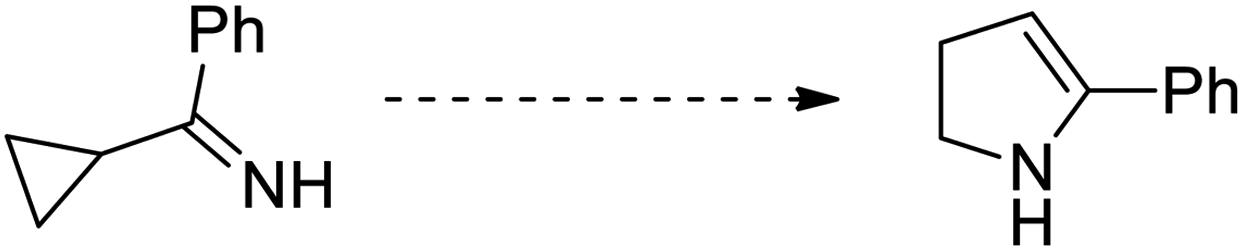DOI:
10.1039/D3RA07410B
(Review Article)
RSC Adv., 2023,
13, 35695-35732
Cloke–Wilson rearrangement: a unique gateway to access five-membered heterocycles
Received
31st October 2023
, Accepted 20th November 2023
First published on 7th December 2023
Abstract
Cyclopropanes are of great synthetic value in heterocyclic chemistry due to their highly reactive nature. They are widely employed to synthesize various biologically active organic compounds. Generally, vinyl, carbonyl, imine, and alkylidene cyclopropanes are utilized as efficient synthetic precursors in organic synthesis. The Cloke–Wilson rearrangement of these activated cyclopropanes is carried out to achieve the synthesis of diverse heterocyclic scaffolds. Various oxygen, nitrogen, and sulfur-containing heterocyclic compounds have been synthesized employing this rearrangement. With time, Cloke–Wilson rearrangement has evolved into a high yielding enantioselective and diastereoselective approach utilizing integrated novel methods. Our review focuses on the recent approaches for Cloke–Wilson rearrangement to synthesize several five-membered heterocycles and its applicability towards the natural product syntheses reported during 2000–2020.
1. Introduction
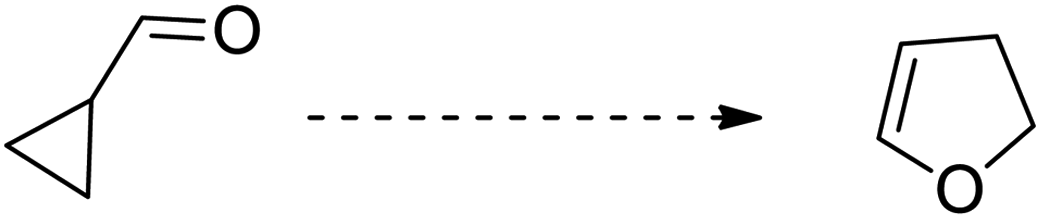
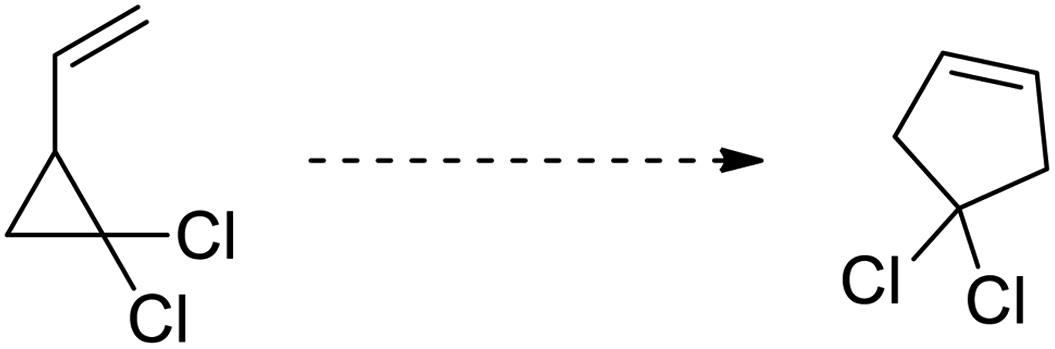
Cyclopropanes are the smallest carbocycles that possess an affluent position in synthetic organic chemistry.1 Cyclopropanes are widely utilized as useful synthetic building blocks owing to the presence of the high π character, intrinsic torsional, and angle strain of about 115 kJ mol−1, which enable them to undergo ring-opening reactions to give diversely functionalized target molecules2,3 Regardless of the ring strain, cyclopropanes are relatively stable and chemically unreactive towards C–C bond cleavage unless polarized and get activated by the introduction of activating groups on the ring.4 Mostly, vinyl, carbonyl, imine, and alkylidene substituents are introduced to enhance the reactivity of cyclopropanes.1,5 The substituted cyclopropanes undergo ring-opening/cyclization reactions to generate five-membered heterocycles, such as vinylcyclopropane to cyclopentene,6 cyclopropyl-carbonyls to dihydrofurans,7 cyclopropylimines to pyrroline,8 and cyclopropyl thioketones to dihydrothiophenes.9 Neureiter was the first chemist to report the synthesis of dichlorocyclopentene by the thermal rearrangement of dichlorovinylcyclopropane above 400 °C.10 After one year, in 1960, vinylcyclopropane was transformed to cyclopentene by thermal rearrangement at 325–500 °C.11 A decade later, Woodward and Hoffman called this rearrangement an example of a [1,3]-sigmatropic reaction.12 Later on, reaction kinetics,13 substituents effect,14 reaction stereochemistry,15 and theoretical calculations16 were thoroughly investigated in organic synthesis.15c The vinylcyclopropane rearrangement is widely applicable in the total synthesis of various organic compounds, such as aphidicolin,17a zizaene,17b hirsutene,17c specionin,17d and salviasperanol.18 Cyclic imines, such as pyrrolines have been employed to synthesize various biologically active organic compounds.19 In 1929, Cloke reported the synthesis of 2-phenylpyrroline hydrochloride by the Cloke–Wilson rearrangement of cyclopropylimine–pyrroline.8a After forty years, Stevens applied it to the synthesis of a variety of alkaloids, such as mesembrine, nicotine, ipalbidine, and septicine.20 In 2011, the synthesis of crispine A and harmicine alkaloids was accomplished via cyclopropylimine rearrangement by Saha and coworkers.21 Many other research groups published the synthesis of pyrrolidine-based alkaloids by this rearrangement.22 Since then, this rearrangement is considered to be an efficient synthetic route to generate several five-membered N-containing heterocycles.23,72–74 In 1947, Wilson reported the thermal rearrangement of cyclopropylcarbaldehyde to dihydrofuran.7a Besides its application in the synthesis of diverse heterocycles,24 this rearrangement is also involved in the total synthesis of various natural products.25 The [3 + 2] cycloaddition reaction of cyclopropanes with aldehydes results in the synthesis of furans,26a but the synthetic route to the furan moiety via the rearrangement of cyclopropyl ketone is a highly efficient and cost-effective protocol.26b The transition metal-catalyzed synthesis of functionalized furans by the ring-opening/closure of cyclopropanyl ketones is another facile and efficacious synthetic pathway.27 The objective of this review is to provide a broad overview of recent methodological development of Cloke–Wilson rearrangement to furnish five-membered O-, N-, and S-heterocycles and its applications in the total synthesis of natural products. To date, no specific review has been published on this particular topic.
2. Chemistry of cyclopropanes
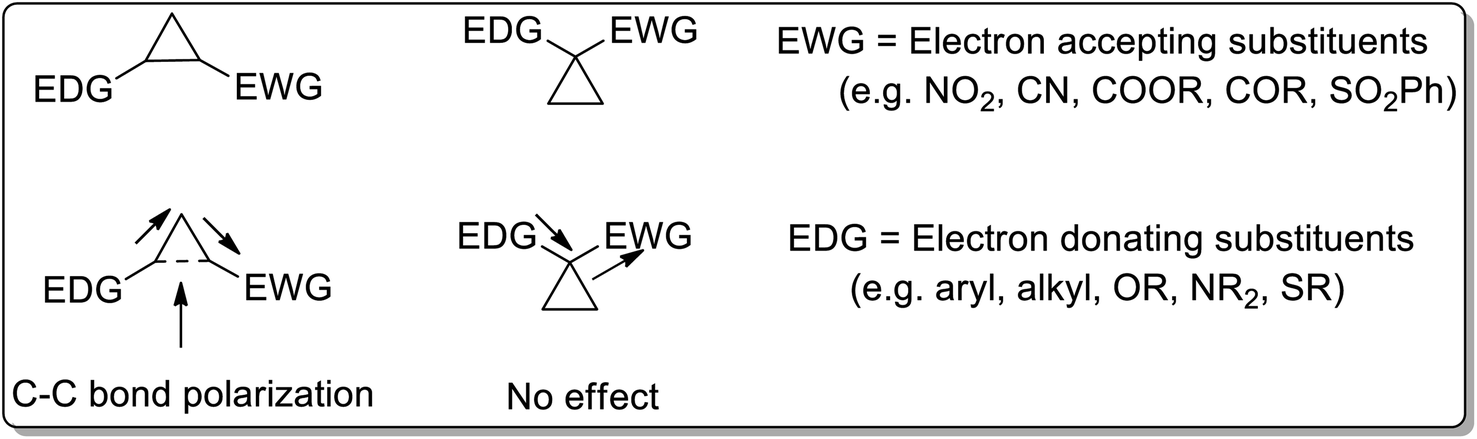
Cyclopropanes are the center of attraction in organic chemistry because of their high π character, intrinsic torsional, and angle strain, which enable them to undergo ring-opening reactions to attain several heterocyclic organic compounds.3 Cyclopropanes are relatively stable and chemically inert towards C–C bond cleavage unless activated.4a The cyclopropane ring gets activated by attaching electron-donating groups (D) and electron-withdrawing groups (A) on the ring. For the activation of the C–C bond, there are two ways of attaching electron-donating groups and electron-withdrawing groups: vicinal positioning and geminal positioning. The push and pull effect (as a result of attached donor and withdrawing substituents at the vicinal position) is responsible for induced polarization between two adjacent C–C atoms. Comparably, electron donating and withdrawing substituents at the geminal position have no effect on C–C bond polarization (Fig. 1).
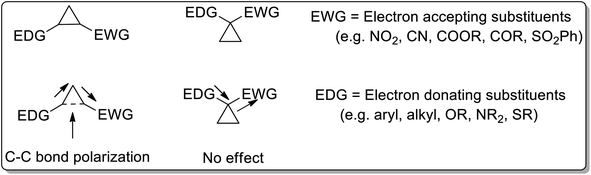 |
| | Fig. 1 Comparison of position of the attached substituents for the activation of the cyclopropyl ring. | |
Cyclopropane ring opening is promoted by induced polarization, which gives rise to the zwitterionic intermediate. Being highly reactive, the generated dipole demonstrates distinctive properties like electrophilic addition reactions, nucleophilic addition reactions, cycloaddition reactions, and rearrangement reactions.28
3. Biological importance of cyclopropanes and five-membered heterocycles
Cyclopropanes are highly reactive three-membered heterocyclic rings, which are structural constituents of various biologically active organic compounds and drugs. For example, tranylcypromine is a cyclopropyl constituting anti-anxiety drug, which is used to relieve stress.29 Similarly, (+)-curacin A,30, (+)-ambruticin S,31 andgrenadamide32 are some of cyclopropyl-based medicinally important naturally occurring compounds that exhibit anti-cancer, anti-fungal, and cannabinoid receptor-binding activities, respectively. The five-membered heterocycles (obtained as a result of Cloke–Wilson rearrangement) are also of significant importance in medicinal chemistry.33a For example, suvorexant is an oxazole-based drug (oxygen containing five-membered heterocycle), which is used to treat sleep disorder.33b A nitrogen-containing five-membered heterocycle, i.e., imidazole, has been found to be active against fungus, bacterial, viral, and cancer cell lines.34a Similarly, purine derivatives have also been found to exhibit cytotoxic potential.34b Moreover, thiophene and its derivatives are sulfur-containing heterocycles that have found applications as anti-bacterial, anti-viral, anti-inflammatory, and anti-cancer agents.35
4. Chronological discovery of cyclopropanes and their heteroatom variants
The chronological discovery of cyclopropanes and their varied heteroatomic rearrangement is of great significance. In 1929, Cloke8a reported the synthesis of 2-phenylpyrroline hydrochloride by cyclopropylimine–pyrroline rearrangement via the vacuum distillation of phenyl cyclopropyl ketimine hydrochloride (Table 1, entry 1). In 1947, Wilson7a reported the heat-catalyzed rearrangement of cyclopropylcarbaldehyde to yield dihydrofuran (Table 1, entry 2). In 1959, Neureiter10 reported the synthesis of dichlorocyclopentene by the rearrangement of dichlorovinylcyclopropane at more than 400 °C (Table 1, entry 3). After a year, in 1960, vinylcyclopropane was found to give cyclopentene via thermal rearrangement11 (Table 1, entry 4). Atkinson and Rees36 reported the thermal rearrangement of vinylaziridines to attain pyrrolines in 1967 (Table 1, entry 5). In 1971, Paladini and Chuche37 reported the synthesis of dihydrofurans via the rearrangement of vinyloxirane (Table 1, entry 6) and, after one year, Lwowski and co-workers38 reported this rearrangement by treating singlet and triplet nitrenes with dienes.
Table 1 Chronological discovery of cyclopropane and its heteroatom variants rearrangement
| Entry |
Main conversion |
Scientist |
| 1 |
 |
Cloke8a |
| 2 |
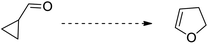 |
Wilson7a |
| 3 |
 |
Neureiter10 |
| 4 |
 |
Vogel, Overberger and Borchert11 |
| 5 |
 |
Atkinson, Rees36 and Lwowski38 |
| 6 |
 |
Paladini and Chuche37 |
5. Cloke–Wilson rearrangement
The highly strained cyclopropyl ketones, cyclopropylimines, and cyclopropyl thioketones are processed via Cloke–Wilson rearrangement to synthesize stable functionalized five-membered heterocycles. This rearrangement proceeds without the addition of an external nucleophile by involving the ring-opening/closing reaction.9,28,39 In 1929, Cloke8a treated cyclopropyl substituted ketones with NH4Cl by applying heat to attain dihydropyrroles. In 1947, a similar kind of transformation was reported by Wilson7a to synthesize dihydrofuran from cyclopropyl carboxaldehyde in the presence of heat. Owing to the requirement of high temperature (200–500 °C) for this strategy, its scope and applications are limited.40 However, many activation methods have been developed including transition metal catalysis,8b,41 photocatalysis,40d,42 cyclopropylcarbaldehyde-promoted activation,43 organocatalysis,44 Lewis acid catalysis,25,54–65 Brønsted acid catalysis,66–74,89,90,93 and Brønsted base catalysis68 to carry out this transformation. Cloke–Wilson rearrangement also finds applications in the total synthesis of natural products.21,25,91–94 Thus, the construction of five-membered O-, N-, and S-heterocycles can be easily achieved via Cloke–Wilson rearrangement.
6. Synthesis of O-heterocycles
6.1. Thermal rearrangement
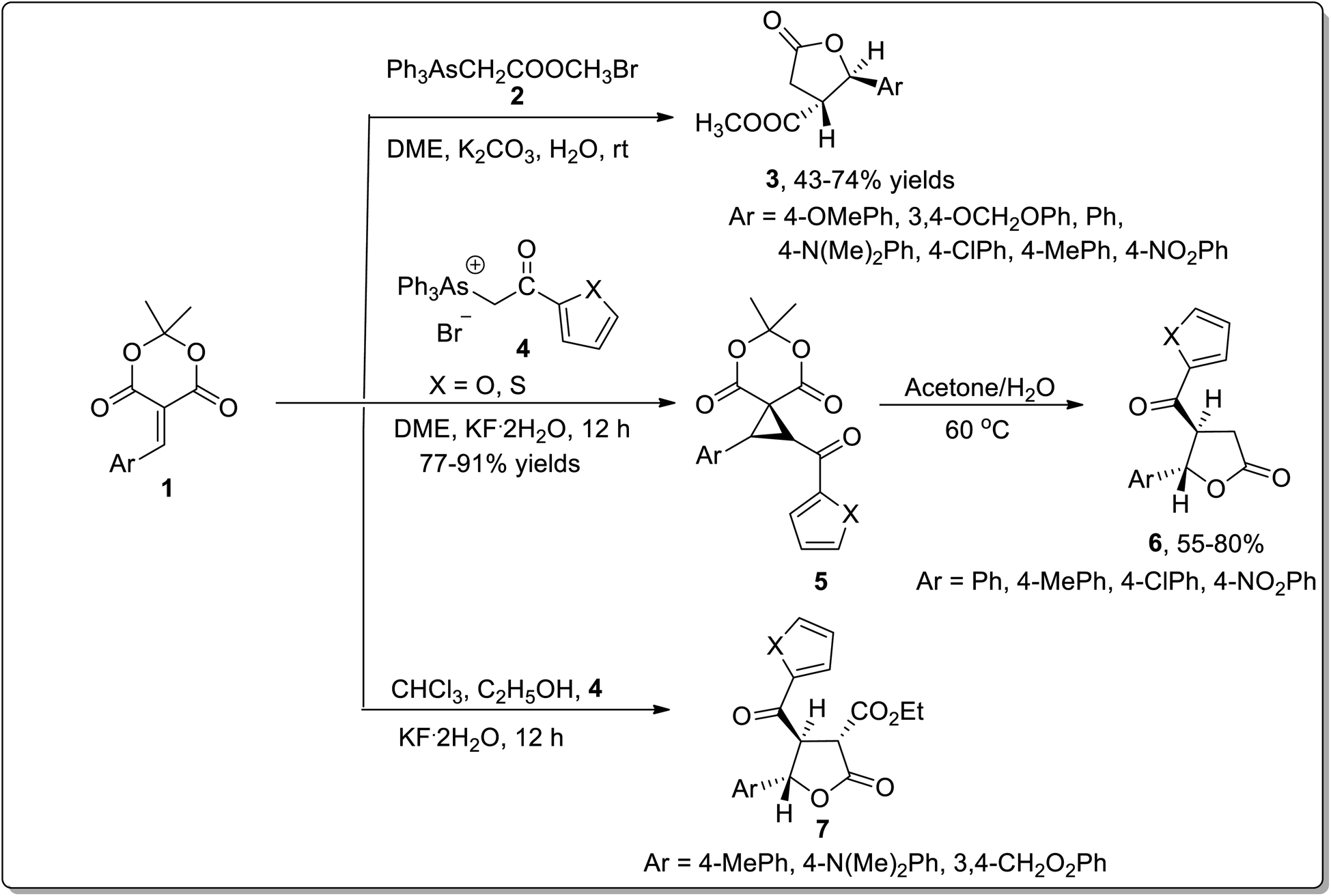
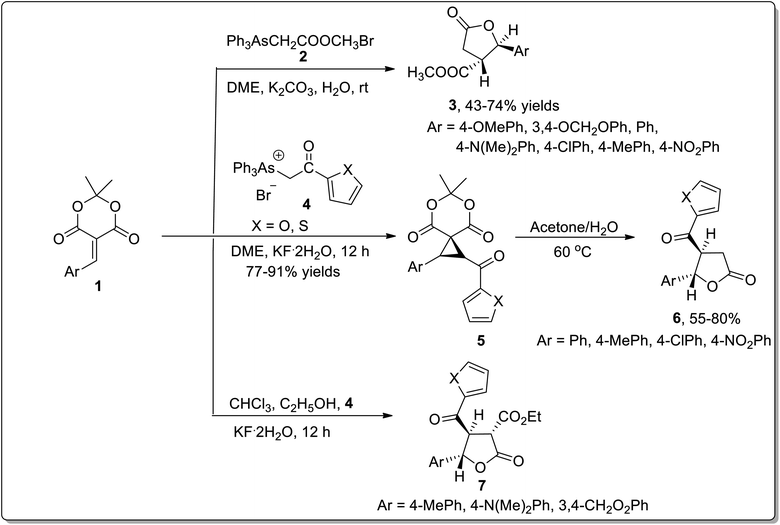
The wide significance of γ-butyrolactones has attracted great interest towards their synthesis. In 2002, Chen45a et al. carried out one-pot reaction between arsonium ylides 2 and substituted electron-deficient olefins 1 to synthesize substituted cyclopropanes or β,γ-disubstituted-γ-butyrolactones 3. In 2008, Wu45b et al. further utilized this rearrangement by reacting arsonium ylides 4 having furoyl or thienoyl functional group with substituted olefins 1 for the synthesis of β,γ-disubstituted butyrolactones 6 and α,β,γ-trisubstituted butyrolactones 7. Elecctron-donating groups bearing olefins 1 (on aryl ring) gave β,γ-disubstituted γ-butyrolactones 6 in good yields via one-pot reaction in moderate reaction conditions. However, a similar reaction resulted in the formation of spirocyclopropyl Meldrum's acids 5 by employing weak electron-donating or electron-withdrawing groups bearing olefins 1. These acids were further transformed to β,γ-disubstituted butyrolactones 6 in the presence of acetone and water at 60 °C. Electron-donating substituents bearing olefins also led to the synthesis of α,β,γ-trisubstituted butyrolactones 7 using chloroform as a solvent in the presence of ethanol. Triphenylarsine was obtained as a by-product and can be reused (Scheme 1).45
 |
| | Scheme 1 Synthesis of substituted butyrolactones 3, 6, and 7 via Cloke–Wilson rearrangement. | |
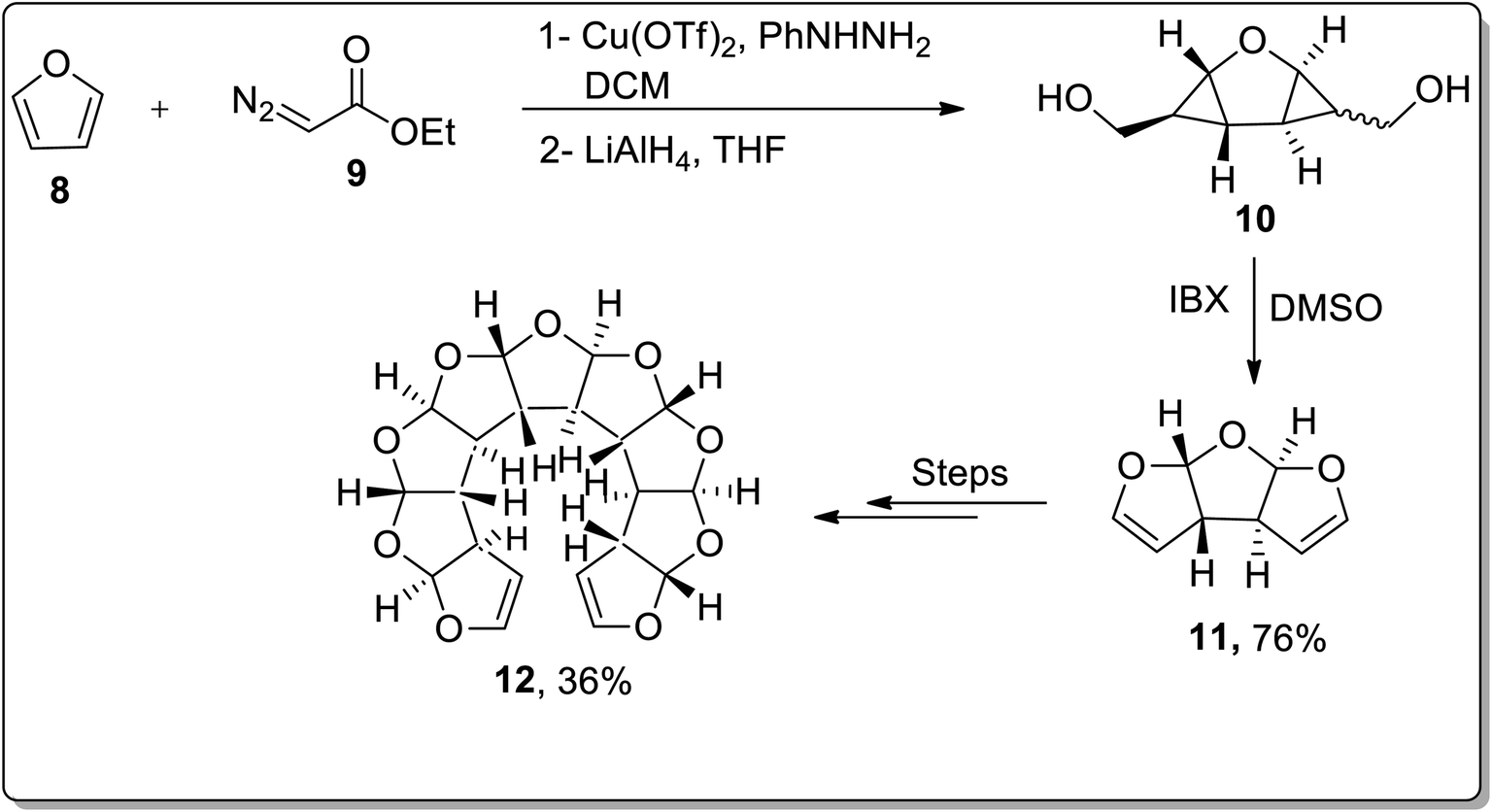
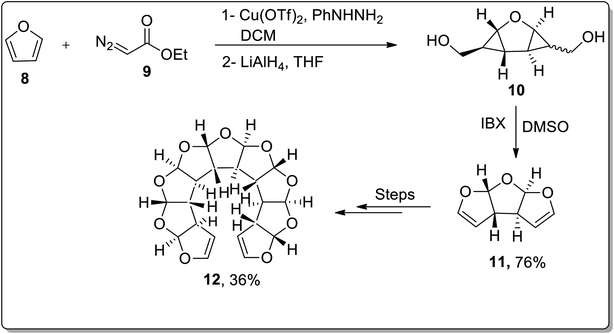
In 2009, the synthesis of anti-fused oligoannelated tetrahydrofuran moieties was carried out by Werz and coworkers from in situ-generated donor–acceptor cyclopropanes as a result of the cyclization reaction. The cyclization was initiated from the Cu(OTf)2-catalyzed double cyclopropanation of furan 8 with ethyl diazoacetate 9 to give a tricyclic bis-cyclopropane. The cyclopropanation reaction was followed by LiAlH4-mediated reduction, which resulted in diol 10. The diol moiety 10 was further subjected to oxidation in the presence of IBX to afford tricyclic bisacetal 11. In order to generate oligoacetal 12 up to a nonacyclic system, this three-step reaction array was repeated thrice (Scheme 2).46
 |
| | Scheme 2 Synthesis of oligoacetal 12 via Cloke–Wilson rearrangement. | |
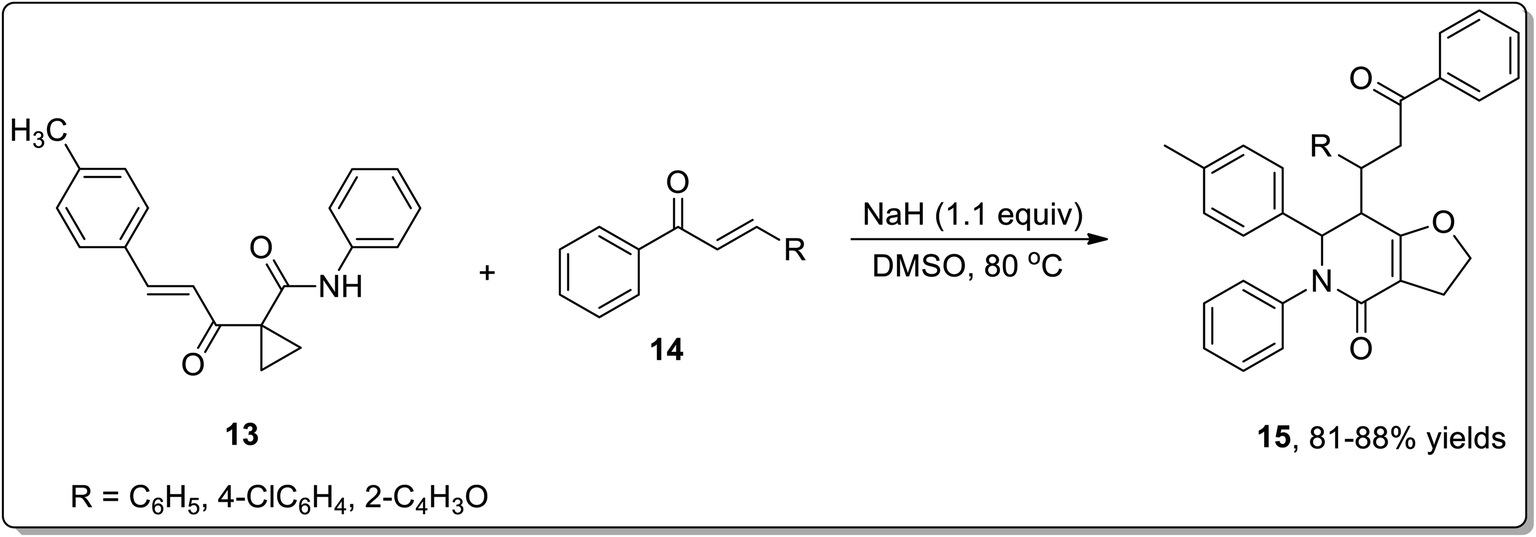
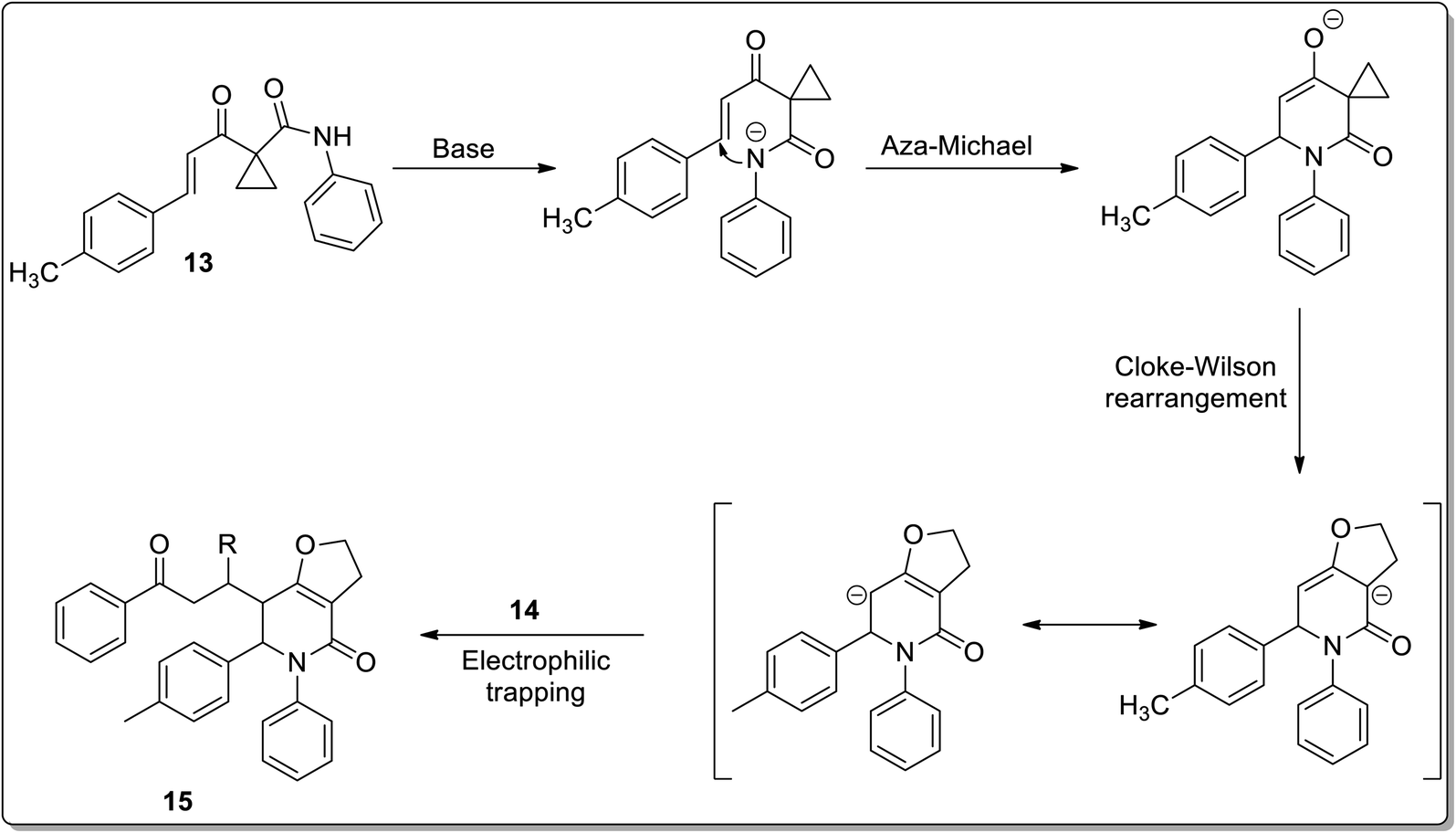
In 2011, Liang and co-workers reported the synthesis of furo[3,2-c]pyridinones 15 by the reaction of 1-cinnamoylcyclopropanecarboxamides 13 with various electrophiles 14 by an aza-oxy-carbanion relay via cascade the aza-Michael addition/Cloke–Wilson rearrangement/transfer of the carbanion and electrophile-confined reactions. This novel and facile synthetic strategy proceeded via non-Brook rearrangement pathway for the synthesis of medicinally important furo[3,2-c]pyridinones 15 in high yields (Schemes 3 and 4).47
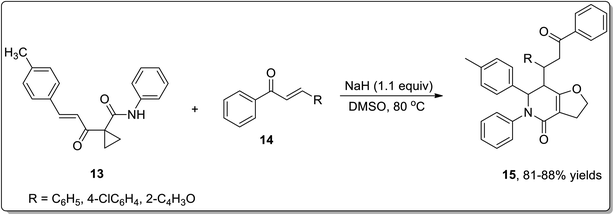 |
| | Scheme 3 Synthesis of furo[3,2-c]pyridinones 15 via Cloke–Wilson rearrangement. | |
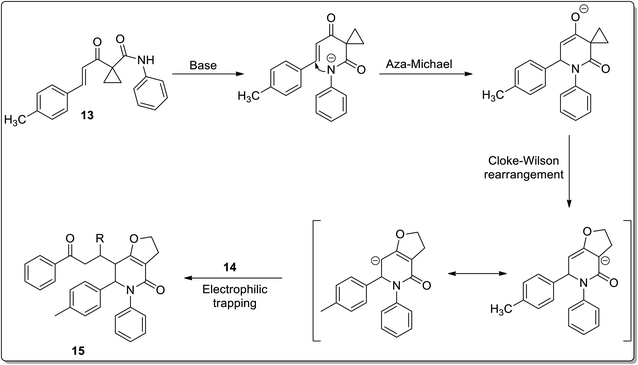 |
| | Scheme 4 Mechanism for the synthesis of furo[3,2-c]pyridinones 15 via Cloke–Wilson rearrangement. | |
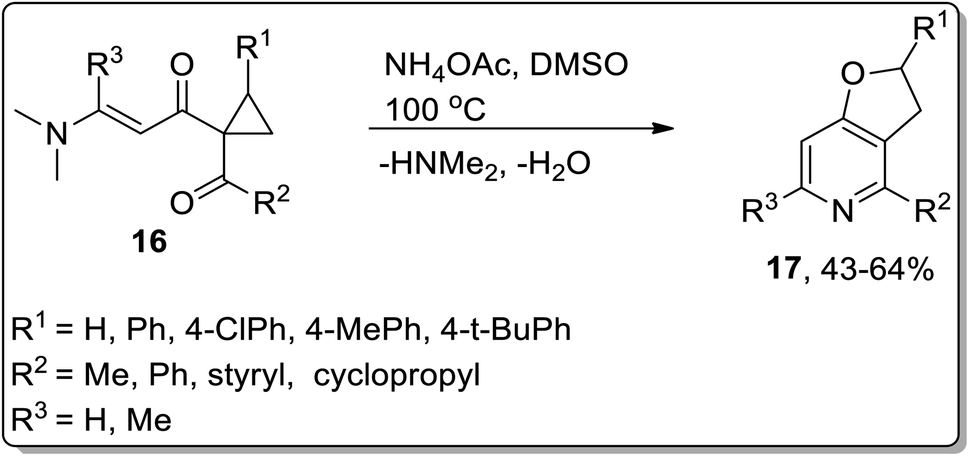
The synthesis of 2,3-dihydrofuro[3,2-c]pyridines 17 (in moderate yields) by employing Cloke–Wilson rearrangement was reported by Dong and coworkers in 2012. The synthesis of target molecules was achieved by treating 1-acyl-1-[(dimethylamino)alkenoyl]cyclopropanes 16 as the electrophilic source, which proceeded via one-pot annulation and ring-expansion reaction. The mechanism of their one-pot synthesis involved the intermolecular addition of ammonia, regioselective thermal Cloke–Wilson rearrangement of cyclopropylketone, intramolecular aza-nucleophilic addition/elimination, and dehydration reaction (Scheme 5).48
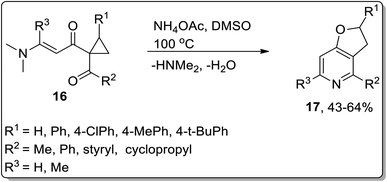 |
| | Scheme 5 Synthesis of 2,3-dihydrofuro[3,2-c]pyridines 17 via Cloke–Wilson rearrangement. | |
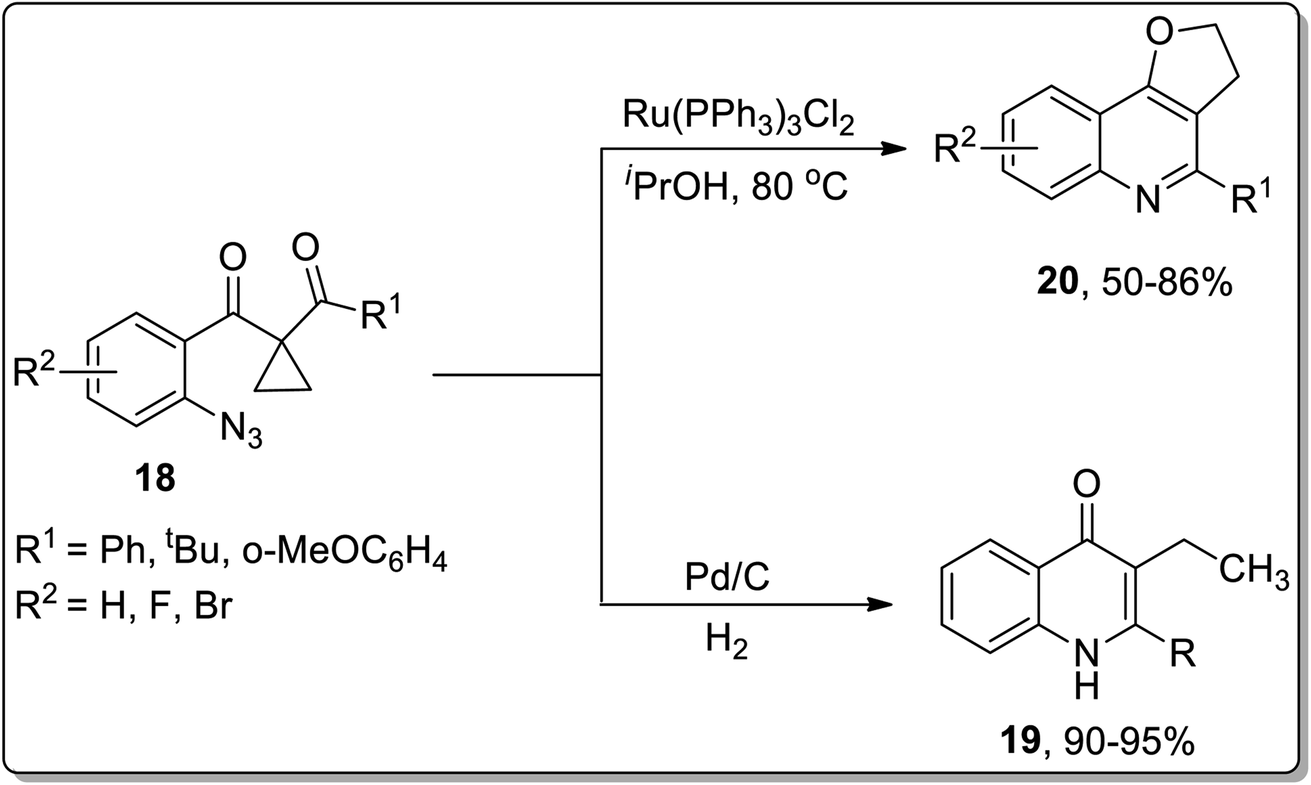
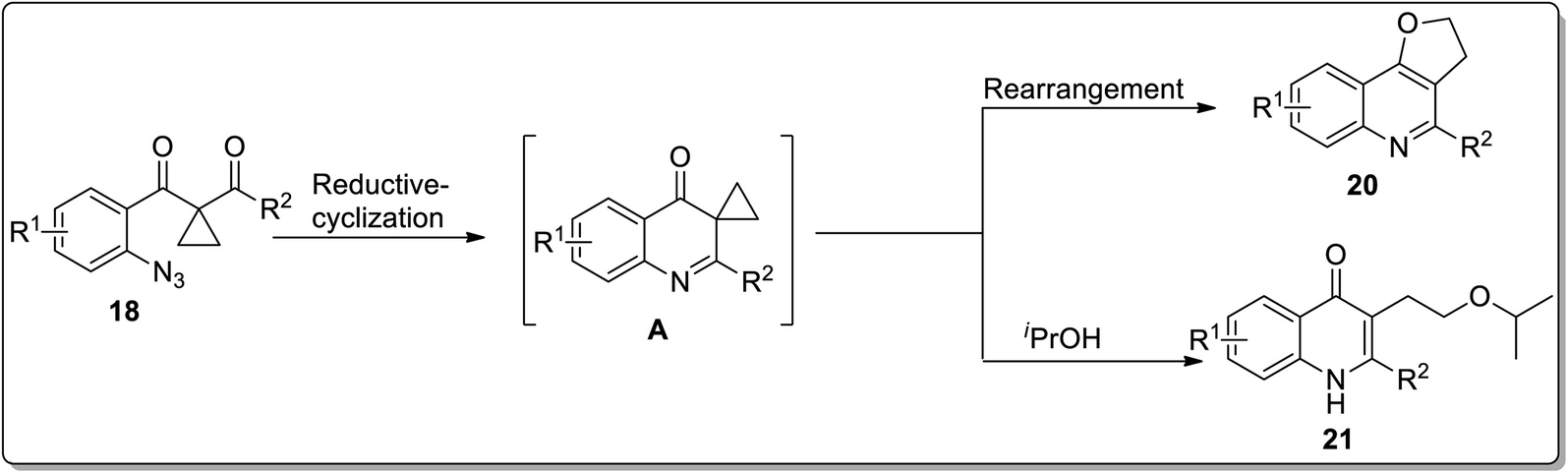
In 2013, the synthesis of substituted dihydrofuroquinolines 20 was established by Ren and co-workers, which proceeded by treating azido-cyclopropyl ketones 18 with a reducing agent, followed by cyclization and rearrangement. The Ru-catalyzed regioselective transformation commenced with the reduction of the azide group upon heating in the presence of the Ru-catalyst in iPrOH to an amine, which was later condensed with a ketone to generate intermediate A. Intermediate A further resulted in the synthesis of dihydrofuroquinolines 20 by Cloke–Wilson rearrangement, while ring-expansion, that took place as a result of the attack of solvent iPrOH on the intermediate A, led to the generation of by-product 21 (Scheme 6).49 Meanwhile, cyclopropyl-substituted scaffolds 18 afforded quinolone derivatives 19 by carrying out the reaction in the presence of hydrogen atmosphere and palladium (supported on charcoal) (Scheme 7).
