
Aqueous seeded RAFT polymerization for the preparation of self-assemblies containing nucleobase analogues†
Miriam
Abad
 ab,
Martina
Nardi
c,
Luis
Oriol
ab,
Milagros
Piñol
*ab and
Eva
Blasco
*de
ab,
Martina
Nardi
c,
Luis
Oriol
ab,
Milagros
Piñol
*ab and
Eva
Blasco
*de
aInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, Zaragoza 50009, Spain. E-mail: mpinol@unizar.es
bDepartamento de Química Orgánica, Facultad de Ciencias, Universidad de Zaragoza, Pedro Cerbuna, 12, Zaragoza 50009, Spain
cInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, Eggenstein-Leopoldshafen 76344, Germany
dOrganic Chemistry Institute, University of Heidelberg, In Neuenheimer Feld 270, 69120, Germany. E-mail: eva.blasco@oci.uni-heidelberg.de
eInstitute for Molecular Systems Engineering and Advanced Materials, University of Heidelberg, In Neuenheimer Feld 225, 69120, Germany
First published on 22nd November 2022
Abstract
Self-assemblies containing the nucleobase analogue 2,6-diacylaminopyridine (DAP) have been successfully prepared for the first time by aqueous seeded RAFT polymerization in high concentrations. For this purpose, a diblock copolymer containing poly(ethylene glycol) (PEG) and DAP polymethacrylate blocks was used as a macro-chain-transfer agent (PEG124-b-PDAP9-CTA) for the polymerization of 2-hydroxypropyl methacrylate (HPMA) in water. From the systematic variation of the degree of polymerization and solid concentration, a phase diagram has been generated that correlates both variables with the morphologies of this new system. Self-assemblies have been characterized by TEM and DLS, observing morphologies from low to high order (from spherical micelles to worms and to vesicles). Self-assembly morphologies are stable for almost a year, except in the case of worms that turn into spherical micelles after a few weeks. In addition, H-bonding supramolecular functionalization of the DAP repeating units during aqueous seeded RAFT polymerization has been examined by functionalization with a cross-linker with four thymine groups. Finally, the loading and the subsequent release of Nile Red have been proven in both supramolecular cross-linked and non-cross-linked self-assemblies.
Introduction
Amphiphilic block copolymers (BCs) are able to self-assemble in aqueous media leading to different structures with sizes at the nanoscale and potential uses in diverse fields including the biomedical one.1,2 The morphology and size of amphiphilic BC self-assemblies are frequently tailored by adjusting the length of each block, their chemical structure and the processing methodology.1 The majority of these self-assembly methodologies, such as thin-film rehydration,3 solvent displacement,4,5 sonication6 or microfluidics,7 are post-polymerization processes which in some cases consist of time-consuming multiple steps and usually provide highly diluted BC dispersions (below 1 wt%). Over the past years, polymerization-induced self-assembly (PISA) has emerged as an attractive one-step alternative where self-assembly occurs during the polymerization, thus avoiding multiple processing or purification steps and also providing highly concentrated self-assembly dispersions (up to 50 wt%) with good colloidal stability. Besides, on adjusting the polymerization conditions, PISA allows access to self-assemblies with morphologies from low to high order (e.g. from spherical micelles to worms and to vesicles).8–10Among the controlled radical polymerizations suitable for PISA, reversible addition–fragmentation chain-transfer (RAFT) polymerization has been the most widely exploited thanks to metal-free conditions, its high tolerance to functional groups and the possibility to obtain well-defined architectures with predetermined molar masses, low dispersities and end-group fidelity.9,11 Commonly, a macro-chain-transfer agent (macro-CTA) is employed for the PISA polymerization to grow a BC that self-assembles in situ. Depending on the solubility of both the macro-CTA and the monomer, emulsion RAFT polymerization,12 dispersion RAFT polymerization13–18 or seeded RAFT polymerization19 is possible. The latter and more recent one uses an amphiphilic BC macro-CTA that self-assembles in the polymerization medium acting as a seed or nanoreactor for a third block chain extension. Depending on the solubility of the monomer employed, and the resultant polymeric chain, seeded RAFT polymerization can be categorized into different types.19–22
Even though PISA self-assemblies are usually stable in the polymerization medium, the conditions of the environment in which they perform their function (i.e. the presence of non-selective solvents, proteins, surfactants or very dilute media) can compromise their stability. Therefore, covalent cross-linking18 either during polymerization, employing divinyl monomers,23–28 or by either chemically29–32 or photochemically33,34 activated post-polymerization processes has been reported to enhance the nanoparticle stability. However, supramolecular or non-covalent cross-linking has been a less explored alternative despite being an easy and more versatile approach. In addition, its dynamic nature can be advantageous for certain applications like drug delivery. Examples of PISA incorporating non-covalent interactions have been described with either electrostatic,35 host–guest36,37 or hydrogen bonding interactions. In particular, hydrogen bonding interactions have been studied not only as cross-linking interactions38,39 but also as a driver for template interactions,14,16,40,41 and even responsible for upper critical solution temperature (UCST) behavior.42
Among suitable monomers for aqueous dispersion RAFT polymerization, 2-hydroxypropyl methacrylate (HPMA) has been widely studied since it is a water-miscible monomer (up to 13 w/v % at room temperature) that forms a water-insoluble polymer.15 PISA polymerization of HMPA has been approached using several macro-CTAs or hydrophilic stabilizer blocks such as poly(glycerol methacrylate),15,23,27–29,43,44 poly(2-(methacryloyloxy)ethyl phosphorylcholine)26 or poly(ethylene glycol) (PEG)13,17,30,33,43,45–48 or even statistical copolymers like poly(glycerol methacrylate-st-glycidyl methacrylate).31,36
For some time now, our group has worked on the synthesis and self-assembly of linear-linear7,49,50 and linear-dendritic51 amphiphilic BCs containing a PEG hydrophilic block and a hydrophobic one bearing the 2,6-diacylaminopyridine (DAP) unit, which is a nucleobase analogue able to form multiple hydrogen bonds, for different purposes. Furthermore, PEG has been recurrently employed in biomedical applications given that it is non-toxic and able to reduce immunogenicity, which enables the extension of in vivo circulation lifetimes of any encapsulated molecules.52 These DAP-containing amphiphilic BCs exhibited excellent self-assembly behavior forming homogeneous dispersions of micelles or vesicles either by solvent-switching or microfluidics, and their potential as nanocarriers has been evaluated using fluorescent probes,7,49–51 or camptothecin49 and naproxen7 as drug models. The cell viability of these systems was confirmed on several cell lines as well as the pH49 and light-stimulated50 release of the cargoes, in the last case using thymine-containing azocompounds as photoresponsive moieties supramolecularly linked to DAP units. Recently, this outstanding self-assembly behavior has been exploited to process hybrid systems consisting of plasmonic Pd(0) nanosheets, grown inside of polymeric micelles, for photothermal therapy under NIR stimulation.7
In order to further exploit the potential of incorporating nucleobase analogues such as DAP units into amphiphilic BCs, this work explores the preparation of highly concentrated aqueous self-assembly dispersions using the PISA methodology and the supramolecular H-bonding functionalization of the formed self-assemblies. Although there are some examples on the dispersion RAFT polymerization of nucleobases, such as the polymerization of adenine- and thymine-containing monomers in chloroform or dioxane by Kang et al.,14,16 to the best of our knowledge neither nucleobase nor nucleobase analogues have been employed in RAFT-PISA in water. One of the reasons is the often poor water solubility of the nucleobase-containing monomers. Thus, a new strategy has been designed herein to integrate DAP units into a BC which is then employed as a macro-CTA for the aqueous seeded RAFT polymerization of HPMA. Moreover, the feasibility of supramolecular functionalization has been demonstrated using a cross-linker with thymine terminal moieties to induce self-assembly and supramolecular cross-linking in one-pot.
