to the German Federal Ministry of Food and Agriculture (Bundesministerium für Ernährung und Landwirtschaft), represented by the FNR (Fachagentur Nachwachsende Rohstoffe) for financial support of the young research group “Renewlysis” (project number 2219NR355).Notes and references
-
A. Behr and T. Seidensticker, Chemistry of Renewables, Springer Nature, Berlin, 2020 Search PubMed
.
-
A. Behr and T. Seidensticker, Einführung in die Chemie nachwachsender Rohstoffe. Vorkommen, Konversion, Verwendung, Springer Spektrum, Berlin, Heidelberg, 2018, DOI:10.1007/978-3-662-55255-1
- U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Oils and fats as renewable raw materials in chemistry, Angew. Chem., Int. Ed., 2011, 50, 3854–3871, DOI:10.1002/anie.201002767
- T. Vanbesien, F. Hapiot and E. Monflier, Hydroformylation of vegetable oils and the potential use of hydroformylated fatty acids, Lipid Technol., 2013, 25, 175–178, DOI:10.1002/lite.201300289
- T. Vanbésien, E. Monflier and F. Hapiot, Hydroformylation of vegetable oils: More than 50 years of technical innovation, successful research, and development, Eur. J. Lipid Sci. Technol., 2016, 118, 26–35, DOI:10.1002/ejlt.201500196
-
S. Krist, G. Buchbauer and C. Klausberger, Lexikon der pflanzlichen Fette und Öle, Springer, Wien, 2008 Search PubMed
- Z. S. Petrović, I. Cvetković, J. Milić, D. Hong and I. Javni, Hyperbranched polyols from hydroformylated methyl soyate, J. Appl. Polym. Sci., 2012, 125, 2920–2928, DOI:10.1002/app.36232
- Z. S. Petrović, I. Cvetković, D. Hong, X. Wan, W. Zhang, T. Abraham and J. Malsam, Vegetable oil-based triols from hydroformylated fatty acids and polyurethane elastomers, Eur. J. Lipid Sci. Technol., 2010, 112, 97–102 CrossRef
-
Dictionary of renewable resources, ed. H. Zoebelein, Wiley-VCH, Weinheim, New York, Chichester, Brisbane, Singapore, Toronto, 2001 Search PubMed
-
A. Thomas, B. Matthäus and H.-J. Fiebig, Fats and Fatty Oils, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2015 Search PubMed
- T. F. H. Roth, A. Kühl, M. L. Spiekermann, H. W. Wegener and T. Seidensticker, Biodiesel as a Sustainable Platform Chemical Enabled by Selective Partial Hydrogenation: Compounds Outplace Combustion?!, ChemSusChem, 2024, e202400036, DOI:10.1002/cssc.202400036
- E. N. Frankel, Methyl 9(10)-formylstearate by selective hydroformylation of oleic oils, J. Am. Oil Chem. Soc., 1971, 48, 248–253, DOI:10.1007/BF02883763
- E. N. Frankel and F. L. Thomas, Selective hydroformylation of polyunsaturated fats with a rhodium-triphenylphosphine catalyst, J. Am. Oil Chem. Soc., 1972, 49, 10–14, DOI:10.1007/BF02545129
- E. N. Frankel, F. L. Thomas and W. K. Rohwedder, Hydroformylation of Methyl Linoleate and Linolenate with Rhodium-Triphenylphosphine Catalyst, Ind. Eng. Chem. Prod. Res. Dev., 1973, 12, 47–53 CrossRef CAS
- J. P. Friedrich, Low-Pressure Hydroformylation of Methyl Oleate with an Activated Rhodium Catalyst, Ind. Eng. Chem. Prod. Res. Dev., 1978, 17, 205–207, DOI:10.1021/i360067a005
- K. F. Muilwijk, P. C. J. Kamer and P. W. N. M. van Leeuwen, A bulky phosphite-modified rhodium catalyst for the hydroformylation of unsaturated fatty acid esters, J. Am. Oil Chem. Soc., 1997, 74, 223–228, DOI:10.1007/s11746-997-0127-8
- P. van Leeuwen and C. F. Roobeek, The hydroformylation of butadiene catalysed by rhodium-diphosphine complexes, J. Mol. Catal., 1985, 31, 345–353, DOI:10.1016/0304-5102(85)85117-8
- S. Bertozzi, N. Campigli, G. Vitulli, R. Lazzaroni and P. Salvadori, Selective hydroformylation of open-chain conjugated dienes promoted by mesitylene-solvated rhodium atoms to give b,y-unsaturated monoaldehydes, J. Organomet. Chem., 1995, 487, 41–45 CrossRef CAS