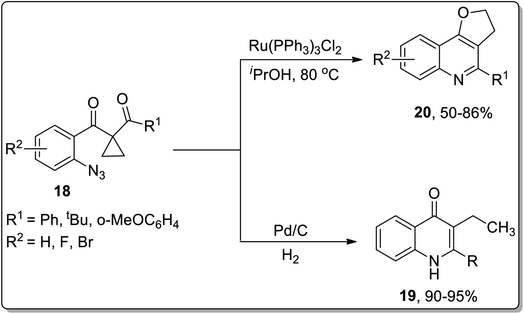 |
| | Scheme 6 Synthesis of dihydrofuroquinolines 20 and quinoline derivatives 19 via Cloke–Wilson rearrangement. | |
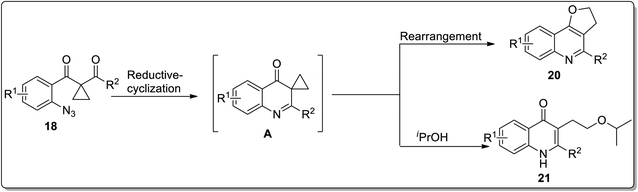 |
| | Scheme 7 Mechanism for the synthesis of dihydrofuroquinolines 20 via Cloke–Wilson rearrangement. | |
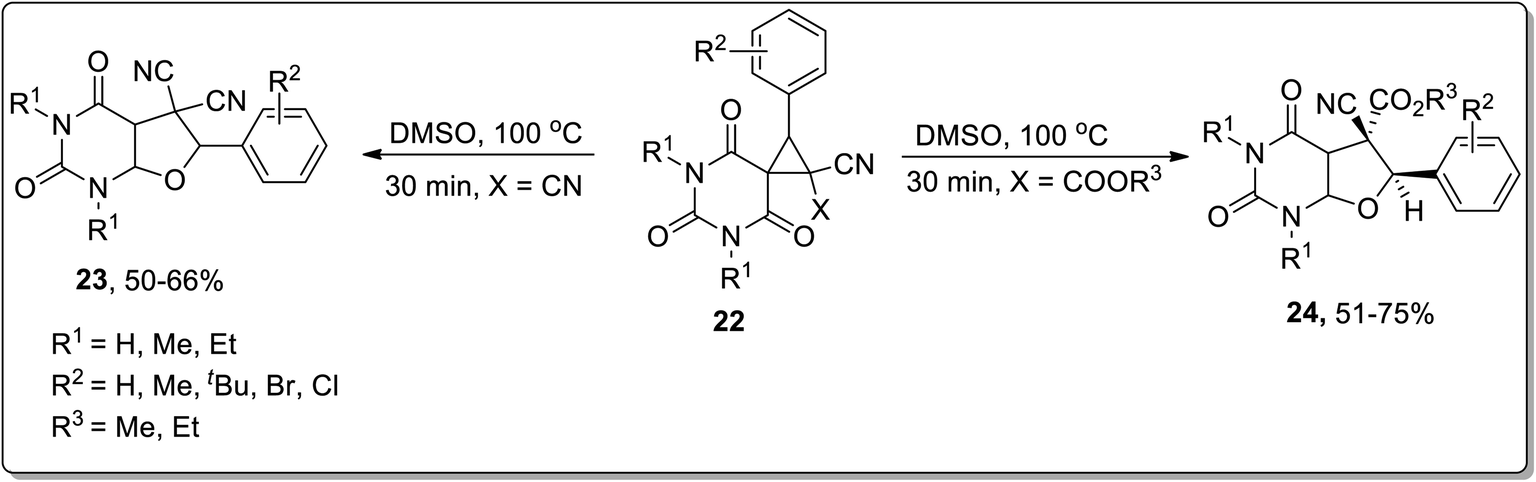
In 2015, Vereshchagin and co-workers carried out thermal Cloke–Wilson rearrangement to synthesize (medicinally essential) substituted furo[2,3-d]pyrimidines 23 and 24 in 50–75% overall yields employing spirocyclic barbiturates 22 as precursors in DMSO at 100 °C. The products were obtained directly from the reaction mixture by water-assisted precipitation without requiring any additional catalyst (Scheme 8).50
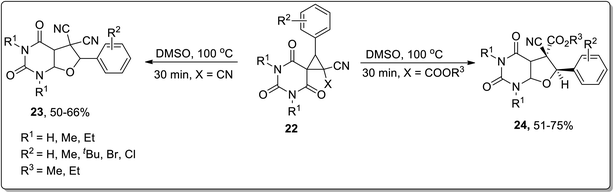 |
| | Scheme 8 Synthesis of substituted furo[2,3-d]pyrimidines 23 and 24 via Cloke–Wilson rearrangement. | |
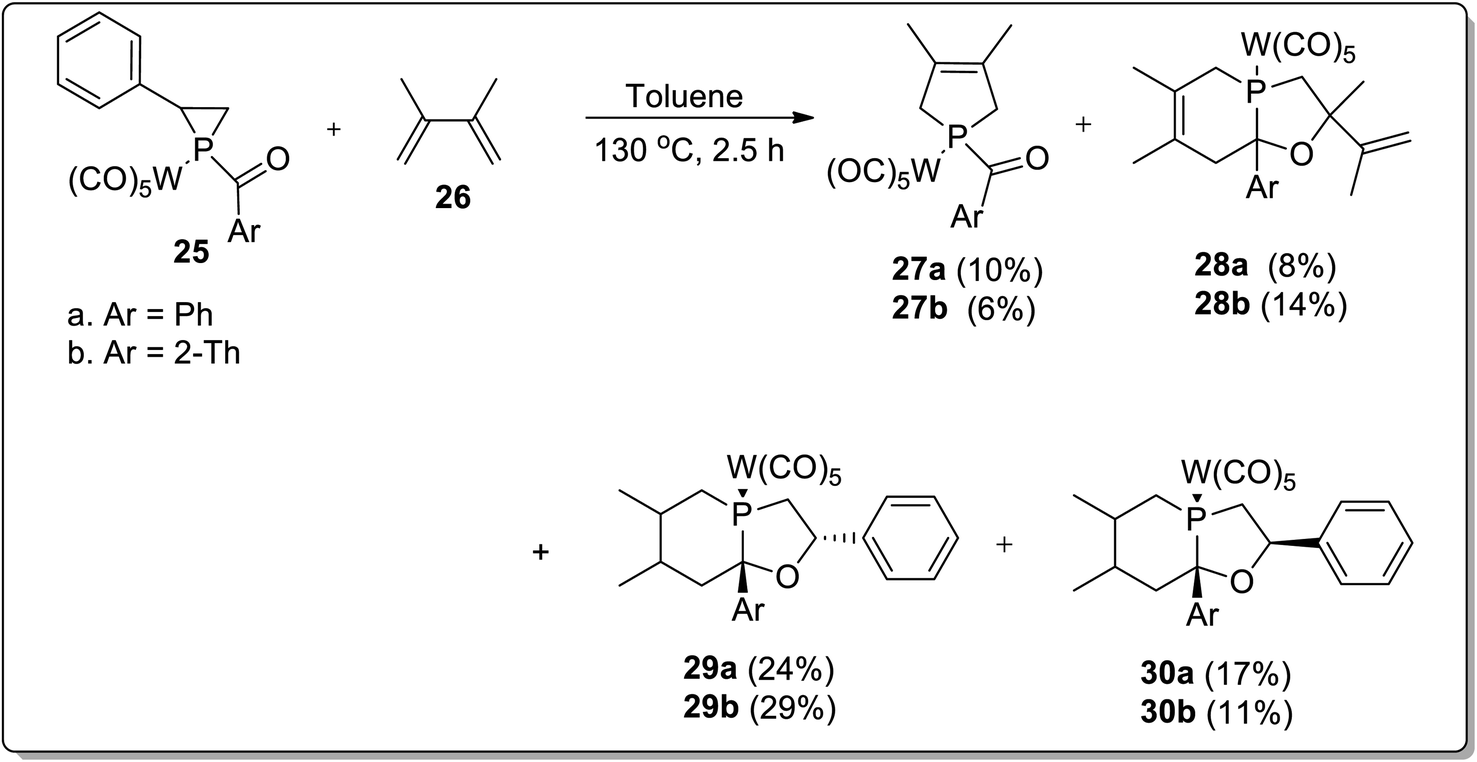
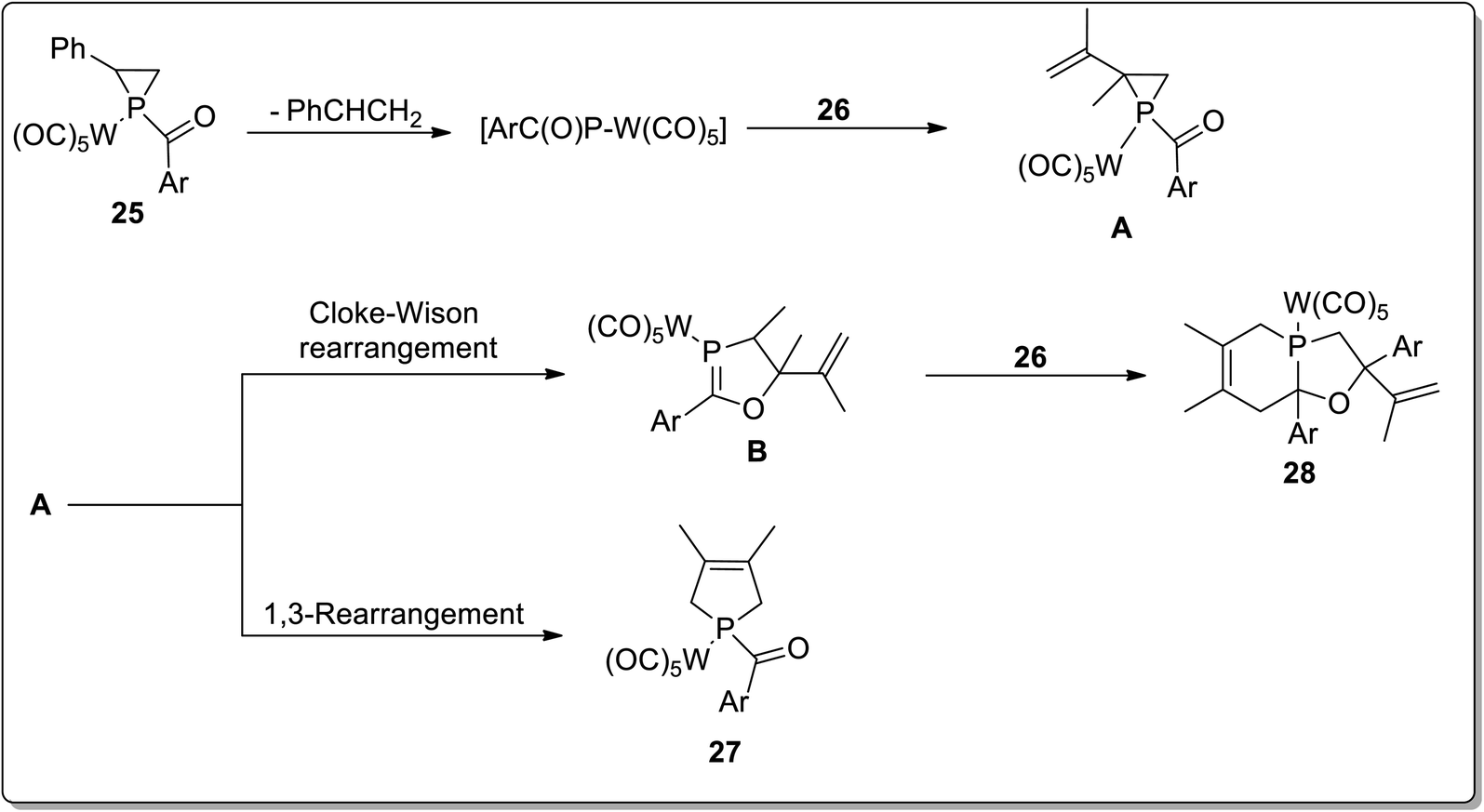
In 2017, Duan and co-workers51 reported the thermolysis of tungsten pentacarbonyl-substituted 1-acylphosphirane complexes 25 and 2,3-dimethylbutadiene 26 at 130 °C in toluene, which generated different complexes (27–30) via Cloke–Wilson rearrangement. Complexes 27 and 28 were synthesized by the transformation of the phosphirane ring to the acylphosphinidene complex, which was further made to react with 2,3-dimethylbutadiene to result in a vinylphosphirane intermediate A. The vinylphosphirane intermediate A was subjected to [1,3] rearrangement to give phospholene 27; meanwhile, it was also converted to oxaphospholene B via Cloke–Wilson rearrangement. Oxaphospholene B was later treated with 2,3-dimethylbutadiene 26 to afford complex 28 (Schemes 9 and 10).
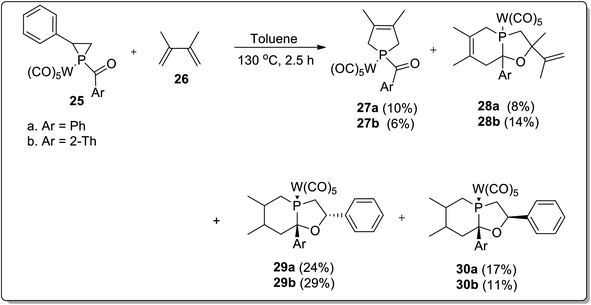 |
| | Scheme 9 Synthesis of different complexes 27–30 via Cloke–Wilson rearrangement. | |
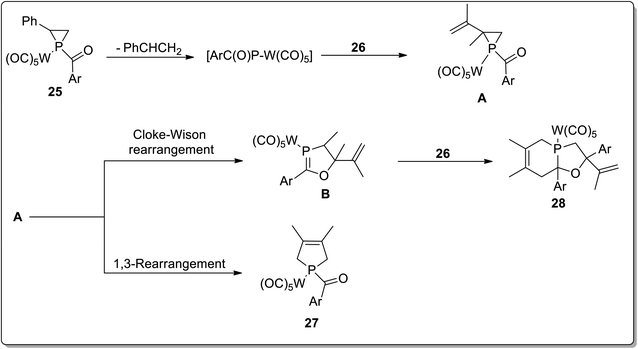 |
| | Scheme 10 Synthesis of complexes 27 and 28 via Cloke–Wilson rearrangement. | |
Furthermore, the tungsten pentacarbonyl-substituted 1-acylphosphirane complex 25 and 2,3-dimethylbutadiene 26 also gave two diastereomeric complexes 29 and 30 by undergoing Cloke–Wilson rearrangement (Scheme 11).51
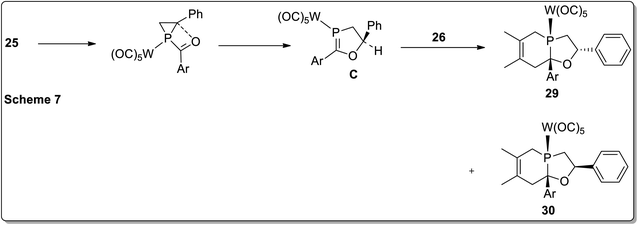 |
| | Scheme 11 Mechanism for the synthesis of complexes 29 and 30 via Cloke–Wilson rearrangement. | |
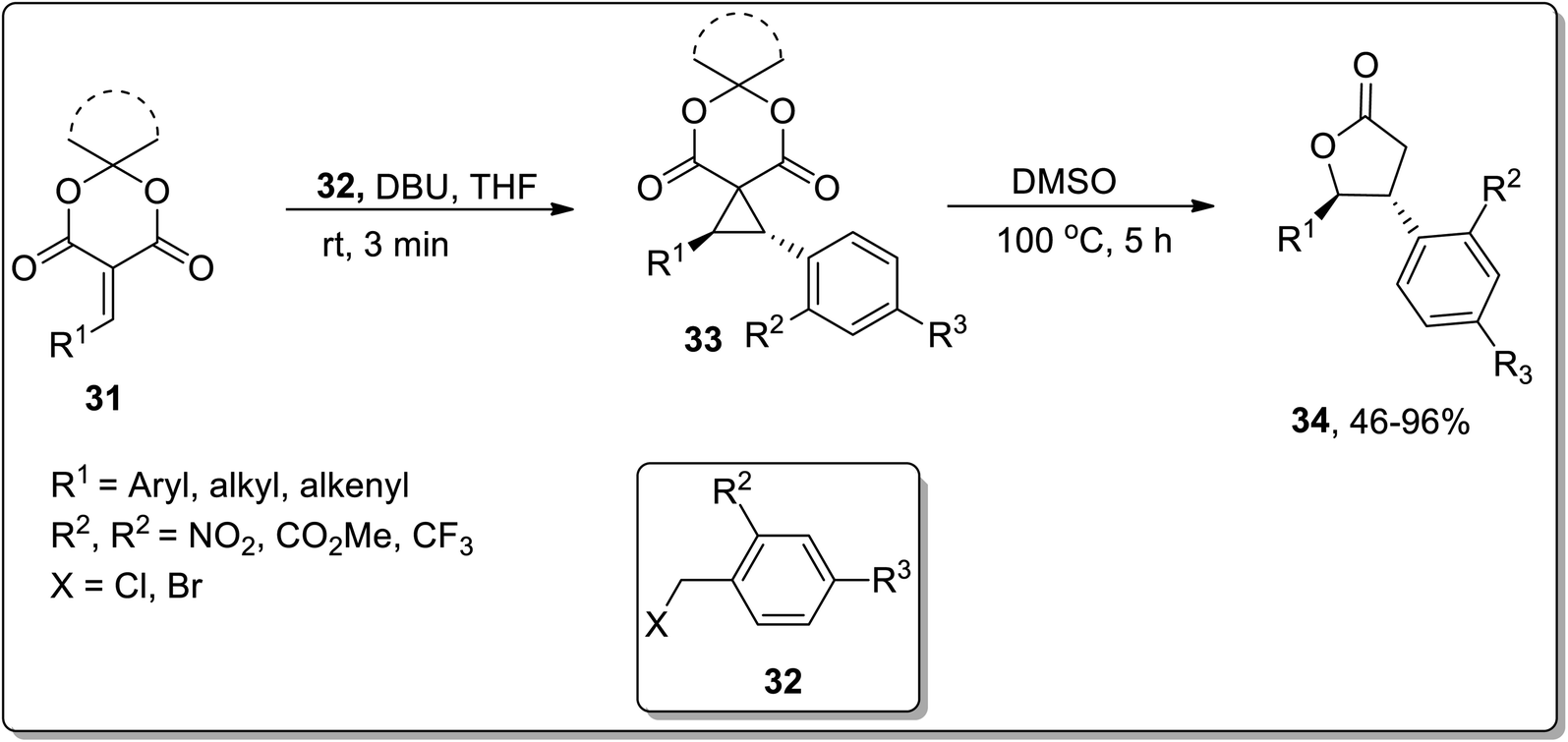
In 2020, Song and co-workers designed the synthesis of trans-β,γ-disubstituted-γ-butyrolactones 34 via an array of stereocontrolled spirocyclopropanation/Cloke–Wilson ring expansion reactions. The first step of this protocol involved the DBU-promoted stereoselective spirocyclopropanation of alkylidene meldrum's acids 31 with benzyl halides 32 at room temperature in THF to generate the transisomeric spirocyclopropyl meldrum's acids 33, followed by DMSO-promoted stereocontrolled thermal decarboxylative Cloke–Wilson ring expansion to result in trans-β,γ-disubstituted-γ-butyrolactones 34 in moderate to good yields (46–96%) with excellent diastereoselectivities. These sequential reactions were tolerated by employing various aromatic and aliphatic olefins and diversely substituted benzyl halides (Scheme 12).52
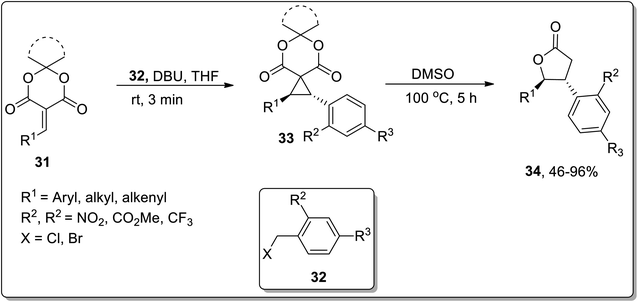 |
| | Scheme 12 Synthesis of substituted trans-β,γ-disubstituted-γ-butyrolactones 34 via Cloke–Wilson rearrangement. | |
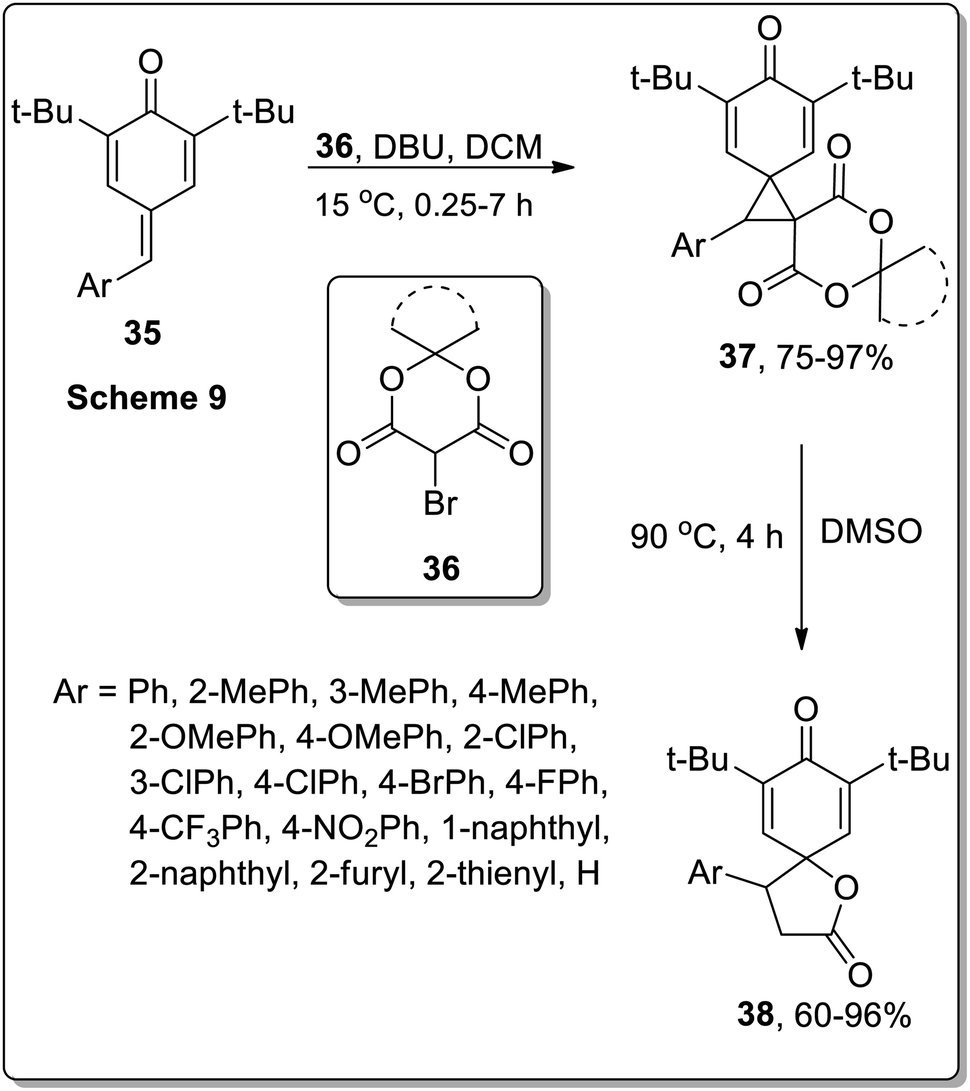
A similar strategy was adopted by Song and co-workers to synthesize spirobutyrolactone para-dienones 38 via the thermal decarboxylative Cloke–Wilson rearrangement of dispirocyclopropanes 37. The synthesis initiated by the spirocyclopropanation of opara-quinone methides 35 with bromo-Meldrum's acids 36, that generated dispirocyclopropanes 37 in good yields (75–97%). The resulting dispirocyclopropanes 37 on subsequent DMSO-mediated thermal decarboxylative Cloke–Wilson rearrangement gave spirobutyrolactones 38 in moderate to good yields (60–96%). These sequential reactions were tolerated by a wide range of substrates. The applicability of these sequential reactions was then tested by gram-scale synthesis under the optimized conditions. The resulting spirobutyrolactones yields were obtained in equivalent yields to the sub-millimole-scale reactions (Scheme 13).53
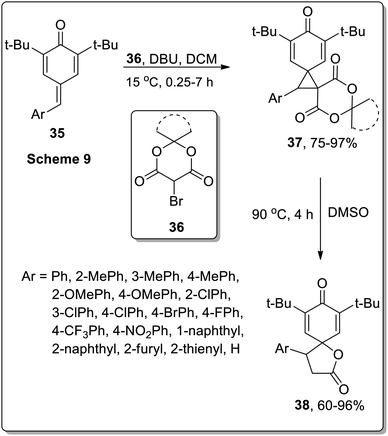 |
| | Scheme 13 Synthesis of spirobutyrolactones 38 via Cloke–Wilson rearrangement. | |
6.2. Photo-catalytic rearrangement
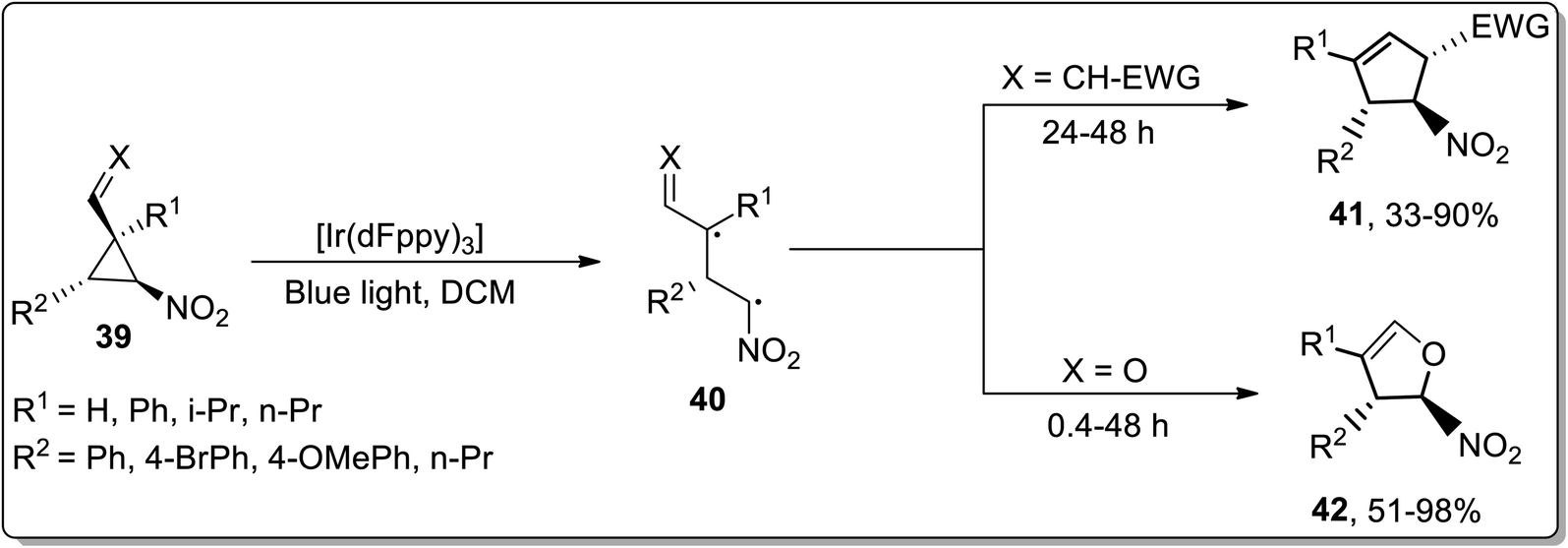
Owing to their applications towards the formation of unusual bonds, photocatalytic reactions have gained remarkable importance. However, these reactions had been rarely employed for the ring-expansion reactions of cyclopropanes. In 2017, Aleman and co-workers utilized a visible-light photocatalyst [Ir(dFppy)3] for the synthesis of enantioenriched dihydrofurans 42 and cyclopentenes 41 via an intramolecular ring expansion of cyclopropane 39. In the presence of heteroatoms such as X = O, the products were obtained as furans derivatives, while cyclopentene derivatives were obtained by treating electron-withdrawing groups bearing cyclopropanes. This methodological development is of great productivity due to the efficient synthesis of products (in good to excellent yields) with excellent enantioselectivity (Scheme 14).42
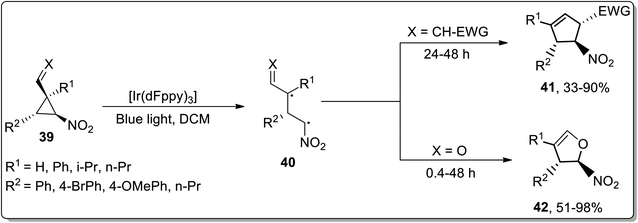 |
| | Scheme 14 Synthesis of dihydrofurans 41 and cyclopentenes 42 via Cloke–Wilson rearrangement. | |
In 2015, Plietker and co-workers demonstrated a base metal complex Bu4N[Fe(CO)3(NO)] (TBA[Fe])-catalyzed Cloke–Wilson rearrangement of substituted cyclopropanes under both thermal and photochemical conditions. The substituted vinyldihydrofuans 44 were obtained in good to excellent yields from the corresponding vinyl cyclopropanes 43. Similarly, the substituted aryldihydrofuans 46 were attained (in good to excellent yields) by reacting cyclopropanes 45 under both thermal (74–99%) and photochemical conditions (40–93%). The iron complex Bu4N[Fe(CO)3(NO)] (TBA[Fe]) showed good reactivity under both the conditions. This iron-catalyst Bu4N [Fe(CO)3(NO)] was observed to exhibit the best conversion at 415 nm (Schemes 15 and 16).40d
 |
| | Scheme 15 Synthesis of substituted vinyldihydrofuans 44 via Cloke–Wilson rearrangement. | |
 |
| | Scheme 16 Synthesis of substituted aryldihydrofuans 46 via Cloke–Wilson rearrangement. | |
6.3. Lewis acid-catalyzed/mediated rearrangement
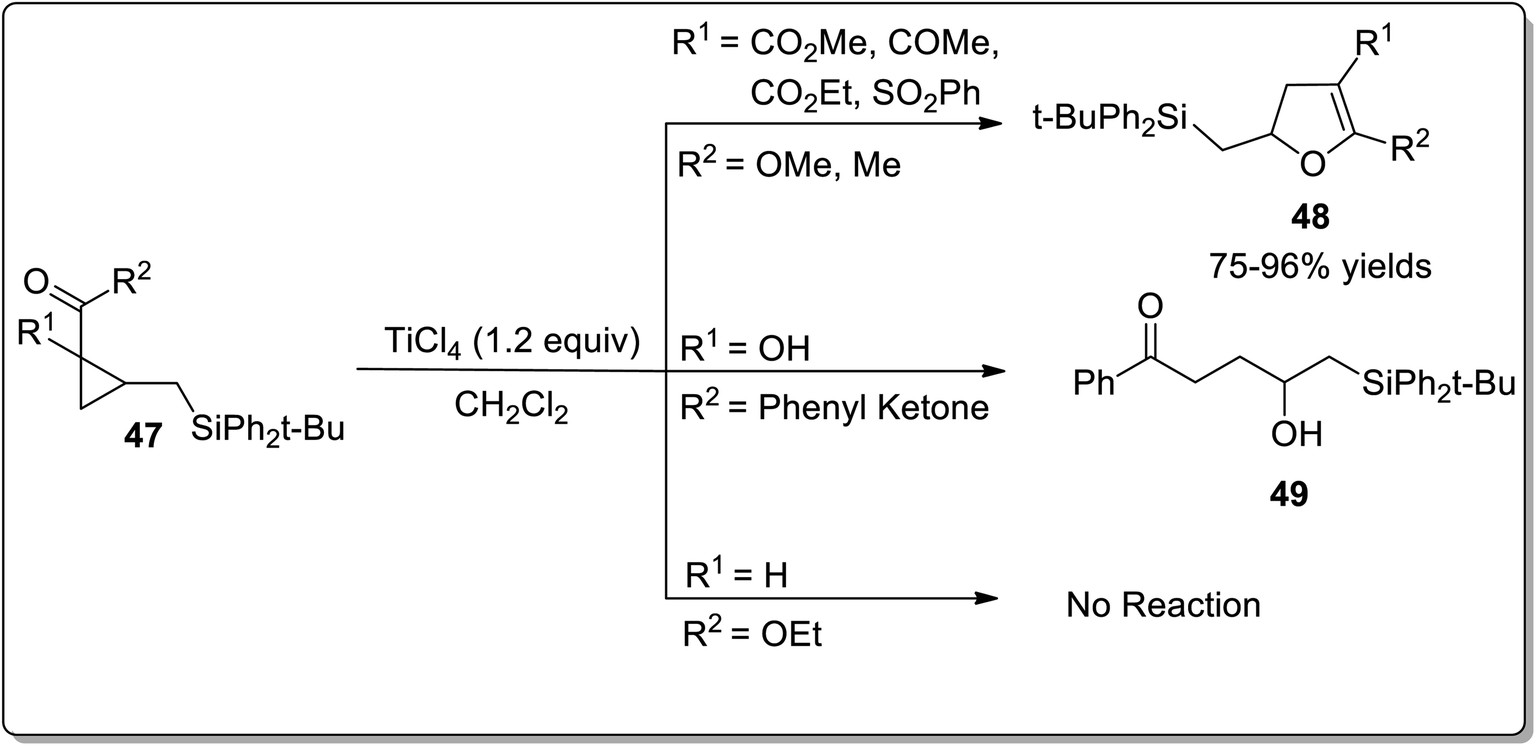
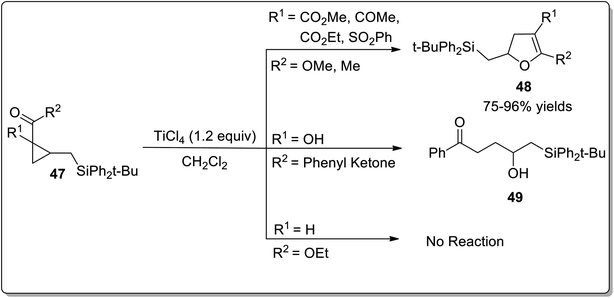
Lewis acids have gained undeniable significance as catalysts in various organic transformations. In 2001, Lewis acid, i.e., TiCl4-mediated Cloke–Wilson rearrangement leading to the synthesis of substituted dihydrofurans 48 was demonstrated by Yadav and Balamurugan in 2001. The synthesis took place by the reaction of electron-withdrawing groups bearing cyclopropanes 47 (acceptor) and (tert-butyldiphenylsilyl)methyl group (donor) with TiCl4 that split the C–C bond of cyclopropane to generate the substituted dihydrofurans 48 in good to excellent yields (75–96%). The cyclopropanes with two electron-withdrawing groups were transformed into dihydrofurans 48. It was observed that use of a single ester group did not proceed to target molecule synthesis due to the inadequate activation of the ring. However, the single phenyl ketone group underwent ring activation and ring-splitting to furnish product 49. In the product, the C–Si bond was retained for further functional group transformations (Scheme 17).43
 |
| | Scheme 17 Synthesis of substituted dihydrofurans 48 via Cloke–Wilson rearrangement. | |
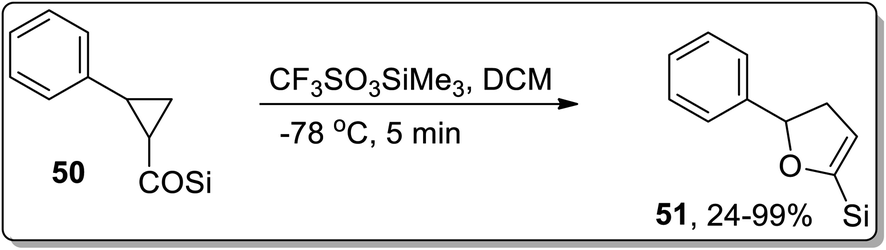
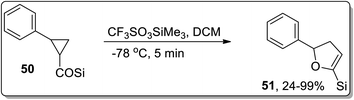
In 2005, Honda and coworkers designed the synthesis of 5-silyl-2,3-dihydrofuran derivatives 51 from cyclopropyl silyl ketones 50 via Lewis acid, i.e., trimethylsilyl trifluoromethanesulfonate (TMSOTf)-mediated Cloke–Wilson ring-expansion reaction. The resulting 5-silyl-2,3-dihydrofuran derivatives 51 possess dual active functionalities (cyclic enol ether and vinylsilane) for use in different organic reactions that involve treatment with electrophilic substrates and Heck reaction. 5-Silyl-2,3-dihydrofuran derivatives 51 were obtained in more than 99% yield (maximum yield) by employing phenyl-substituted cyclopropyl silyl ketones 50 (Scheme 18).54
 |
| | Scheme 18 Synthesis of 5-silyl-2,3-dihydrofuran derivatives 51 via Cloke–Wilson rearrangement. | |
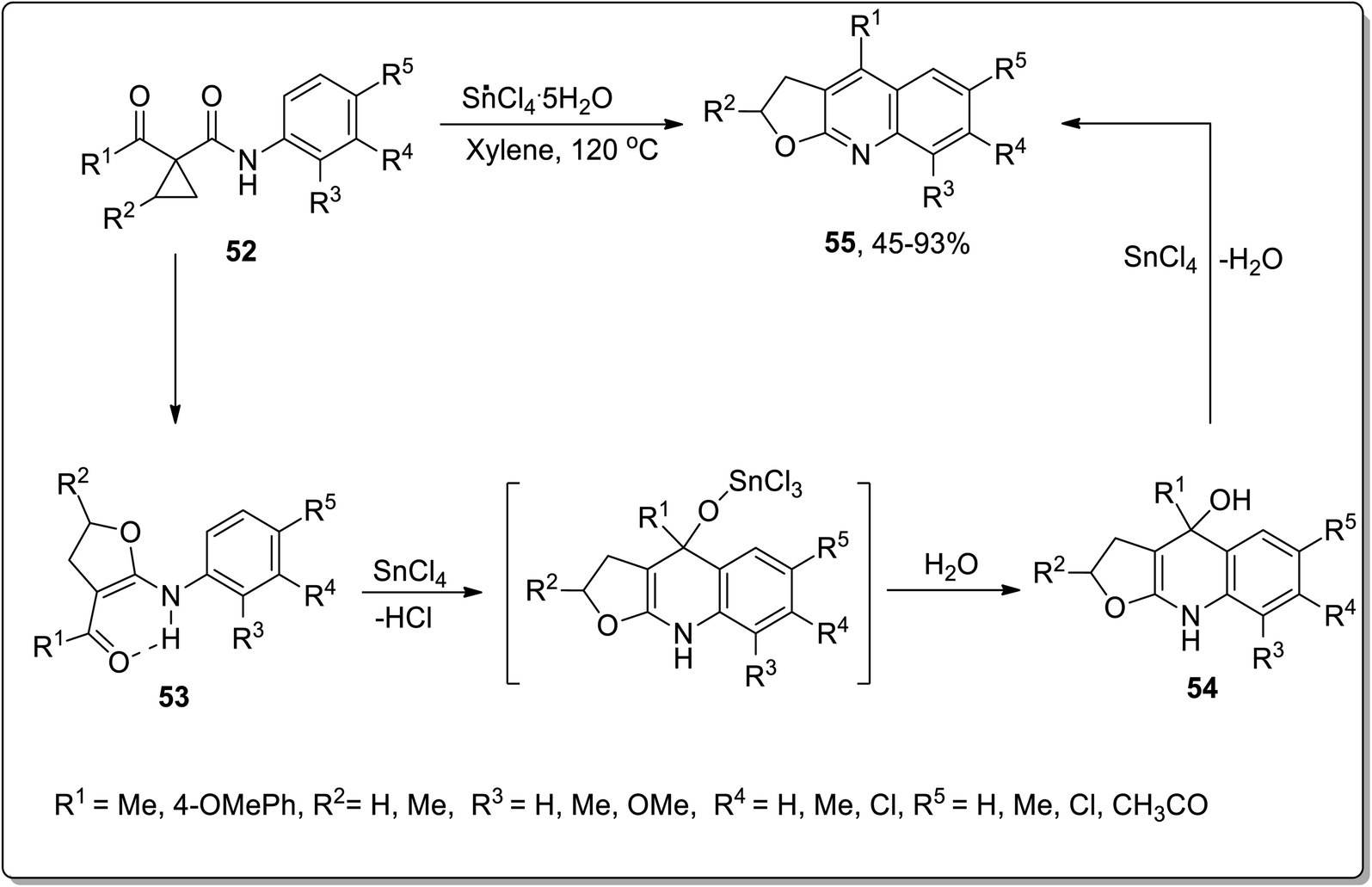
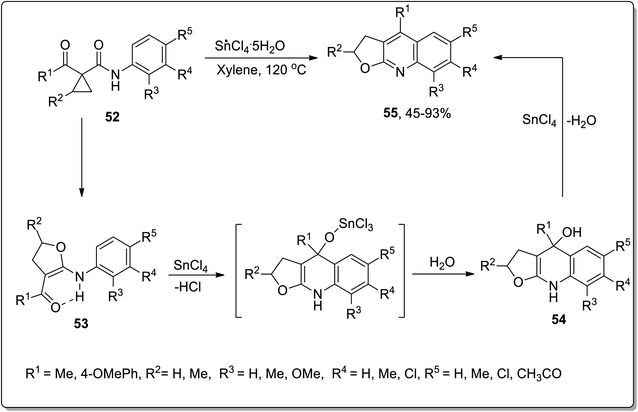
In 2007, Zhang and coworkers carried out the synthesis of furoquinoline derivatives 55 from highly-activated cyclopropanes 52, in which the dihydrofuran moiety was attained by the Lewis acid, i.e., SnCl4-mediated Cloke–Wilson ring-expansion of cyclopropyl ketone. The synthesis initiated with the ring opening, followed by its conversion to a new fused ring, that resulted in intermediate 54; then, subsequent Combes-type annulation with the elimination of water furnished the furoquinoline derivatives 55. Easily accessible reagents, broad substrate scope, high chemoselectivity and regioselectivity are the key features of this synthetic strategy (Scheme 19).55
 |
| | Scheme 19 Synthesis of furoquinoline derivatives 55 via Cloke–Wilson rearrangement. | |
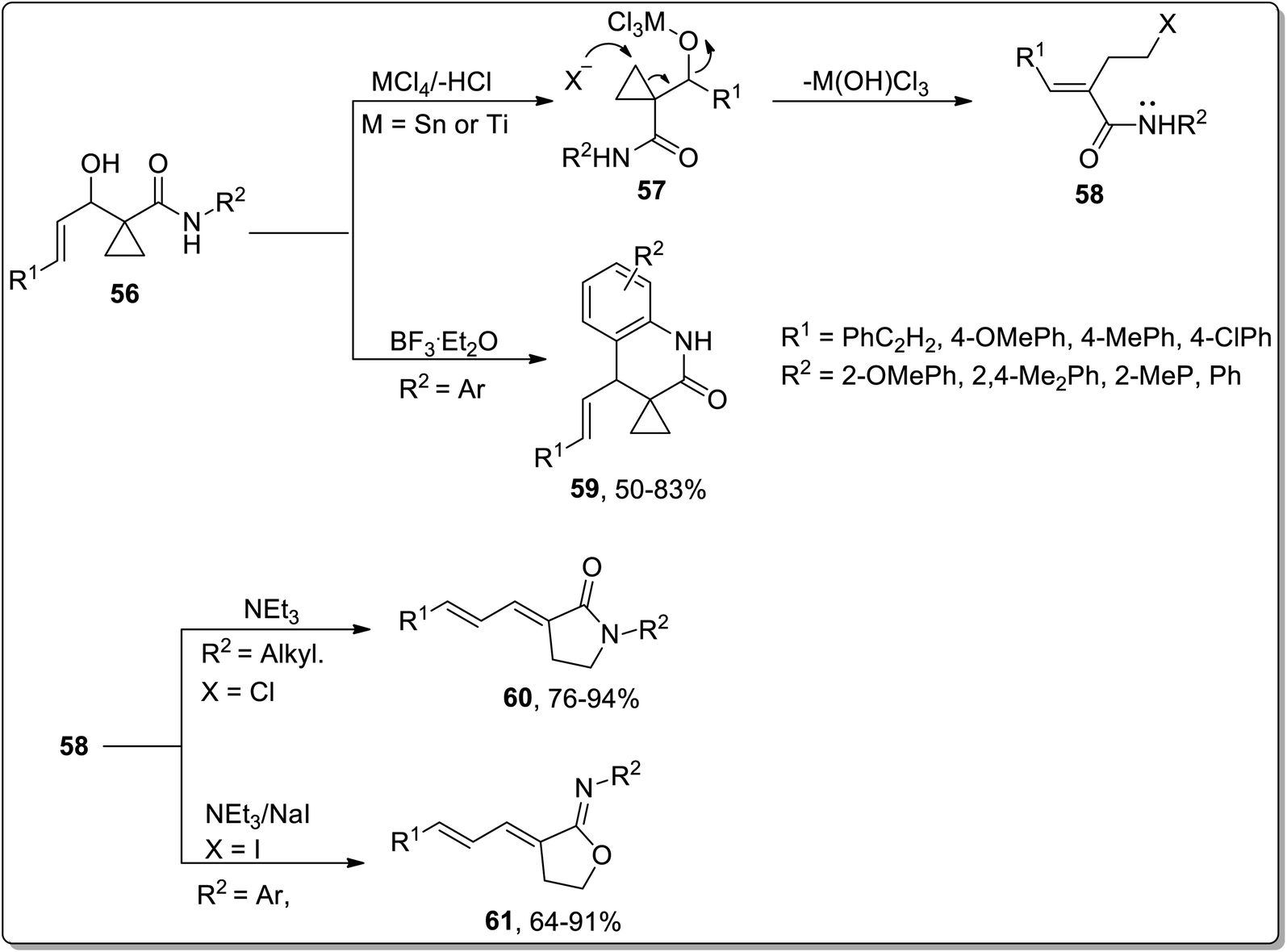
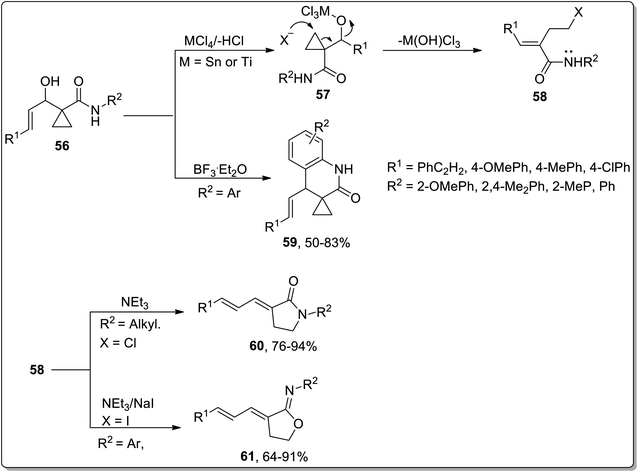
In 2007, Zhang and co-workers designed the synthesis of γ-lactams 59, γ-iminolactones 60, and dihydroquinolin-2-ones 61 from β-hydroxymethylcyclopropanylamides 56. β-Hydroxymethylcyclopropanylamides 56 underwent boron trifluoride diethyl etherate-mediated intramolecular Friedel–Crafts alkylation to produce dihydroquinolin-2-one 59 in 50–83% yields. On the other side, the Lewis acid, i.e., SnCl4/NaI or TiCl4-catalyzed reaction generated intermediate 58 via ring opening, which furnished γ-lactams 60 or γ-iminolactones 61 by the employment of either an intramolecular N-annulation or O-annulation. Bulky groups such as aryl group-bearing reagents preferably proceeded via O-annulation, while relatively small groups like methyl, benzyl, and n-propyl moved forward by N-annulation. Furthermore, N-annulation was carried out with great efficacy (even without sodium iodide) due to the more nucleophilic tendency of nitrogen in comparison to oxygen (Scheme 20).56