Results and discussion
Synthesis of DAP-containing macro-CTA and self-assembly behavior
The direct incorporation of the DAP-containing methacrylic monomer in an aqueous RAFT polymerization system was not obvious, since it is poorly soluble in water and also in several tested water/miscible organic solvent mixtures (see the ESI†). To face this challenge, a different approach was adopted where PEG-CTA is chain extended with DAP to form a BC macro-CTA subsequently employed for aqueous RAFT polymerization of HPMA (Scheme 1). Indeed, HPMA is commercialized as a mixture of 2-hydroxypropyl methacrylate and 1-hydroxypropan-2-yl methacrylate; so, strictly speaking, the resulting polymeric block is a statistical copolymer of the two isomers.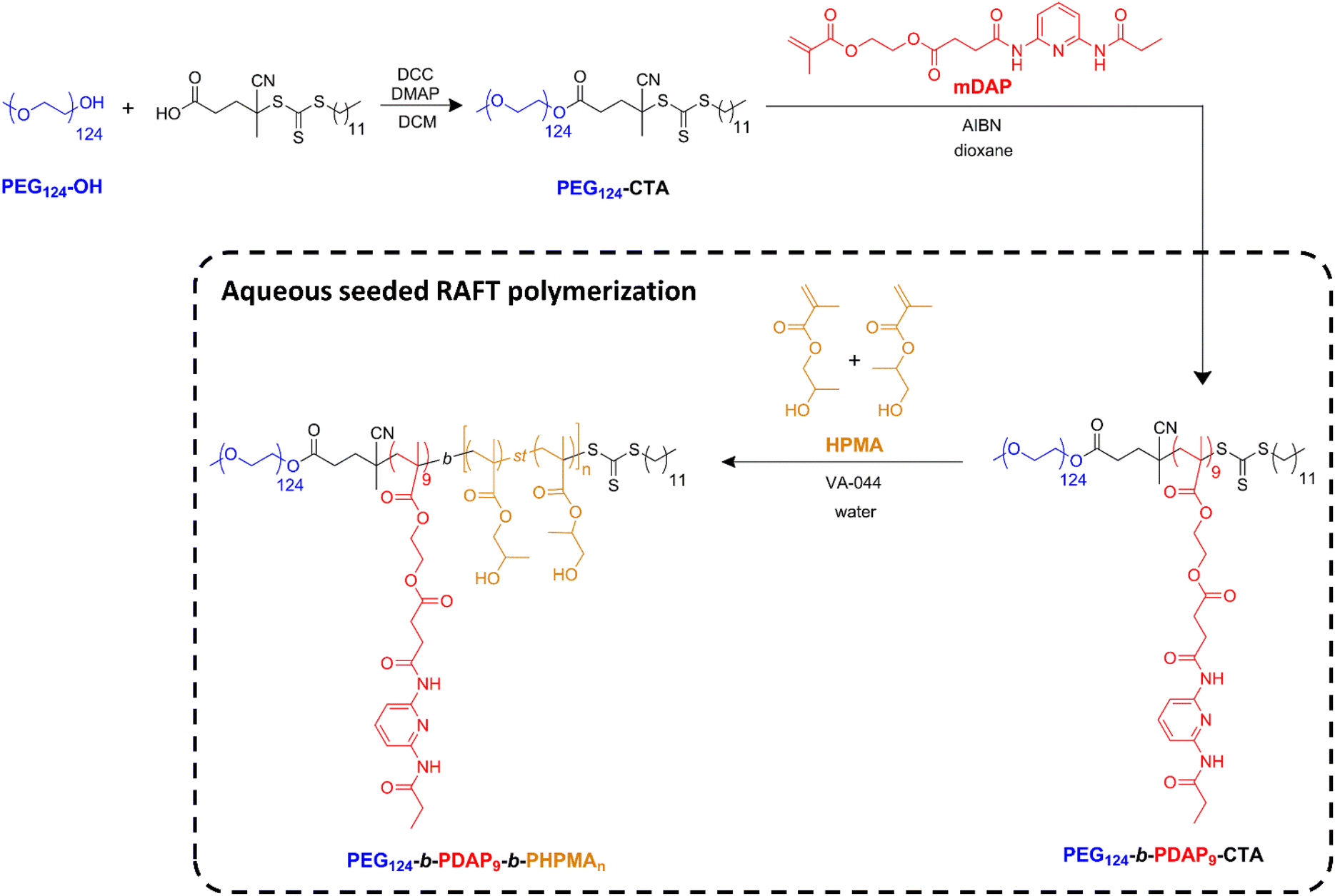 | ||
| Scheme 1 Synthesis of the PEG124-b-PDAP9-CTA macro-CTA by RAFT polymerization, and PEG124-b-PDAP9-b-PHPMAn by aqueous seeded RAFT polymerization. | ||
In this way, a diblock copolymer macro-CTA with a PEG and a PDAP blocks was first synthesized in two steps (Scheme 1) by RAFT polymerization. Commercial PEG monomethyl ether (PEG124-OH, 5450 g mol−1) was first conjugated with 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid by a Steglich esterification reaction to obtain the corresponding PEG124-CTA. This chain-transfer agent was chosen since it is suitable for (meth)acrylate polymerization.53,54PEG124-CTA was characterized by GPC (Fig. S1†) and the successful incorporation of the CTA was verified by 1H NMR spectroscopy (Fig. S2†). In a second step, the BC PEG124-b-PDAP9-CTA was prepared by RAFT polymerization (Scheme 1) of the methacrylic monomer mDAP from PEG124-CTA.55 Based on our experience, the target degree of polymerization of the PDAP block was limited to 10 to ensure an adequate water compatibility of the resulting BC macro-CTA. The experimental degree of polymerization and average molar mass (Mn) of the resulting diblock copolymer were determined by 1H NMR spectroscopy (Fig. S3†) as 9 and 9440 g mol−1, respectively. The polymer was also characterized by GPC and a low polydispersity was found (Đ = 1.18) (Fig. S1†).