- G. Liu and M. Garland, The competitive and non-competitive hydroformylation of conjugated dienes starting with tetrarhodium dodecacarbonyl. An in-situ high-pressure infrared spectroscopic study, J. Organomet. Chem., 2000, 608, 76–85 CrossRef CAS
- S. Schmidt, P. Deglmann and P. Hofmann, Density Functional Investigations of the Rh-Catalyzed Hydroformylation of 1,3-Butadiene with Bisphosphite Ligands, ACS Catal., 2014, 4, 3593–3604, DOI:10.1021/cs500718v
- C. M. Foca, H. J. V. Barros, E. N. dos Santos, E. V. Gusevskaya and J. Carles Bayón, Hydroformylation of myrcene: metal and ligand effects in the hydroformylation of conjugated dienes, New J. Chem., 2003, 27, 533, 10.1039/b207947j
- H. J. V. Barros, C. C. Guimarães, E. N. dos Santos and E. V. Gusevskaya, Rhodium-Catalyzed Hydroformylation of Isoprene: Unusual Accelerating Effects of Phosphorus Ligands and Gas Pressure, Organometallics, 2007, 26, 2211–2218, DOI:10.1021/om060994n
- H. J. V. Barros, J. G. Da Silva, C. C. Guimarães, E. N. dos Santos and E. V. Gusevskaya, Hydroformylation of Monoterpenic Polyenes: Effect of the Conjugation of Double Bonds on Reactivity, Organometallics, 2008, 27, 4523–4531, DOI:10.1021/om800451t
- E. R. Nelsen, A. C. Brezny and C. R. Landis, Interception and characterization of catalyst species in rhodium bis(diazaphospholane)-catalyzed hydroformylation of octene, vinyl acetate, allyl cyanide, and 1-phenyl-1,3-butadiene, J. Am. Chem. Soc., 2015, 137, 14208–14219, DOI:10.1021/jacs.5b09858
- N. Herrmann, D. Vogelsang, A. Behr and T. Seidensticker, Homogeneously Catalyzed 1,3-Diene Functionalization – A Success Story from Laboratory to Miniplant Scale, ChemCatChem, 2018, 10, 5342–5365, DOI:10.1002/cctc.201801362
- A. Guo, D. Demydov, W. Zhang and Z. Petrovic, Polyols and Polyurethanes from Hydroformylation of Soybean Oil, J. Polym. Environ., 2002, 10, 49–52 CrossRef CAS
- P. Kandanarachchi, A. Guo, D. Demydov and Z. Petrovic, Kinetics of the hydroformylation of soybean oil by ligand-modified homogeneous rhodium catalysis, J. Am. Oil Chem. Soc., 2002, 79, 1221–1225, DOI:10.1007/s11746-002-0631-2
- P. Kandanarachchi, A. Guo and Z. Petrovic, The hydroformylation of vegetable oils and model compounds by ligand modified rhodium catalysis, J. Mol. Catal. A: Chem., 2002, 184, 65–71, DOI:10.1016/S1381-1169(01)00420-4
- P. W. N. M. Leeuwen and C. F. Roobeek, Hydroformylation of less reactive olefins with modified rhodium catalysts, J. Organomet. Chem., 1983, 258, 343–350 CrossRef
- T. Jongsma, G. Challa and P. W. N. M. Leeuwen, A mechanistic sutdy of rhoidum tri(o-t-butylphenyl)phosphite complexes as hydroformylation catalysts, J. Organomet. Chem., 1991, 421, 121–128 CrossRef CAS
- A. van Rooy, E. N. Orij, P. C. J. Kamer, F. van den Aardweg and P. W. N. M. Leeuwen, Hydroformylation of Oct-1-ene with Extremely High Rates using Rhoidum Catalysts containing Bulky Phosphites, J. Chem. Soc., Chem. Commun., 1991, 1096–1097 RSC
- A. A. Dabbawala, H. C. Bajaj and R. V. Jasra, Rhodium complex of monodentate phosphite as a catalyst for olefins hydroformylation, J. Mol. Catal. A: Chem., 2009, 302, 97–106, DOI:10.1016/j.molcata.2008.12.002
- A. Polo, E. Fernandez, C. Claver and S. Castillon, Hydroformylation of Glucal Derivatives with Rhodium Catalysts. Crucial Influence of the Auxiliary Ligand Tris(ortho-tert-butylphenyl) Phosphite, J. Chem. Soc., Chem. Commun., 1992, 639–640 RSC
-
M. Beller, Catalytic carbonylation reactions, Springer, Berlin, Heidelberg, 2006, DOI:10.1007/b105253