 |
| | Scheme 20 Synthesis of γ-lactams 59, γ-iminolactones 60, and dihydroquinolin-2-ones 61 via Cloke–Wilson rearrangement. | |
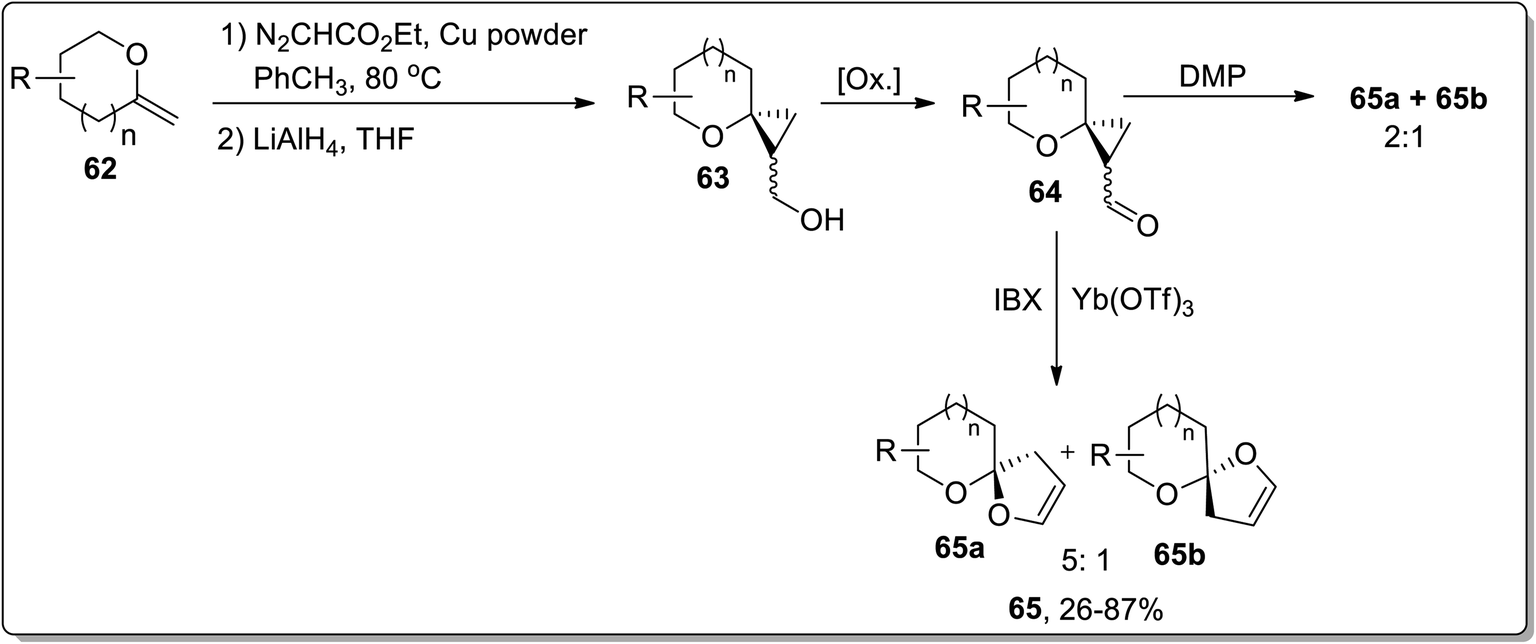
In 2009, Werz and co-workers demonstrated the synthesis of [n,5]-spiroketals (n = 5, 6) 65 from exocyclic enol ethers 62 by a three-step methodology: Cu-mediated cyclopropanation of exocyclic enol ethers 62 to spiroannelated cyclopropanes formation (bearing an ester group), the reduction of an ester group with LiAlH4 generated respective alcohols 63, subsequent IBX-oxidation of hydroxyl functionality along with the Lewis acid Yb(OTf)3-catalyzed Cloke–Wilson rearrangement of cyclopropyl ketone moiety 64 afforded [n,5]-spiroketals 65. The product investigation unveiled the instability of the configuration of the spirocenter, synthesized by utilizing only IBX or IBX/Yb(OTf)3-mediated ring expansion. During this transformation, the thermodynamically more stable spiroketal is favored. These results indicated that this reaction did not proceed through a concerted mechanism, while the main reaction pathway was supposed to advance forward through a zwitterionic intermediate formation, which would permit the erosion of stereochemistry at the spirocenter. On the other hand, the use of Dess–Martin periodinane (DMP) preserved the stereochemistry at the spirocenter (Scheme 21).57
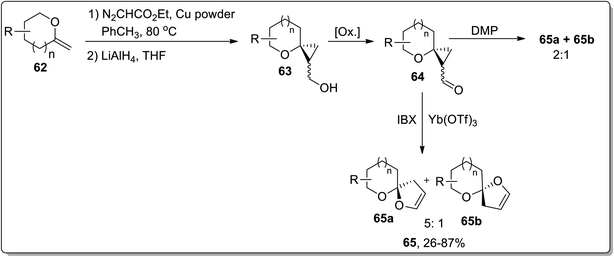 |
| | Scheme 21 Synthesis of [n,5]-spiroketals (n = 5, 6) 65 via Cloke–Wilson rearrangement. | |
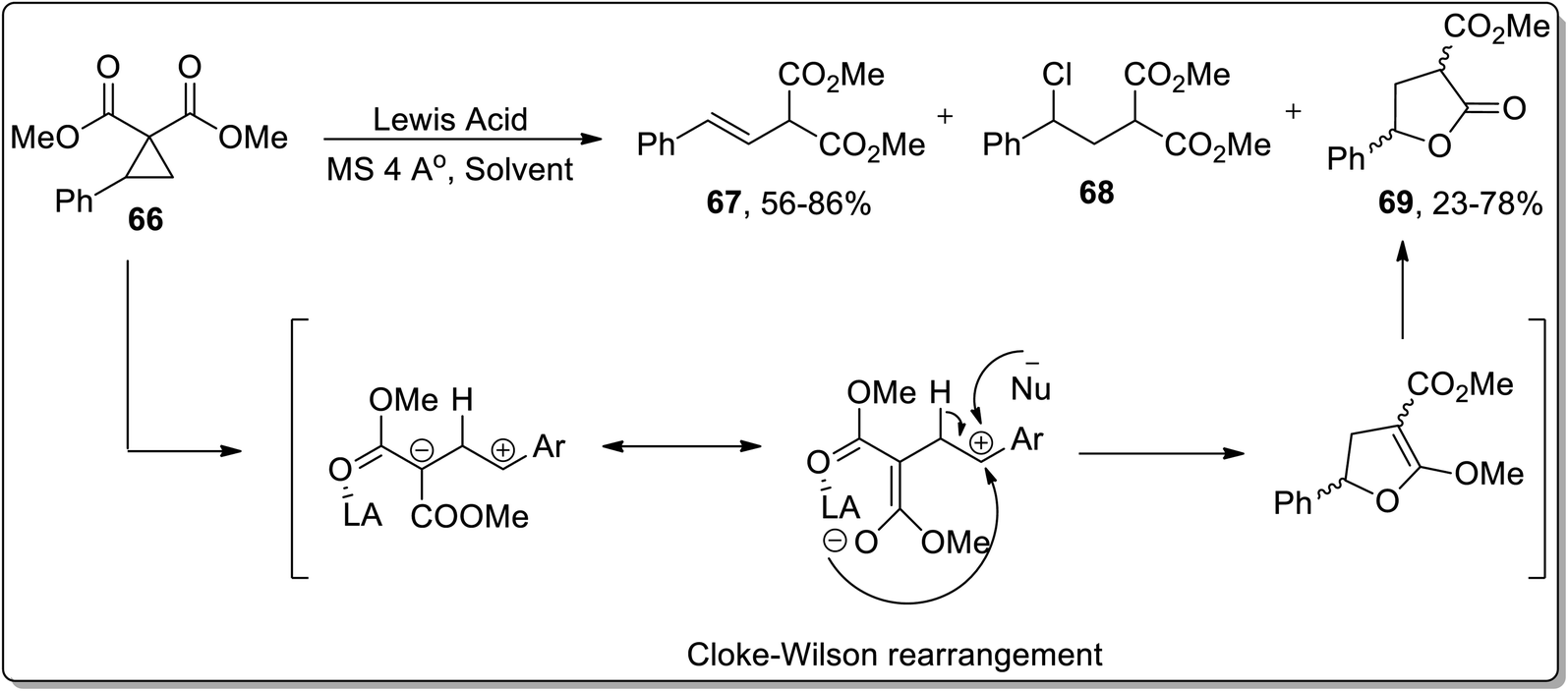
The Lewis acid, i.e., TMSOTf-mediated isomerization of cyclopropanes such as 2-arylcyclopropane-1,1-dicarboxylates 66 was reported by Melnikov and coworkers in 2010. The Lewis-acid promoted ring opening gave rise to three varied products, i.e., E-styrylmalonates 67, substituted chloropropane 68, and γ-butyrolactone 69. Boron trifluoride dietyl etherate-mediated ring expansion resulted in the synthesis of γ-butyrolactone 69 in 78% yield in the presence of phenyl chloride. Meanwhile, 81% yield of γ-butyrolactone 69 was achieved using SnCl4 and dichloromethane; however, Sn(OTf)2/CH3NO2-promoted isomerization led to 77% yield of 69. Cyclopropane 66 resulted in dihydrofuran formation via Cloke–Wilson rearrangement, which was subsequently subjected to acetal hydrolysis to afford γ-butyrolactone 69 in 23–78% yields (Scheme 22).58
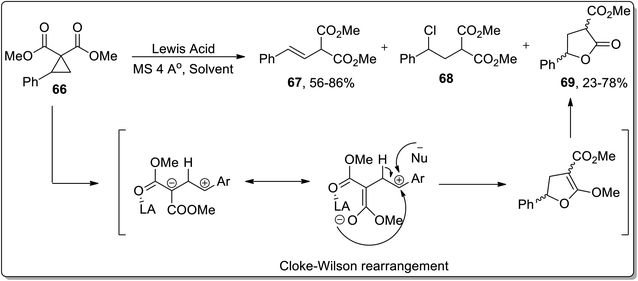 |
| | Scheme 22 Synthesis of E-styrylmalonates 67, substituted chloropropane 68, and γ-butyrolactone 69 via Cloke–Wilson rearrangement. | |
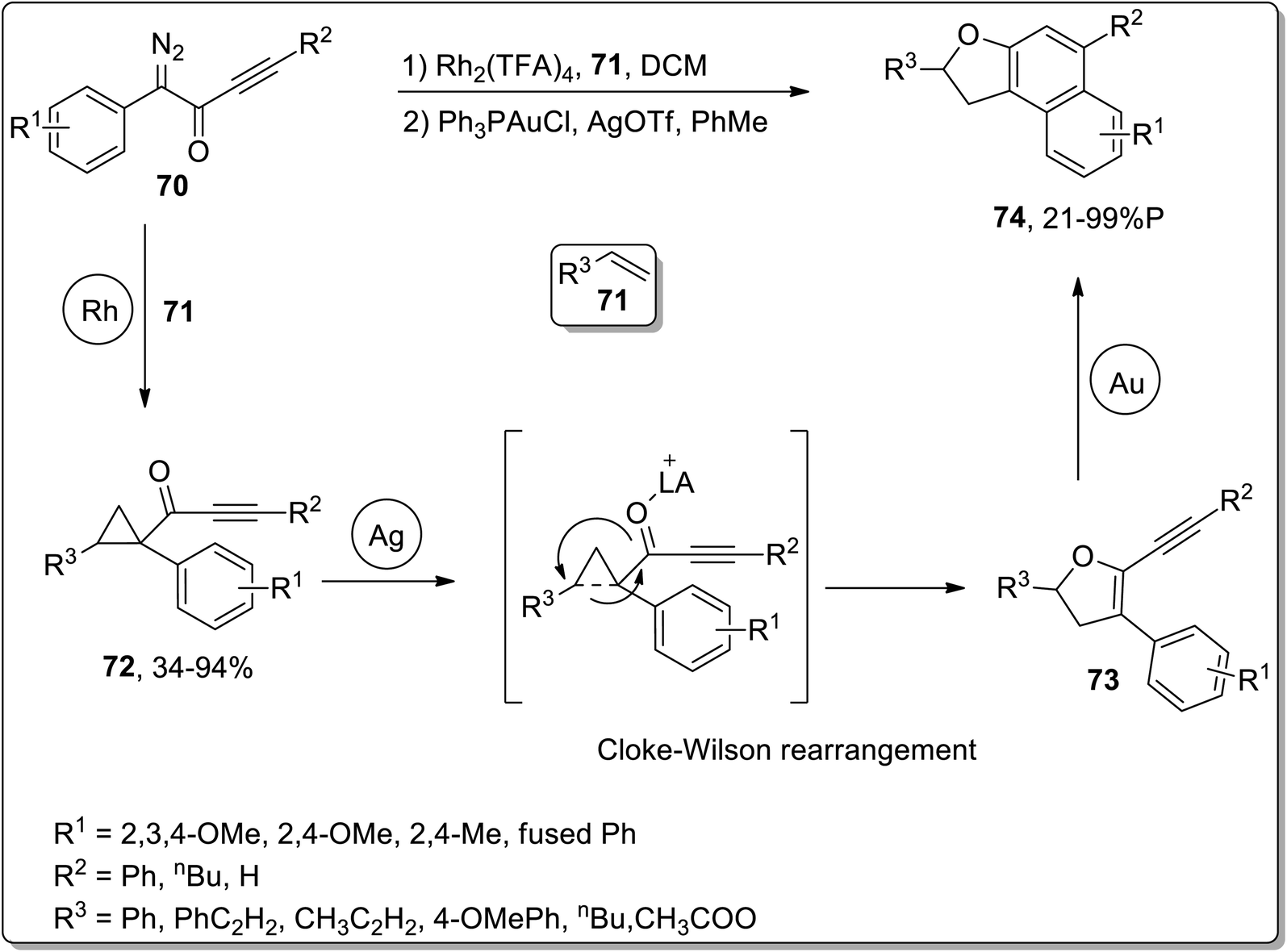
In 2011, Davies and co-workers reported the synthesis of polycyclic benzo-fused dihydrofurans 74 by three sequential rhodium, silver, and gold-catalyzed reactions. The cascade reactions commenced with the generation of cyclopropyl ketones 72 (in fair to excellent yields) by treating alkenes 71 and diazoketones 70 in the presence of rhodium catalyst. The sequential AgOTf-catalyzed ring-expansion of cyclopropyl ketones 72 resulted in the formation of dihydrofurans 73, followed by Au-catalyzed benzannulation to achieve benzo-fused dihydrofurans 74 in low to excellent yield range (21–99%). For these cascade reactions, a one-pot strategy was also implemented, which involved dichloromethane-involving Rh-catalyzed cyclopropanation. After bringing the solution to room temperature, silver and gold catalysts were then added along with toluene. This one-pot protocol furnished polycyclic benzo-fused dihydrofurans 74 in good yields (Scheme 23).59
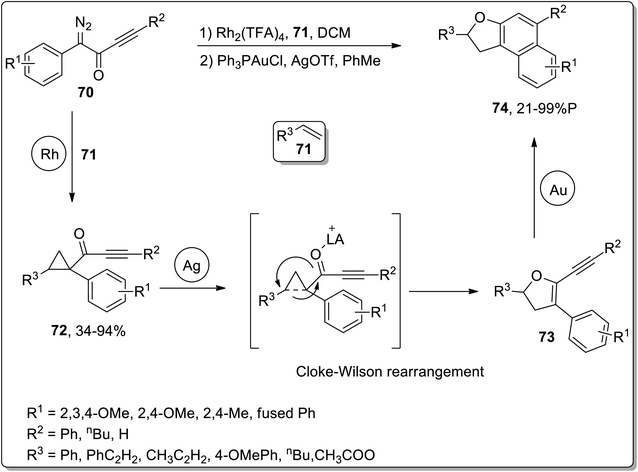 |
| | Scheme 23 Synthesis of benzo-fused dihydrofurans 74 via Cloke–Wilson rearrangement. | |
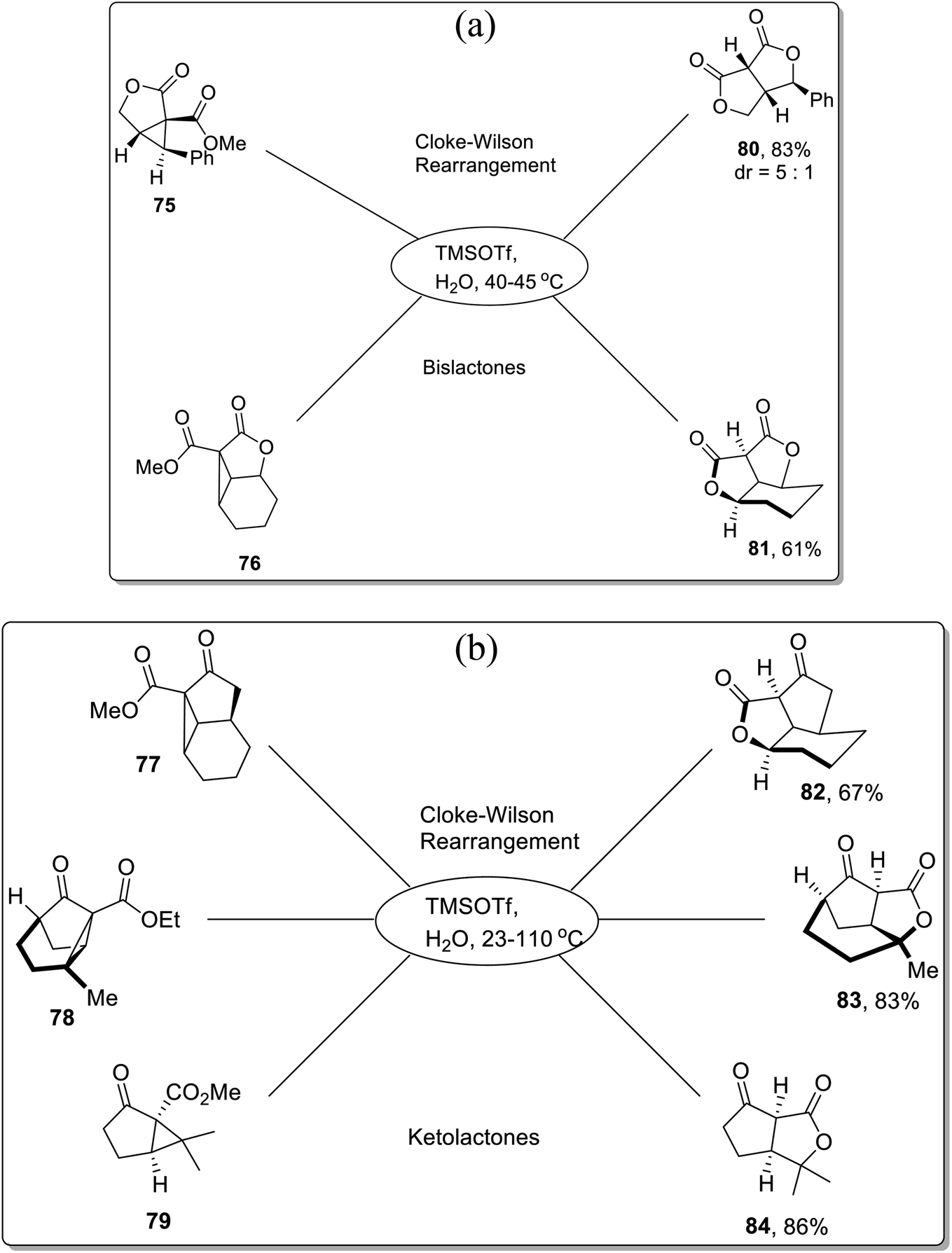
In 2013, Corey and colleagues demonstrated the synthesis of bislactones 80–81 and ketolactones 82–84 in 61–86% (moderate to good yields) via Lewis acid, i.e., TMSOTf-mediated Cloke–Wilson rearrangement of cyclopropyl esters. The donor–acceptor cyclopropanes 75–79 were treated with TMSOTf in aqueous conditions using 2-nitropropane as the solvent within the temperature range of 23–110 °C. The TMSOTf-promoted cleavage of cyclopropane ring was followed by oxygen atom integration to the ester functionality, which ultimately generated the lactones (80–84) after subsequent hydrolysis (Scheme 24a and b).60
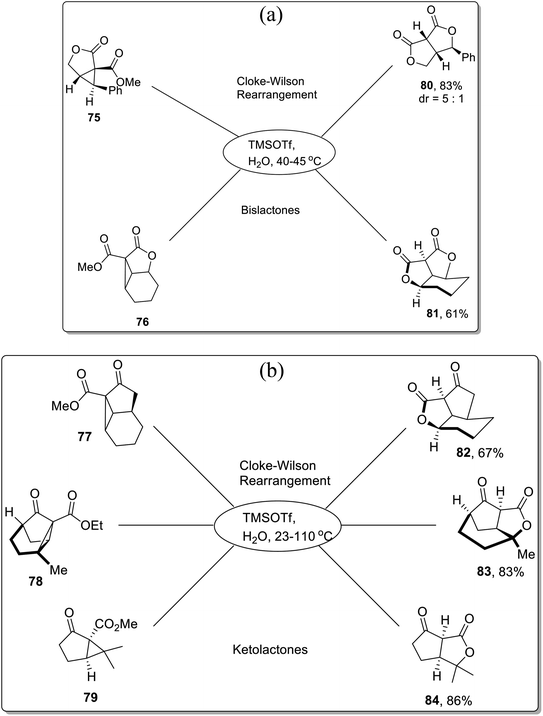 |
| | Scheme 24 (a) Synthesis of bislactones 80–81 via Cloke–Wilson rearrangement. (b) Synthesis of ketolactones 82–84 via Cloke–Wilson rearrangement. | |
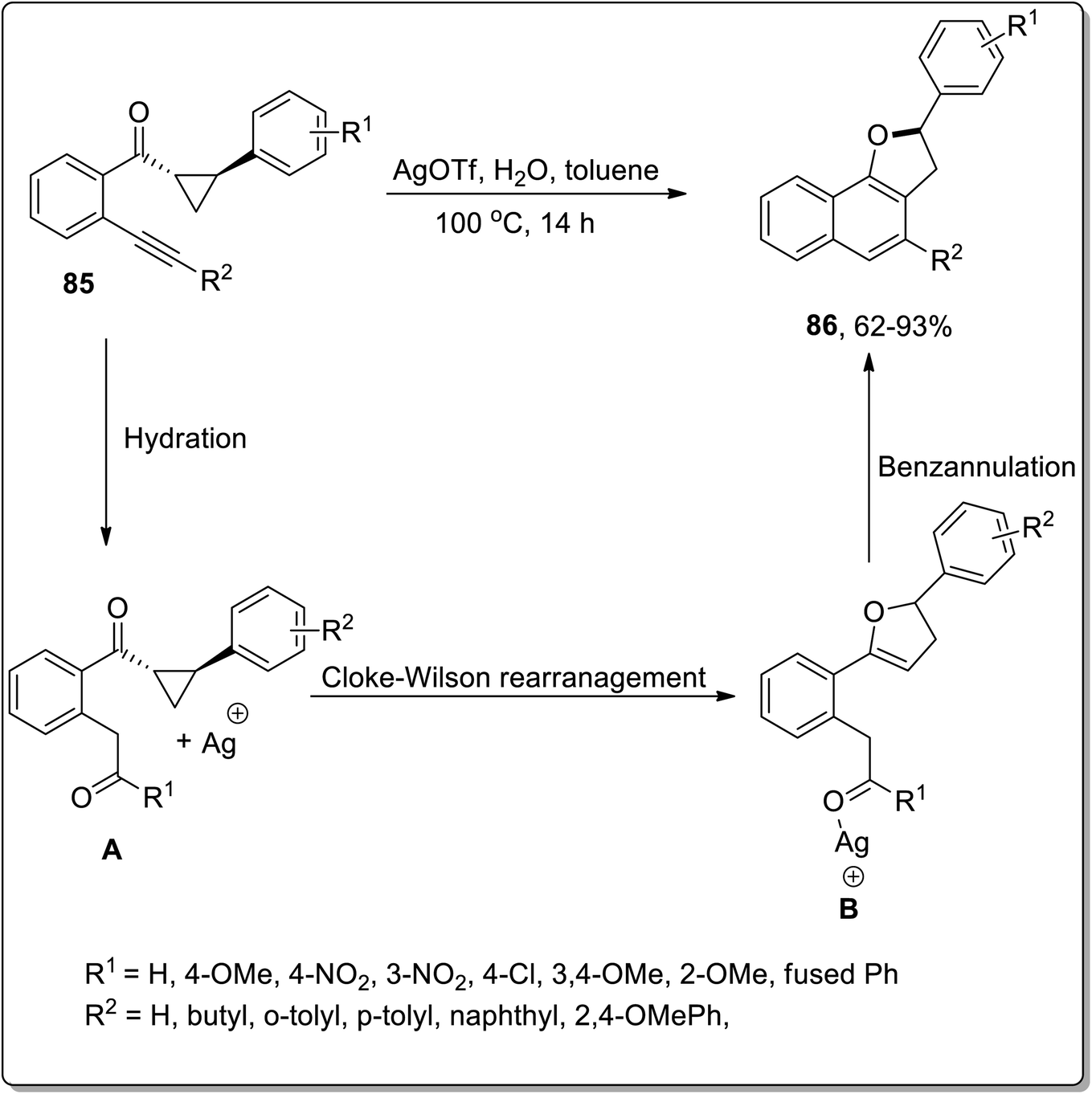
In 2016, Katukojvala and coworkers designed the substituted 2,3-dihydronaphthofurans 86, employing silver (Ag)-promoted intramolecular transannulation of ((2-alkynyl)aryl)cyclopropyl ketones 85. This transformation proceeded with the hydration of the alkyne, followed by the regioselective ring-expansion of cyclopropyl ketone to yield substituted benzofuran heterocycles B via Cloke–Wilson rearrangement, and finally benzannulation took place to yield the target molecules 86. AgOTf (as an efficient Lewis acid) enhanced the −ve inductive effect of carbonyl group and thus carried out the conversion of cyclopropyl ketone into the dihydrofuran skeleton. This transformation could not proceed with terminal alkynes. This is a direct way to synthesize the tricyclic core structures, which are biologically important scaffolds and are main constituents of various natural products (Scheme 25).61
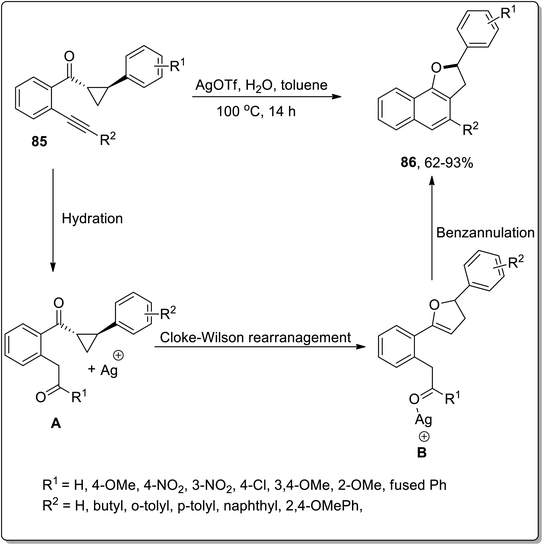 |
| | Scheme 25 Synthesis of substituted 2,3-dihydronaphthofurans 86 via Cloke–Wilson rearrangement. | |
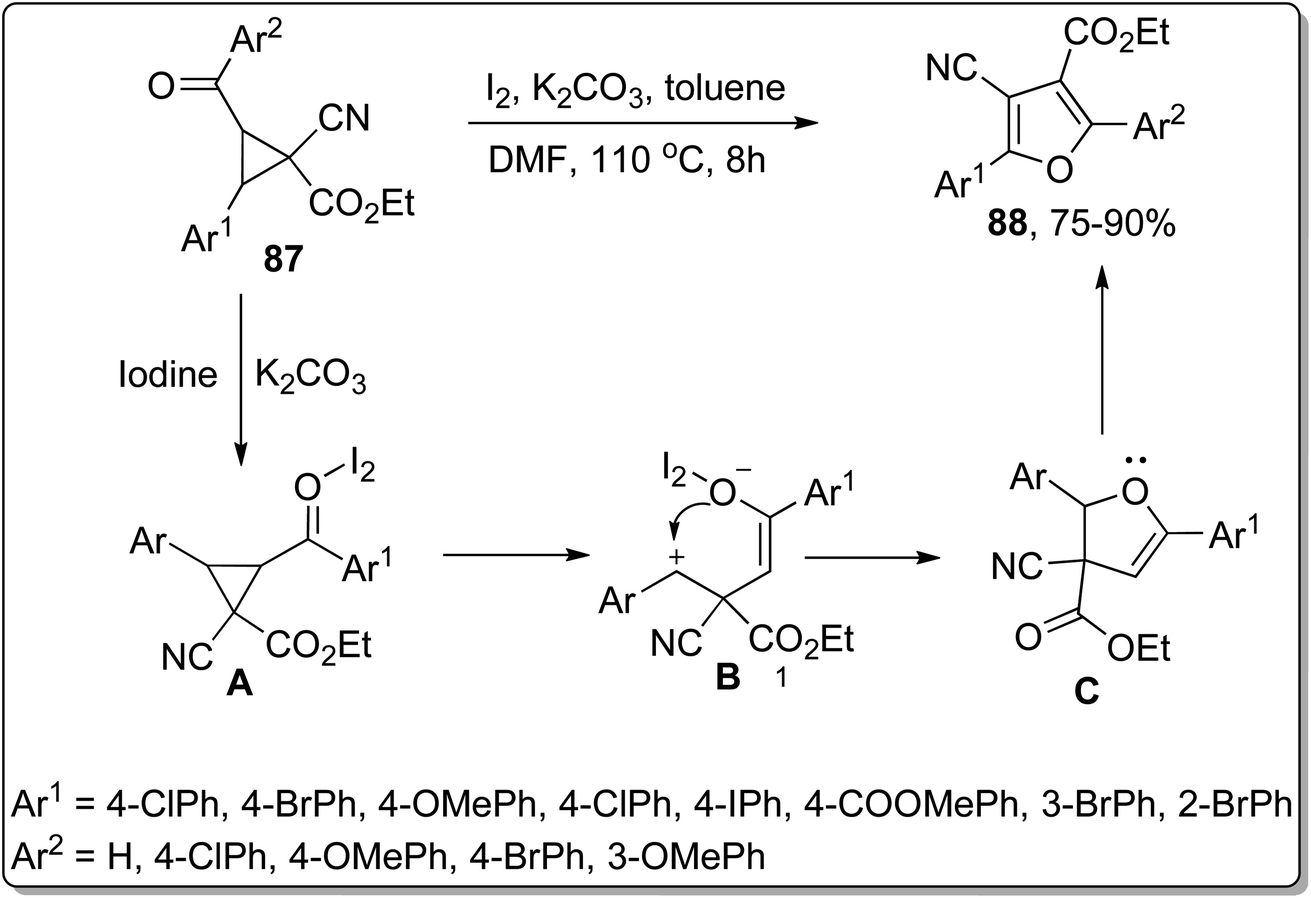
In 2017, Wang and co-workers employed Cloke–Wilson rearrangement to synthesize 4-cyanofuran-3-carboxylate derivatives 88 by subjecting 1-cyanocyclopropane-1-carboxylates 87 to iodine and potassium carbonate-promoted ring cleavage, followed by cyclization and subsequent rearrangement. In this transformation, iodine behaved as a Lewis acid and enhanced the electrophilic character of the carbonyl group, thereby generating an iodine–carbonyl complex A. Then, the base-mediated opening of ring, followed by tautomerization, gave intermediate B. Later on, intermediate B was treated with phenylmethylium ion via intramolecular nucleophilic coupling to afford intermediate C, which upon further intramolecular nucleophilic addition, iodination, tautomerism, and elimination of HI resulted in 4-cyanofuran-3-carboxylate derivatives 89. This protocol can be immensely employed for the synthesis of diverse organic compounds owing to easily accessible starting reagents and high yields of target molecules. Moreover, various medicinally important polysubstituted furancarboxylates are envisioned to be synthesized utilizing this protocol (Scheme 26).62
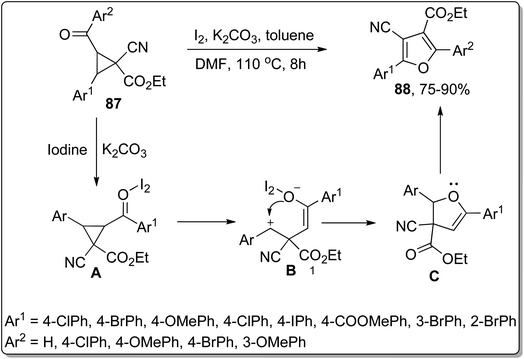 |
| | Scheme 26 Synthesis of 4-cyanofuran-3-carboxylate derivatives 88 via Cloke–Wilson rearrangement. | |
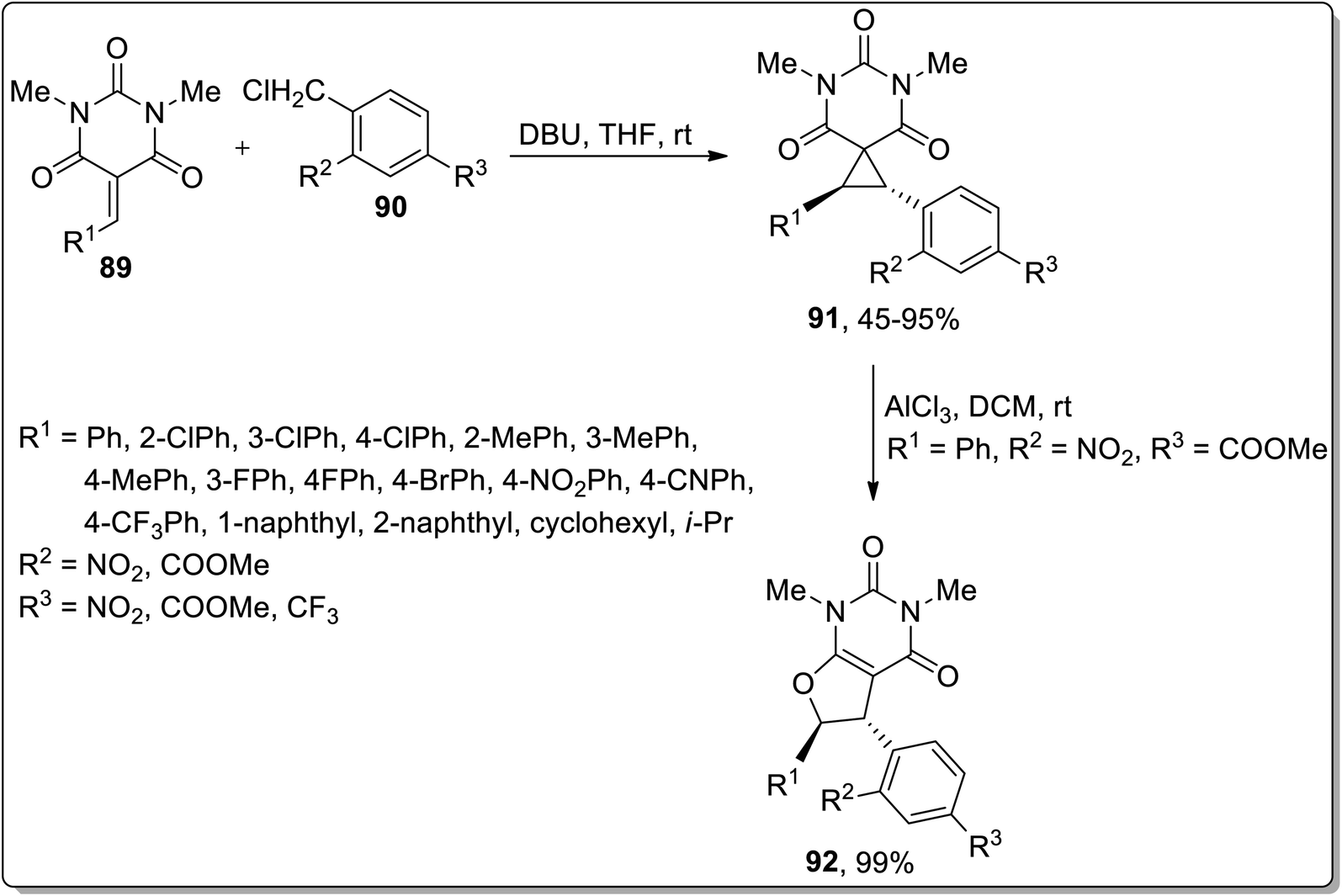
In 2019, Song et al. demonstrated the DBU-mediated stereospecific expansion of spirocyclopropyl compounds to synthesize spirobarbiturate-cyclopropanes 92 obtained by treating barbiturate-based olefins 90 with substituted benzyl chlorides 91 at room temperature in tetrahydrofuran. As a result, moderate to high yields (45–95%) of spirobarbiturate-based cyclopropanes 92 were afforded with remarkable diastereoselectivity ratio (dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The next step involved the procurement of dihydrofuran-based pyrimidinedione frameworks 93 in 99% yield via AlCl3-mediated Cloke–Wilson rearrangement. This synthetic protocol proceeded with the retention of anti-configuration, entailing a wide substrate scope, thereby providing a series of diversely-substituted target molecules (Scheme 27).63
:1). The next step involved the procurement of dihydrofuran-based pyrimidinedione frameworks 93 in 99% yield via AlCl3-mediated Cloke–Wilson rearrangement. This synthetic protocol proceeded with the retention of anti-configuration, entailing a wide substrate scope, thereby providing a series of diversely-substituted target molecules (Scheme 27).63
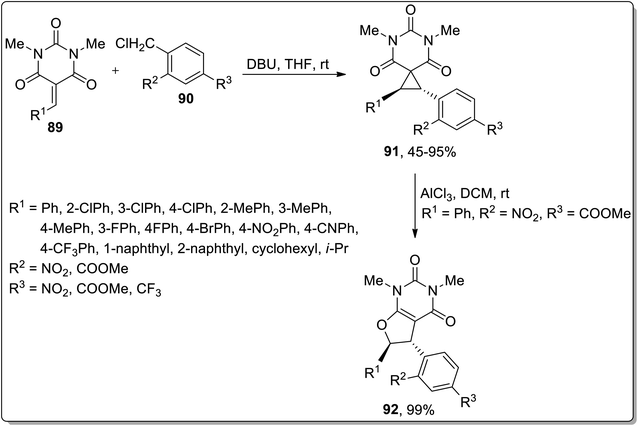 |
| | Scheme 27 Synthesis of spirobarbiturate-cyclopropanes 92 via Cloke–Wilson rearrangement. | |
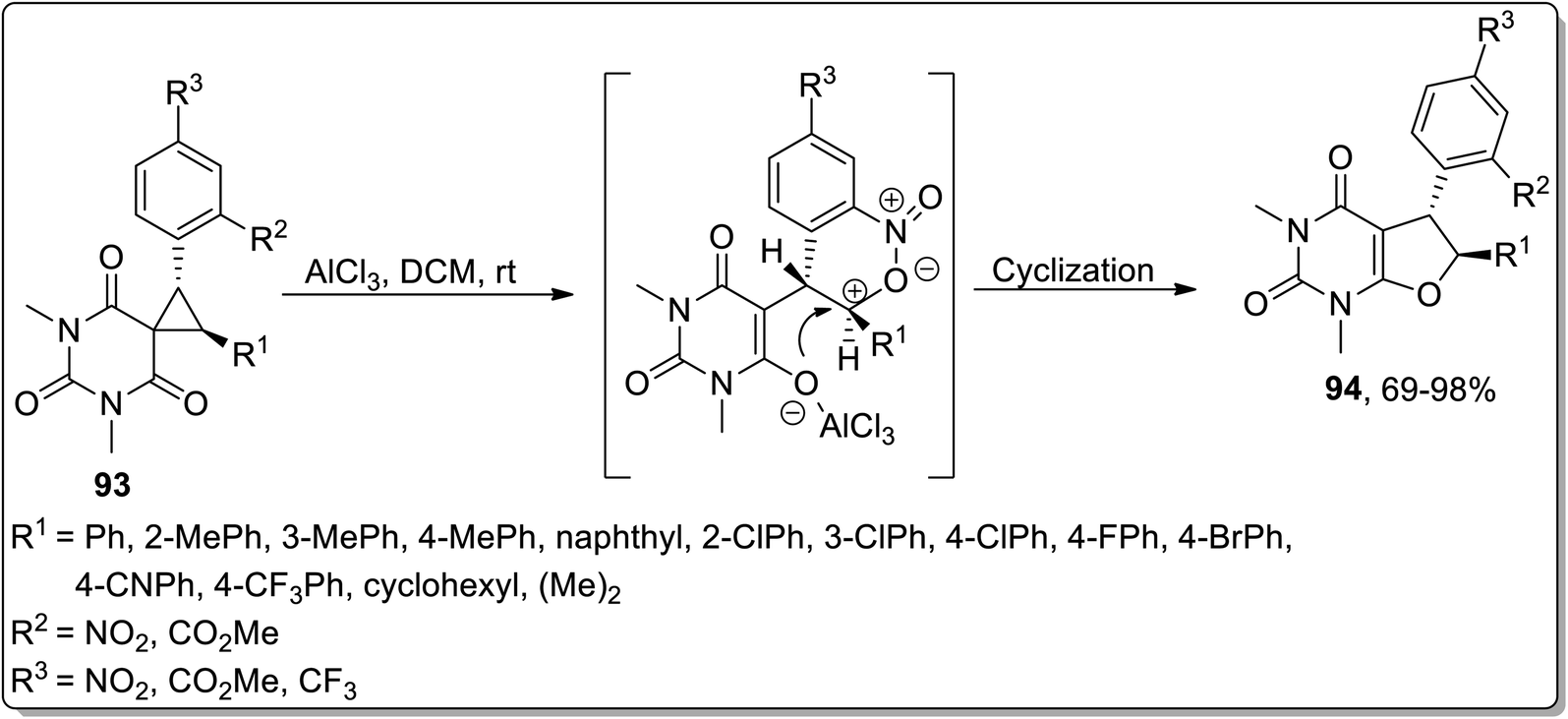
Another Lewis acid, i.e., AlCl3-mediated Cloke–Wilson rearrangement, was established by Song and coworkers (in 2019) to generate substituted 5,6-dihydrofuro[2,3-d]pyrimidines 94 from spirocyclopropyl barbiturates 93. A wide range of diversely-substituted spirocyclopropanes were endured by treating them at room temperature. The efficiency of the reaction was observed to be unaltered due to the electronic properties and place of attachment of various substituents on the phenyl ring. In consequence, 69–98% (good to excellent yields) of furan-based pyrimidines 94 were achieved with the retention of anti-configuration (Scheme 28).64
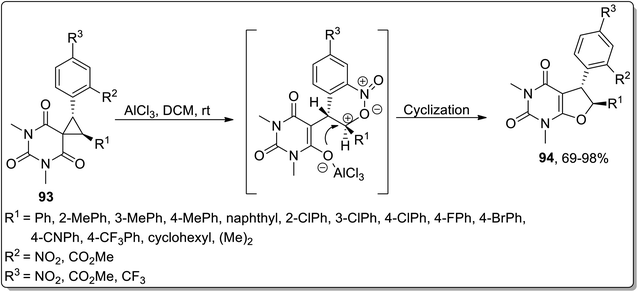 |
| | Scheme 28 Synthesis of substituted 5,6-dihydrofuro[2,3-d]pyrimidines 94 via Cloke–Wilson rearrangement. | |
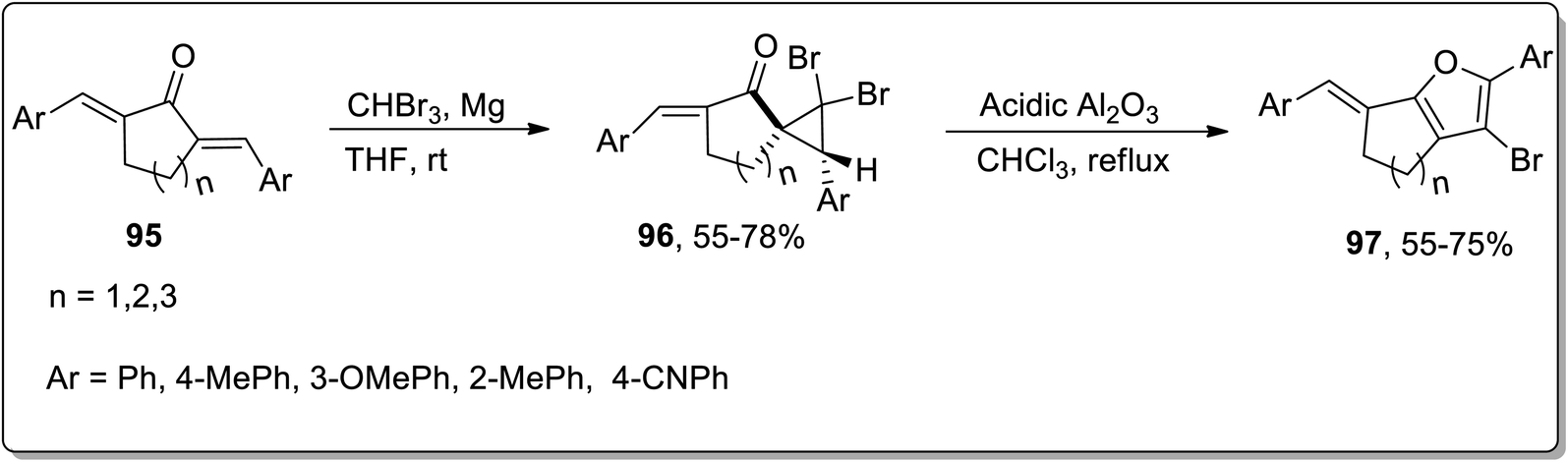