The resulting BC, PEG124-b-PDAP9-CTA, was directly dispersed in water forming transparent solutions up to 10 g of polymer per 100 mL of water. Due to its amphiphilic character, the possible self-assembly in water was evaluated by transmission electron microscopy (TEM) (Fig. 1) and dynamic light scattering (DLS) (Fig. S4†). Short worms coexisting with a small population of spherical micelles of 15 ± 4 nm diameter were observed by TEM at 0.1 g per 100 mL polymer concentration (Fig. 1a). The self-assembly behavior of PEG124-b-PDAP9-CTA in mixtures of water and HPMA was also studied simulating the aqueous RAFT polymerization conditions (T = 50 °C). Two mixtures containing [HPMA]/[PEG124-b-PDAP9-CTA] = 100 and 300 molar ratios at a solid content of 10 g in 100 mL of water were evaluated and the values corresponded to the molar ratios and concentration feed in the aqueous RAFT polymerization (see below). The TEM images indicated that worms were shortened, and spherical micelles of 15 ± 4 nm diameters were predominant in the first mixture (Fig. 1b). However, by increasing the concentration of HPMA, it was found that the worms disappeared and spherical micelles were larger in size (27 ± 10 nm) and dispersity (Fig. 1c). DLS measurements of the self-assemblies were also performed at the polymerization temperature (50 °C) and it was observed that the presence of HPMA decreased the apparent Dh (Fig. S4†). Therefore, when the macro-CTA and HPMA were dispersed together in water, the initial PEG124-b-PDAP9-CTA preferentially formed spherical micelles.
 | ||
| Fig. 1 (a) TEM images of self-assemblies formed in water by PEG124-b-PDAP9-CTA at 0.1 g per 100 mL. TEM images of self-assemblies formed in water for (b) the [HPMA]/[PEG124-b-PDAP9-CTA] = 100 ratio and (c) [HPMA]/[PEG124-b-PDAP9-CTA] = 300 ratio at a content of 10 g of solids in 100 mL of water. | ||
Aqueous seeded RAFT polymerization and morphological characterization of self-assemblies
Since it is expected that the CTA group is embedded inside the micelle core (hydrophobic part) after self-assembly, chain extension to form the third HMPA block (PHMPA) should occur mainly inside the micelles. To check this HMPA migration, 1H NMR spectra were recorded for HPMA and a mixture of HPMA and PEG124-b-PDAP9-CTA, both in D2O (Fig. S5†). An attenuation in the signals was observed for the mixture of HPMA and PEG124-b-PDAP9-CTA compared to the signals of HPMA. This fact suggested a HPMA migration into PEG124-b-PDAP9-CTA self-assemblies as reported in the literature for other systems.56 Therefore, the self-assemblies would act as seeds for the polymerization of the water soluble HPMA monomer. Thus, polymerization of HMPA using the macro-CTA PEG124-b-PDAP9-CTA can be properly considered as a seeded dispersion RAFT polymerization in water.19The aqueous seeded RAFT polymerization of HMPA using PEG124-b-PDAP9-CTA (Scheme 1) was initiated by 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044) at 50 °C. A 0.3 molar ratio between the initiator and macro-CTA ([VA-044]/[PEG124-b-PDAP9-CTA]) was employed. This ratio was previously reported as optimum for aqueous RAFT-PISA of HMPA using PEG124-CTA to obtain well defined copolymers with relatively low dispersity and minimal homopolymer contamination.13 Polymerization was maintained for 5 h.
The influence of the polymer concentration and the length of the hydrophobic segments on the self-assembling properties was investigated by performing three series of experiments, x = 1, 2 and 3, where the target degree of polymerization given by the [HPMA]/[PEG124-b-PDAP9-CTA] ratio was approx. n = 100, 200 and 300, respectively. For each series, polymerization was performed at different solid concentrations (c = 10, 15, 20 and 25 expressed as grams of solids per 100 mL of water). 25 g per 100 mL was determined to be the maximum concentration at which PEG124-b-PDAP9-CTA was well dispersed in water before precipitation. The experiments were coded as c − x, where c refers to the solid concentration and x refers to the series of polymers. In all cases very high HPMA conversion values (≥98%) were obtained (Table S1†), as calculated by 1H NMR (Fig. S6†), using DMF as an internal standard.17
Polymers were analyzed by GPC (Fig. 2, S7 and Table S1†). GPC traces of the prepared polymers showed similar average molar masses for BCs with PHPMA of comparable length prepared at different polymer concentrations (Fig. 2). The dispersity values were in all cases between 1.31 and 1.45, higher than that of PEG124-b-PDAP9-CTA (Đ = 1.18). Nonetheless, it was observed that combining high target PHPMA degrees of polymerization and low solid concentrations favored the appearance of a small shoulder at higher elution times. GPC measurements were carried out at different polymer concentrations to dismiss the presence of self-assemblies in the eluent. Thus, shoulders should be most likely attributed to the presence of residual PEG124-b-PDAP9-CTA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 44,46 due to a relative low degree of uncontrolled polymerization at higher PHPMA degrees of polymerization and lower solid concentration.
44,46 due to a relative low degree of uncontrolled polymerization at higher PHPMA degrees of polymerization and lower solid concentration.