-
Hydroformylation. Fundamentals, processes, and applications in organic systhesis, ed. A. Börner and R. Franke, Wiley-VCH, Weinheim, 2016, DOI:10.1002/9783527677931
- E. Benetskiy, S. Lühr, M. Vilches-Herrera, D. Selent, H. Jiao, L. Domke, K. Dyballa, R. Franke and A. Börner, Rhodium-Catalyzed Nonisomerizing Hydroformylation of Methyl Oleate Applying Lactame-Based Phosphoramidite Ligands, ACS Catal., 2014, 4, 2130–2136, DOI:10.1021/cs500274n
- N. Herrmann, J. Bianga, T. Gaide, M. Drewing, D. Vogt and T. Seidensticker, Aqueous biphasic hydroformylation of methyl oleate: a green solvent-only strategy for homogeneous catalyst recycling, Green Chem., 2019, 21, 6738–6745, 10.1039/C9GC02868D
- J. Bianga, N. Herrmann, L. Schurm, T. Gaide, J. M. Dreimann, D. Vogt and T. Seidensticker, Improvement of Productivity for Aqueous Biphasic Hydroformylation of Methyl 10-Undecenoate: A Detailed Phase Investigation, Eur. J. Lipid Sci. Technol., 2020, 122, 1900317, DOI:10.1002/ejlt.201900317
- C. Kubis, M. König, B. N. Leidecker, D. Selent, H. Schröder, M. Sawall, W. Baumann, A. Spannenberg, A. Brächer, K. Neymeyr, R. Franke and A. Börner, Interplay between Catalyst Complexes and Dormant States: In Situ Spectroscopic Investigations on a Catalyst System for Alkene Hydroformylation, ACS Catal., 2023, 13, 5245–5263, DOI:10.1021/acscatal.2c06320
- A. Bara-Estaún, C. L. Lyall, J. P. Lowe, P. G. Pringle, P. C. J. Kamer, R. Franke and U. Hintermair, Mapping catalyst activation, turnover speciation and deactivation in Rh/PPh 3-catalysed olefin hydroformylation, Catal. Sci. Technol., 2022, 12, 5501–5516, 10.1039/D2CY00312K
- E. B. Walczuk, P. C. J. Kamer and P. W. N. M. van Leeuwen, Dormant States of Rhodium Hydroformylation Catalysts: Carboalkoxyrhodium Complex Formed from Enones in the Alkene Feed, Angew. Chem., 2003, 115, 4813–4817, DOI:10.1002/ange.200351884
- R. Crous, M. Datt, D. Foster, L. Bennie, C. Steenkamp, J. Huyser, L. Kirsten, G. Steyl and A. Roodt, Rhodium hydride formation in the presence of a bulky monophosphite ligand: a spectroscopic and solid-state investigation, Dalton Trans., 2005, 1108–1116, 10.1039/b416917d
- R. J. Harris, J. Park, T. A. F. Nelson, N. Iqbal, D. C. Salgueiro, J. Bacsa, C. E. MacBeth, M.-H. Baik and S. B. Blakey, The Mechanism of Rhodium-Catalyzed Allylic C-H Amination, J. Am. Chem. Soc., 2020, 142, 5842–5851, DOI:10.1021/jacs.0c01069
- P. Roesle, L. Caporaso, M. Schnitte, V. Goldbach, L. Cavallo and S. Mecking, A comprehensive mechanistic picture of the isomerizing alkoxycarbonylation of plant oils, J. Am. Chem. Soc., 2014, 136, 16871–16881, DOI:10.1021/ja508447d
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed information is given on the chemicals used, analysis and experimental procedures. Further experimental results are also given. See DOI: https://doi.org/10.1039/d4cy00839a |
| ‡ For completeness, it should be noted that Rh6(CO)12 species may form under the choosen conditions. This can subsequently lead to the formation of a highly active HRh(CO)4 species, which may be a contributor to the observed activity. However, the fact that the addition of ligand always results in higher activities than in the experiment without ligand-addition is an reliable indication that the catalysis is mainly carried out by the phosphine/phosphite coordinated species. For a detailed study on the active species in rhodium-catalyzed hydroformylation with phosphines and phosphites, the reader is recommended to consult ref. 39 and 40. |
| This journal is © The Royal Society of Chemistry 2024 |