In 2020, Namboothiri and colleagues illustrated the synthesis of fused furans 97 from alkylidenecycloalkanones 95 by utilizing cyclopropanation reaction and Cloke–Wilson rearrangement in their sequential two-step methodology. Initially, substituted alkylidenecycloalkanones were made to react via diastereoselective dibromocyclopropanation using magnesium and tribromomethane in THF at room temperature to synthesize dibromocyclopropyl ketones 96. Cyclopropyl ketone 96 was further subjected to acidic Al2O3-mediated regioselective ring expansion by Cloke–Wilson rearrangement reaction by refluxing in chloroform. The regioselective fused furan derivatives 97 were furnished in 55–75% yields. Various alkylidenecycloalkanones derived from tetralone, indanone, and benzosuberone were subjected to the optimal conditions that produced 2-aryl-3-bromofurans in good yields, validating the wide substrate tolerant nature of this protocol (Scheme 29).65
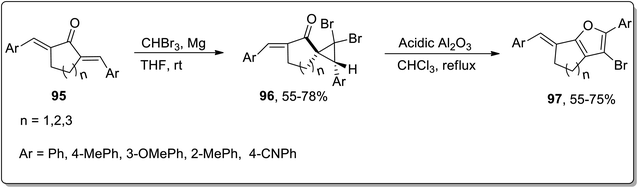 |
| | Scheme 29 Synthesis of furan derivatives 97 via Cloke–Wilson rearrangement. | |
6.4. Brønsted acid-catalyzed/mediated rearrangement
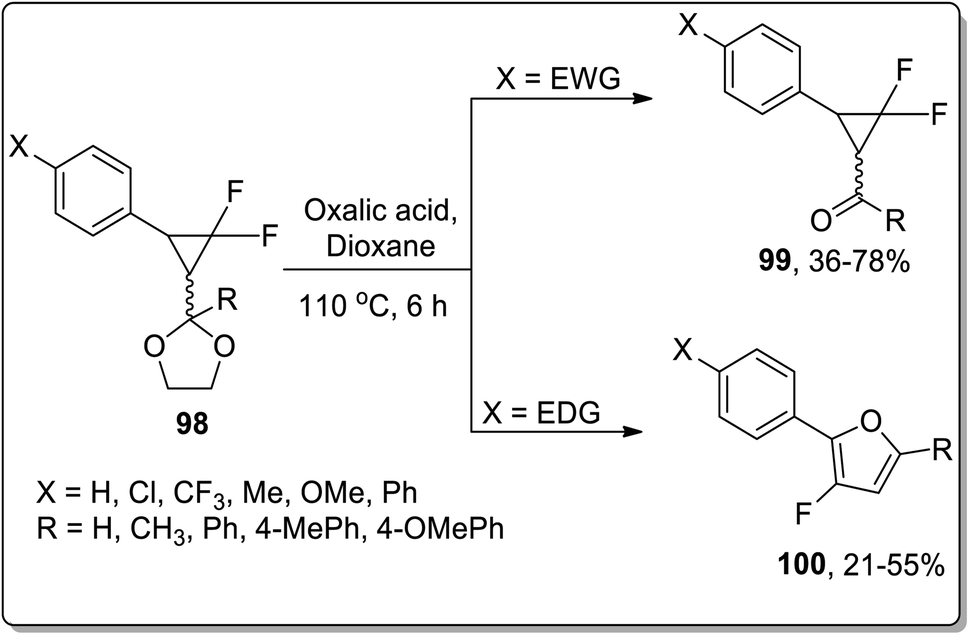
Since the advent of the 21st century, Brønsted acids have been efficiently employed as catalysts to synthesize numerous organic compounds.66a In 2003, Chen and Xu reported the synthesis of substituted cyclopropyl ketones 99 and substituted fluorofurans 100 from gem-difluorocyclopropyl acetals and ketals 98, which were obtained by the [1 + 2] cycloaddition of difluorocarbene and α,β-unsaturated aromatic aldehydes and ketones. These fluorinated scaffolds 98 were further subjected to acidic hydrolysis to give fluorofuran derivatives 100 via Brønsted acid-mediated intramolecular Cloke–Wilson ring rearrangement. However, the electron withdrawing-substituted compounds furnished substituted cyclopropyl ketones 99. The product ratio was observed to be dependent on the electronic properties of the substituent on the β-phenyl ring, i.e., the electron-donating groups bearing reagents resulted in the synthesis of furan derivatives 100 via Cloke–Wilson rearrangement, while electron-withdrawing groups-bearing compounds gave normal ketones 99 (Scheme 30).66b
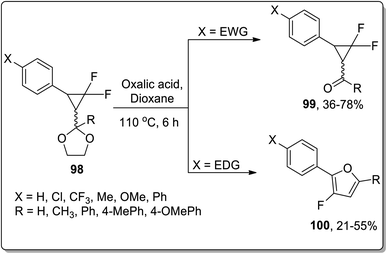 |
| | Scheme 30 Synthesis of fluorofuran derivatives 100 via Cloke–Wilson rearrangement. | |
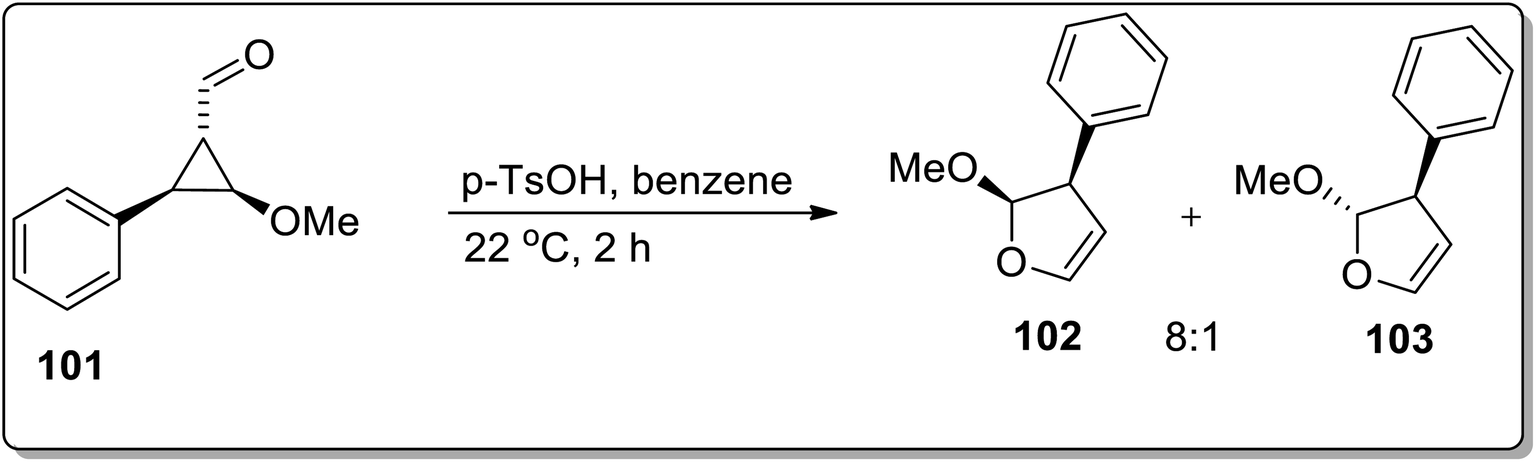
Another Brønsted-acid mediated Cloke–Wilson rearrangement was processed by Baines and co-workers, which resulted in the diastereoselective synthesis of 2-methoxy-3-phenyl-2,3-dihydrofurans 102 and 103. For this purpose, trans-2-methoxy-3-phenylcyclopropane carbaldehyde 101 was subjected to Cloke–Wilson rearrangement in p-toluenesulfonic acid and benzene. This transformation afforded a 8:1 cis and trans mixture of methoxy-3-phenyl-2,3-dihydrofuran 102 and 103 (Scheme 31).67
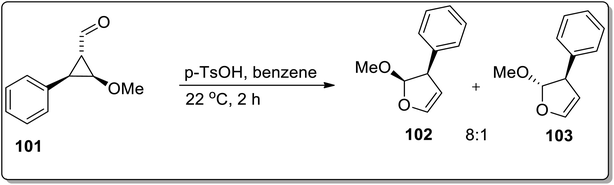 |
| | Scheme 31 Synthesis of methoxy-3-phenyl-2,3-dihydrofuran 102 and 103 via Cloke–Wilson rearrangement. | |
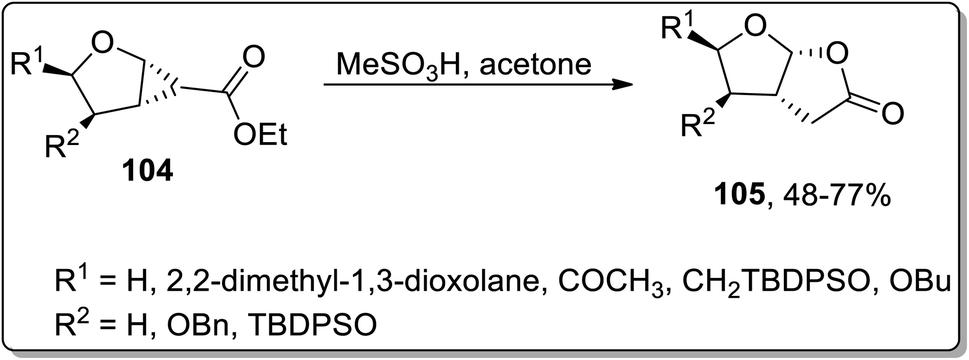
In 2004, Theodorakis and co-workers reported another Brønsted acid, i.e., methanesulfonic acid-catalyzed Cloke–Wilson rearrangement, which led to the synthesis of fused tetrahydrofuran-γ-lactone 105. This protocol involved the one-pot ring extension of cyclopropanes 104 using acetone as the solvent, which yielded required the bicyclic compounds in moderate to good yields. However, no product formation was observed employing the sp2 hybridized α-carbon atom bearing cyclopropyl ring as the precursor (Scheme 32).68
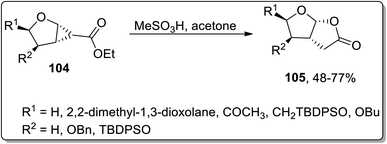 |
| | Scheme 32 Synthesis of fused tetrahydrofuran-γ-lactone 105 via Cloke–Wilson rearrangement. | |
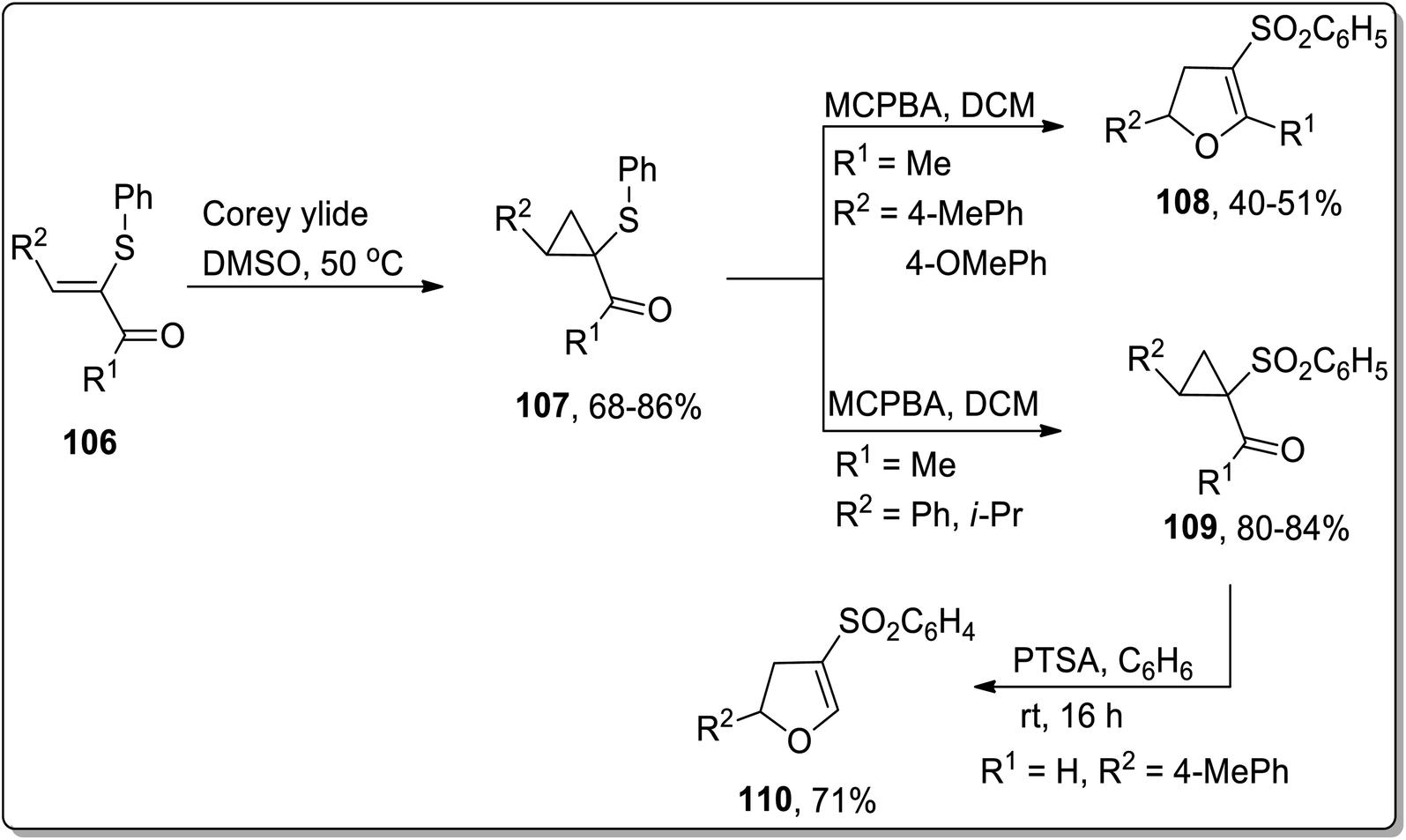
In 2005, Piras and coworkers designed the synthesis of 2,4,5-tri-substituted 2,3-dihydrofurans 109 and cyclopropyl sulfones 108. Initially, the reaction of enone 106 with Corey ylide gave cyclopropylsulfides 107, which upon oxidation with MCPBA (meta-chloroperoxybenzoic acid) resulted in the generation of either cyclopropyl sulfones 109 or 2,4,5-tri-substituted 2,3-dihydrofurans 108. In the formation of 2,3-dihydrofurans, MCPBA has been observed to play the role of an oxidant as well as Brønsted acid, thereby facilitating ring opening and closure. The formation of target molecules was interpreted to be dependent upon the nature of the attached substituents. The cyclopropyl ring with only strong electron-donating substituents favored the formation of 2,4,5-tri-substituted 2,3-dihydrofurans 108 (Scheme 33). Similarly, under the mild reaction conditions (using PTSA in benzene at room temperature), Cloke–Wilson rearrangement of cyclopropyl sulfone 109 furnished 2,3-dihydrofurans 110 (Scheme 33).69
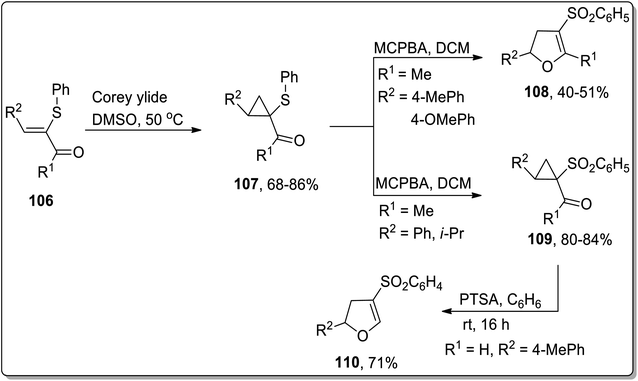 |
| | Scheme 33 Synthesis of 2,3-dihydrofurans 110 and cyclopropyl sulfones 108 via Cloke–Wilson rearrangement. | |
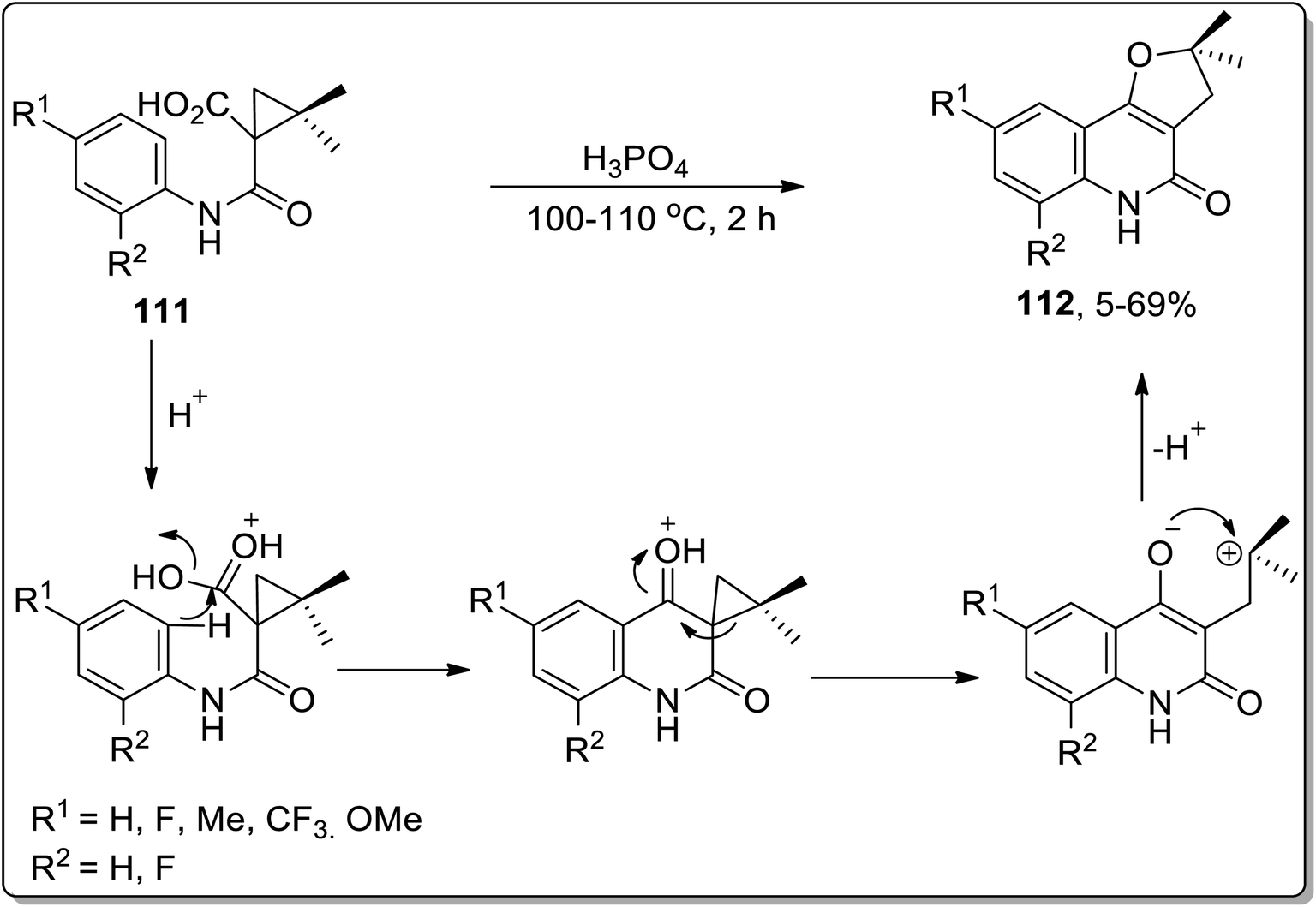
In 2006, angular dihydrofuroquinoline derivatives 112 were obtained by subjecting 1-(phenylcarbamoyl)cyclopropane-1-carboxylic acid derivatives 111 to Cloke–Wilson ring-enlargement of cyclopropyl ketone by Su et al. In this transformation, phosphoric acid protonated the carbonyl oxygen regioselectively, which facilitated quinolinone ring formation by intramolecular Friedel–Crafts reaction. The synthesis of the furan ring is highly affected by the altered electron density on the involved intermediate. This one-step procedure gave access to angular dihydrofuroquinoline derivatives 112 in moderate yields (Scheme 34).70
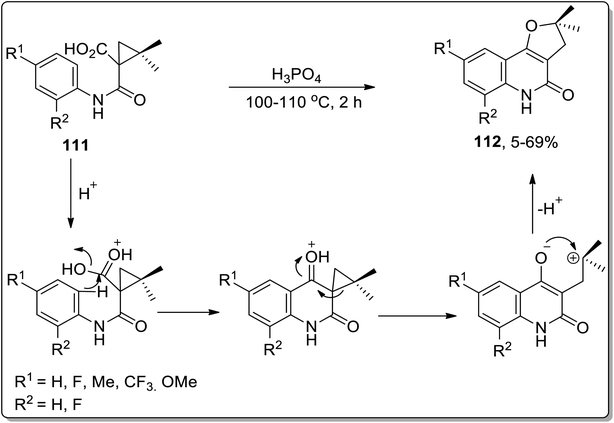 |
| | Scheme 34 Synthesis of dihydrofuroquinoline derivatives 112 via Cloke–Wilson rearrangement. | |
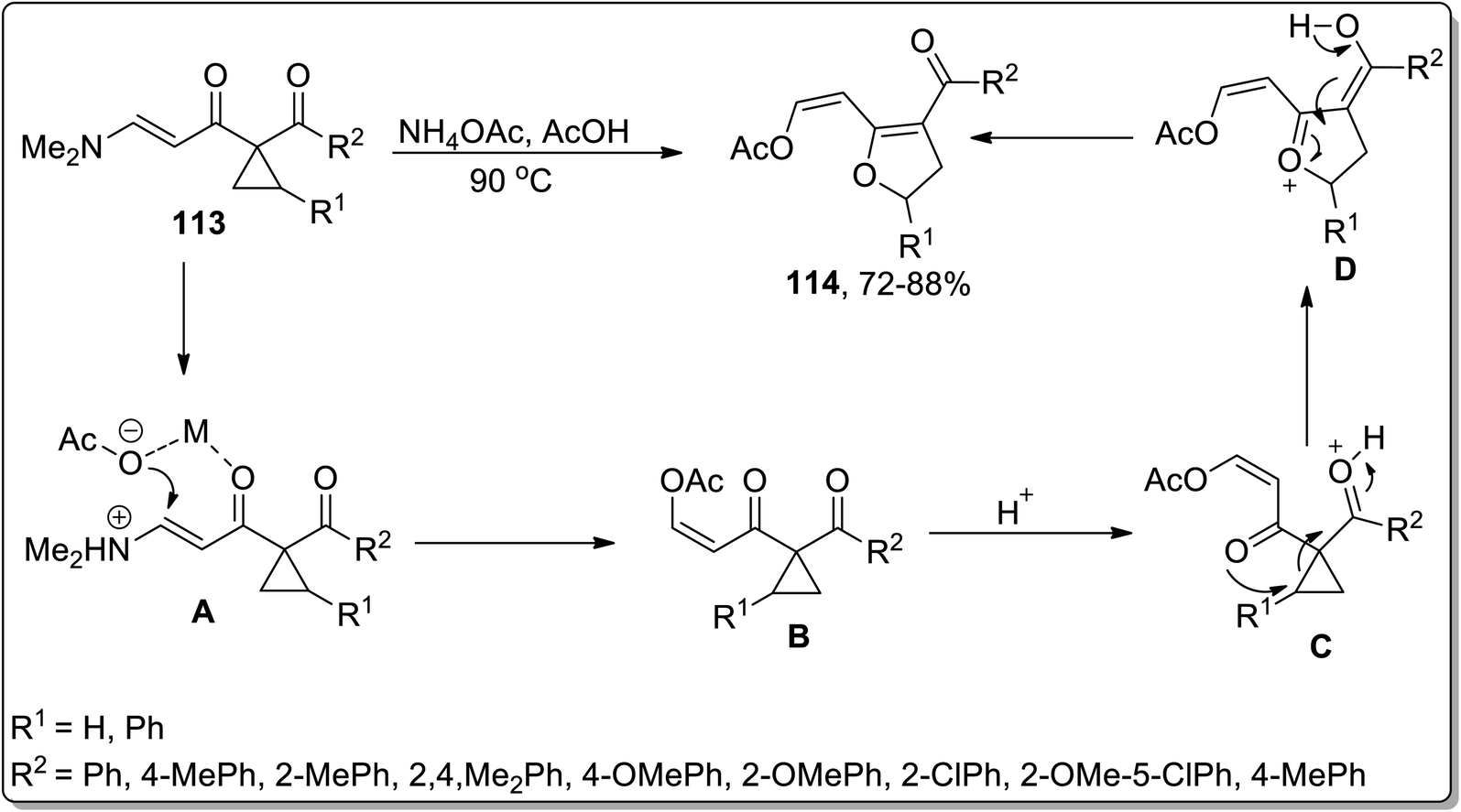
In 2008, Dong et al. designed the synthesis of substituted 2,3-dihydrofurans 114 via ammonium acetate-mediated Cloke–Wilson ring-enlargement of activated cyclopropanes 113. This transformation commenced with the N-protonation and H-bonding interaction of ammonium acetate to intermediate A formation, which later on gave rise to intermediate B by employing oxa-Michael addition and elimination reaction; finally, upon regioselective ring-enlargement, 2,3-dihydrofurans 114 were attained in good yields. These substituted dihydrofurans 114 were then made to undergo an intramolecular annulation reaction to produce the corresponding 5-aryl-2,3-dihydrofuro[ 3,2-c]pyridin-4(5H)-ones in efficient yields (Scheme 35).71
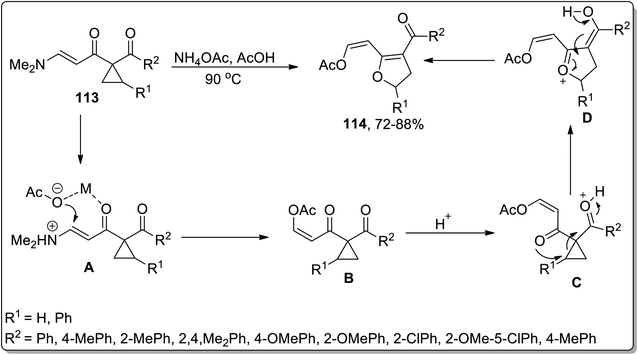 |
| | Scheme 35 Synthesis of substituted dihydrofurans 114 via Cloke–Wilson rearrangement. | |
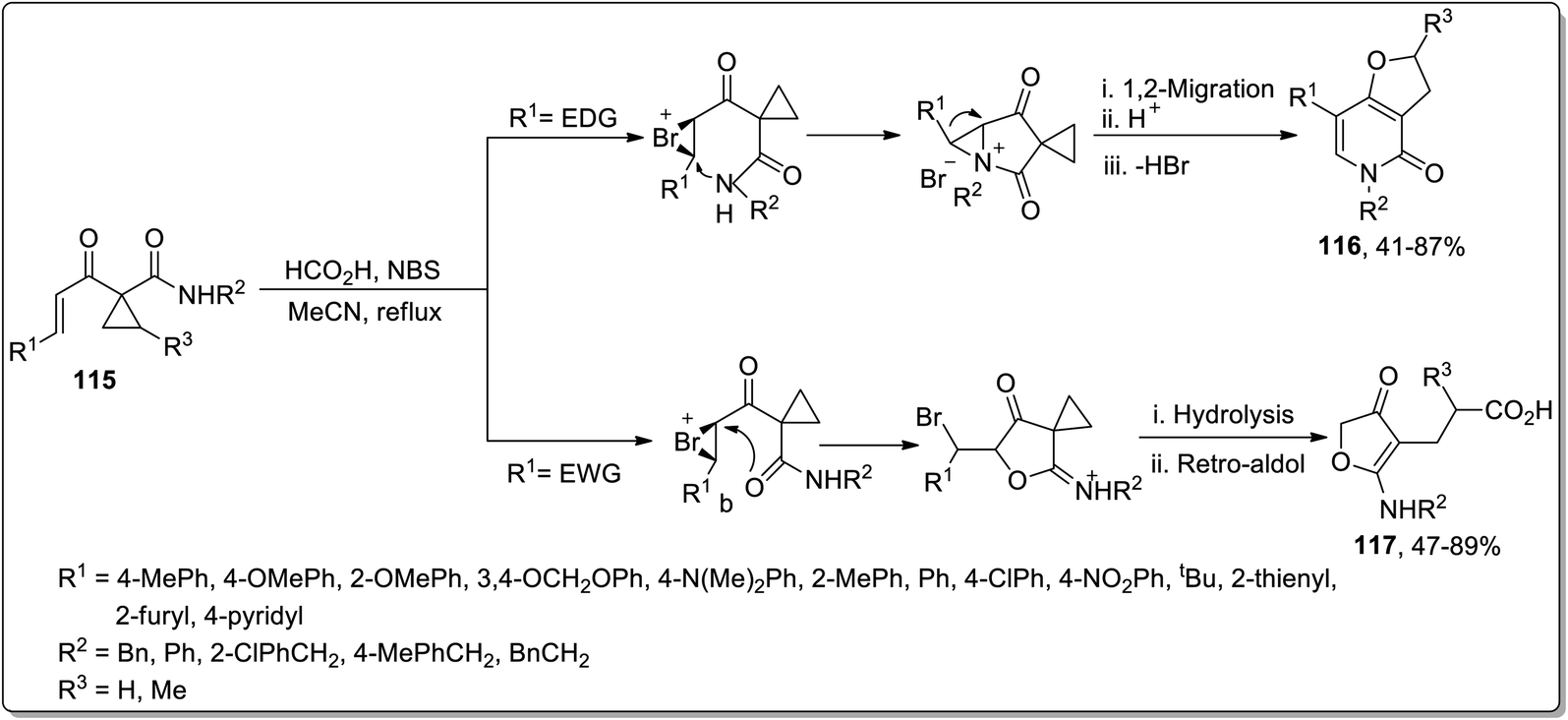
In 2011, Liang and co-workers designed the carboxylic acid-catalyzed synthesis of dihydrofuropyridinones 116 and 3(2H)-furanones 117 from 1-alkenoylcyclopropane carboxamides 115 by halonium-initiated tandem reaction. The synthesis initiated with the formation of halonium ion intermediate A, which either followed an intramolecular oxa-cyclization (Path a), deprotonation, 1,2-migration, β-hydride elimination, cyclopropane ring opening, and recyclization to produce dihydrofuropyridinones 116 or halo-oxa-cyclization (Path b), cyclopropyl ring opening, and retro-aldol reaction to afford 3(2H)-furanones 117. These products were produced in efficient yields with high chemo- and regioselectivity, and the substituent appended on the enone framework decided the type of the resulting product. The groups with positive inductive effect resulted in dihydrofuropyridinones 116 while 3(2H)-furanones 117 were generated by groups with negative inductive effect (Scheme 36).72
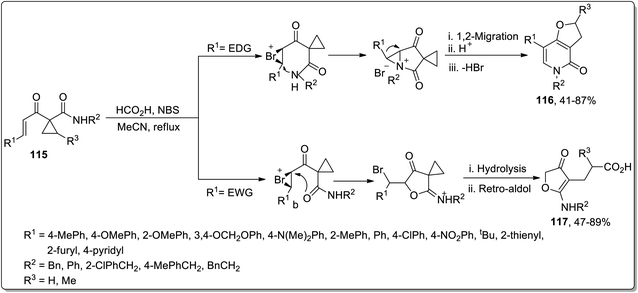 |
| | Scheme 36 Synthesis of dihydrofuropyridinones 116 and 3(2H)-furanones 117 via Cloke–Wilson rearrangement. | |
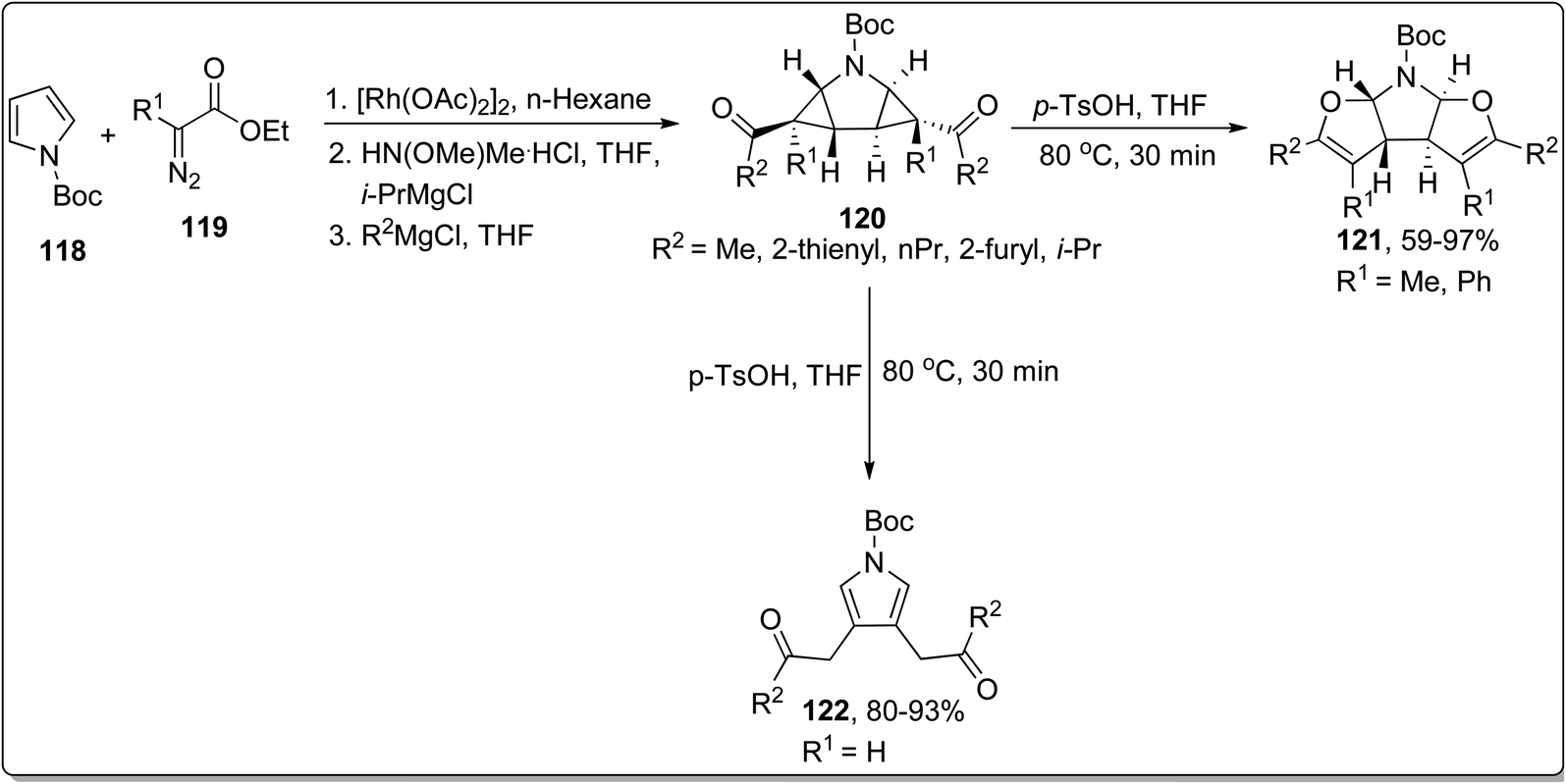
In 2013, Werz and co-workers designed the synthesis of N,O-bisacetals 121 from the corresponding pyrrolidine substrate 120 (prepared by treating N-Boc protected pyrroles 118 with functionalized imines 119) via Cloke–Wilson rearrangement reaction. The rearrangement was proceeded in the presence of p-TsOH (Brønsted-acid catalyst) in THF at 80 °C in fair to efficient yields by substituting R1 as methyl or phenyl. However, as a result of substituting R1 as H, the cyclopropane ring-opening and rapid aromatization took place to produce pyrroles 122 in good to excellent yields (Scheme 37).73
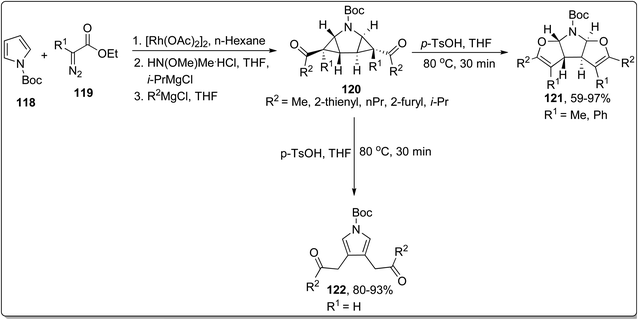 |
| | Scheme 37 Synthesis of N,O-bisacetals 121 and pyrroles 122 via Cloke–Wilson rearrangement. | |
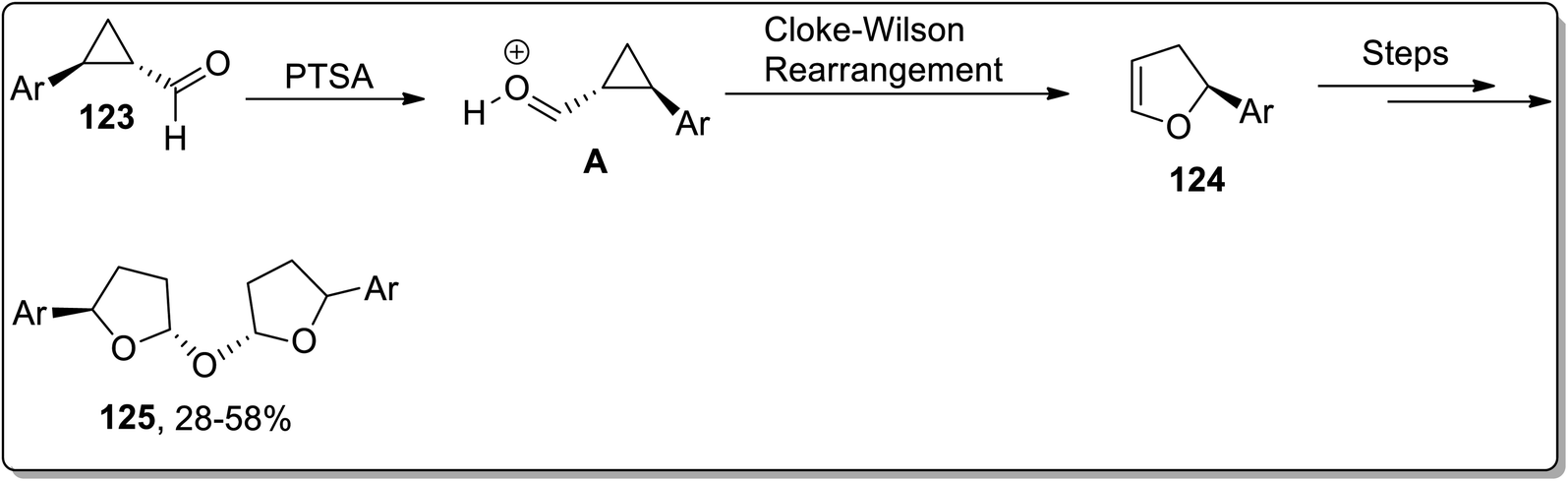
In 2020, Banerjee and coworkers designed the metal-free synthesis of oxybis(2-aryltetrahydrofuran) derivatives 125 from cyclopropane carbaldehydes 123 by a facile three-steps methodology: the Cloke–Wilson rearrangement, which gave 124 intermediate, followed by hydration and finally dimerization. This transformation was accomplished by exploiting inexpensive Brønsted acid-catalyst (PTSA) in an open environment (Scheme 38).74
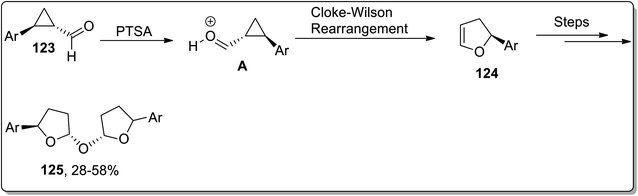 |
| | Scheme 38 Synthesis of oxybis(2-aryltetrahydrofuran) derivatives 125 via Cloke–Wilson rearrangement. | |
6.5. Brønsted base-mediated rearrangement
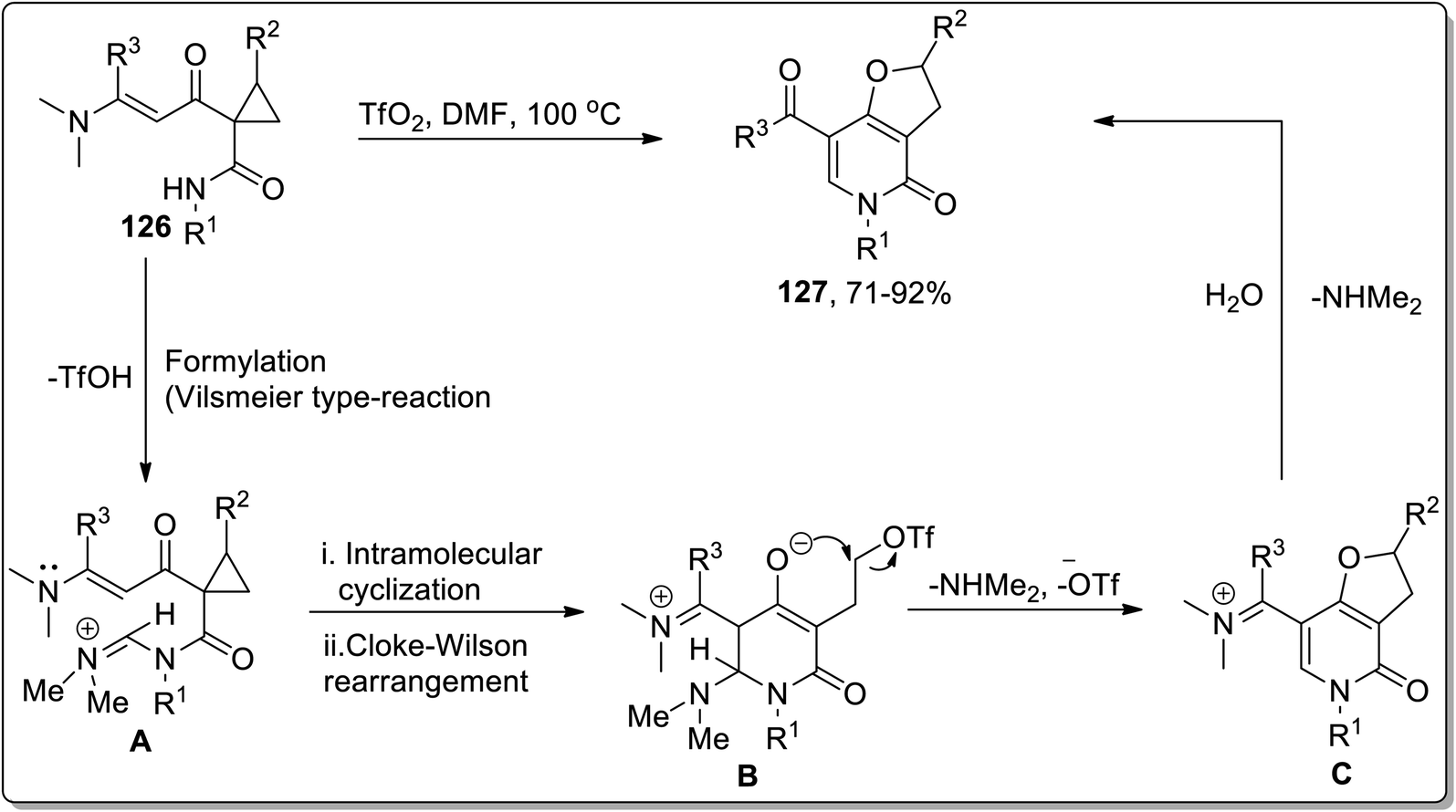
In 2012, Dong and coworkers carried out the synthesis of dihydrofuran-based pyridinones 127 by treating functionalized cyclopropanes 126 in the presence of triflic anhydride (Tf2O) in dimethylformamide. This reaction commenced from the activation of DMF with TfO2 to produce Vilsmeier-type reagent that formylated (Vilsmeier-type reaction) substrate 126 to produce iminium salt intermediate A, followed by intramolecular cyclization to generate intermediate B, which further underwent an intramolecular Cloke–Wilson ring extension promoted by trifluoromethanesulfonate anion (TfO−) to produce iminium intermediate C, which upon treatment with water finally gave target molecules 127 in good to excellent yields (Scheme 39).75
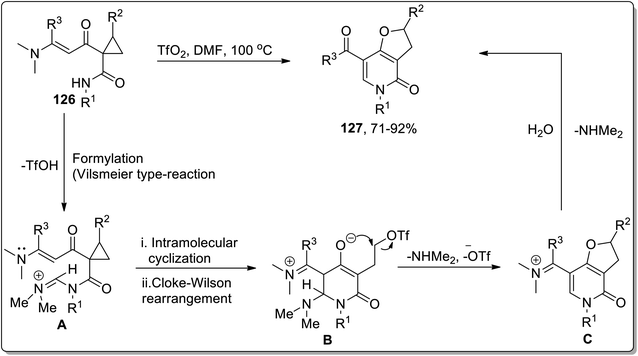 |
| | Scheme 39 Synthesis of dihydrofuran-based pyridinones 127 via Cloke–Wilson rearrangement. | |
6.6. Transition metals-catalyzed rearrangement
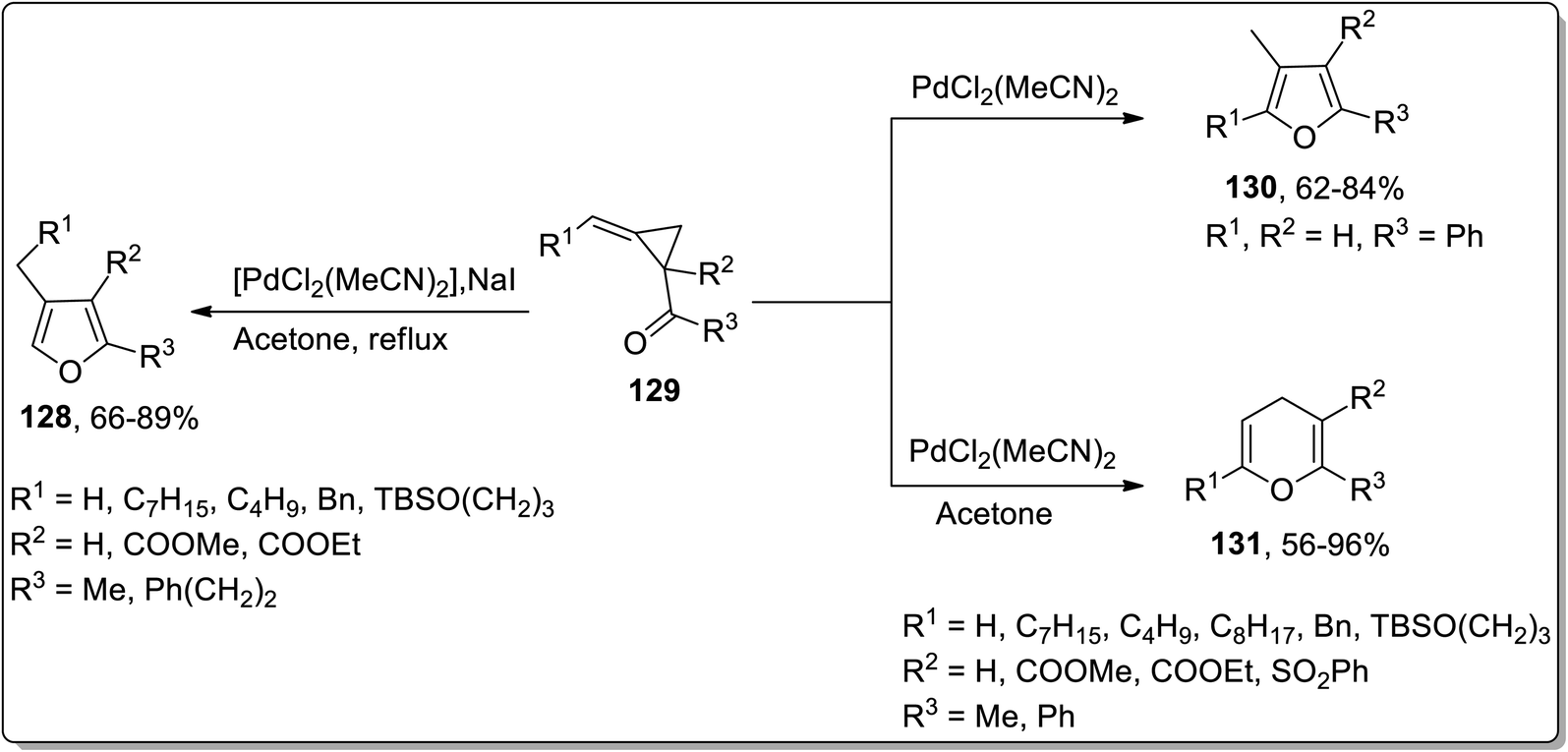