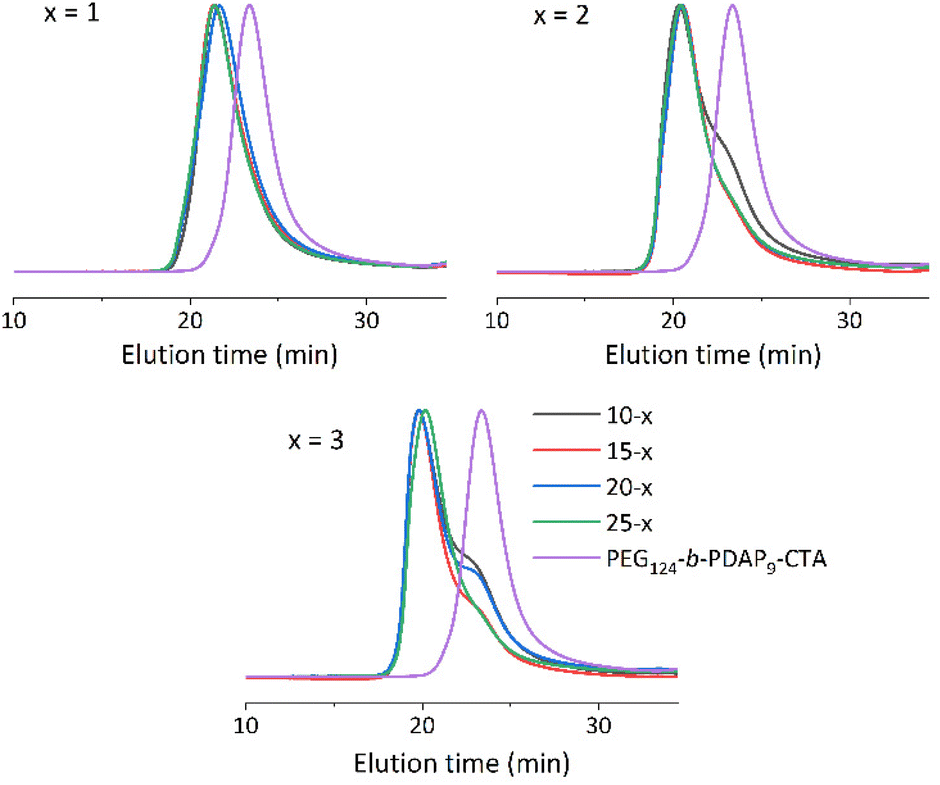 | ||
| Fig. 2 GPC chromatograms of series x = 1–3, polymerized at c = 10, 15, 20 and 25 solid concentrations. Eluent: DMF (LiBr 50 mM). Detector: UV (290 nm). | ||
The morphology of the self-assemblies prepared by aqueous seeded RAFT polymerization was identified by TEM and cryo-TEM while the average hydrodynamic diameters (Dh) and polydispersity indexes (PDI) were determined by DLS (Table 1). The resulting phase diagram is shown in Fig. 3.
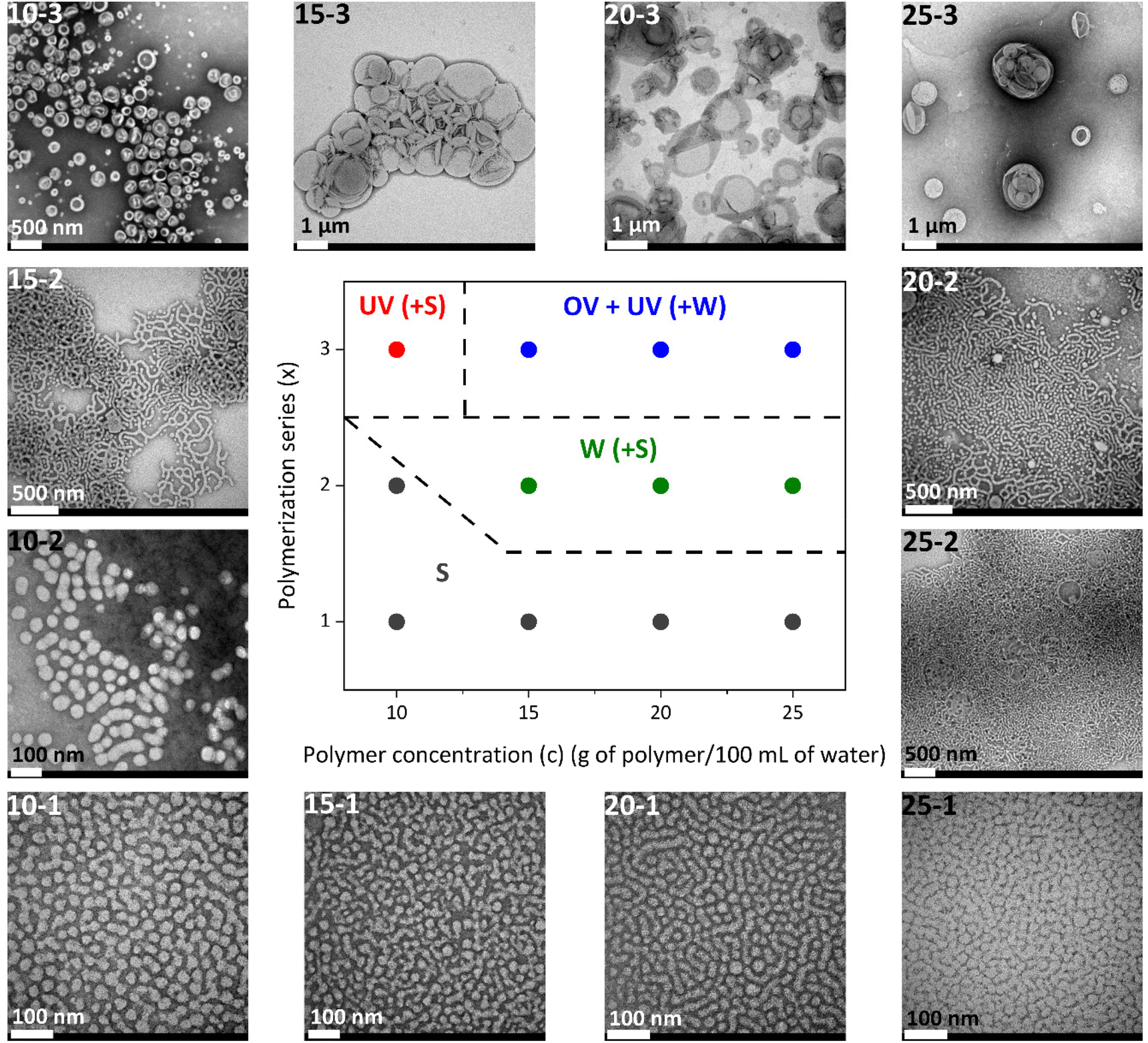 | ||
| Fig. 3 Phase diagram of PEG124-b-PDAP9-b-PHPMAn self-assembly dispersions prepared and accompanying TEM images (S = spheres or spherical micelles; W = worms; UV = unilamellar vesicles; and OV = oligolamellar vesicles). | ||
| Sample | M n (kDa) | Hydrophilic/hydrophobic ratiob | D h (PDI)c (nm) | TEM sized (nm) | Morphology |
|---|---|---|---|---|---|
| a Average number molar mass (Mn) calculated considering for PEG124-b-PDAP9-CTAMn = 9440 g mol−1 (estimated by 1H NMR) and the PHPMA degree of polymerization. b Hydrophobic/hydrophilic weight percentage ratio estimated considering the PEG block as the hydrophilic part and the PDAP9-b-PHPMAn segment as the hydrophobic part. c Average Dh values and PDI provided by Zetasizer software, from the intensity particle size distribution recorded by DLS. d TEM size data were reported as the mean ± SD (standard deviation) of a 150 measurements histogram. e Mean size and SD could not be reported since the histogram obtained did not fit to a Gaussian curve. | |||||
| 10-1 | 26.0 | 21/79 | 39 ± 9 (0.02) | 25 ± 6 | Spherical micelles |
| 10-2 | 40.3 | 13/87 | 70 ± 20 (0.07) | 44 ± 9 | Spherical micelles |
| 10-3 | 58.2 | 9/91 | 164 ± 57 (0.10) | 55–350e | Unilamellar vesicles |
| 15-1 | 24.6 | 22/78 | 40 ± 11 (0.06) | 29 ± 7 | Spherical micelles |
| 15-2 | 41.0 | 13/87 | 326 ± 178 (0.29) | 33 ± 6 | Worms (+spheres) |
| 15-3 | 57.9 | 9/91 | 883 ± 422 (0.19) | 220–1750e | Oligolamellar vesicles |
| 20-1 | 25.5 | 21/79 | 38 ± 12 (0.10) | 21 ± 6 | Spherical micelles |
| 20-2 | 40.7 | 13/87 | 436 ± 169 (0.37) | 32 ± 9 | Worms (+spheres) |
| 20-3 | 56.5 | 10/90 | 949 ± 489 (0.24) | 100–1500e | Oligolamellar vesicles |
| 25-1 | 24.5 | 22/78 | 41 ± 11 (0.05) | 25 ± 6 | Spherical micelles |
| 25-2 | 40.2 | 14/86 | 883 ± 551 (0.47) | 37 ± 8 | Worms (+spheres) |
| 25-3 | 54.7 | 10/90 | 906 ± 540 (0.34) | 95–1500e | Oligolamellar vesicles |
For series x = 1, having an approx. target polymerization degree of n = 100, turbid but fluid homogeneous dispersions of micelles were obtained for all tested polymerization concentrations (i.e. from c = 10 to 25 g solids per 100 mL of water) with average Dh values around 40 nm which were larger than those of PEG124-b-PDAP9-CTA, and low PDI (<0.1). Taking these observations as a starting point, transitions to more ordered morphologies from spherical micelles-to-worms-to-vesicles were detected on increasing both the length of the PHPMA block, i.e., the hydrophobic content of the final BCs, and the total solid concentration.
For instance, when fixing the solid concentration at c = 10 g per 100 mL but increasing the target degree of polymerization, an increase of the turbidity of the collected dispersions was observed with the naked eye. Accordingly, for 10-2 spherical homogeneous micelles of a larger size were self-assembled, with the average Dh increasing from 39 ± 9 nm for 10-1 to 70 ± 20 nm for 10-2 (Fig. S8†). Then, unilamellar vesicles were identified by cryo-TEM for 10-3 (Fig. 4) with sizes ranging from 55 to 350 nm (average Dh = 164 ± 57 nm) and a membrane thickness of 21 ± 2 nm. For 10-3, the remaining spherical micelles were also observed by TEM.