Considering the wide-utilization of catalytic property of transition metals in organic synthesis,76a–c Cloke–Wilson rearrangement has also been observed to be carried out employing various transition metals-based catalysts. Palladium-based catalysts are widely employed in numerous organic reactions.77 In 2003 and 2004, Ma and Zhang reported the synthesis of tetra-substituted furans 130 and polysubstituted 4H-pyrans 131 by the isomerization of alkylidene cyclopropyl ketones 129. The Cloke–Wilson rearrangement of substituted cyclopropane 129 in the presence of PdCl2(MeCN)2 yielded the tetra-substituted furans 130. Meanwhile, 4H-pyrans 131 were obtained by carrying out regioselective chloropalladation of alkylidene with PdCl2, followed by β-decarbopalladation, and an intramolecular insertion of double bond with the elimination of PdCl2 (Scheme 40).41b
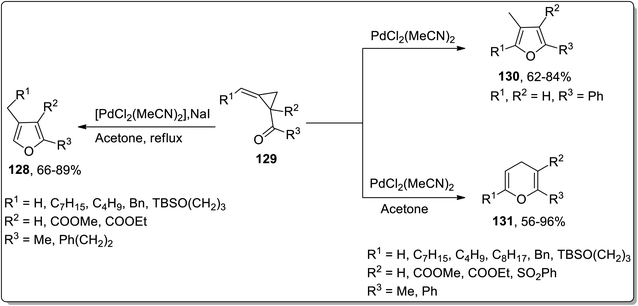 |
| | Scheme 40 Synthesis of tetra-substituted furans 130 and polysubstituted 4H-pyrans 131 via Cloke–Wilson rearrangement. | |
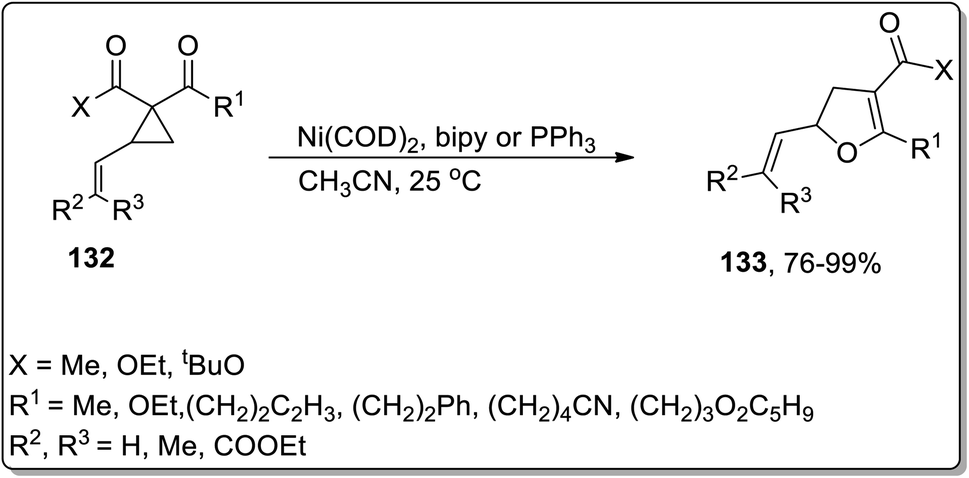
In 2006, Bowman and Johnson reported the Ni-catalyzed Cloke–Wilson ring enlargement of vinyl cyclopropyl ketones 132 to accomplish the synthesis of substituted dihydrofurans 133 in acetonitrile. The substituted dihydrofurans 133 were obtained in good to excellent yields. In order to ensure the chirality transfer, enantioenriched substituted dihydrofurans were synthesized in excellent yield by keeping the original configuration intact at the vinyl-substituted chiral center. This novel methodology is useful due to low catalyst loading and short reaction time along with various functional groups tolerance (Scheme 41).7a
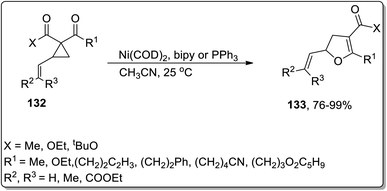 |
| | Scheme 41 Synthesis of substituted dihydrofurans 133 via Cloke–Wilson rearrangement. | |
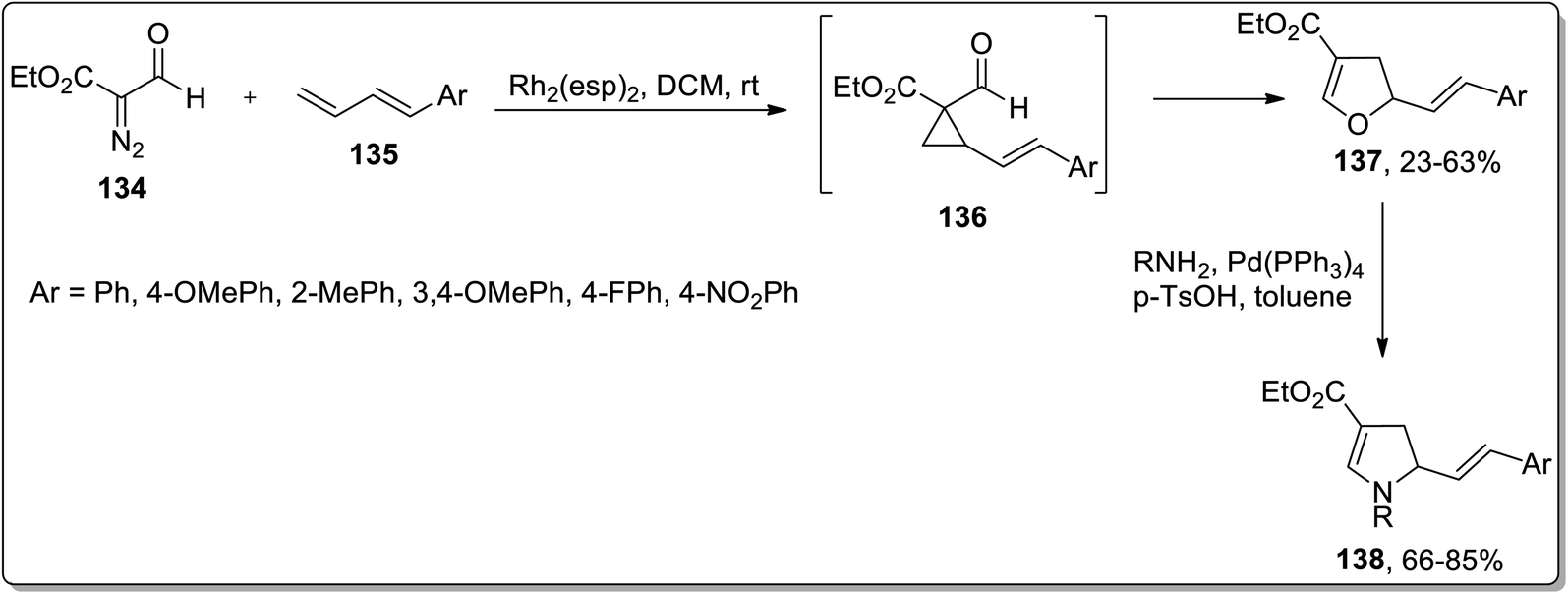
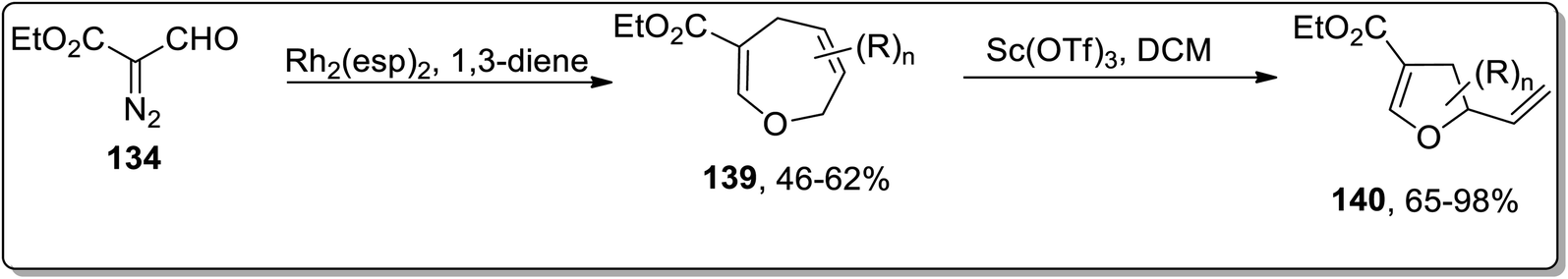
In 2018, Piotrowski and Kerr reported the rhodium-mediated cyclopropanation of 1,3-dienes 135 with ethyl 2-formyldiazoacetate 134, which was proceeded further via Cloke–Wilson rearrangement to give dihydrofurans 137 or dihydrooxepines 138. The Rh-catalyzed cascade cyclopropanation and Cloke–Wilson rearrangement of aryl-substituted butadienes 135 with ethyl 2-formyldiazoacetate 134 gave dihydrofurans 137 in moderate yields, while the dihydrooxepine 139 scaffolds were obtained when the simple 1,3-butadiene was used instead. Furthermore, dihydrofurans 137 were converted to dihydropyrroles 138 through Pd-catalyzed oxygen to nitrogen transposition (Scheme 42).78 Dihydrooxepines 139 were subjected to Lewis-acid, i.e., Sc(OTf)3-induced rearrangement involving 1,3-oxygen migration to attain dihydrofurans 140 in moderate to excellent yields (Scheme 43).41a
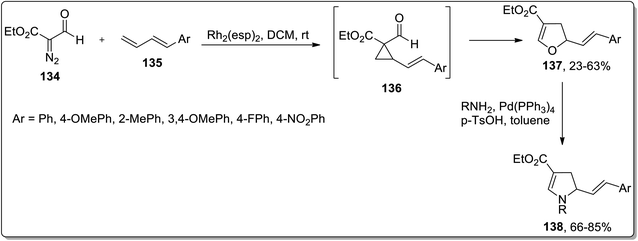 |
| | Scheme 42 Synthesis of dihydropyrroles 138 via Cloke–Wilson rearrangement. | |
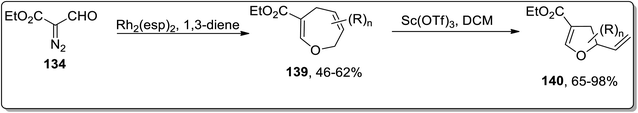 |
| | Scheme 43 Synthesis of dihydrofurans 140 via Cloke–Wilson rearrangement. | |
6.7. Organo-catalytic/mediated rearrangement
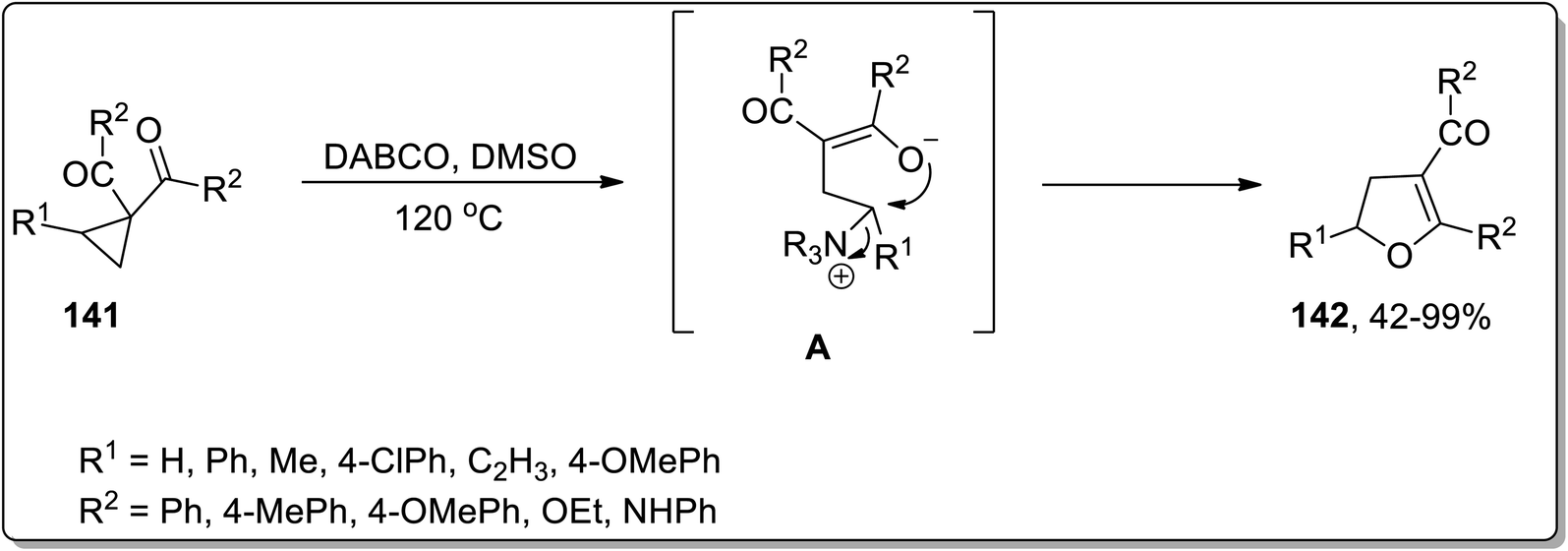
In 2017, Xu and co-workers designed the development of 2,3-dihydrofurans 142 by an organo-catalyzed DABCO-promoted Cloke–Wilson ring enlargement of cyclopropyl ketones 141. In this transformation, the nucleophilic attack of DABCO on cyclopropane led to the formation of an enolate intermediate A, which further underwent cyclization to produce the desired products 142 and released the catalyst. These established conditions for the rearrangement reaction (such as DABCO in DMSO at 120 °C) are compatible for a wide array of substrates, thereby giving the desired products in fair to excellent yields with complete regioselectivity (Scheme 44).44a
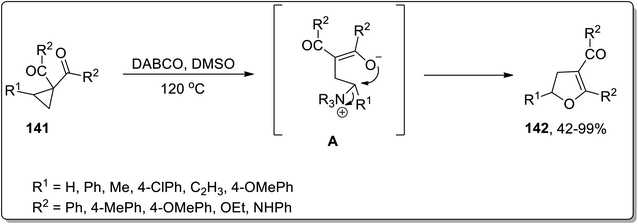 |
| | Scheme 44 Synthesis of 2,3-dihydrofurans 142 via Cloke–Wilson rearrangement. | |
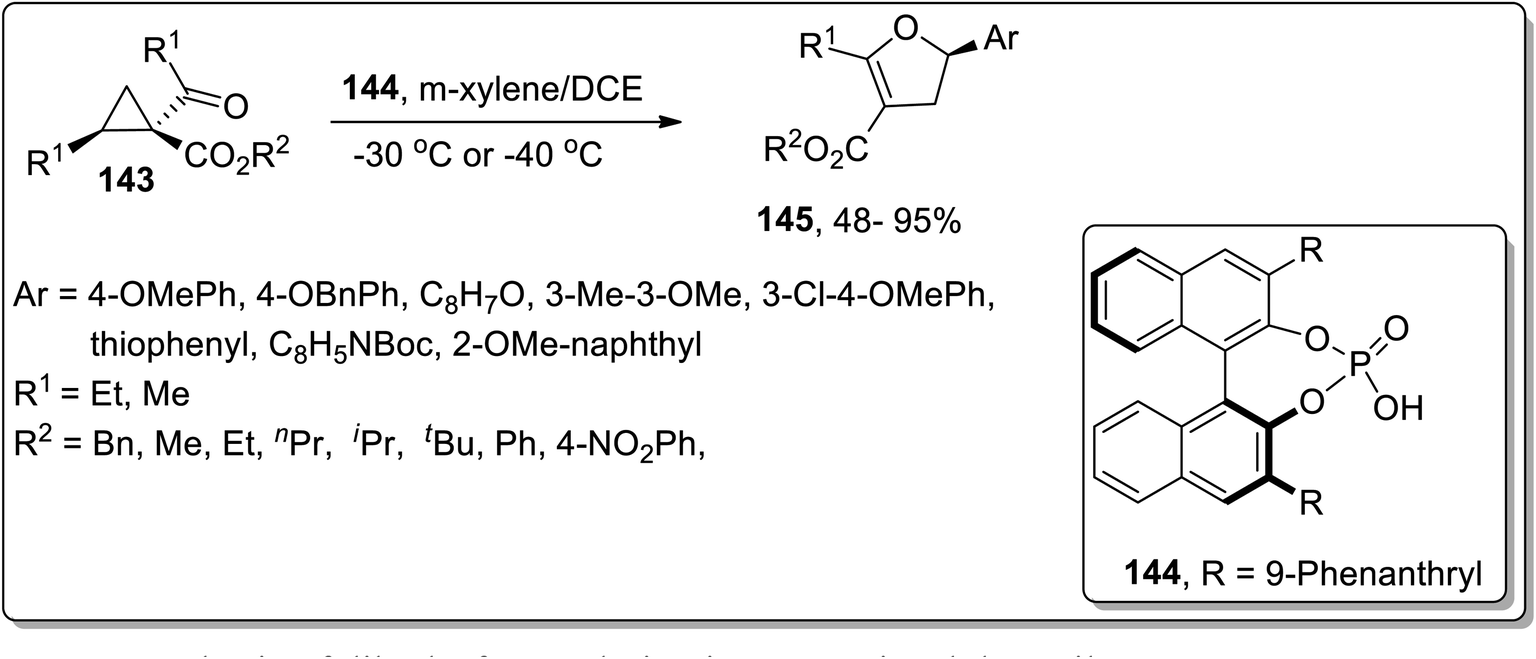
In 2018, Vicario and co-workers discussed the chiral phosphoric acid-catalyzed enantioselective Cloke–Wilson rearrangement of substituted cyclopropanes 143 to achieve the synthesis of dihydrofuran scaffolds 145. During this transformation, the D–A cyclopropanes 143 were activated by chiral phosphoric acid-catalyst 144 that promoted the cleavage of the ring to result in carbocationic intermediate, which upon further cyclization gave dihydrofuran derivatives 145 in fair to excellent yields. The established conditions are suitable to get the corresponding dihydrofurans in high yields with remarkable enantioselectivity. The size of the R3-substituent significantly affected the product yield, such that in the presence of sterically hindering tert-butyl ester, the reaction did not take place (Scheme 45).44b
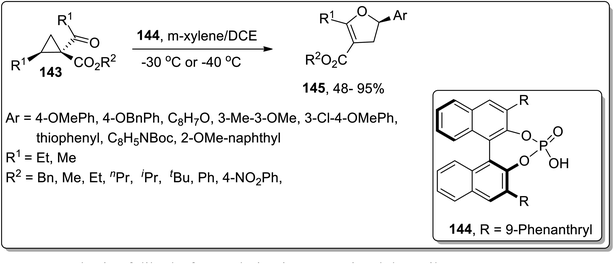 |
| | Scheme 45 Synthesis of dihydrofurans derivatives 145 via Cloke–Wilson rearrangement. | |
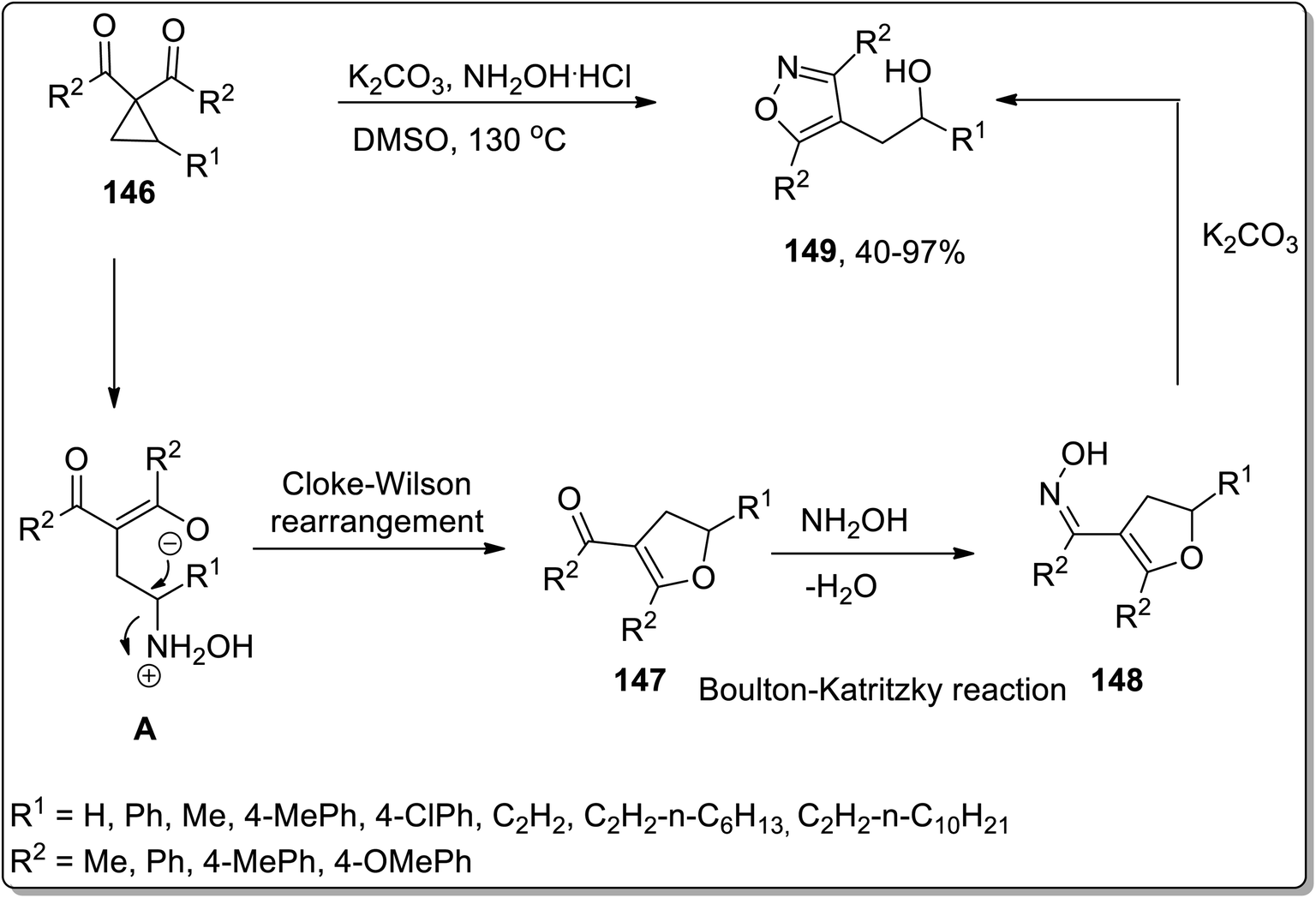
In 2018, Xu and co-workers reported the hydroxylamine-mediated one-pot cascade Cloke–Wilson rearrangement/Boulton–Katritzky reaction of cyclopropylketones 146 to afford the synthesis of fully substituted isoxazoles 149 utilizing hydroxylamine as a catalyst for Cloke–Wilson rearrangement. However, in the Boulton–Katritzky reaction, it was employed as the reactant. Substituted isoxazoles 149 were obtained in fair to excellent yields. This strategy was also applied to synthesize pyrazoles (biologically active scaffolds79) and tricyclic heterocycles; both were accomplished in efficient yields (Scheme 46).44c
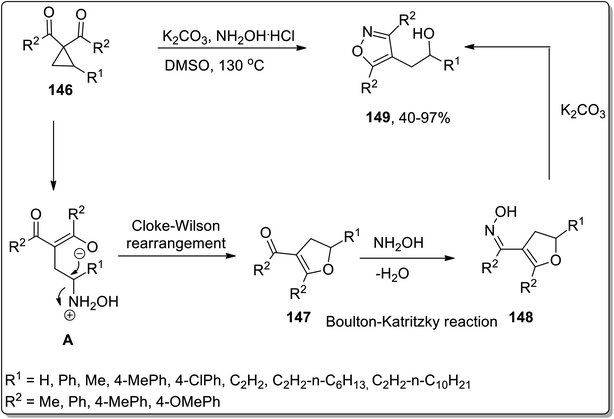 |
| | Scheme 46 Synthesis of substituted isoxazoles 149 via Cloke–Wilson rearrangement. | |
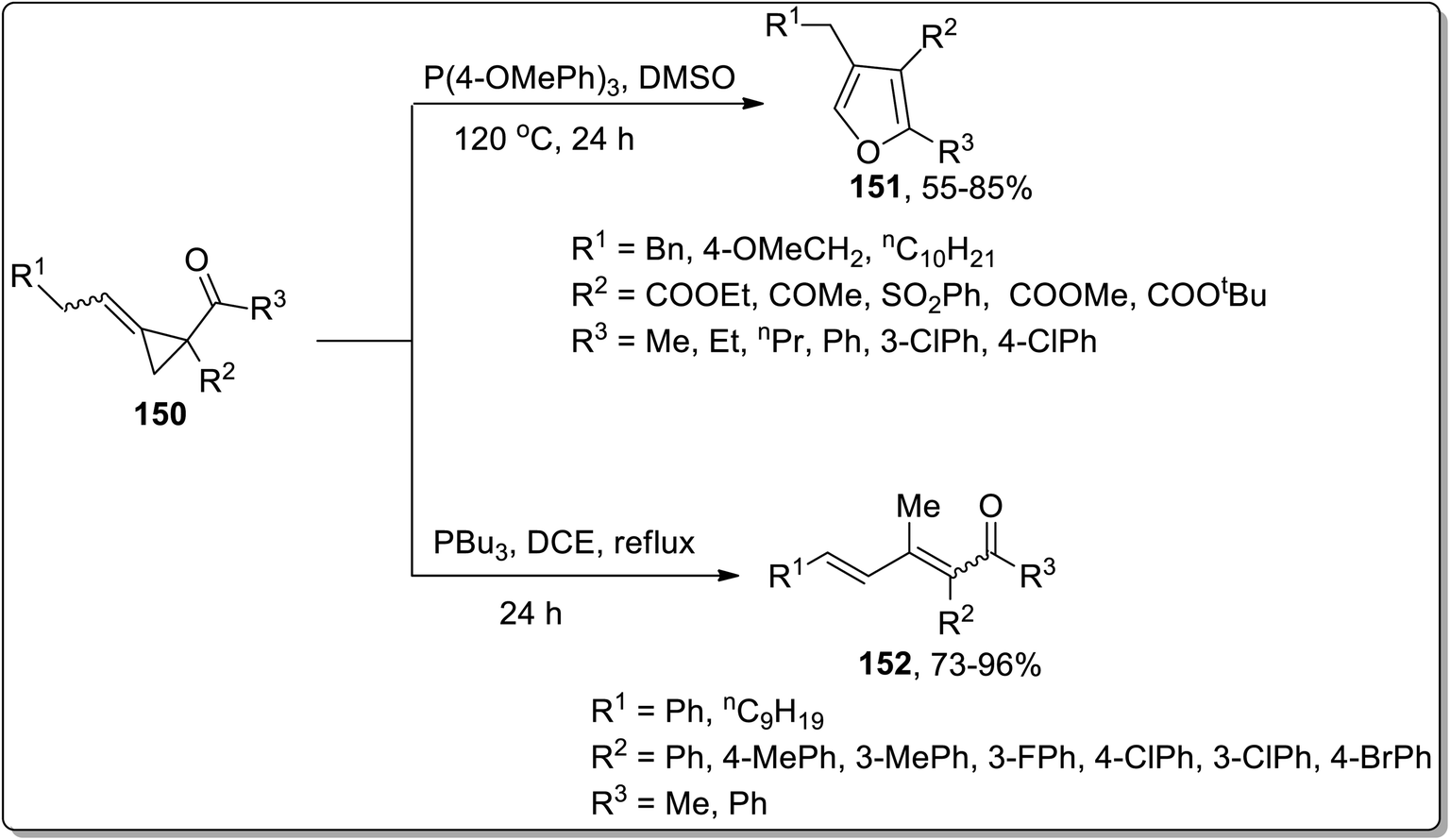
In 2019, Xu and co-workers performed the phosphine-catalyzed synthesis of 2,3,4-trisubstituted furans 151, fully substituted furans, and 1,2,4-trisubstituted dienones 152 from electron-deficient alkylidenecyclopropanes 150. In this transformation, the initial nucleophilic attack of phosphine cleaved the alkylidenecyclopropane ring 150, followed by the subsequent release of phosphine via intramolecular SN2 reaction to generate 2,3,4-trisubstituted furans 151 (when R2 was employed as the electron withdrawing group). However, using aryl groups as R2 substitution, the intramolecular 1,4-proton transfer reaction was processed, followed by the 1,4-elimination of the phosphonium to achieve heat-resistant 1,2,4-trisubstituted dienones 152 (Scheme 47).44d
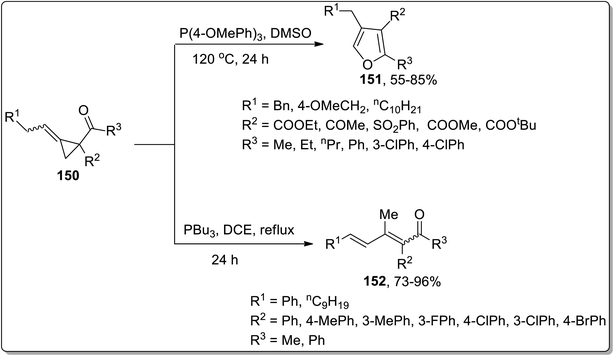 |
| | Scheme 47 Synthesis of 2,3,4-trisubstituted furans 151 via Cloke–Wilson rearrangement. | |
7. Synthesis of N-heterocycles
7.1. Thermal rearrangement
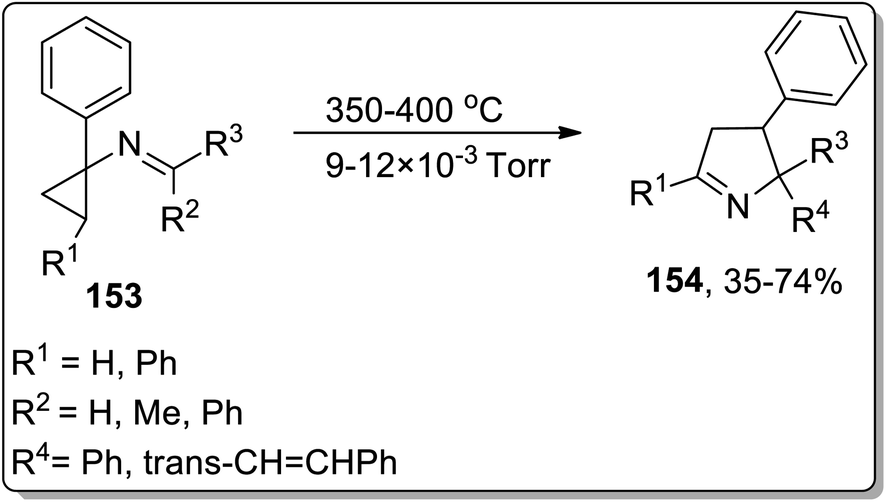
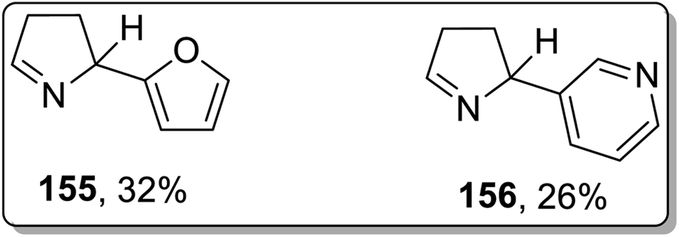
In 2002, Campos and co-workers proposed the thermal Cloke–Wilson rearrangement of N-cyclopropylimines 153 to generate 1-pyrroline derivatives 154. The reaction proceeded successfully within the temperature range of 350–400 °C regiospecifically in the presence of various substituents, i.e., alkyl, alkenyl, and aryl. Furan derivative 155 and nornicotine precursor 156 were also synthesized in 32% and 26% yields, respectively, at 350 °C under 20 torr pressure (Scheme 48 and Fig. 2).8c
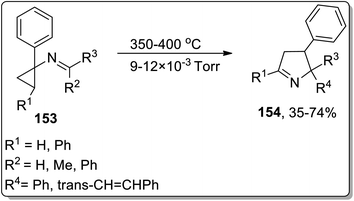 |
| | Scheme 48 Synthesis of 1-pyrroline derivatives 154 via Cloke–Wilson rearrangement. | |
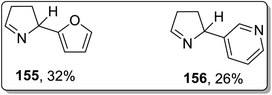 |
| | Fig. 2 Structures of furan derivative 155 and nornicotine precursor 156. | |
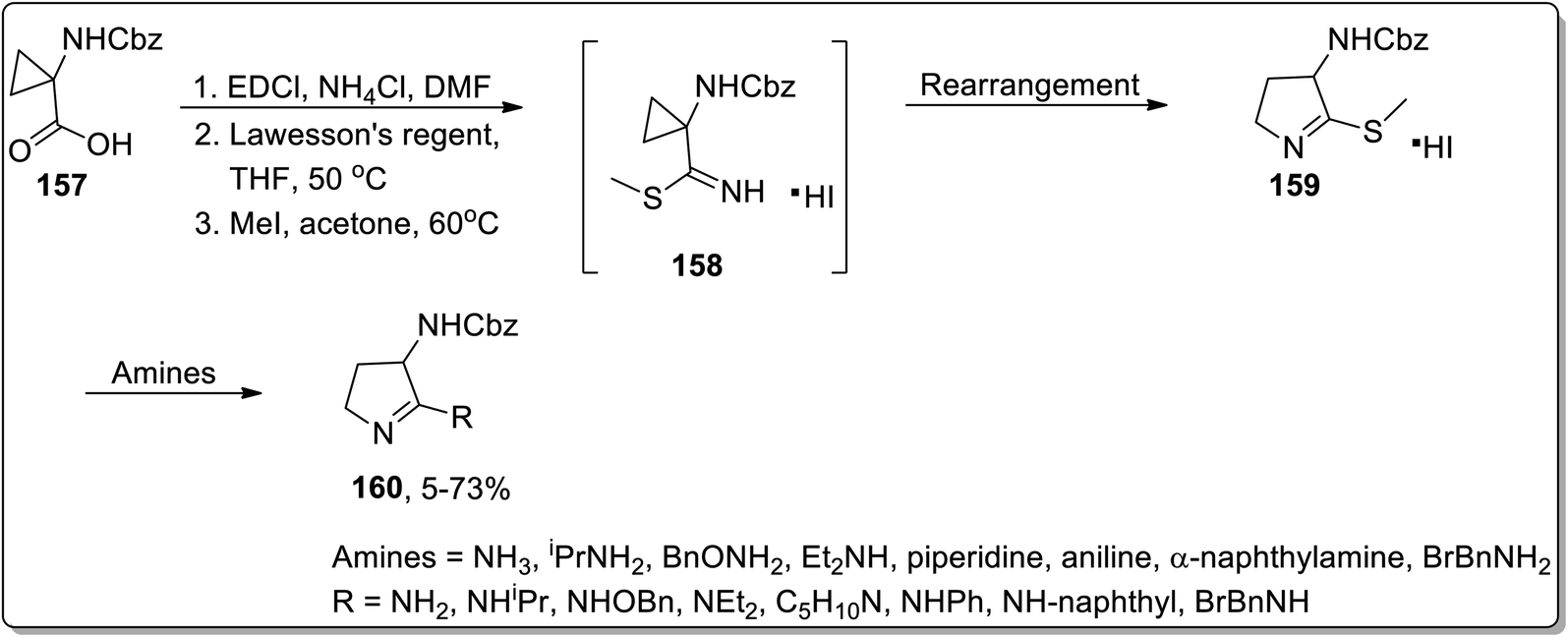
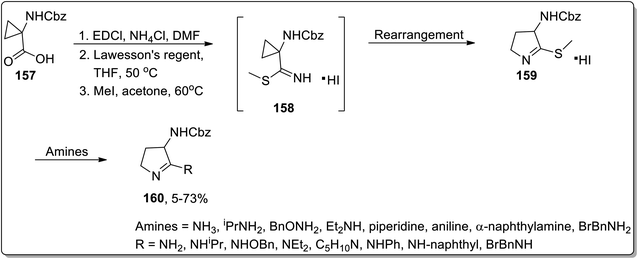
In 2003, Kuduk and coworkers designed the synthesis of 2,3-diaminodihydropyrroles 160 by thermal cyclopropyl thioimidate 157 rearrangement. 2,3-Diaminodihydropyrroles 160 formation was achieved by carrying out three steps, which involved the preparation of imidate 158, rearrangement, and displacement of amine. The Cloke–Wilson rearrangement of thiomethylimidate cyclopropane 157 was the key step in the developed protocol, leading to the formation of pyrrolothiomethylimidate intermediate 160. Diverse range of amines such as ammonia, primary amines, and weakly nucleophilic amine reacted efficiently with pyrroloimidate 159 (Scheme 49).80
 |
| | Scheme 49 Synthesis of 2,3-diaminodihydropyrroles 160 via Cloke–Wilson rearrangement. | |
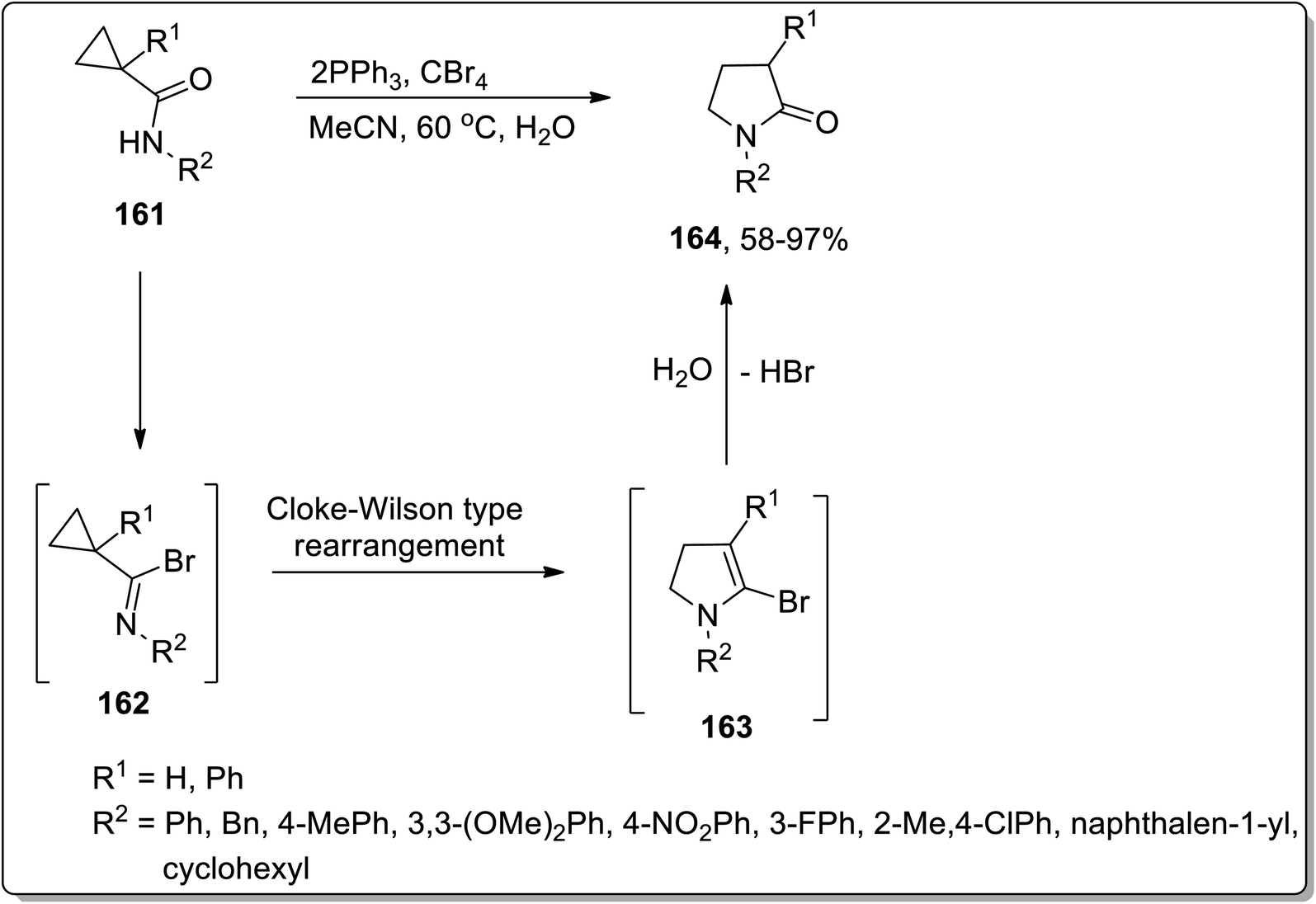
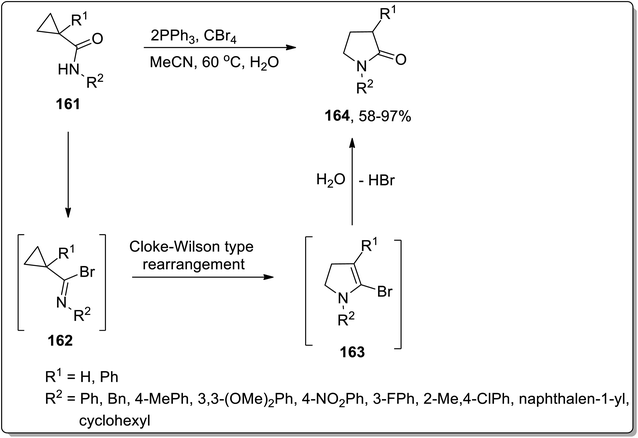
In 2005, Shi and Yang reported the Cloke–Wilson rearrangement of cyclopropyl amides 161 to generate N-substituted pyrrolidin-2-ones 164 in good to excellent yields via the ring expansion of in situ generated imidoyl halides intermediate 162. Intermediate 162, consisting of two electron withdrawing groups, facilitated the ring expansion via thermal Cloke–Wilson-type rearrangement to generate the next intermediate 163, which upon the elimination of bromine by hydroxyl group yielded N-substituted pyrrolidin-2-ones 164 (Scheme 50).81
 |
| | Scheme 50 Synthesis of N-substituted pyrrolidin-2-ones 164 via Cloke–Wilson rearrangement. | |
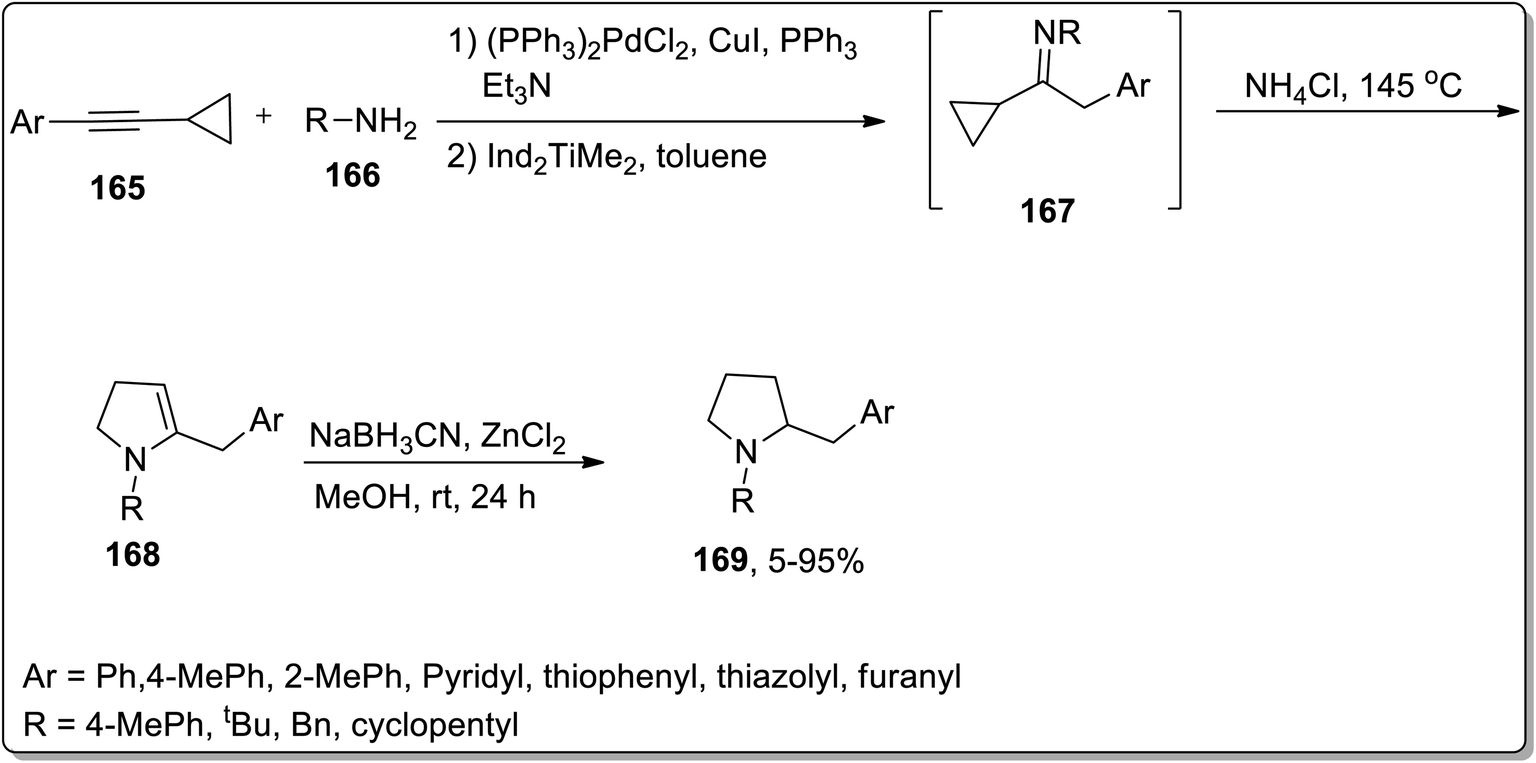
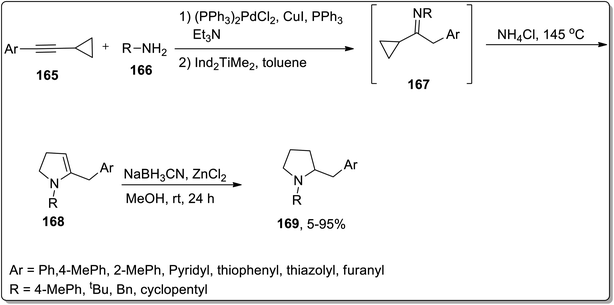
In 2009, Doye and coworkers designed the one-pot generation of N-substituted 2-(arylmethyl)pyrrolidines 169 in good yields by treating aryl-substituted cyclopropylalkynes 165 and primary amines 166 via Cloke–Wilson rearrangement. The reaction was initiated with the generation of cyclopropylimine 167 via the regioselective hydroamination of the aryl-substituted cyclopropyl moiety using[Ind2TiMe2] as the catalyst. Later on, intermediate 167 underwent heat-induced Cloke–Wilson rearrangement to produce 2-pyrrolines 168 by employing ammonium chloride catalyst. 2-Pyrrolines 168 were further reduced with NaBH3CN/ZnCl2 to give the target functionalized pyrrolidines 169 with up to 90% yields (Scheme 51).82
 |
| | Scheme 51 Synthesis of functionalized pyrrolidines 169 via Cloke–Wilson rearrangement. | |
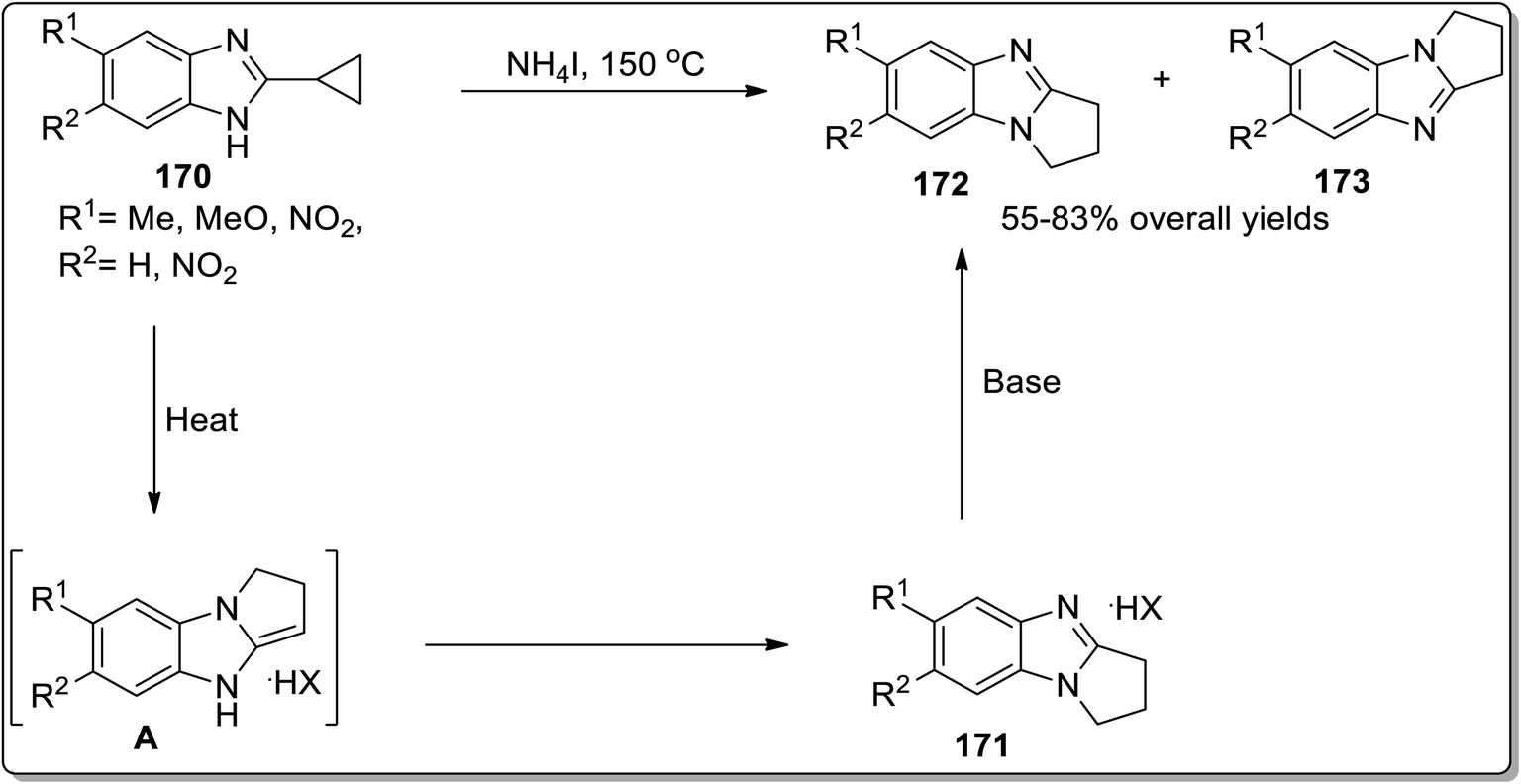
Another example of thermally-induced Cloke–Wilson rearrangement was reported by Tomilov and colleagues to attain the synthesis of condensed azoles 172 and 173 in moderate to good yields from cyclopropylazoles 170 by exploiting the neat conditions. Benzopyrroloimidazoles 174 and benzopyrrolothiazolium salts 175 were afforded in comparatively good yields utilizing this synthetic approach (Scheme 52 and Fig. 3).22b
 |
| | Scheme 52 Synthesis of condensed azoles 172 and 173 via Cloke–Wilson rearrangement. | |
 |
| | Fig. 3 Structures of benzopyrroloimidazoles 174 and benzopyrrolothiazolium salts 175. | |
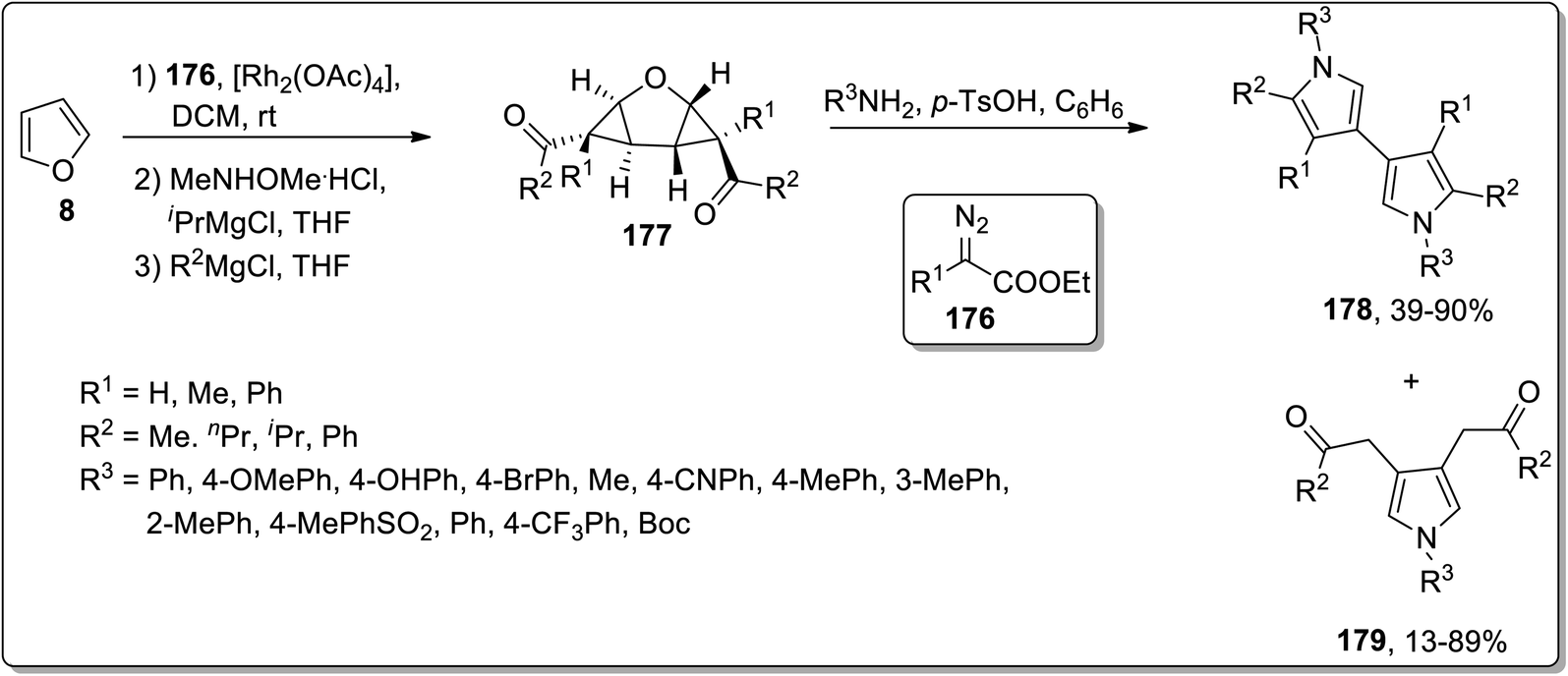
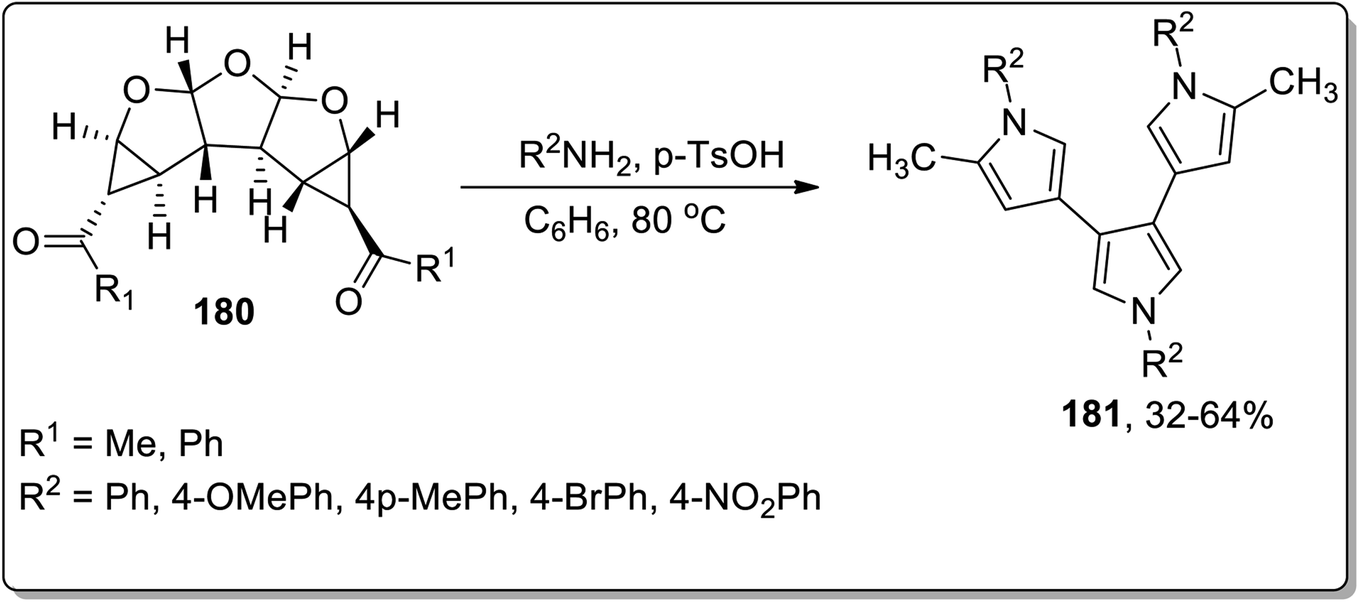
In 2012, Werz and coworkers demonstrated the synthesis of bispyrroles 178 and oligopyrroles 179. For this purpose, diketones-substituted cyclopropanes with furan core 181 were obtained from furan 8 by treating it with substituted imines 176, followed by reaction with Weinreb amides to yield tricyclic diketones 177. Diketones 177 were then treated with substituted amines and p-TsOH in benzene to afford bispyrroles 178 and 179 with up to 90% yields via Cloke–Wilson rearrangement. When extended oligoacetalic diketones 180 were used as starting materials, oligopyrroles 181 were accessed in a single step in moderate yields. With the increasing number of pyrrole units, the synthetic yield of target molecules went downhill due to the intrinsic instability of these oligopyrroles (Schemes 53 and 54).83
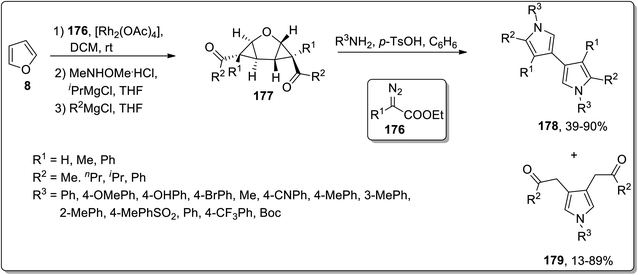 |
| | Scheme 53 Synthesis of bispyrroles 178 and oligopyrroles 179 via Cloke–Wilson rearrangement. | |
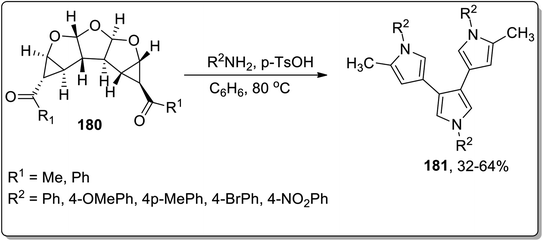 |
| | Scheme 54 Synthesis of oligopyrroles 181 via Cloke–Wilson rearrangement. | |
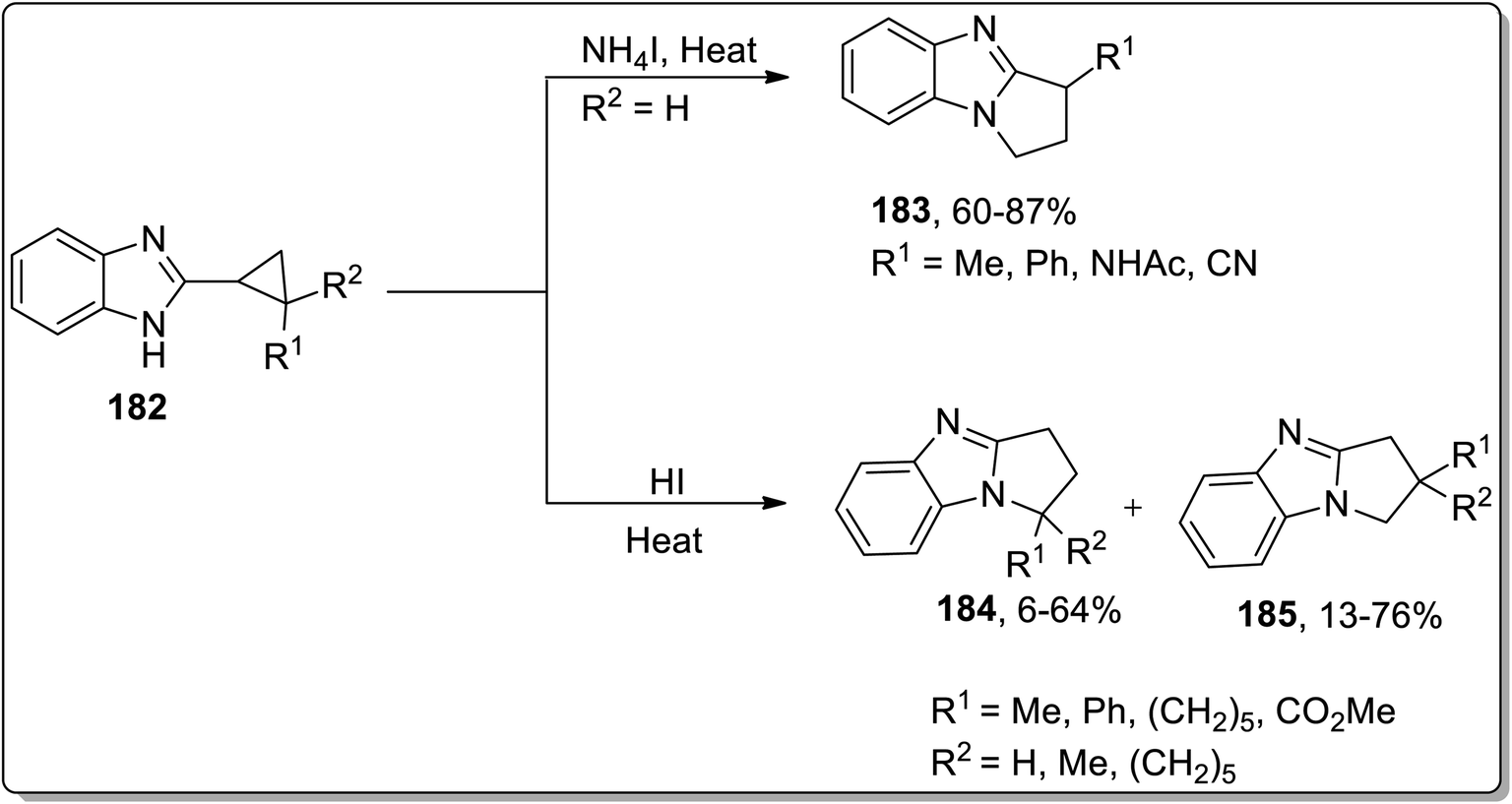
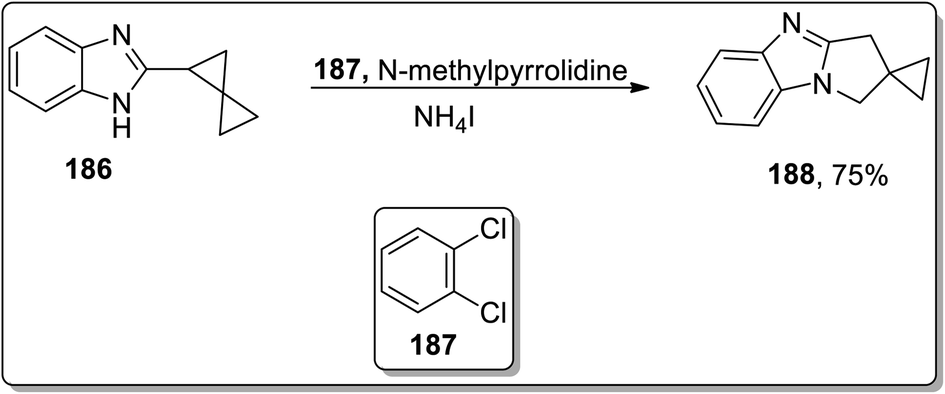
In 2013, Tomilov and coworkers performed the synthesis of substituted pyrrolidine-based benzimidazoles 183 via thermal Cloke–Wilson cyclopropyliminium rearrangement. The 2-cyclopropylbenzimidazoles 182 bearing substituent at the C1 of the cyclopropane ring resulted in C3-substituted 2,3-dihydropyrrolo[1,2-a]benzimidazoles 183, while the substituent at position 2 of cyclopropane 184 led to the formation of a mixture of isomers 184 and 185 because the cyclopropane ring-opening proceeded via the cleavage of less or more substituted bonds. The ratio of both the isomers yield was observed to be dependent upon the polarity of the solvent. The polar solvent promoted the ring cleavage of the more substituted bond while weakly polar solvents cleaved the less substituted bond. In contrast, 2-spiropentylbenzimidazole 186 rearrangement furnished product 188, placing the spiro-cyclopropane fragment at the C2 of pyrrolo-benzimidazoles 188 regioselectively (Schemes 55 and 56).84
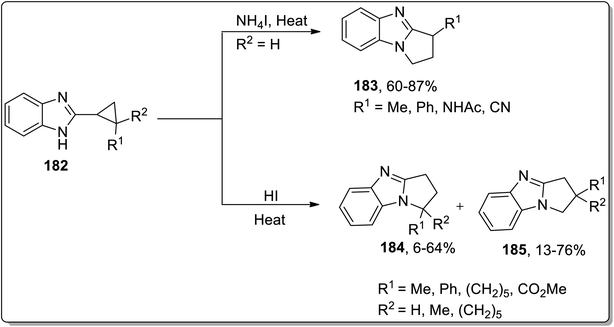 |
| | Scheme 55 Synthesis of benzimidazoles 183, mixture of isomers 184 and 185 via Cloke–Wilson rearrangement. | |
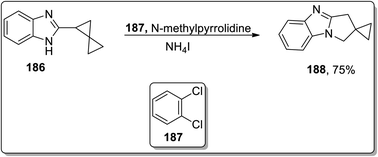 |
| | Scheme 56 Synthesis of pyrrolo-benzimidazoles 188 via Cloke–Wilson rearrangement. | |
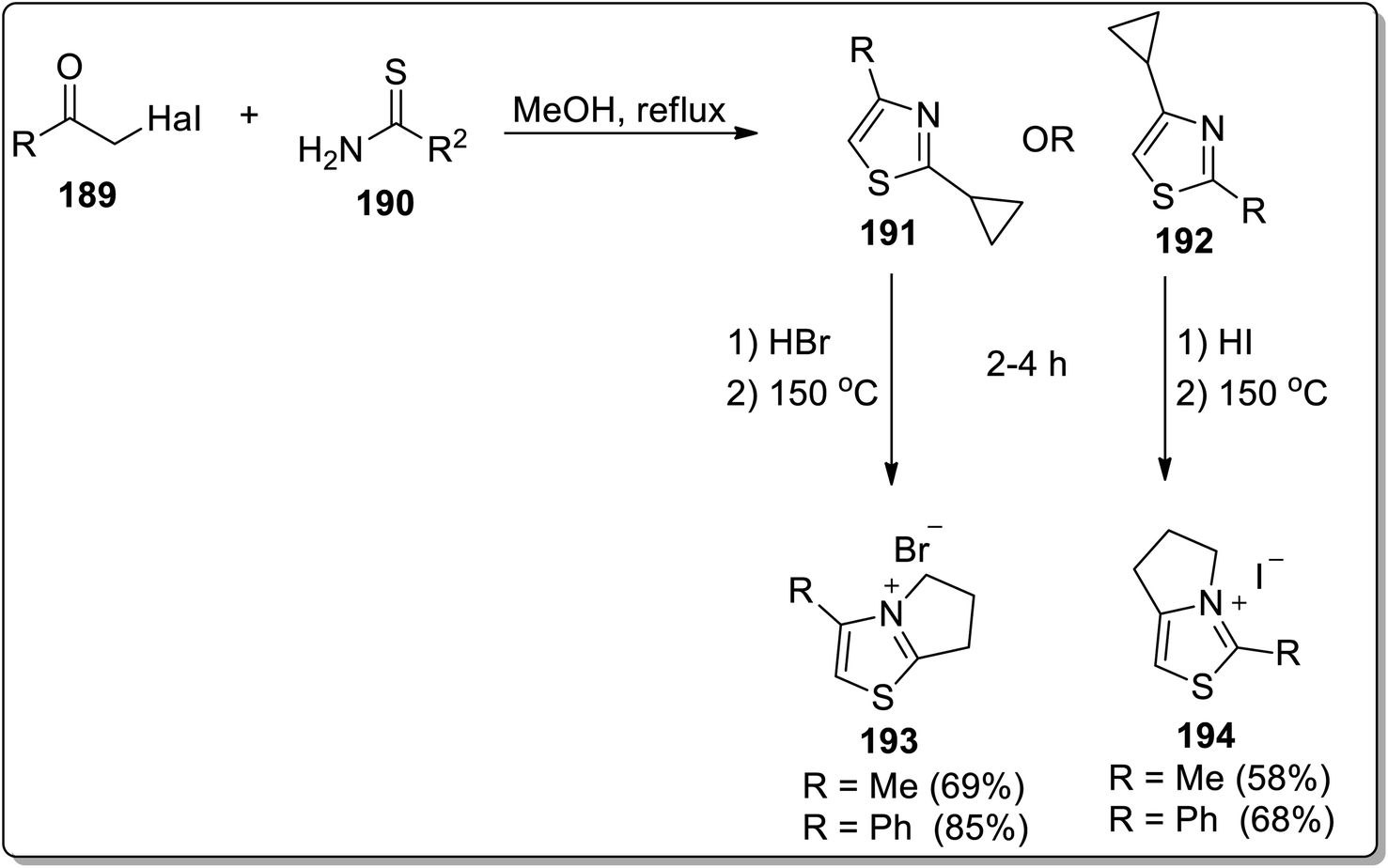
In 2013, Tomilov and colleagues designed the synthesis of fused heterocycles, i.e., pyrrolo-thiazolium bromides 193 and pyrrolo-thiazolium iodides 194 by subjecting 2-cyclopropylthiazole 191/192 with hydrobromides/iodides, respectively, followed by heat-induced Cloke–Wilson cyclopropyliminium rearrangement reaction at 150 °C (Scheme 57).85
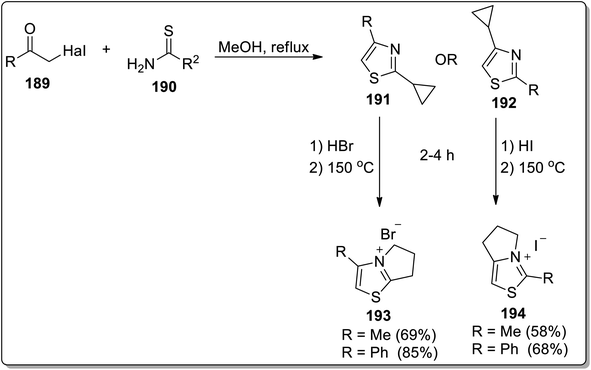 |
| | Scheme 57 Synthesis of pyrrolo-thiazolium bromides 193 and pyrrolo-thiazolium iodides 194 via Cloke–Wilson rearrangement. | |
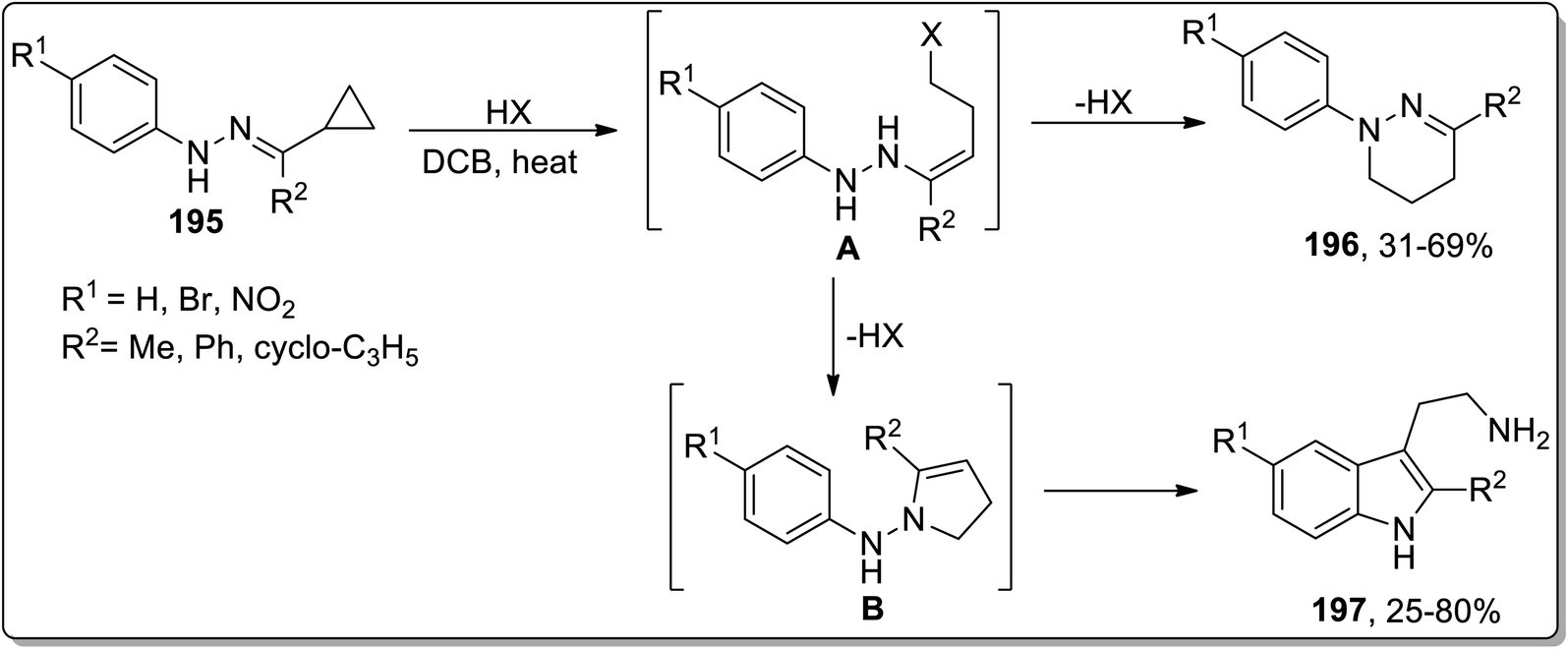
Another thermal Cloke–Wilson rearrangement was reported by Tomilov and colleagues to synthesize a mixture of two possible products, i.e., a six-membered tetrahydropyridazines 196 and tryptamines 197 by reacting cyclopropylketone arylhydrazones 195. The protonation at nitrogen atom facilitated the ring-opening of cyclopropane to generate halides A that either followed ring-closure, thereby giving tetrahydropyridazines 196, or followed Cloke rearrangement to generate pyrroline intermediate B. The intermediate B upon subsequent domino Steve–Grandberg rearrangement furnished tryptamines 197 in 25–80% yields. The ratio of the product decided the type of the precursor hydrazones (Scheme 58).86
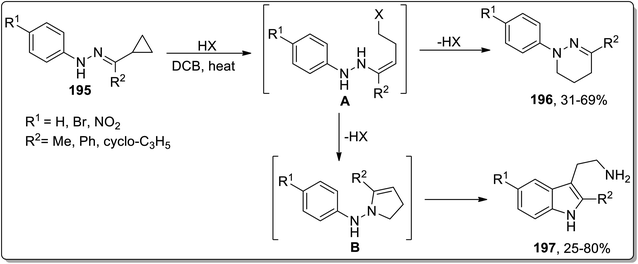 |
| | Scheme 58 Synthesis of tetrahydropyridazines 196 and tryptamines 197 via Cloke–Wilson rearrangement. | |
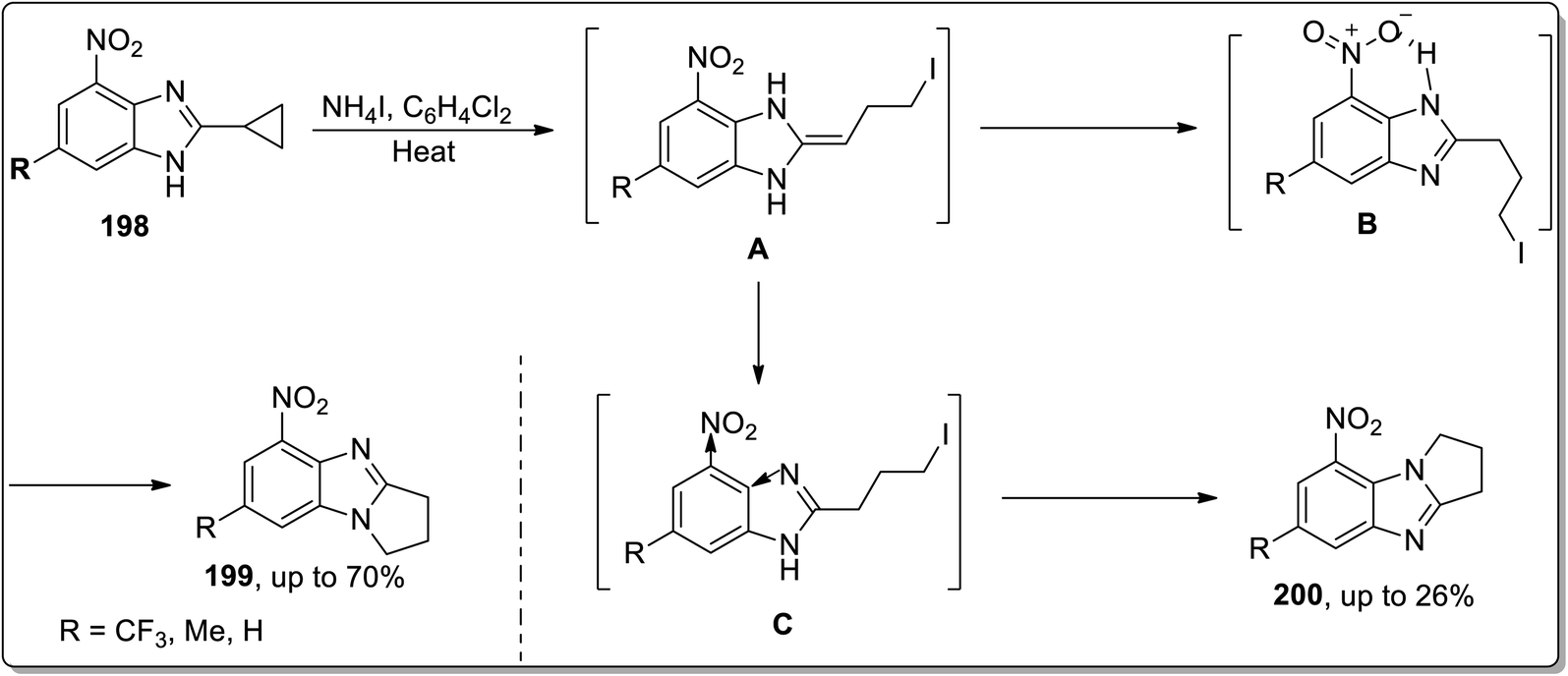
In 2014, Tomilov and co-workers depicted the regioselective synthesis of two isomers of dihydro-pyrrolo-benzimidazoles 199 and 200 by the thermally-promoted Cloke–Wilson reaarangement of benzimidazole-substituted cyclopropyl scaffolds 198. These isomers were obtained by dominating one isomer over the other. Initially, cyclopropyl ring opening gave intermediate A, which was immediately transformed to intermediate C and B by proton migration. Later, both were cyclized to give 200 and 199, respectively. Intermediate B was stabilized as a result of H-bonding interaction with the nitro group, which ensured its higher concentration in the reaction than another isomer. On the other hand, the −ve inductive effect of the nitro group is responsible for the lower nucleophilicity of nearby nitrogen and thus decreased the rate of cyclization of intermediate C. This transformation was found to be highly regioselective due to the thermodynamic, electronic, and steric effects (Scheme 59).8d
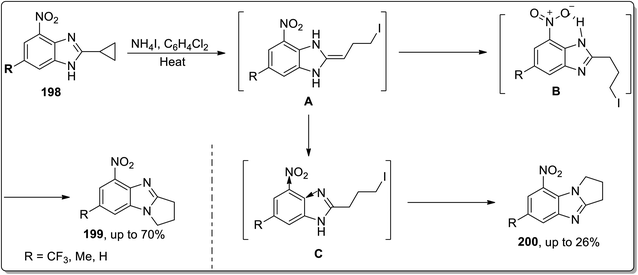 |
| | Scheme 59 Synthesis of dihydro-pyrrolo-benzimidazoles 199 and 200 via Cloke–Wilson rearrangement. | |
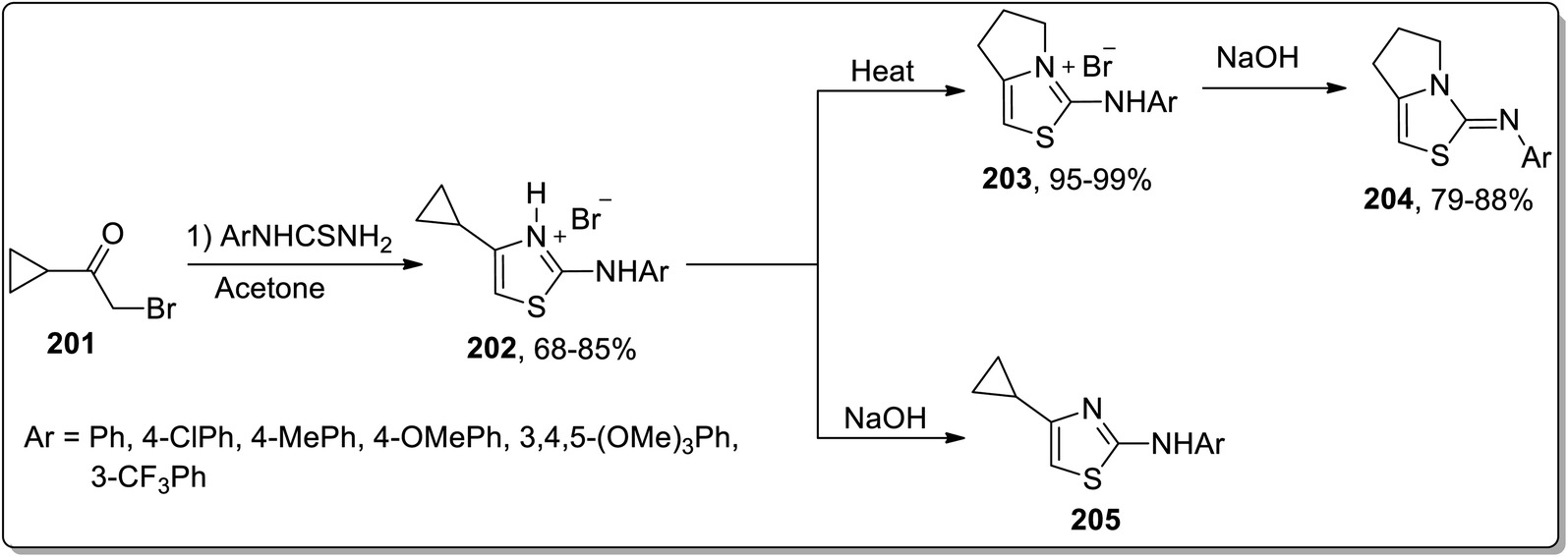
In 2016, Samet and co-workers demonstrated another solvent-free, thermally-promoted Cloke–Wilson rearrangement, leading to the synthesis of fused 2-aryliminothiazolines 204 and cyclopropyl-substituted aminothiazole 205. The hydromides 202 were obtained in a single step by the Hantzch reaction of aryl-substituted thiourea and cyclopropyl ketone 201. They were then treated in two ways. In route 1, they were subjected to Cloke–Wilson rearrangement using heat, which melted to produce pyrrolo-thiazolium bromides 203 in excellent yields, which upon treatment with sodium hydroxide resulted in the elimination of hydrogen bromide, thereby giving iminothiazoline derivatives 204 in good yields. In route 2, hydrobromides 202 were subjected to treatment with sodium hydroxide directly to afford thiazole derivatives 205 (Scheme 60).87
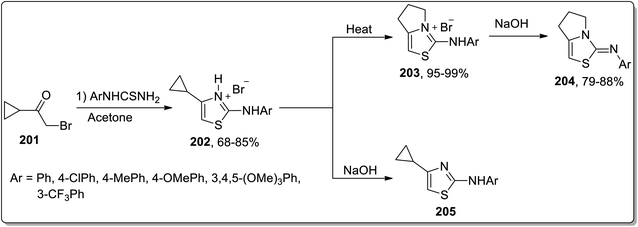 |
| | Scheme 60 Synthesis of iminothiazoline derivatives 204 via Cloke–Wilson rearrangement. | |
7.2. Photo-chemical rearrangement
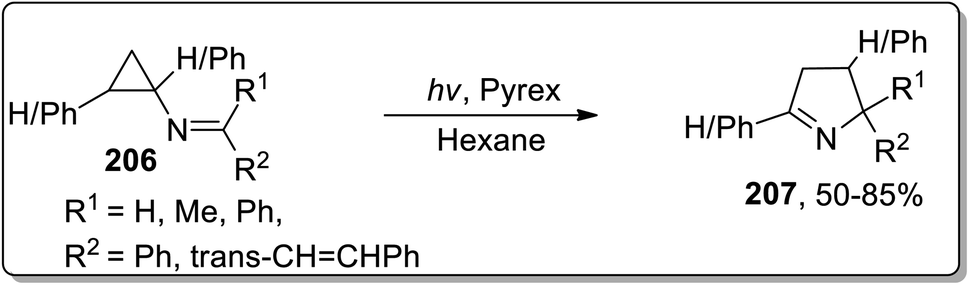
In 2001, Campos and coworkers proposed the synthesis of 1-pyrrolines 207 by photochemical the Cloke–Wilson rearrangement of N-cyclopropylimines 206 in hexane. N-Cyclopropylimines 206 were irradiated via pyrex glass whose absorption was noted to be 290 nm. The product distribution was not generally affected by varying the substituents (Scheme 61).8b
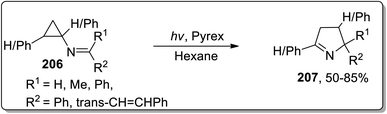 |
| | Scheme 61 Synthesis of 1-pyrrolines 207 via Cloke–Wilson rearrangement. | |
7.3. Brønsted acid-catalyzed/mediated synthesis
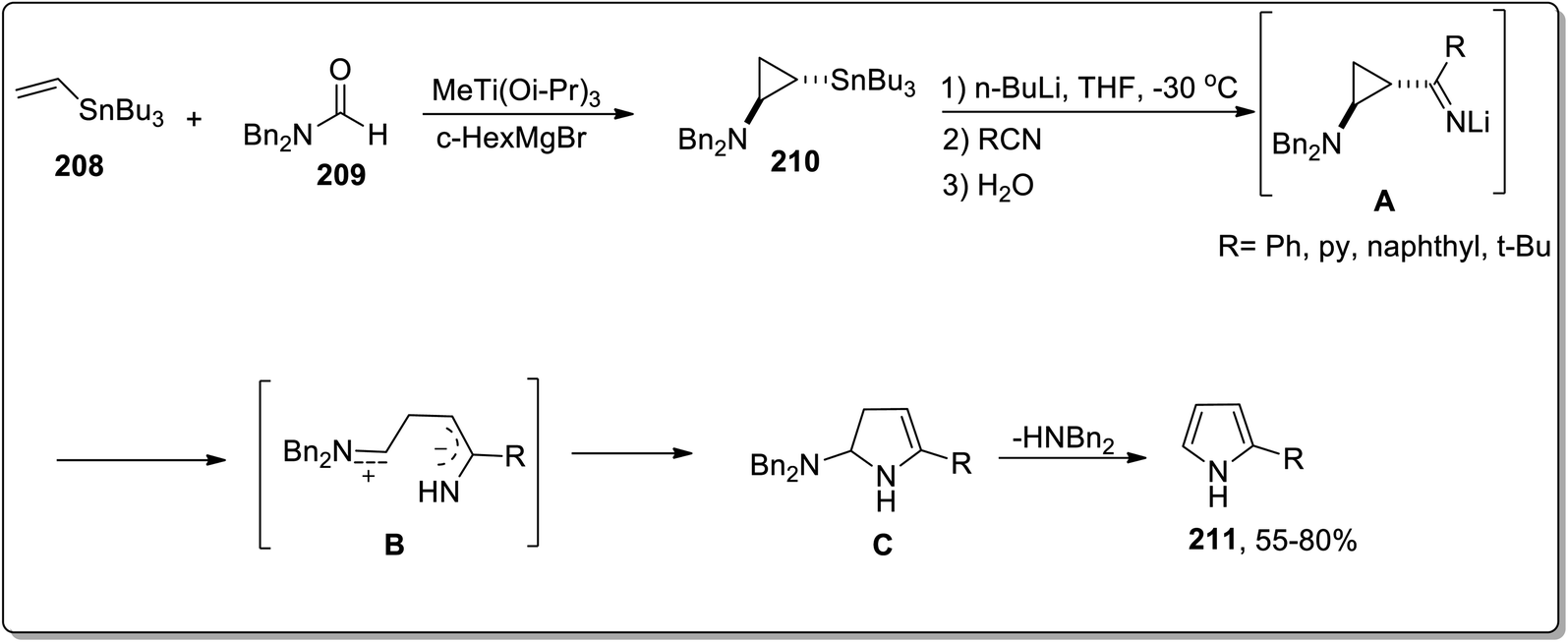
In 2006, Meijere and co-workers designed the one-pot synthesis of pyrrole derivatives 211 by the treatment of 2-lithiated N,N-dibenzylcyclopropylamines 210 (obtained by reacting substituted alkenes 208 with aldehydes 209) with nitriles via the formation of N,N-dibenzylaminocyclopropyl ketimines intermediate A. The ketimines intermediate A in the presence of AcOH underwent protonation to give intermediate B, which was then readily cyclized to an amino-2,3-dihydropyrrole C, followed by the removal of dibenzylamine to generate the substituted pyrroles 215 in moderate to good yields (Scheme 62).88
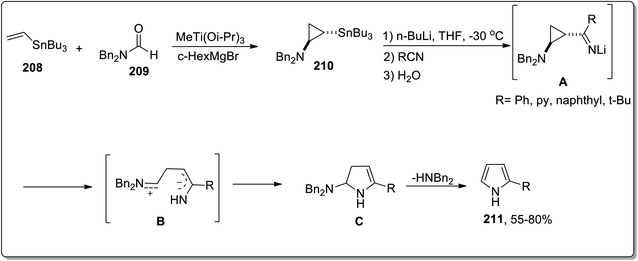 |
| | Scheme 62 Synthesis of pyrrole derivatives 211 via Cloke–Wilson rearrangement. | |
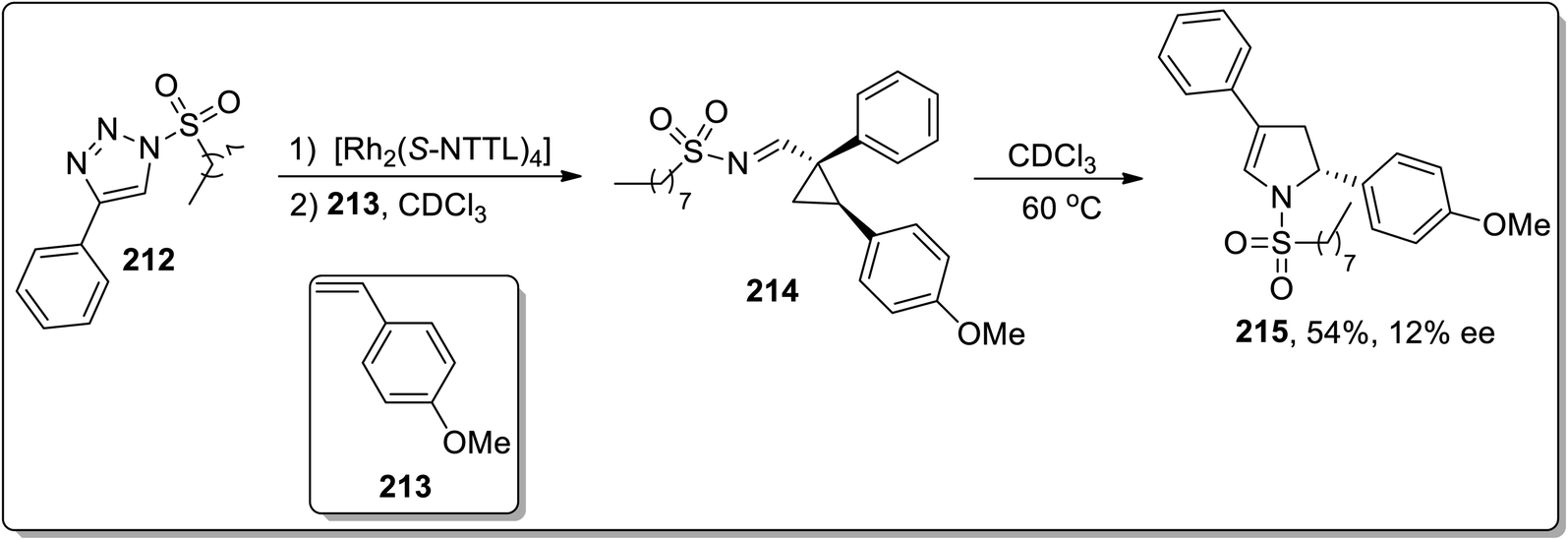
In 2014, Fokin and colleagues designed the one-pot synthesis of enantioenriched 2,3-dihydropyrroles 215 by treating triflated triazoles 212 (which were prepared within the reaction) and olefins 213 involving formation of the triflated cyclopropylaldimine intermediate 214. In this transformation, the 2,6-di-tert-butyl-4-methylpyridinium triflate was exploited as a source of the Brønsted acid, which facilitated the rearrangement of triflated cyclopropylaldimine intermediate 214 to enantioenriched 2,3-dihydropyrroles 215 (Scheme 63).78 This methodology was further explored to achieve the synthesis of 2,3-dihydropyrrole 217 by chiral phosphoric acid-catalyzed cyclopropylimine rearrangement in toluene. When cyclopropylaldimine was heated by employing rhodium catalyst or under catalyst-free conditions, no product formation was observed. This indicated that heat and Rh catalyst did not play any role in cyclopropylaldimine intermediate 214 to dihydropyrrole conversion; actually, the Brønsted acid facilitated this conversion (Scheme 64).89a
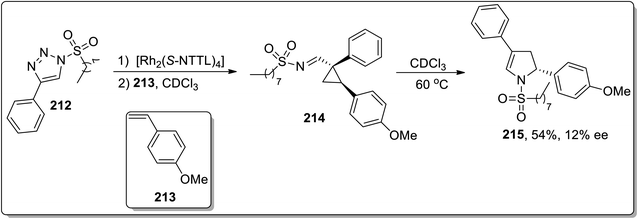 |
| | Scheme 63 Synthesis of 2,3-dihydropyrroles 215 via Cloke–Wilson rearrangement. | |
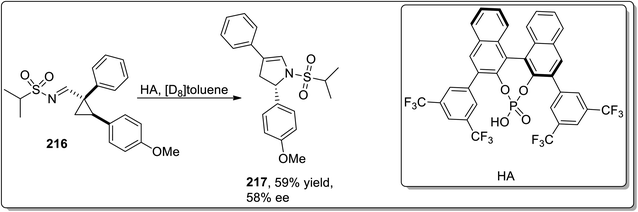 |
| | Scheme 64 Synthesis of 2,3-dihydropyrrole 217 via Cloke–Wilson rearrangement. | |
8. Synthesis of S-heterocycles
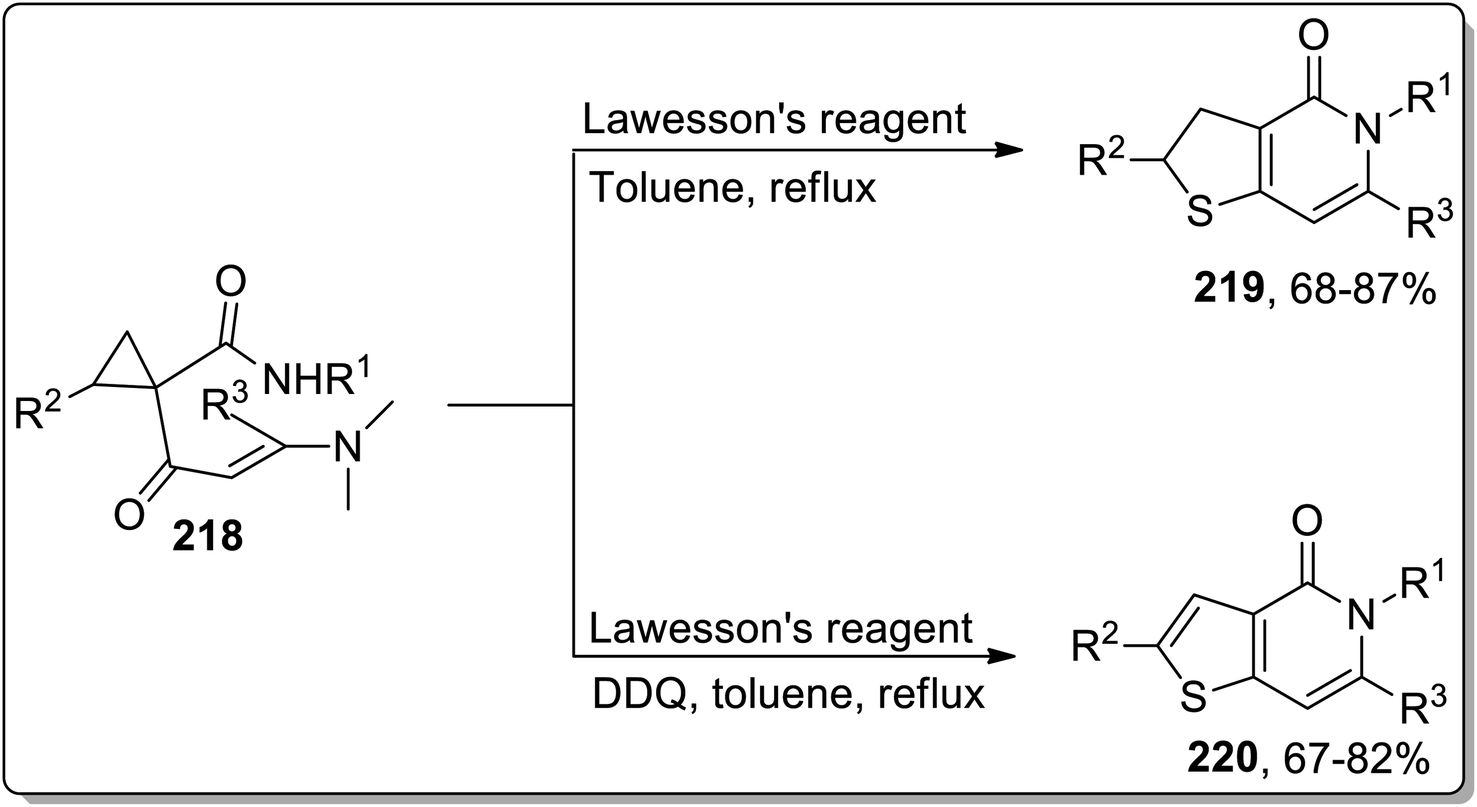
Sulfur-containing heterocycles are of wide medicinal significance and are synthesized by several methods.89b In 2011, the synthesis of S-heterocycles, i.e., thieno-pyridine derivatives 219 and 220, were accomplished by Dong and coworkers via a tandem reaction of dimethylaminopropenoyl cyclopropanes 218 with Lawesson's reagent. This one-pot methodology involved sequential regioselective thionation, thermal Cloke–Wilson rearrangement, and intramolecular aza-cyclization reactions. Dihydrothieno[3,2-c]pyridinones 219 were obtained by utilizing Lawesson's reagent in toluene, while the thieno[3,2-c]pyridin-4(5H)-ones 220 were obtained in moderate to good yields utilizing this protocol in the presence of oxidant, i.e., DDQ (Scheme 65).9a