 | ||
| Fig. 4 Cryo-TEM images from PEG124-b-PDAP9-b-PHPMAn dispersions of series x = 3, obtained at different solid concentrations (c = 10, 15, 20 and 25). | ||
Considering higher solid concentrations (c = 15, 20 and 25 g per 100 mL) and high target degree of polymerization (series x = 2 and 3), opaque white viscous dispersions with intermediate or even more complex and mixed self-assembly morphologies were produced. For 15-2, 20-2 and 25-2, worms of approx. 35 nm cross section were predominantly detected in TEM images with some residual spherical micelles. The high viscosity of dispersions with a soft gel like appearance can probably be associated with a network formation due to entanglements of the worms.57 By DLS (Fig. S8†), broad size distributions with large PDI values were registered. Nonetheless, DLS data should be handled with caution for worm-like particles since Dh values are estimated under the assumption that the particles exist in solution as compact spheres.
For samples of series x = 3, more complex morphologies were observed by TEM (Fig. 3) and cryo-TEM (Fig. 4) above a concentration of 10 g per 100 mL, consisting of a mixture of unilamellar and oligolamellar vesicles, or vesicles within vesicles, with sizes ranging from a few hundred nm to a few μm and 25–30 nm membrane thickness. Vesicles with sizes above 1 μm appeared as broken or folded even in cryo-TEM. The folding of such large vesicles in cryo-TEM is not unusual since the vitrified ice layer should only be around a few hundred nm. In addition, some worms and intermediate morphologies of worm-to-vesicle transitions such as wormballs and jellyfish-like geometries were observed (Fig. S9†).15
Despite the presence of PDAP blocks, the results obtained from the morphological characterization of PEG124-b-PDAP9-b-PHPMAn self-assembly dispersions prepared by aqueous seeded RAFT polymerization were in accordance with the ones from BCs prepared by aqueous dispersion RAFT polymerization, using PEG as the macro-CTA and HPMA as the monomer.13,17
The stability of the dispersions upon storage was evaluated by DLS up to 11 months (Fig. S10†). Those dispersions with self-assembled spherical micelles (10-1, 15-1, 20-1, 25-1 and 10-2) and unilamellar vesicles (10-3) were stable as concluded from the registered DLS curves, with only a slightly increase in average Dh and PDI for samples 10-1 and 15-1 due to some aggregation of the micelles as observed by TEM (Fig. S11†) after 11 months. For those samples containing oligolamellar vesicles or vesicles within vesicles (15-3, 20-3 and 25-3) only slight variations were observed along the time. However, a clear evolution was observed after 6 weeks for dispersions containing worms (15-2, 20-2 and 25-2) with a remarkable decrease of the apparent Dh. However, DLS measurements taken at the 12th week were similar to those of the 6th week (Fig. S12a†). After 12 weeks, TEM images showed a large population of spherical micelles coexisting with short worms (Fig. S12b†). This suggests the worm dissociation into cylindrical and spherical micelles, which might be more thermodynamically stable morphologies.13,43 No significant further evolution of the samples was observed after 11 months.