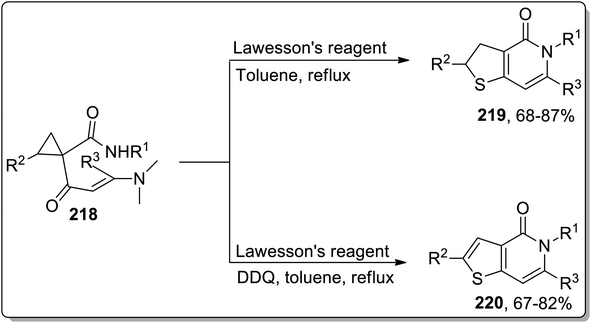 |
| | Scheme 65 Synthesis of thieno-pyridine derivatives 219 and 220 via Cloke–Wilson rearrangement. | |
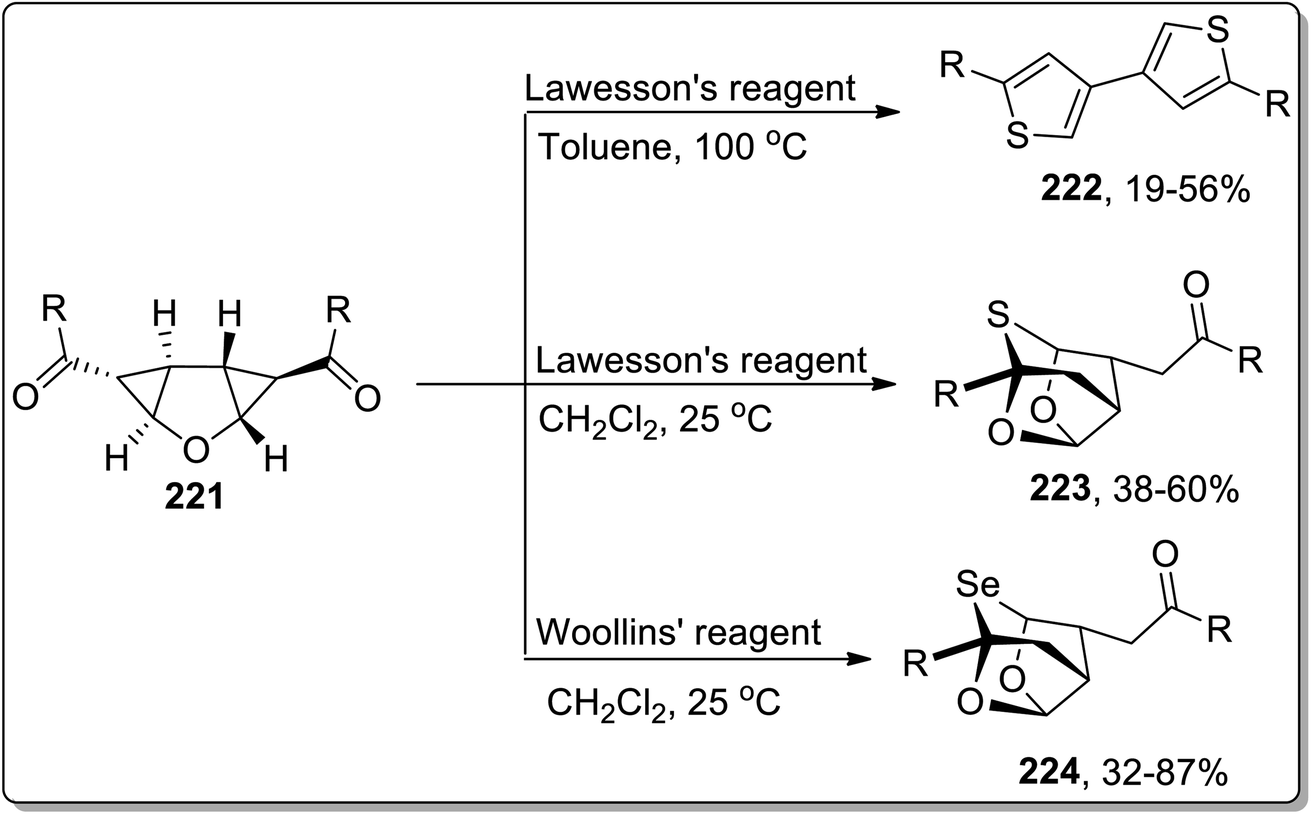
In 2013, Werz and co-workers designed the synthesis of 3,3′-linked bisthiophenes 222. This reaction commenced by treating diketones-substituted cyclopropanes with the furan core in the presence of Lawesson's reagent that produced thioketones, which further underwent thermal Cloke–Wilson ring enlargement, followed by the loss of water to afford 3,3′-linked bisthiophenes 222 up to 56% yields. Intriguingly, when the reaction was carried out employing electron-withdrawing aromatic substituents or at room temperature in CH2Cl2, the “cage-like” structures 223 with a bridging sulfur moiety were obtained in fair yields. Similarly, the high yields of selenium-containing “cage-like” products 224 were managed utilizing Woollins' reagent (Scheme 66).9b
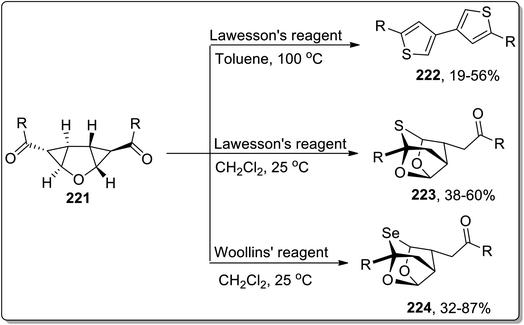 |
| | Scheme 66 Synthesis of bisthiophenes 222, 223, and 224 via Cloke–Wilson rearrangement. | |
9. Application in total synthesis
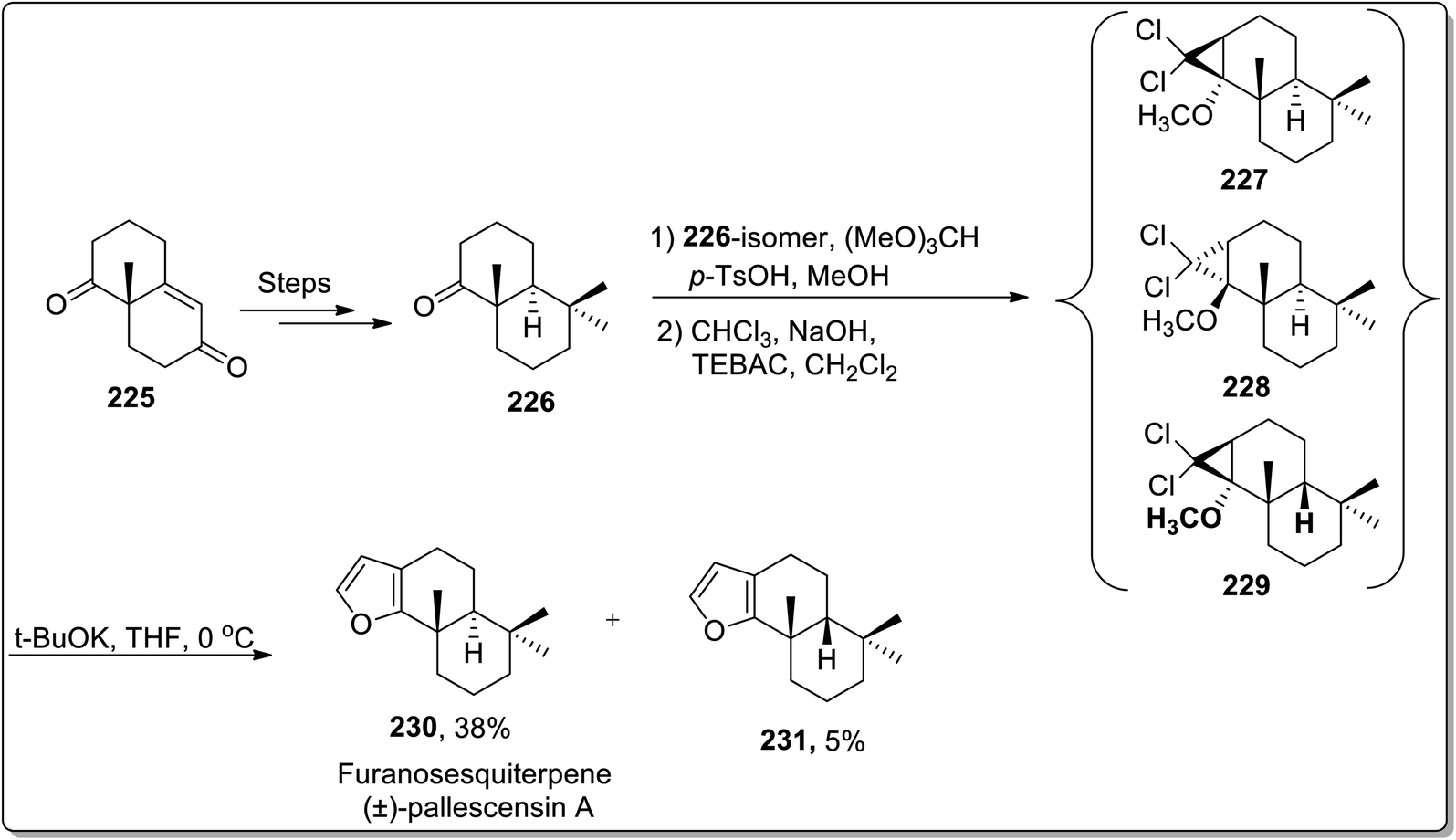
The total synthesis of natural products generally involves various named reactions.90 Cloke–Wilson rearrangement has also found its applications towards the synthesis of various naturally-occurring organic compounds. In 2006, Banwell and co-workers designed the total synthesis of the furanosesquiterpene (±)-pallescensin A 230 by employing Cloke–Wilson rearrangement as the key step. Their synthetic route was initiated by the synthesis of decalone 226, followed by an array of three steps: the reaction of decalone 226 with (MeO)3CH in MeOH exploiting p-TSOH catalyst gave methyl enol ether along with the precursor ketone. Later on, the reaction mixture was treated with dichlorocarbene, which was generated by Makosza's phase transfer conditions, thereby furnishing precursor ketone in 27% with a mixture of dichlorocyclopropanes 227–229. This mixture of dichlorocyclopropanes was made to undergo Cloke–Wilson rearrangement to afford the chromatographically separable mixture of (±)-pallescensin A 230 in 38% yield with its cis-isomer 231 in 5% yield. This synthetic methodology was successfully applied for the conversion of readily available cyclopropanes to annulated furans (Scheme 67).91
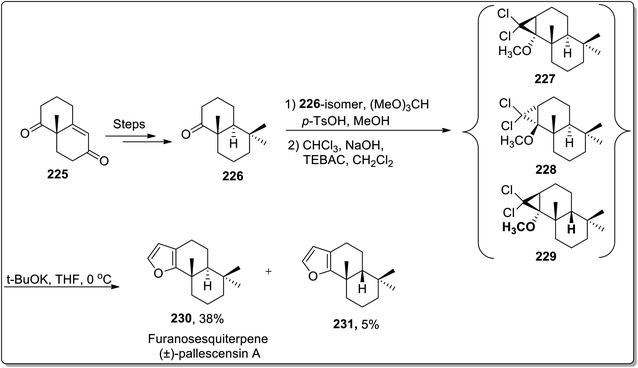 |
| | Scheme 67 Total synthesis of furanosesquiterpene (±)-pallescensin A 230 via Cloke–Wilson rearrangement. | |
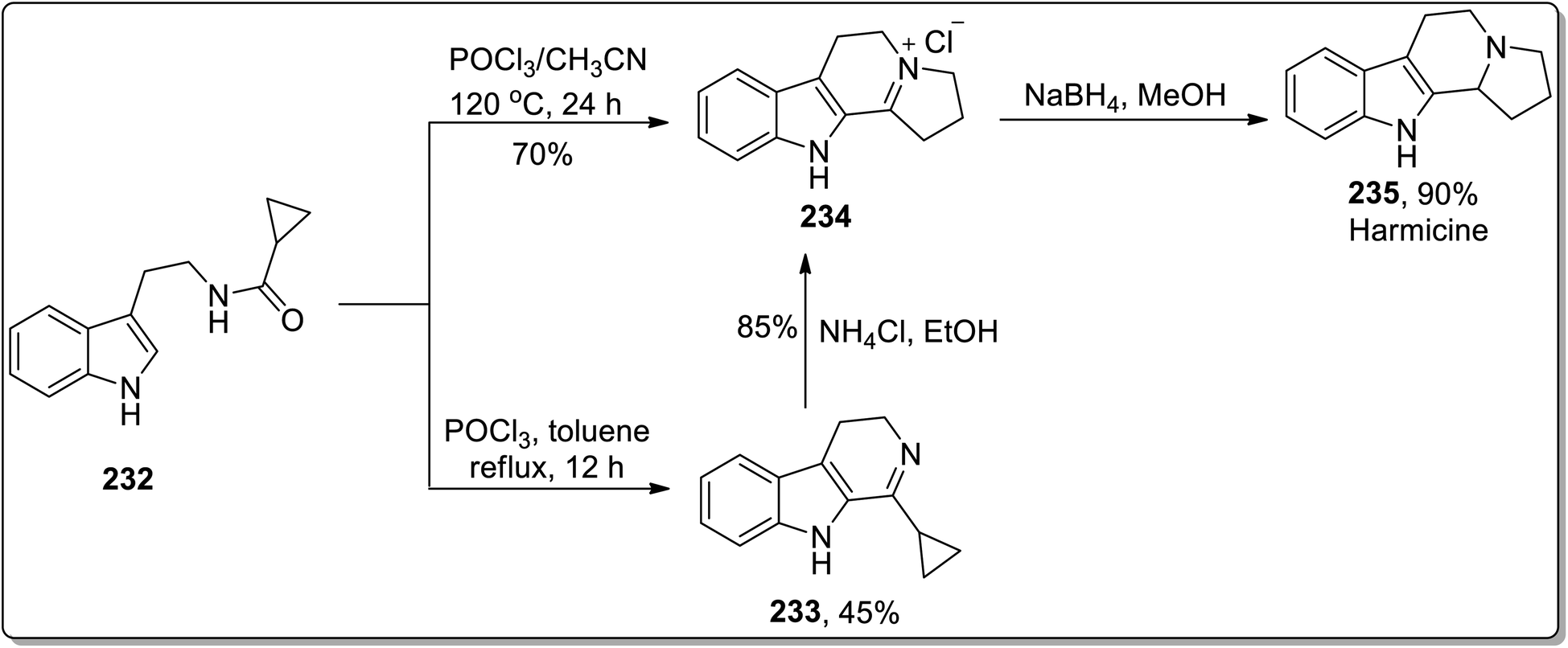
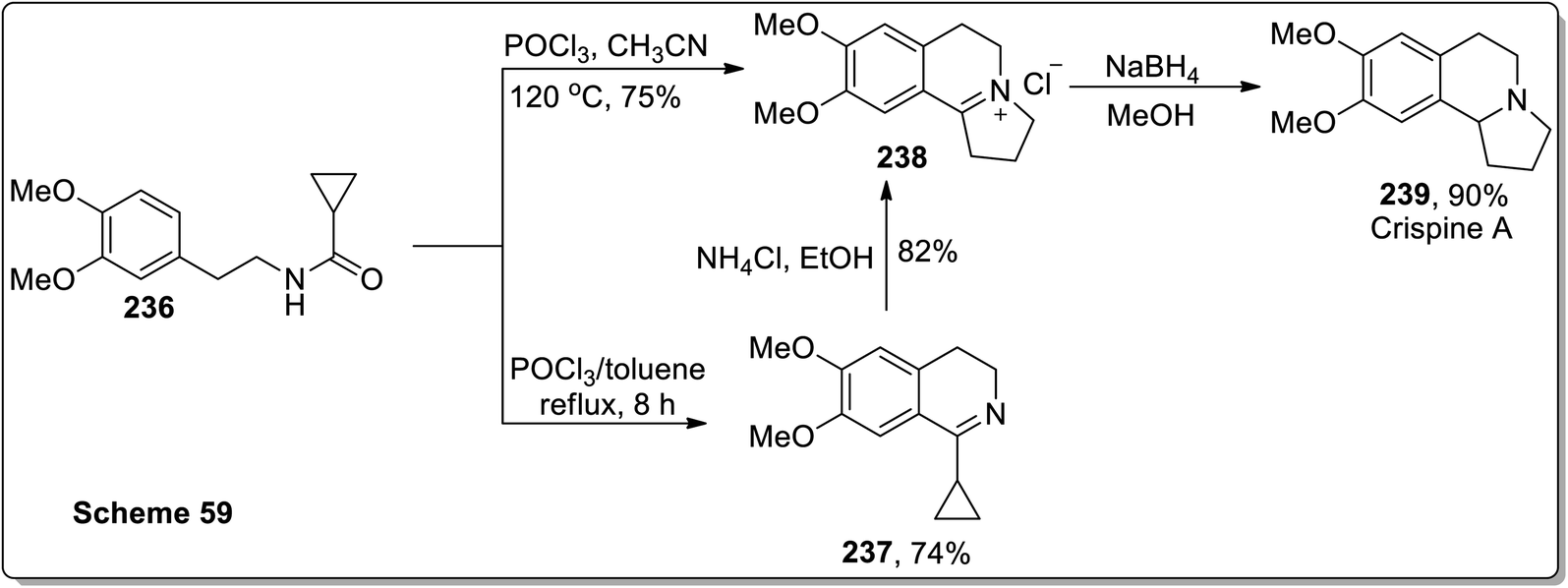
In 2011, Patro and coworkers performed the synthesis of antileishmania compound, i.e., (±)-harmicine 235 and cytotoxic alkaloid, i.e., (±)-crispine A 239 by sequential tandem cyclization, Bischler Napieralski reaction, and Cloke–Wilson rearrangement (cyclopropylimine rearrangement) reaction, followed by reduction with NaBH4. The reaction of substituted cyclopropanecarboxamide 232 with POCl3 in toluene resulted in the synthesis of compound 233 in 45% yield after 12 h. Compound 233 was then subjected to Cloke–Wilson rearrangement by treating it with NH4Cl in refluxing ethanol to afford compound 234 in 85% yield. The reaction of compound 232 was also carried out with POCl3 in acetonitrile at 120 °C, which gave compound 234 in 70% yield, which upon reduction with NaBH4 in methanol at 0 °C furnished racemic harmicine 235 in 90% yield. Similarly, crispine A 239 was synthesized from N-(3,4-dimethoxyphenethyl)cyclopropanecarboxamide 236 in 90% yield (Schemes 68 and 69).21
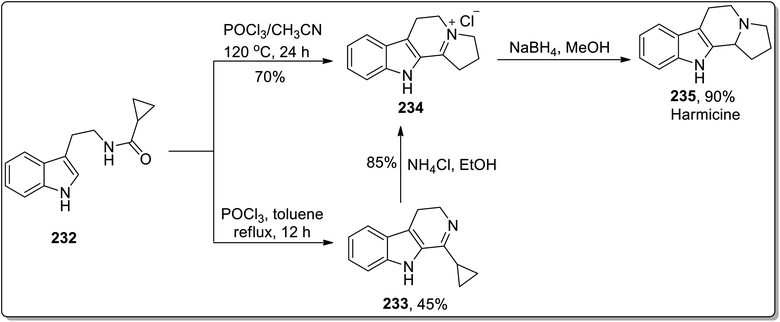 |
| | Scheme 68 Total synthesis of harmicine 235 via Cloke–Wilson rearrangement. | |
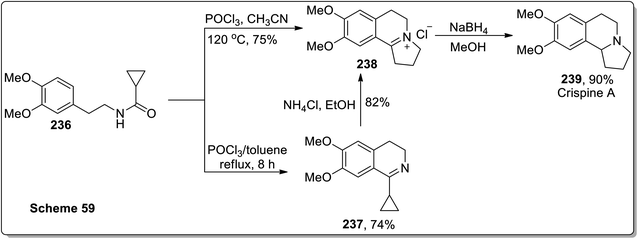 |
| | Scheme 69 Total synthesis of crispine A 239 via Cloke–Wilson rearrangement. | |
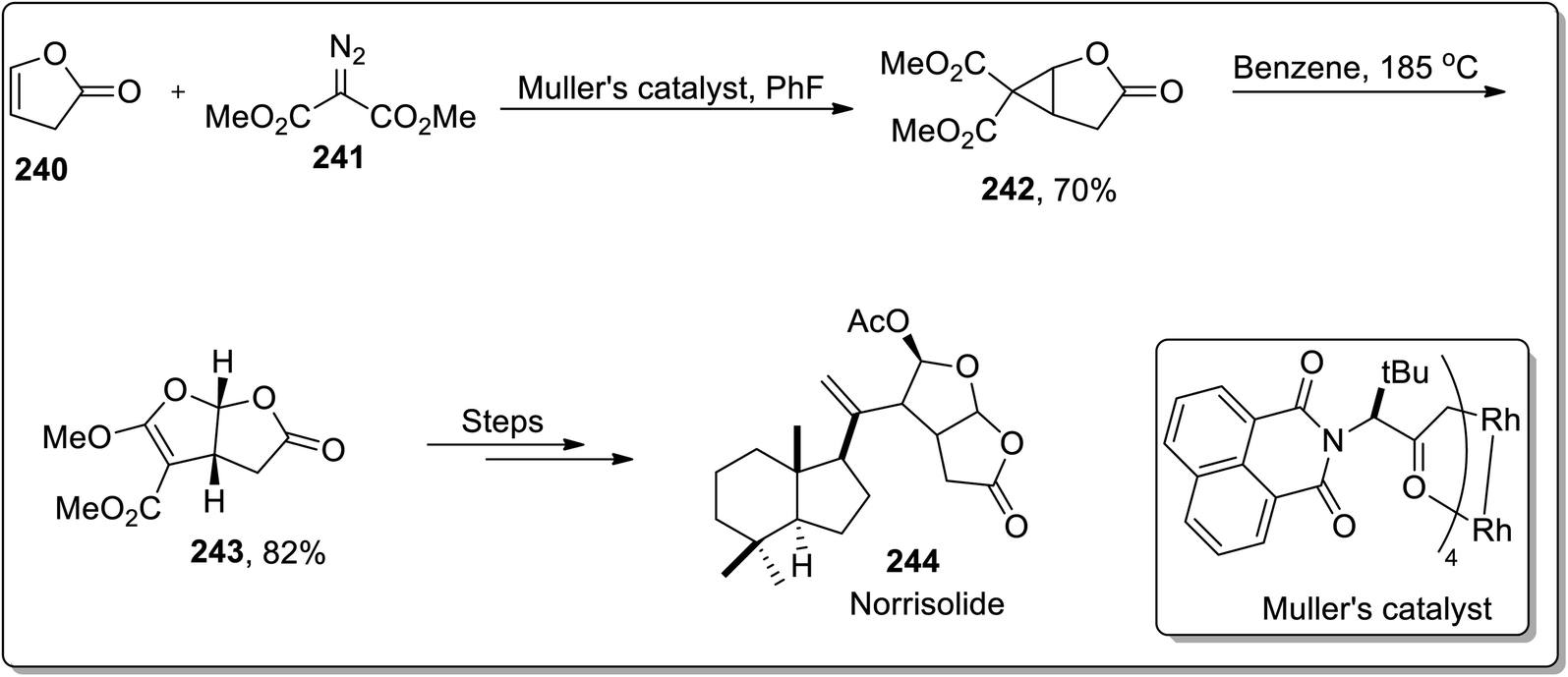
In 2012, Snapper and Granger reported the total synthesis of (+)-norrisolide 244 in which the tetrahydrofurofuranone side chain was synthesized by sequential enantioselective cyclopropanation, which proceeded further by thermal Cloke–Wilson rearrangement, which was a key step in their synthetic methodology. Further, Müller's catalyst promoted the cyclopropanation reaction between lactone 240 and dimethyl-2-diazomalonate 241 to produce D–A cyclopropane 242, which underwent cyclopropane ring-opening when heated in benzene, accompanied by ring closure to the corresponding bicyclic furan derivative 243. These furan derivatives 243 were treated in numerous steps to synthesize the target molecule. The total synthesis of (+)-norrisolide 244 consisted of total 14 steps with 1.7% overall yield (Scheme 70).92
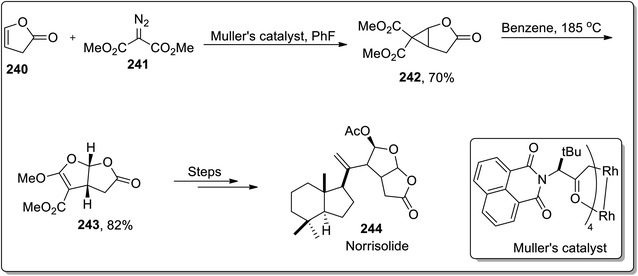 |
| | Scheme 70 Total synthesis of (+)-norrisolide 244 via Cloke–Wilson rearrangement. | |
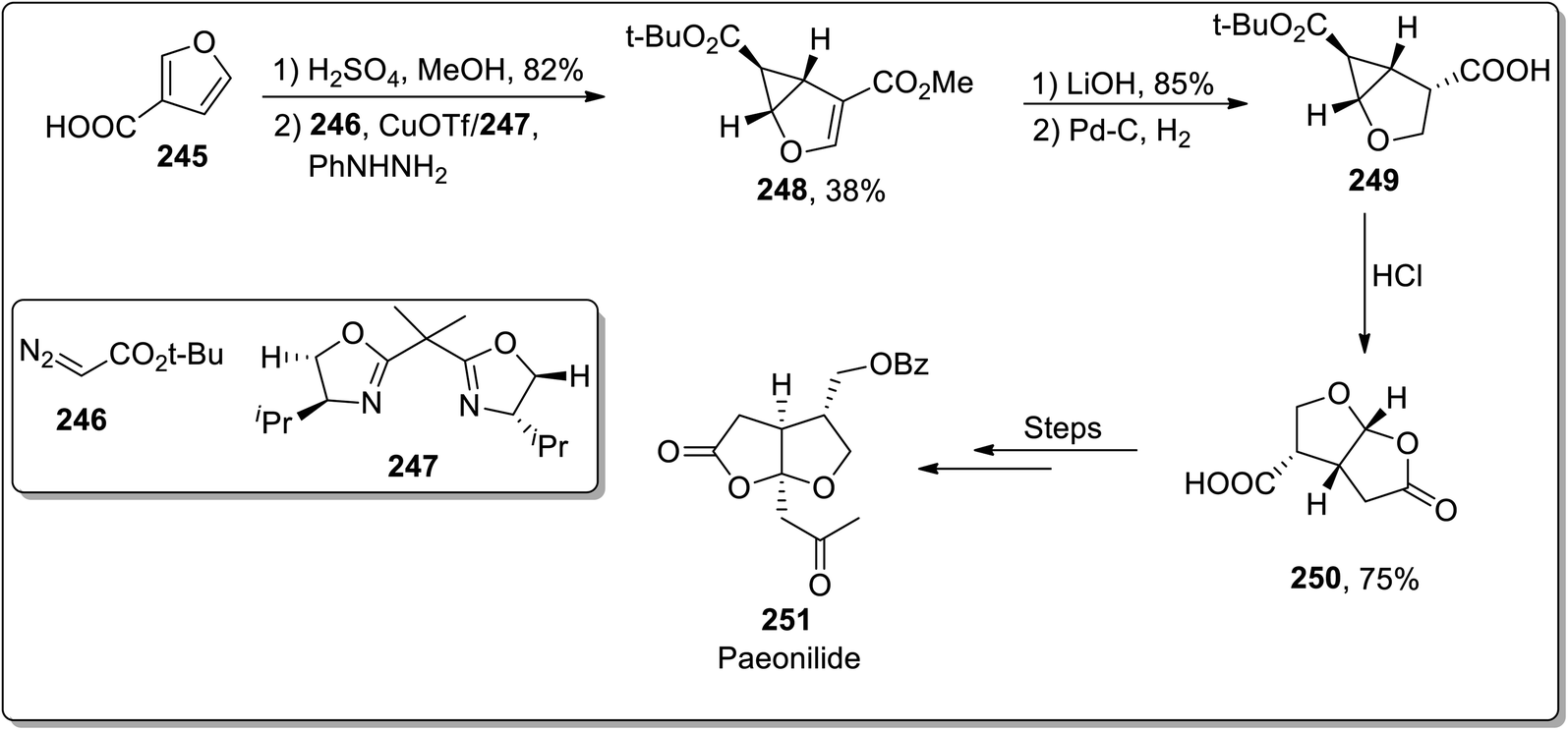
In 2012, Reiser and Harrar reported the enantioselective synthesis of the natural product (−)-paeonilide 251 from the inexpensive starting material furan-3-carboxylic acid 245. The total synthesis was initiated with the synthesis of furan-substituted ester from acid 245, which was then subjected to treatment with t-butyl diazoacetate 246 via stereoselective intermolecular cyclopropanation in the presence of copper(I) trifluoromethanesulfonate and bis-oxazoline ligand 247. This led to the generation of cyclopropane 248, which upon chemoselective hydrolysis and catalytic hydrogenation gave acid 249. The Brønsted acid-mediated cyclopropane ring cleavage/closure provided lactone 250, which was proceeded further by the incorporation of the side chain. The total synthesis of (−)-paeonilide 251 consisted of a sequence of 12 steps with 4.4% overall yield (Scheme 71).93
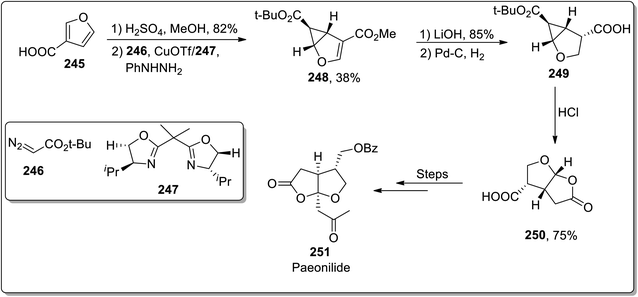 |
| | Scheme 71 Total synthesis of paeonilide 251 via Cloke–Wilson rearrangement. | |
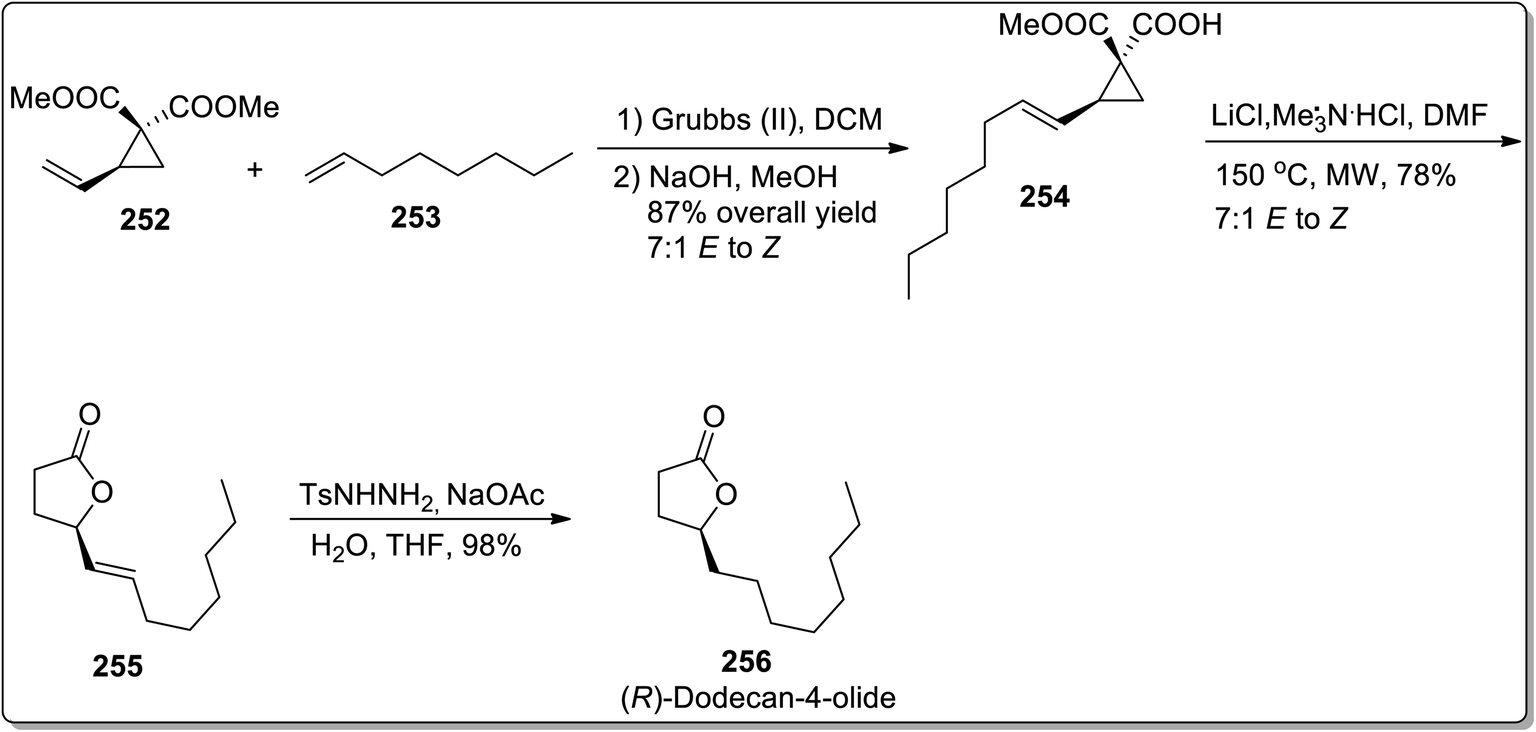
In 2013, Kerr and co-workers reported the facile approach for the synthesis of substituted butanolides 256 by carrying out cyclopropane hemimalonate 254 cyclization/dealkoxycarbonylation. Cyclopropane hemimalonates 254 (synthesized by subjecting vinyl substituted cyclopropane 252 and octene 253 to the conditions similar to cross-metathesis reaction) were rapidly converted to butanolides 255 with the retention of stereochemistry by exploiting microwave irradiation and inorganic salt, i.e., LiCl. This synthetic route was successfully applied for the synthesis of naturally-occurring (R)-dodecaolide 256 with 67% overall yield (Scheme 72).94
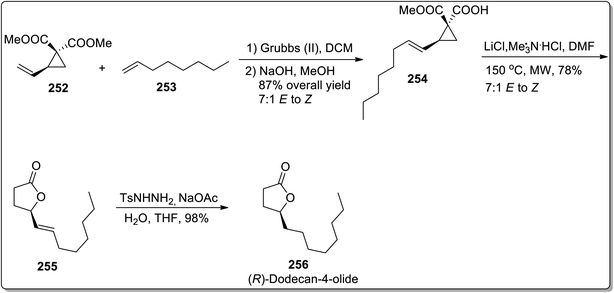 |
| | Scheme 72 Total synthesis of (R)-dodecaolide 256 via Cloke–Wilson rearrangement. | |
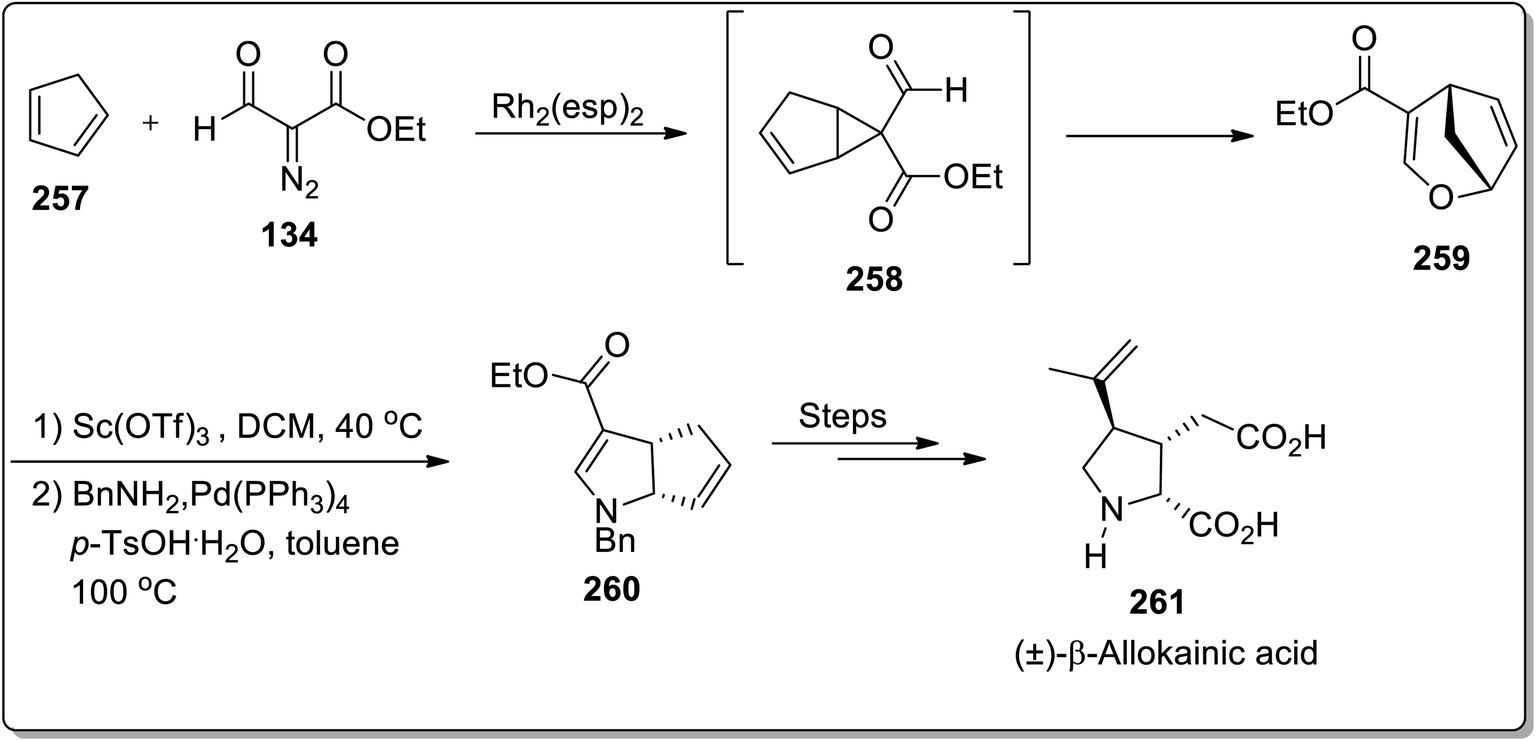
In 2019, Piotrowski and Kerr reported the total synthesis of kainoid alkaloid, i.e., (±)-β-allokainic acid 261, which was obtained in 60% yield (3.5% overall yield). During this transformation, the key step involved the Lewis acid-promoted Cloke–Wilson rearrangement of 258 (achieved by the reaction of cyclopentadiene 257 with substituted diazo compound 134) to the formation of the substituted 2,5-dihydrooxepine ring 259, followed by the transition metal-induced transformations to produce pyrrolidine core 260, which was suitable for the generation of a target molecule 261 (Scheme 73).25
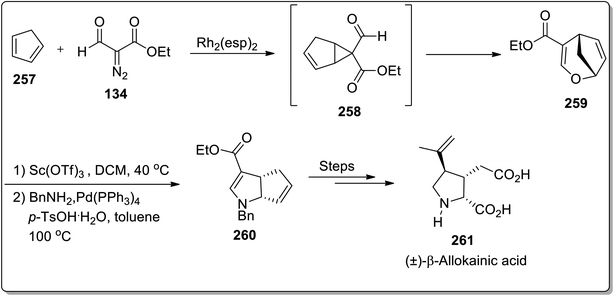 |
| | Scheme 73 Total synthesis of (±)-β-allokainic acid 261 via Cloke–Wilson rearrangement. | |
10. Conclusions
Herein, a detailed summary of recent methodological advances of Cloke–Wilson rearrangement along with its applications in the total synthesis of natural products has been presented, thereby covering the literature reported in the last two decades. Cyclopropanes are three carbon-containing privileged heterocycles, widely employed as building blocks in synthetic organic chemistry utilizing Cloke–Wilson rearrangement. The high angular and torsional strain of cyclopropanes along with the induced polarization of the ring due to the attached donor–acceptor substituents are the main factors, leading to their high reactivity in organic synthesis. Various activated cyclopropanes, i.e., carbonyl and imine-substituted cyclopropyls, have been extensively utilized to synthesize various oxygen and nitrogen-based five-membered heterocycles. Similarly, Cloke–Wilson rearrangement also finds use in the synthesis of sulfur-containing heterocycles. Moreover, the total synthesis of various natural products also utilizes this rearrangement as a key step. The requirement of high temperature was considered to be the limitation of Cloke–Wilson rearrangement. To address this drawback, various novel and efficient methods have been adopted for the activation of cyclopropanes to attain several five-membered heterocycles with broad substrate scope. These novel methods involve transition-metal catalysis, photocatalysis, organo-catalysis, Lewis acid catalysis, Brønsted acid catalysis, and Brønsted base catalysis. This detailed review is expected to motivate synthetic chemists to further advance regarding Cloke–Wilson rearrangement to unveil its synthetic potential in heterocyclic chemistry.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) H.-U. Reissig and Z. Rappoport, The Chemistry of the Cyclopropyl Group, John Wiley & Sons, Chichester, 1987, pp. 375–443 Search PubMed;
(b) A. de Meijere, Carbocyclic Three- and Four Membered Ring Compounds, in Houben-Weyl Methods of Organic Chemistry, Thieme, Stuttgart, 1997 Search PubMed;
(c) H. N. C. Wong, M. Y. Hon, C. W. Tse, Y. C. Yip, J. Tanko and T. Hudlicky, Use of cyclopropanes and their derivatives in organic synthesis, Chem. Rev., 1989, 89, 165–198 CrossRef CAS.