Functionalization of the DAP-containing self-assemblies with a thymine cross-linker
As a next step, the ability of DAP-containing self-assemblies to be functionalized by supramolecular recognition of thymine derivatives mediated by a triple H-bonding was exploited (Fig. 5a). In particular, a molecule containing four thymine units (T4) (Fig. 5b) was employed as a cross-linker to improve the stability of the self-assemblies in water. It is expected that the incorporation of T4 plays a key role in potential application of these systems as nanocarriers.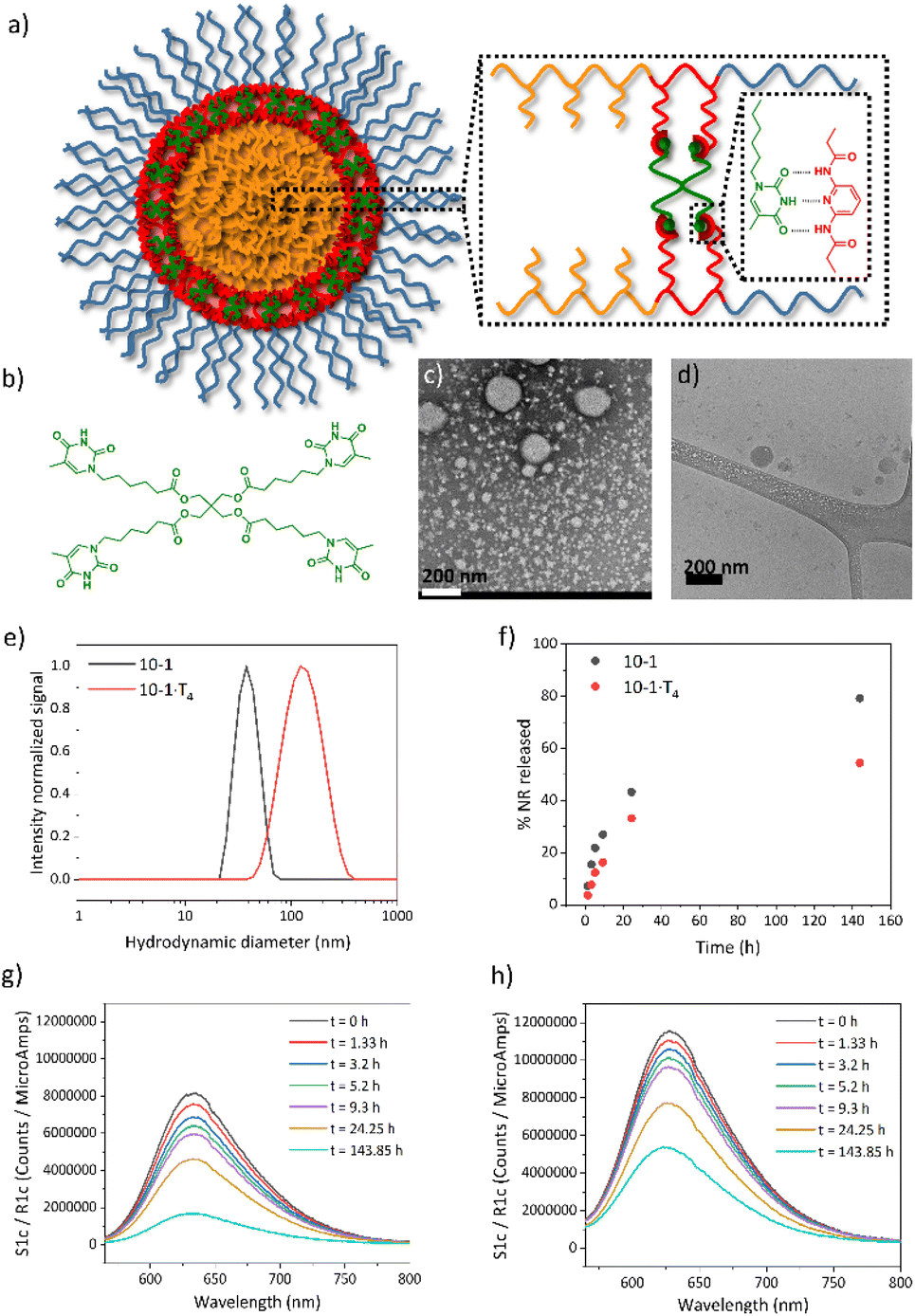 | ||
| Fig. 5 (a) Supramolecular recognition through a triple H-bond between DAP units (red) from PEG124-b-PDAP9-b-PHPMAn and thymine moieties (green) of the cross-linker (T4). (b) Chemical structure of the cross-linker (T4). (c) TEM and (d) cryo-TEM images of 10-1·T4 self-assemblies. (e) Intensity particle size distributions recorded by DLS of 10-1 and 10-1·T4 self-assembly dispersions. (f) Release kinetics curves of NR encapsulated into 10-1 and 10-1·T4. Fluorescence spectra of NR loaded (g) 10-1 and (h) 10-1·T4 self-assemblies at different dialysis times. | ||
In order to incorporate the thymine derivative T4 into the system, one-pot seeded polymerization and supramolecular cross-linking were carried out. In particular, we performed an analogous experiment on a 10-1 sample targeting a degree of polymerization of approx. 100 (series x = 1) and a final polymer concentration c = 10 (resulting material coded 10-1·T4) using a T4:PEG124-b-PDAP9-CTA molar ratio of 2.2:1 (i.e. a 1:1 thymine:DAP ratio). Because T4 is not soluble in water, it was incorporated into a small amount of DMF employed as an internal standard in the polymerization process (see details in the Experimental section). Prior to seeded aqueous polymerization, the influence of T4 on the initial self-assemblies of PEG124-b-PDAP9-CTA and PEG124-b-PDAP9-CTA and HPMA mixture was tested by TEM (Fig. S13†) observing that T4 induced the transformation to shorter worms and more spherical micelles in the first case while only spherical micelles were observed if HPMA was present. Polymerization was conducted to a high conversion (98%; Table S1†) similar to the 10-1 sample.
This time the envisaged self-assembly and supramolecular cross-linking resulted in a milky appearance dispersion for 10-1·T4 in contrast to the translucent dispersion collected for 10-1. GPC analysis (Fig. S14†) gave a low dispersity value, Đ = 1.31, in analogy to 10-1. Notwithstanding, GPC curves show two polymer distributions, one of them at the same elution time as the corresponding non-cross-linked sample, and another at higher elution time. This suggests that the HPMA polymerization is hindered to some extent by the presence of T4. By TEM (Fig. 5c), spherical micelles with a diameter of 21 ± 3 nm, smaller in size than the corresponding non-cross-linked ones, were observed, and they tend to aggregate. These micelles coexist with big spheres having sizes between 125 and 200 nm which seemed to have a compact and solid core as was suggested by cryo-TEM (Fig. 5d). These big spheres’ contribution in DLS shifted the mean size distribution to a higher Dh value (Dh = 137 ± 55 nm) (Fig. 5e). Therefore, the presence of T4 during the polymerization seemed to affect the polymerization and consequently the morphology of the self-assemblies formed.
Having established the conditions for the preparation of supramolecular cross-linked self-assemblies in water, the influence of cross-linking on the encapsulation and release of small molecular cargoes was investigated. Nile Red (NR) was selected as a hydrophobic molecular fluorescent probe. First, NR was encapsulated by diffusion in 10-1 and 10-1·T4 samples. NR loaded dispersions were characterized by DLS and TEM (Fig. S15†). By DLS only one size distribution was observed in both cases, very similar to the corresponding plain samples. Also by TEM, similar morphologies were observed with only a slight increase in the average size for 10-1 + NR, from 25 to 29 nm when compared to 10-1. As was seen by fluorescence spectroscopy (Fig. 5g and h), a higher amount of NR was encapsulated into 10-1·T4 than into 10-1, around 1.4 times more. To evaluate the NR release, both dispersions were dialyzed against a large water volume following NR fluorescence evolution as it is related to the NR still encapsulated. As can be seen in Fig. 5f, NR release was slower for 10-1·T4 than for 10-1, which evidences the higher stability of supramolecular cross-linked self-assemblies compared to non-cross-linked ones.
Experimental
Materials
Poly(ethylene glycol) monomethyl ether (PEG124-OH) with a molar mass of 5000 g mol−1 was purchased from Polysciences (Polysciences Europe GmbH, Hirschberg an der Bergstrasse, Germany) and its commercially reported average molar mass and polydispersity were checked by 1H NMR (degree of polymerization = 124; Mn = 5450 g mol−1; Fig. S16†) and by mass spectrometry (Mn determined using Polytools Bruker software for the analysis of mass spectrometry data = 5320 g mol−1). 4-Cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (97%) (CTA) was purchased from Merck (Merck KGaA, Darmstadt, Germany) and used as received. The methacrylate monomer mDAP was synthesized following previously reported procedures.55 2,2′-Azobis(2-methylpropionitrile) (AIBN) was purchased from Merck and recrystallized in ethanol before use. Dioxane was purchased from Merck and passed through a basic alumina column prior to use. 2,2′-Azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044) was purchased from TCI (TCI Europe N.V., Zwijndrecht, Belgium). The monomer 2-hydroxypropyl methacrylate (97%, a mixture of isomers 2-hydroxypropyl methacrylate and 1-hydroxypropan-2-yl methacrylate, HPMA) was purchased from Merck and passed through a basic alumina column to remove the inhibitor prior to use. A four thymine-terminal group supramolecular cross-linker (T4) was synthesized following previously reported procedures.51 Chloroform-d3 (CDCl3, 99.8 atom % D), methanol-d4 (MeOD, 99.8 atom % D) and water-d2 (D2O, 99.9% D) were purchased from Merck and utilized as solvents for NMR measurements. Other reagents were purchased from Merck and used as received.Characterization techniques and instrumentation
1H NMR, either in CDCl3, MeOD or D2O, was performed using a Bruker Avance III 300 spectrometer (Bruker, Billerica, MA, USA) operating at 300 MHz proton frequency, or in a Bruker Avance III 400 Prodigy spectrometer operating at a 400 MHz proton frequency, at 25 °C using standard pulse sequences. Chemical shifts are given in ppm relative to TMS, and the solvent residual peak was used as an internal reference. MALDI-TOF mass spectrometry was performed on a Microflex Bruker mass spectrometer, employing dithranol as the matrix. Gel permeation chromatography (GPC) was performed using a Water Alliance 2695 liquid chromatography system equipped with a UV-vis-Waters 2998 Photodiode Array detector and two column Styragel (HR1, 100 Å and HR3, 1000 Å) from Waters (5 μm, 7.8 × 300 mm) using DMF HPLC grade, with 50 mM LiBr as the eluent (0.5 mL min−1) applying a calibration with polystyrene standards. Samples were prepared taking an aliquot of the polymer dispersion that was first lyophilized and then dissolved in the eluent (DMF (LiBr 50 mM)).Dynamic Light Scattering (DLS) measurements were carried out in a Malvern Instrument Nano Zs (Malvern, Worcestershire, UK) using a He–Ne laser with a 633 nm wavelength and a detector angle of 173° at 25 °C. The aqueous self-assembly dispersions were measured at an approximately 1.0 mg mL−1 concentration, and hydrodynamic diameters (Dh) were given as an average of three measurements on each sample to ensure reproducibility. Transmission Electron Microscopy (TEM) analysis was performed using a FEI Tecnai T20 microscope (FEI Company, Waltham, MA, USA) operating at 200 kV. TEM samples were prepared adding 10 μL of each self-assembly dispersion at an approximately 1.0 mg mL−1 concentration on a continuous carbon film-copper grid, and the excess was removed by capillarity using filter paper. Then, the grids were stained with uranyl acetate (1% aqueous solution), removing the excess again by capillarity using filter paper. The grids were dried overnight under vacuum. Cryogenic Transmission Electron Microscopy (Cryo-TEM) analysis was performed in a JEM-2011 microscope (Jeol Europe SAS, Croissy-sur-Seine, France) operating at 80 kV. For cryo-TEM, holey carbon film-copper grids previously ionized using a plasma cleaner were employed. A droplet was placed onto the grid, and sample vitrification was automatically performed using FEI Vitrobot and liquid ethane. Samples were kept under liquid nitrogen with a Gatan TEM cryo-holder (FEI Company). Fluorescence emission spectra were obtained using a Fluorolog FL3-11 spectrometer (HORIBA ABX SAS, Madrid, Spain), with a Front Face accessory.
Macro-CTA synthesis
Poly(ethylene glycol) monomethyl ether (PEG124-OH) (1.48 g, 0.3 mmol), 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (0.35 g, 0.87 mmol) and 4-(dimethylamino)pyridine (0.03 g, 0.24 mmol) were dissolved in dry dichloromethane (14 mL) under an Ar atmosphere. After the solution was cooled to 0 °C, N,N′-dicyclohexylcabodiimide (0.27 g, 1.3 mmol) was added. The solution was stirred at 0 °C for 1 h, and then at room temperature for 24 h. The white precipitate was filtered off and the solvent was removed under vacuum. The crude product was precipitated twice in cold diethyl ether. The solid was centrifuged and dried. Poly(ethylene glycol) methyl ether 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl] pentanoate (PEG124-CTA) was obtained as a yellow powder. Yield: 90%. FT-IR (KBr disk, cm−1): 2885, 1970, 1761, 1467, 1360, 1342, 1280, 1242, 1150, 1106, 1060, 963, 947, 842. 1H NMR (400 MHz, CDCl3) δ (ppm): 4.25 (t, J = 4.8 Hz, 2H), 3.63 (s, 514H), 3.37 (s, 3H), 3.31 (t, J = 7.4 Hz, 2H), 2.66 (m, 2H), 2.57–2.31 (2H), 1.87 (s, 3H), 1.68 (qu, J = 4.7 Hz, 2H) 1.43–1.19 (20H), 0.87(t, J = 6.8 Hz, 3H). GPC (DMF): Mn = 22605 g mol−1; Đ = 1.07.