- J. Salaün, Cyclopropane derivatives and their diverse activities, Small Ring Compounds in Organic Synthesis VI, Springer, 2000, pp. 1–67 Search PubMed.
-
(a) M. S. Gordon, Ring strain in cyclopropane, cyclopropene, silacyclopropane, and silacyclopropene, J. Am. Chem. Soc., 1980, 102, 7419–7422 CrossRef CAS;
(b) R. D. Bach and O. Dmitrenko, Strain energy of small ring, hydrocarbons, Influence of C–H bond dissociation energies, J. Am. Chem. Soc., 2004, 126, 4444–4452 CrossRef CAS;
(c) A. D. Meijere, Bonding properties of cyclopropane and their chemical consequences, Angew. Chem., Int. Ed., 1979, 18, 809–826 CrossRef.
-
(a) H.-U. Reissig and E. Hirsch, Donor-Acceptor Substituted Cyclopropanes: Synthesis and Ring Opening to 1, 4-Dicarbonyl Compounds, Angew. Chem., Int. Ed., 1980, 19, 813–814 CrossRef;
(b) H.-U. Reissig, Lewis-acid-promoted additions of carbonyl compounds to donor-acceptor substituted cyclopropanes: a new synthesis of 2, 3-dihydrofurane derivatives, Tetrahedron Lett., 1990, 22, 2981–2984 CrossRef;
(c) H.-U. Reißi, Donor-acceptor-substituted cyclopropanes: Versatile building blocks in organic synthesis, Small Ring Compounds in Organic Synthesis III, Springer, 1988, pp. 73–135 Search PubMed.
-
(a) P. Binger and H. M. Büch, Cyclopropenes and methylenecylopropanes as multifunctional reagents in transition metal catalyzed reactions, Small Ring Compounds in Organic Synthesis II, Springer, 1987, pp. 77–15 Search PubMed;
(b) I. Nakamura and Y. Yamamoto, Transition metal-catalyzed reactions of methylenecyclopropanes, Adv. Synth. Catal., 2002, 344, 111–129 CrossRef CAS;
(c) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Heterocycles from alkylidenecyclopropanes, Chem. Rev., 2003, 103, 1213–1270 CrossRef CAS PubMed;
(d) M. Rubin, M. Rubina and V. Gevorgyan, Recent advances in cyclopropene chemistry, Synthesis, 2006, 8, 1221–1245 Search PubMed;
(e) F. D. Simone and J. Waser, Cyclization and cycloaddition reactions of cyclopropyl carbonyls and imines, Synthesis, 2009, 20, 3353–3374 Search PubMed.
-
(a) E. M. Mil'vitskaya, A. V. Tarakanova and A. F. Plate, Thermal Rearrangements of Vinylcyclopropanes, Russ. Chem. Rev., 1976, 45, 938–958 Search PubMed;
(b) Z. Goldschmidt and B. Crammer Vinylcyclopropane rearrangements, Chem. Soc. Rev., 1988, 17, 229–267 RSC;
(c) E. Piers and B. M. Trost, Comprehensive Organic Synthesis, Pergamon Press, New York, 1991, vol. 5, pp. 971–998 Search PubMed;
(d) J. E. Baldwin, Thermal rearrangements of vinylcyclopropanes to cyclopentenes, Chem. Rev., 2003, 103, 1197–1212 CrossRef CAS PubMed;
(e) T. Hudlicky and J. W. Reed, From discovery to application: 50 years of the vinylcyclopropane–cyclopentene rearrangement and its impact on the synthesis of natural products, Angew. Chem., Int. Ed., 2010, 49, 4864–4876 CrossRef CAS PubMed.
-
(a) C. L. Wilson, Reactions of Furan Compounds. VII. Thermal Interconversion of 2, 3-Dihydrofuran and Cyclopropane Aldehyde, J. Am. Chem. Soc., 1947, 69, 3002–3004 CrossRef CAS;
(b) R. K. Bowman and J. S. Johnson, Nickel-catalyzed rearrangement of 1-acyl-2-vinylcyclopropanes. A mild synthesis of substituted hydrocarbons, Org. Lett., 2006, 8, 573–576 CrossRef CAS PubMed;
(c) Y. Zhu, M. Zhang, T. Li and X. Song, AlCl3-Promoted Stereospecific Cloke-Wilson Rearrangement of Spirocyclopropyl Barbiturates for the Synthesis of Substituted Dihydrofuro [2, 3-d] pyrimidines, ChemistrySelect, 2019, 4, 10838–10842 CrossRef CAS;
(d) P. Kumar, R. Kumar and P. Banerjee, Accessing Dihydro-1, 2-oxazine via Cloke–Wilson-Type Annulation of Cyclopropyl Carbonyls: Application toward the Diastereoselective Synthesis of Pyrrolo [1, 2-b][1, 2] oxazine, J. Org. Chem., 2020, 85, 6535–6550 CrossRef CAS PubMed.
-
(a) J. B. Cloke, The Formation of Pyrrolines From Gamma-Chloropropyl and Cyclopropyl Ketimines, J. Am. Chem. Soc., 1929, 51, 1174–1187 CrossRef CAS;
(b) P. J. Campos, A. Soldevilla, D. Sampedro and M. A. Rodriguez, N-Cyclopropylimine-1-pyrroline rearrangement. A novel photochemical reaction, Org. Lett., 2001, 3, 4087–4089 CrossRef CAS PubMed;
(c) P. J. Campos, A. Soldevilla, D. Sampedro and M. A. Rodriguez, Simple and versatile synthesis of 1-pyrroline derivatives through thermal rearrangement of N-cyclopropylimines, Tetrahedron Lett., 2002, 43, 8811–8813 CrossRef CAS;
(d) R. Salikov, D. N. Platonov, D. L. Lipilin, A. E. Frumkin and Y. V. Tomilov, The cyclopropyliminium rearrangement of 2-cyclopropyl-4-nitrobenzimidazoles, Russ. Chem. Bull., 2014, 63, 765–769 CrossRef CAS.
-
(a) P. Huang, R. Zhang, Y. Liang and D. Dong, Lawesson's reagent-initiated domino reaction of aminopropenoyl cyclopropanes: synthesis of thieno [3, 2-c] pyridinones, Org. Biomol. Chem., 2012, 10, 639–1644 Search PubMed;
(b) J. Kaschel, C. D. Schmidt, M. Mumby, D. Kratzert, D. Stalke and D. B. Werz, Donor–acceptor cyclopropanes with Lawesson's and Woollins' reagents: formation of bisthiophenes and unprecedented cage-like molecules, Chem. Commun., 2013, 49, 4403–4405 RSC.
- N. Neureiter, Pyrolysis of 1,1-Dichloro-2-vinylcyclopropane Synthesis of 2-Chlorocyclopentadiene, J. Org. Chem., 1959, 24, 2044–2046 CrossRef CAS.
-
(a) E. Vogel and E. Kleine kohlenstoffringe, Angew. Chem., 1960, 72, 4–26 CrossRef CAS;
(b) C. G. Overberger and A. E. Borchert, Novel thermal rearrangements accompanying acetate pyrolysis in small ring systems, J. Am. Chem. Soc., 1960, 82, 1007–1008 CrossRef CAS;
(c) C. G. Overberger and A. E. Borchert, Ionic Polymerization. XVI. Reactions of 1-Cyclopropylethanol-Vinylcyclopropane1-3, J. Am. Chem. Soc., 1960, 82, 4896–4899 CrossRef CAS.
- R. B. Woodward and R. Hoffmann, The Conservation of Orbital Symmetry, Angew Chem. Int. Ed. Engl., 1969, 8, 781–853 CrossRef CAS.
- R. H. De Wolfe, Comprehensive Chemical Kinetics, ed. C. H. Bamford and C. F. H. Tipper, Elsevier, New York, 1973, vol. 9, p. 417 Search PubMed.
-
(a) J. M. Impson and H. G. Richey, The effects of methoxyl and phenyl substituents on the thermal rearrangements of vinylcyclopropane, Tetrahedron Lett., 1973, 14, 2545–2548 CrossRef;
(b) G. McGaffin, B. Grimm, U. Heinecke, H. Michaelson and A. de Meijer, On the Substituent Effects of the Thermal Ethenylcyclopropane-to-Cyclopentene Rearrangement: Gas-Phase Kinetics of Ethoxy-, Methylthio-and Trimethylsilyl-Substituted Ethenylcyclopropanes, Eur. J. Org Chem., 2001, 18, 3559–3573 CrossRef.
-
(a) R. H. Newman-Evans, R. J. Simon and B. K. Carpenter, The influence of intramolecular dynamics on branching ratios in thermal rearrangements, J. Org. Chem., 1990, 55, 695–711 CrossRef CAS;
(b) L. A. Asuncion and J. E. Baldwin, Stereochemistry of the Thermal Isomerizations of (1S, 2R)-1-(E-Styryl)-2-phenylcyclopropane to 3, 4-Diphenylcyclopentenes, J. Am. Chem. Soc., 1995, 117, 10672–10677 CrossRef CAS;
(c) W. E. Doering and E. A. Barsa, Fate of diradicals in the caldera: stereochemistry of thermal stereomutation and ring enlargement in cis-and trans-1-cyano-2 (E)-propenylcyclopropanes, J. Am. Chem. Soc., 2004, 126, 12353–12362 CrossRef PubMed.
-
(a) E. R. Davidson and J. J. Gajewski, Calculational evidence for lack of intermediates in the thermal unimolecular vinylcyclopropane to cyclopentene 1, 3-sigmatropic shift, J. Am. Chem. Soc., 1997, 119, 10543–10544 CrossRef CAS;
(b) K. N. Houk, M. Nendel, O. Wiest and J. W. Storer, The Vinylcyclopropane−Cyclopentene Rearrangement: A Prototype Thermal Rearrangement Involving Competing Diradical Concerted and Stepwise Mechanisms, J. Am. Chem. Soc., 1997, 119, 10545–10546 CrossRef CAS.
-
(a) E. Piers and J. Banville, Five-membered ring annelation via thermal rearrangement of β-cyclopropyl-α,β-unsaturated ketones: a new total synthesis of (±)-zizaene, J. Chem. Soc., Chem. Commun., 1979, 24, 1138–1140 RSC;
(b) T. Hudlicky, T. M. Kutchan, S. R. Wilson and D. T. Mao, Total synthesis of (±)-hirsutene, J. Am. Soc., 1980, 102, 6351–6353 CrossRef CAS;
(c) T. Hudlicky and M. Natchus, Chemoenzymic enantiocontrolled synthesis of (-)-specionin, J. Org. Chem., 1992, 57, 4740–4746 CrossRef CAS;
(d) G. Majetich and J. Grove, Total synthesis of (−)-salviasperanol, Org. Lett., 2008, 10, 85–87 CrossRef CAS.
- G. Majetich, G. Zou and J. Grove, Total Synthesis of (−)-Salviasperanol, Org. Lett., 2008, 10, 85–87 CrossRef CAS.
- G. Dannhardt and W. Kiefer, 1-Pyrrolines (3,4-dihydro-2H-pyrroles) as a template for new drugs, Arch. Pharm., 2001, 334, 183–188 CrossRef CAS.
- R. V. Stevens, General methods of alkaloid synthesis, Acc. Chem. Res., 1977, 10, 193–198 CrossRef CAS.
- S. Saha, C. V. R. Reddy and B. Patro, Facile two-step synthesis of crispine A and harmicine by cyclopropylimine rearrangement, Tetrahedron Lett., 2011, 52, 4014–4016 CrossRef CAS.
-
(a) H. H. Wasserman and R. P. Dion, Acid-catalyzed thermolysis of dicyclopropyl ketimines. Substituent effects on the course of the rearrangement, Tetrahedron Lett., 1983, 24, 3409–3412 CrossRef CAS;
(b) R. K. Boeckman and M. A. Walters, Advances in Heterocyclic Natural Product Synthesis, ed. W. H. Pearson, JAI Press, Greeenwich, 1990, vol. 1, pp. 1–40 Search PubMed;
(c) Y. V. Tomilov, D. N. Platonov, A. E. Frumkin, D. L. Lipilin and R. F. Salikov, Synthesis of condensed heterocycles via cyclopropylimine rearrangement of cyclopropylazoles, Tetrahedron Lett., 2010, 51, 5120–5123 CrossRef CAS , and references therein..
- A. Soldevilla, D. Sampedro, P. J. Campos and M. A. Rodriguez, The N-cyclopropylimine-1-pyrroline photorearrangement as a synthetic tool: Scope and limitations, J. Org. Chem., 2005, 70, 6976–6979 CrossRef CAS.
-
(a) Y. Zhang, Z. Chen, Y. Xiao and J. Zhang Y, Rh(I)-Catalyzed Regio-and Stereospecific Carbonylation of 1-(1-Alkynyl) cyclopropyl Ketones: A Modular Entry to Highly Substituted 5, 6-Dihydrocyclopenta [c] furan-4-ones, Chem. - Eur. J., 2009, 15, 5208–5211 CrossRef CAS;
(b) Y. Zhang, F. Liu and J. Zhang, Catalytic Regioselective Control in the Diastereoselective 1,3-Dipolar Cycloaddition Reactions of 1-(1-Alkynyl) cyclopropyl Ketones with Nitrones, Chem. - Eur. J., 2010, 16, 6146–6150 CrossRef CAS;
(c) Y. Bai, W. Tao, J. Ren and Z. Wang, Lewis Acid Catalyzed Intramolecular [4+ 2] and [3+ 2] Cross-Cycloaddition of Alkynylcyclopropane Ketones with Carbonyl Compounds and Imines, Angew. Chem., 2012, 124, 4188–4192 CrossRef;
(d) N. Satam, S. Nemu, G. N. Gururaja and I. N. N. Namboothiri, Substrate-oriented selectivity in the Mg-mediated conjugate addition of bromoform to electron-deficient alkenes, Org. Biomol. Chem., 2020, 18, 5697–5707 RSC.
- M. L. Piotrowski and M. A. Kerr, Synthesis of (±)-β-Allokainic Acid, Eur. J. Org Chem., 2019, 20, 3122–3126 CrossRef.
-
(a) J. Sabbatani and N. Maulide, Temporary generation of a cyclopropyl oxocarbenium ion enables highly diastereoselective donor–acceptor cyclopropane cycloaddition, Angew. Chem., Int. Ed., 2016, 55, 6780–6783 CrossRef CAS;
(b) J. Liu, W. Ye, X. Qing and C. Wang, Solvent-Free DABCO-Mediated [3+2] Cycloadditions of Donor–Acceptor Cyclopropanes with Aldehydes: Strategy for Synthesis of Fully Substituted Furans, J. Org. Chem., 2016, 81, 7970–7976 CrossRef CAS PubMed;
(c) M. Miao, H. Xu, M. Jin, Z. Chen, J. Xu and H. Ren, 1,2-Gold carbene transfer empowers regioselective synthesis of polysubstituted furans, Org. Lett., 2018, 20, 3096–3100 CrossRef CAS PubMed.
- F. De Simone and J. Waser, Cyclization and cycloaddition reactions of cyclopropyl carbonyls and imines, Synthesis, 2009, 20, 3353–3374 Search PubMed.
- M. A. Cavitt, L. H. Phun and S. France, Intramolecular donor–acceptor cyclopropane ring-opening cyclizations, Chem. Soc. Rev., 2014, 43, 804–818 RSC.
- A. Burger and W. L. Yost, Arylcycloalkylamines. I. 2-Phenylcyclopropylamine, J. Am. Chem. Soc., 1948, 70, 2198–2201 CrossRef CAS.
- Z. Chang, N. Sitachitta, J. V. Rossi, M. A. Roberts, P. M. Flatt, J. Jia, D. H. Sherman and W. H. Gerwick, Biosynthetic Pathway and Gene Cluster Analysis of Curacin A, an Antitubulin Natural Product from the Tropical Marine Cyanobacterium Lyngbya majuscula, J. Nat. Prod., 2004, 67, 1356–1367 CrossRef CAS.
- S. Hanessian, T. Focken, X. Mi, R. Oza, B. Chen, D. Ritson and R. Beaudegnies, Total Synthesis of (+)-Ambruticin S: Probing the Pharmacophoric Subunit, J. Org. Chem., 2010, 75, 5601–5618 CrossRef CAS PubMed.
- N. Sitachitta and W. H. Gerwick, Grenadadiene and Grenadamide, Cyclopropyl-Containing Fatty Acid Metabolites from the Marine Cyanobacterium Lyngbya majuscula, J. Nat. Prod., 1998, 61, 681–684 CrossRef CAS.
-
(a) M. D. Delost, D. T. Smith, B. J. Anderson and J. T. Njardarson, From oxiranes to oligomers: Architectures of US FDA approved pharmaceuticals containing oxygen heterocycles, J. Med. Chem., 2018, 61, 10996–11020 CrossRef CAS;
(b) S. Faiz, A. F. Zahoor, M. Ajmal, S. Kamal, S. Ahmad, A. M. Abdelgawad and M. E. Elnaggar, Design, synthesis, antimicrobial evaluation, and laccase catalysis effect of novel benzofuran–oxadiazole and benzofuran–triazole hybrids, J. Heterocycl. Chem., 2019, 56, 2839–2852 CrossRef CAS.
-
(a) M. Hossain and A. K. Nanda, A review on heterocyclic: synthesis and their application in medicinal chemistry of imidazole moiety, Science, 2018, 6, 83–94 CAS;
(b) I. Shahzadi, A. F. Zahoor, A. Rasul, N. Rasool, Z. Raza, S. Faisal, B. Parveen, S. Kamal, M. Zia-ur-Rehman and F. M. Zahid, Synthesis, anticancer, and computational studies of 1, 3, 4-oxadiazole-purine derivatives, J. Heterocycl. Chem., 2020, 57, 2782–2794 CrossRef CAS.
- S. Pathania, R. K. Narang and R. K. Rawal, Role of sulphur-heterocycles in medicinal chemistry: An update, Eur. J. Med. Chem., 2019, 180, 486–508 CrossRef CAS.
- R. S. Atkinson and C. W. Rees, A vinylaziridine to pyrroline rearrangement, Chem. Commun., 1967, 23, 1232a RSC.
- J. C. Paladini and J. Chuche, Rearrangement thermique d'epoxydes vinyliques, Tetrahedron Lett., 1971, 12, 4383–4386 CrossRef.
- A. Mishra, S. N. Rice and W. Lwowski, Singlet and triplet nitrenes. III. Addition of carbethoxynitrene to 1,3-dienes, J. Org. Chem., 1968, 33, 481–486 CrossRef CAS.
-
(a) H.-U. Reissig and R. Zimmer, Donor-acceptor-substituted cyclopropane derivatives and their application in organic synthesis, Chem. Rev., 2003, 103, 1151–1196 CrossRef CAS;
(b) T. F. Schneider, J. Kaschel and D. B. Werz, A new golden age for donor–acceptor cyclopropanes, Angew. Chem., Int. Ed., 2014, 53, 5504–5523 CrossRef CAS;
(c) K. R. Babu, X. Hu and S. Xu, Lewis base catalysis based on homoconjugate addition: rearrangement of electron-deficient cyclopropanes and their derivatives, Synlett, 2020, 31, 117–124 CrossRef CAS.
-
(a) T. F. Schneider and D. B. Werz, Ring-Enlargement Reactions of Donor− Acceptor-Substituted Cyclopropanes: Which Combinations are Most Efficient?, Org. Lett., 2011, 13, 1848–1851 CrossRef CAS;
(b) E. Gopi and I. N. N. Namboothiri, Synthesis of fused bromofurans via Mg-mediated dibromocyclopropanation of cycloalkanone-derived chalcones and Cloke–Wilson rearrangement, J. Org. Chem., 2013, 78, 910–919 CrossRef CAS PubMed;
(c) A. N. Vereshchagin, M. N. Elinson, E. O. Dorofeeva, O. O. Sokolova, I. S. Bushmarinov and M. P. Egorov, Stereoselective synthesis of medicinally relevant furo [2,3-d] pyrimidine framework by thermal rearrangement of spirocyclic barbiturates, RSC Adv., 2015, 5, 94986–94989 RSC;
(d) C.-H. Lin, D. Pursley, J. E. M. N. Klein, J. Teske, J. A. Allen, F. Rami, A. Köhn and B. Plietker, Non-decarbonylative photochemical versus thermal activation of Bu4N[Fe(CO)3 (NO)]–the Fe-catalyzed Cloke–Wilson rearrangement of vinyl and arylcyclopropanes, Chem. Sci., 2015, 6, 7034–7043 RSC.
-
(a) M. L. Piotrowski and M. A. Kerr, Tandem Cyclopropanation/Vinylogous Cloke–Wilson Rearrangement for the Synthesis of Heterocyclic Scaffolds, Org. Lett., 2018, 20, 7624–7627 CrossRef CAS PubMed;
(b) S. Ma and J. Zhang, Tuning the Regioselectivity in the Palladium (II)-Catalyzed Isomerization of Alkylidene Cyclopropyl Ketones: A Dramatic Salt Effect, Angew. Chem., Int. Ed., 2003, 42, 2183–2187 Search PubMed;
(c) S. Ma, L. Lu and J. Zhang, Catalytic regioselectivity control in ring-opening cycloisomerization of methylene-or alkylidenecyclopropyl ketones, J. Am. Chem. Soc., 2004, 126, 9645–9660 CrossRef CAS PubMed.
- J. Luis-Barrera, V. Laina-Martin, T. Rigotti, F. Peccati, X. Solnas-Monfort, M. Sodupe, R. R. Mas-Ballesta, M. Liras and J. Aleman, Visible-Light Photocatalytic Intramolecular Cyclopropane Ring Expansion, Angew. Chem., Int. Ed., 2017, 56, 7826–7830 CrossRef CAS.
- V. K. Yadav and R. Balamurugan, cyclopropylcarbaldehyde -Assisted Ring Opening of Donor− Acceptor Substituted Cyclopropanes. An Expedient Entry to Substituted Dihydrofurans, Org. Lett., 2001, 3, 2717–2719 CrossRef CAS.
-
(a) J. Zhang, Y. Tang, W. Wei, Y. Wu, Y. Li, J. Zhang, Y. Zheng and S. Xu, Organocatalytic Cloke–Wilson Rearrangement: DABCO-Catalyzed Ring Expansion of Cyclopropyl Ketones to 2, 3-Dihydrofurans, Org. Lett., 2017, 19, 3043–3046 CrossRef CAS PubMed;
(b) A. Ortega, R. Manzano, U. Uria, L. Carrillo, E. Reyes, T. Tejero, P. Merino and J. L. Vicario, Catalytic Enantioselective Cloke–Wilson Rearrangement, Angew. Chem., 2018, 130, 8357–8361 CrossRef;
(c) W. Wei, Y. Tang, Y. Zhou, G. Deng, Z. Liu, J. Wu, Y. Li, J. Zhang and S. Xu, Recycling Catalyst as Reactant: A Sustainable Strategy To Improve Atom Efficiency of Organocatalytic Tandem Reactions, Org. Lett., 2018, 20, 6559–6563 CrossRef CAS PubMed;
(d) X. He, Y. Tang, Y. Wang, J.-B. Chen, S. Xu, J. Dou and Y. Li, Phosphine-Catalyzed Activation of Alkylidenecyclopropanes: Rearrangement to Form Polysubstituted Furans and Dienones, Angew. Chem., Int. Ed., 2019, 58, 10698–10702 CrossRef CAS.
-
(a) Y. Chen, W. Ding, W. Cao and C. Lu, Stereoselective Synthesis of trans-β-Methoxycarbonyl-γ-Aryl-γ-Butyrolactones, Synth. Commun., 2002, 32, 1953–1960 CrossRef CAS;
(b) X. Wu, W. Cao, H. Zhang, J. Chen, H. Jiang, H. Deng, M. Shao, J. Zhang and H. Chen, Highly stereoselective synthesis of β, γ-disubstituted and α,β,γ-trisubstituted butyrolactones, Tetrahedron, 2008, 64, 10331–10338 CrossRef CAS.
- T. F. Schneider, J. Kaschel, B. Dittrich and D. B. Werz, Anti-Oligoannelated THF Moieties: Synthesis via Push− Pull Substituted Cyclopropanes, Org. Lett., 2009, 11, 2317–2320 CrossRef CAS.
- F. Liang, S. Lin and Y. Wei, Aza-Oxy-Carbanion Relay via Non-Brook Rearrangement: Efficient Synthesis of Furo [3, 2-c] pyridinones, J. Am. Chem. Soc., 2011, 133, 1781–1783 CrossRef CAS.
- P. Huang, R. Zhang, Y. Li and D. Dong, [5C+1N] Annulations: Two Novel Routes to Substituted Dihydrofuro [3,2-c] pyridines, Org. Lett., 2012, 14, 5196–5199 CrossRef CAS.
- W. Yang, L. Xu, Z. Chen, L. Zhang, M. Miao and H. Ren, Ru-catalyzed synthesis of dihydrofuroquinolines from azido-cyclopropyl ketones, Org. Lett., 2013, 15, 1282–1285 CrossRef CAS.
- A. N. Vereshchagin, M. N. Elinson, E. O. Dorofeeva, O. O. Sokolova, I. S. Bushmarinov and M. P. Egorov, Stereoselective synthesis of medicinally relevant furo [2, 3-d] pyrimidine framework by thermal rearrangement of spirocyclic barbiturates, RSC Adv., 2015, 5, 94986–94989 RSC.
- R. Tian, C. Zhang, Y. Xu, C. Liu and Z. Duan, The chemistry of 1-acylphosphirane complexes: a phosphorus analogue of the Cloke–Wilson rearrangement, Chem. - Eur. J., 2017, 23, 13006–13009 CrossRef CAS PubMed.
- M. Zhang, T. Li, C. Cui, X. Song and J. Chan, Stereoselective sequential spirocyclopropanation/Cloke–Wilson rearrangement reactions for synthesis of trans-β, γ-disubstituted γ-butyrolactones using alkylidene meldrum's acid and benzyl halides, J. Org. Chem., 2020, 85, 2266–2276 CrossRef CAS PubMed.
- T. Li, D. Yan, C. Cui, X. Song and J. Chang, A thermal decarboxylative Cloke–Wilson rearrangement of dispirocyclopropanes derived from para-quinone methides and bromo-Meldrum's acids: an approach to synthesize spirobutyrolactone para-dienones, Org. Chem. Front., 2020, 7, 2682–2688 RSC.
- M. Honda, T. Naitou, H. Hoshino, S. Takagi, M. Segi and T. Nakajima, An efficient synthesis of 5-silyl-2,3-dihydrofurans via acid-catalyzed ring-enlargement of cyclopropyl silyl ketones and their functionalization, Tetrahedron Lett., 2005, 46, 7345–7348 CrossRef CAS.
- Z. Zhang, Q. Zhang, S. Sun, T. Xiong and Q. Liu, Domino Ring-Opening/Recyclization Reactions of Doubly Activated Cyclopropanes as a Strategy for the Synthesis of Furoquinoline Derivatives, Angew. Chem., Int. Ed., 2007, 46, 1726–1729 CrossRef CAS.
- T. Xiong, Q. Zhang, Z. Zhang and Q. Liu, A Divergent Synthesis of γ-Iminolactones, Dihydroquinolin-2-ones, and γ-Lactames from β-Hydroxymethylcyclopropanylamides, J. Org. Chem., 2007, 72, 8005–8009 CrossRef CAS PubMed.
- C. Brand, G. Rauch, M. Zanoni, B. Dittrich and D. B. Werz, Synthesis of [n,5]-spiroketals by ring enlargement of donor-acceptor-substituted cyclopropane derivatives, J. Org. Chem., 2009, 74, 8779–8786 CrossRef CAS.
- A. O. Chagarovskiy, O. A. Ivanova, E. R. Rakhmankulov and E. M. Budynina, Lewis Acid-Catalyzed Isomerization of 2-Arylcyclopropane-1,1-dicarboxylates: A New Efficient Route to 2-Styrylmalonates, Adv. Synth. Catal., 2010, 352, 3179–3184 CrossRef CAS.
- H. Wang, J. R. Denton and H. M. L. Davies, Sequential rhodium-, silver-, and gold-catalyzed synthesis of fused dihydrofurans, Org. Lett., 2011, 13, 4316–4319 CrossRef CAS.
- T. R. Newhouse, P. S. J. Kaib, A. W. Gross and E. J. Corey, Versatile Approaches for the Synthesis of Fused-Ring γ-Lactones Utilizing Cyclopropane Intermediates, Org. Lett., 2013, 15, 1591–1593 CrossRef CAS PubMed.
- S. G. Dawande, M. Harode, J. Kalepu and S. Katukojvala, Ag (I)-catalyzed intramolecular transannulation of enynone tethered donor–acceptor cyclopropanes: a new synthesis of 2,3-dihydronaphtho [1,2-b] furans, Chem. Commun., 2016, 52, 13699–13701 RSC.
- J.-M. Liu, X.-Y. Liu, X.-S. Qing, T. Wang and C.-D. Wang, I2/K2CO3-promoted ring-opening/cyclization/rearrangement/aromatization sequence: A powerful strategy for the synthesis of polysubstituted furans, Chin. Chem. Lett., 2017, 28, 458–462 CrossRef CAS.
- Y. Zhu, S. Zhao, M. Zhang, X. Song and J. Chang, Diastereoselective Synthesis of Spirobarbiturate-Cyclopropanes through Organobase-Mediated Spirocyclopropanation of Barbiturate-Based Olefins with Benzyl Chlorides, Synthesis, 2019, 51, 899–906 CrossRef CAS.
- Y. Zhu, M. Zhang, T. Li and X. Song, AlCl3-Promoted Stereospecific Cloke-Wilson Rearrangement of Spirocyclopropyl Barbiturates for the Synthesis of Substituted Dihydrofuro [2,3-d] pyrimidines, ChemistrySelect, 2019, 4, 10838–10842 CrossRef CAS.
-
(a) E. Gopi and I. N. N. Namboothiri, Synthesis of fused bromofurans via Mg-mediated dibromocyclopropanation of cycloalkanone-derived chalcones and Cloke–Wilson rearrangement, J. Org. Chem., 2013, 78, 910–919 CrossRef CAS PubMed;
(b) N. Satam, S. Nemu, G. N. Gururaja and I. N. N. Namboothiri, Substrate-oriented selectivity in the Mg-mediated conjugate addition of bromoform to electron-deficient alkenes, Org. Biomol. Chem., 2020, 18, 5697–5707 RSC.
-
(a) T. Akiyama and K. Mori, Stronger Brønsted Acids: Recent Progress, Chem. Rev., 2015, 115, 9277–9306 CrossRef CAS PubMed;
(b) W. Xu and Q.-Y. Chen, A novel approach of cycloaddition of difluorocarbene to α, β-unsaturated aldehydes and ketones: synthesis of gem-difluorocyclopropyl ketones and 2-fluorofurans, Org. Biomol. Chem., 2003, 1, 1151–1156 RSC.
- M. S. Samuel, H. A. Jenkins, D. W. Hughes and K. M. Baines, Mechanistic studies of the addition of carbonyl compounds to tetramesityldisilene and tetramesitylgermasilene, Organometallics, 2003, 22, 603–1611 CrossRef.
- C. Kim, T. Brady, S. H. Kim and E. A. Theodorakis, Synthesis of Fused Tetrahydrofuran-γ-lactone Motifs via One-Pot Ring Expansion of Cyclopropane Rings, Synth. Commun., 2004, 34, 1951–1965 CrossRef CAS.
- A. M. Bernard, A. Frongia, P. P. Piras, F. Secci and M. Spiga, Regioselective synthesis of trisubstituted 2, 3-dihydrofurans from donor-acceptor cyclopropanes or from reaction of the corey ylide with α-sulfenyl-, α-sulfinyl-, or α-sulfonylenones, Org. Lett., 2005, 7, 4565–4568 CrossRef CAS PubMed.
- J. Su, J. Xiong, S. Liang, G. Qiu, X. Feng, H. Teng, L. Wu and X. Hu, Concise synthesis of the angular dihydrofuroquinoline alkaloids via cyclopropane opening in the presence of polyphosphoric acid, Synth. Commun., 2006, 36, 693–699 CrossRef CAS.
- R. Zhang, Y. Liang, G. Zhou, K. Wang and D. Dong, Ring-enlargement of dimethylaminopropenoyl cyclopropanes: An efficient route to substituted 2, 3-dihydrofurans, J. Org. Chem., 2008, 73, 8089–8092 CrossRef CAS PubMed.
- Y. Wei, S. Lin, J. Zhang, Z. Niu, Q. Fu and F. Liang, Halonium-initiated electrophilic cascades of 1-alkenoylcyclopropane carboxamides: efficient access to dihydrofuropyridinones and 3(2H)-furanones, Chem. Commun., 2011, 47, 12394–12396 RSC.
- J. Kaschel, T. F. Schneider, P. Schirmer, C. Maaß, D. Stalke and D. B. Werz, Rearrangements of Furan-, Thiophene-and N-Boc-Pyrrole-Derived Donor-Acceptor Cyclopropanes: Scope and Limitations, Eur. J. Org Chem., 2013, 2013, 4539–4551 CrossRef CAS.
- R. Dey, S. Rajput and P. Banerjee, Metal-free domino Cloke–Wilson rearrangement-hydration-dimerization of cyclopropane carbaldehydes: A facile access to oxybis (2-aryltetrahydrofuran) derivatives, Tetrahedron, 2020, 76, 131080 CrossRef CAS.
- P. Huang, N. Zhang, R. Zhang and D. Dong, Vilsmeier-Type Reaction of Dimethylaminoalkenoyl Cyclopropanes: One-Pot Access to 2,3-Dihydrofuro [3,2-c] pyridin-4(5H)-ones, Org. Lett., 2012, 14, 370–373 CrossRef CAS.
-
(a) Y. Bai, J. Chen and S. C. Zimmerman, Designed transition metal catalysts for intracellular organic synthesis, Chem. Soc. Rev., 2018, 47, 1811–1821 RSC;
(b) R. Akhtar, A. F. Zahoor, N. Rasool, M. Ahmad and K. G. Ali, Recent trends in the chemistry of Sandmeyer reaction: a review, Mol. Diversity, 2022, 26, 1837–1873 CrossRef CAS;
(c) I. Ojima, M. Tzamarioudaki, Z. Li and R. J. Donovan, Transition metal-catalyzed carbocyclizations in organic synthesis, Chem. Preview, 1996, 96, 635–662 CAS.
- S. Noreen, A. F. Zahoor, S. Ahmad, I. Shahzadi, A. Irfan and S. Faiz, Novel chiral ligands for palladium-catalyzed asymmetric allylic alkylation/asymmetric Tsuji-Trost reaction: a review, Curr. Org. Chem., 2019, 23, 1168–1213 CrossRef CAS.
- P. Huang, N. Zhang, R. Zhang and D. Dong, Vilsmeier-Type Reaction of Dimethylaminoalkenoyl Cyclopropanes: One-Pot Access to 2,3-Dihydrofuro [3,2-c] pyridin-4(5H)-ones, Org. Lett., 2012, 14, 370–373 CrossRef CAS PubMed.
- H. Aziz, A. F. Zahoor and S. Ahmad, Pyrazole bearing molecules as bioactive scaffolds: A review, J. Chil. Chem. Soc., 2020, 65, 4746–4753 CrossRef CAS.
- S. D. Kuduk, C. Ng, R. K. Chang and M. G. Bock, Synthesis of 2, 3-diaminodihydropyrroles via thioimidate cyclopropane rearrangement, Tetrahedron Lett., 2003, 44, 1437–1440 CrossRef CAS.
- Y.-H. Yang and M. Shi, Ring-expanding reaction of cyclopropyl amides with triphenylphosphine and carbon tetrahalide, J. Org. Chem., 2005, 70, 8645–8648 CrossRef CAS PubMed.
- K. Graebe, B. Zwafelink and S. Doye, One-Pot Procedure for the Synthesis of N-Substituted 2-(Arylmethyl) pyrrolidines from 1-Aryl-2-cyclopropylalkynes and Primary Amines by a Hydroamination/Cyclopropylimine Rearrangement/Reduction Sequence, Eur. J. Org Chem., 2009, 2009, 5565–5575 CrossRef.
- J. Kaschel, T. F. Schneider, D. Kratzert, D. Stalke and D. B. Werz, Domino Reactions of Donor–Acceptor-Substituted Cyclopropanes for the Synthesis of 3, 3′-Linked Oligopyrroles and Pyrrolo [3, 2-e] indoles, Angew. Chem., Int. Ed., 2012, 51, 11153–11156 CrossRef CAS.
- R. F. Salikov, D. N. Platonov, A. E. Frumkin, D. L. Lipilin and Y. V. Tomilov, Synthesis of 2, 3-dihydro-1H-pyrrolo [1, 2-a] benzimidazoles via the cyclopropyliminium rearrangement of substituted 2-cyclopropylbenzimidazoles, Tetrahedron, 2013, 69, 3495–3505 CrossRef CAS.
- Y. V. Tomilov, R. F. Salikov, D. N. Platinov, D. L. Lipilin and A. E. Frumkin, The cyclopropyliminium rearrangement of cyclopropylthiazoles, Mendeleev Commun., 2013, 1, 22–23 CrossRef.
- R. F. Salikov, A. Y. Belyy and Y. V. Tomilov, The rearrangement of cyclopropylketone arylhydrazones. Synthesis of tryptamines and tetrahydropyridazines, Tetrahedron Lett., 2014, 55, 5936–5939 CrossRef CAS.
- A. V. Samet, E. A. Silyanova, M. N. Semenova, V. A. Karnoukhova and V. V. Semenov, An Efficient Synthesis of Fused 2-Aryliminothiazolines via a Solvent-Free Cyclopropyliminium Rearrangement, ChemistrySelect, 2016, 1, 2373–2376 CrossRef CAS.
- C. Tanguy, P. Bertusa, J. Szymoniak, O. V. Larionovb and A. de Meijere, Convenient access to 2-arylpyrroles from 2-lithio-N, N-dibenzylcyclopropylamine and nitriles, Synlett, 2006, 2006, 2339–2341 CrossRef.
-
(a) S. W. Kwok, L. Zhang, N. P. Grimster and V. V. Fokin, Catalytic Asymmetric Transannulation of NH-1, 2, 3-Triazoles with Olefins, Angew. Chem., 2014, 126, 3520–3524 CrossRef;
(b) S. Ahmad, A. F. Zahoor, S. A. R. Naqvi and M. Akash, Recent trends in ring opening of epoxides with sulfur nucleophiles, Mol. Diversity, 2018, 22, 191–205 CrossRef CAS PubMed.
- S. Tabassum, A. F. Zahoor, S. Ahmad, R. Noreen, S. G. Khan and H. Ahmad, Cross-coupling reactions towards the synthesis of natural products, Mol. Diversity, 2021, 1–43 Search PubMed.
- J. S. Foot, A. T. Phillis, P. P. Sharp, A. C. Willis and M. G. Banwell, Dichlorocarbene adducts of alkyl enol ethers as precursors to furans: application to a total synthesis of the furanosesquiterpene (±)-pallescensin A, Tetrahedron Lett., 2006, 47, 6817–6820 CrossRef CAS.
- K. Granger and M. L. Snapper, Concise synthesis of norrisolide, Eur. J. Org. Chem., 2012, 2012, 2308–2311 CrossRef CAS.
- K. Harrar and O. Reiser, Enantioselective synthesis of (-)-paeonilide, Chem. Commun., 2012, 48, 3457–3459 RSC.
- H. K. Grover, M. R. Emmett and M. A. Kerr, γ-Substituted butanolides from cyclopropane hemimalonates: An expedient synthesis of natural (R)-dodecan-4-olide, Org. Lett., 2013, 15, 4838–4841 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Freeha Hafeezac,
Irum Shahzadia and
Rabia Akhtarad
*a,
Freeha Hafeezac,
Irum Shahzadia and
Rabia Akhtarad