PEG124-CTA (0.52 g, 0.10 mmol), monomer mDAP (0.36 g, 0.95 mmol), and AIBN (2.35 mg, 0.014 mmol) were added to a crimp vial and dissolved in dioxane (2.2 mL). The resulting solution was purged for 20 min with Ar and then placed into a preheated oil bath at 75 °C. After 6 h, the reaction mixture was cooled down with an ice bath and exposed to air. The crude product was precipitated twice in cold diethyl ether. The solid was centrifuged and dried under vacuum overnight. PEG124-b-PDAP9-CTA was obtained as a slight yellow powder. Yield: 83%. Conversion: 93%. FT-IR (KBr disk, cm−1): 3318, 2885, 1965, 1734, 1695, 1585, 1517, 1448, 1359, 1343, 1282, 1243, 1150, 1114, 1061, 964, 947, 842, 803. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.97–8.23 (19H), 7.96–7.50 (29H), 4.42–4.05 (37H), 3.83–3.43 (528H), 3.37 (3H), 2.89–2.59 (37H), 2.44–2.27 (19H), 1.34–0.78 (65H). GPC (DMF): Mn = 32910 g mol−1; Đ = 1.18.
Seeded aqueous dispersion RAFT polymerization
The following representative protocol was used for the synthesis of 25-1. PEG124-b-PDAP9-CTA (0.075 g, 8.0 μmol), HPMA (0.12 g, 0.83 mmol), and VA-044 (0.8 mg, 2.5 μmol) were added into a 10 mL crimp vial and dissolved in distilled water (0.78 mL). Then, DMF (26 μL, 0.3 μmol) was added as an internal standard for conversion calculations by 1H NMR. In the case of one-pot PISA and supramolecular cross-linking, T4 was previously dissolved in this DMF adjusting its concentration to have a stoichiometric 1:1 thymine–DAP ratio in the final dispersion; since T4 is able to interact with four DAP moieties, and considering that each polymer chain has an average of 9 DAP units, the calculated T4:PEG124-b-PDAP9-CTA molar ratio was approx. 2.2:1. The mixture was degassed by bubbling Ar for 15 min. After this time, the vial was placed into a preheated oil bath at 50 °C. After 5 h, the vial was opened to air and allowed to cool down to room temperature. Conversion: >99%. 1H NMR (400 MHz, MeOD) δ (ppm): 7.80–7.60 (29H), 4.40–4.09 (37H), 4.07–3.75 (281H), 3.68–3.51 (650H), 3.36 (s, 3H), 2.83–2.69 (39H), 2.48–2.37 (19H), 2.09–1.82 (160H), 1.37–0.82 (827H).
Nile Red encapsulation and release
100 μL of NR solution in DCM (0.035 mg mL−1) were added to two flasks and then the solvent was evaporated. Afterwards, 11 mL of 10-1·T4 diluted dispersion (1.04 mg mL−1) was added into one of those flasks and 10.32 mL of a 10-1 diluted dispersion (1.02 mg mL−1) was added to the other one. In this way, the same polymer concentration, without taking into account T4, (0.957 mg mL−1) was achieved. The resulting dispersions were stirred for 24 h at room temperature to reach equilibrium before the fluorescence was measured (t = 0). After that, 0.5 mL of each solution were placed into a dialysis cup (Slide-A-Lyzer MINI Dialysis Device, 3.5 K MWCO, 0.5 mL; Thermo Fisher Scientific) and dialyzed against Milli-Q® water (14 mL) at room temperature. At a given time, the sample was removed from the dialysis cup, fluorescence was measured and then it was placed back into the dialysis cup. The emission spectra of NR were recorded from 565 to 800 nm while being excited at 550 nm.Conclusions
Highly concentrated aqueous self-assembly dispersions containing a non-water soluble nucleobase analogue (DAP) have been successfully prepared for the first time employing PISA methodology. To enable aqueous polymerization, an amphiphilic diblock copolymer composed of a PEG and a PDAP blocks was first synthesized by RAFT polymerization. This copolymer was easily dispersed in water and successfully used as the macro-CTA for the aqueous seeded RAFT polymerization of HPMA.By changing the length of the PHPMA block (series x = 1, 2 and 3) and the solid concentration (from 10 to 25 g per 100 mL), a phase diagram of these new PEG124-b-PDAP9-b-PHPMAn formulations has been constructed. TEM and DLS studies indicated that the aqueous seeded RAFT polymerization of HPMA led to aqueous self-assembly dispersions with diverse morphologies (spherical micelles, worms and vesicles), which depended on the length of the PHPMA block and solid concentration. The resulting self-assembly dispersions remain almost stable at least for 11 months, except in the case of worms’ morphology, in which a transition to spherical micelles was progressively observed.
Incorporation of nucleobase analogue DAP units allows the supramolecular functionalization with thymine complementary derivatives by triple H-bonding. This possibility has been initially evaluated by incorporating a cross-linking agent with four terminal thymine groups (T4) during aqueous seeded RAFT polymerization of HPMA. It was found that T4 influenced the polymerization and the self-assembly morphologies. In addition, NR was successfully encapsulated as a hydrophobic molecular fluorescent probe by diffusion in both non-cross-linked and supramolecular cross-linked self-assemblies. For the latter, a larger amount of NR could be encapsulated as well as a slower release was also observed. These preliminary results suggest a higher stability of the supramolecular cross-linked self-assemblies, evidencing the potential use of the system as nanocarriers.
Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
LO and MP acknowledge the Ministerio de Economía y Competitividad “Programa Excelencia” (MINECO)-FEDER (project grant number MAT2017-84838-P) and “Programa Generación de Conocimiento” (MICINN)-FEDER (project grant number PID2021-126132NB-I00), “Fondo Social Europeo and Gobierno de Aragón” and “Fondo Europeo de Desarrollo Regional” (E47-17R, FEDER 2014-2020 “Construyendo Europa desde Aragón”). MA Acknowledges Gobierno de Aragón and Fondo Social Europeo for her Ph.D. grant. EB acknowledges the Excellence Cluster “3D Matter Made to Order” (EXC-2082/1-390761711) and the Carl Zeiss Foundation through the “Carl-Zeiss-Foundation-Focus@HEiKA” as well as the support by the Helmholtz program “Materials Systems Engineering” (MSE) at the Karlsruhe Institute of Technology for general support. The authors acknowledge the “Centro de Química y Materiales de Aragón (CEQMA)” for the NMR facilities and the use of Electron Microscopy facilities of the “Laboratorio de Microscopías Avanzadas (LMA)” of the Universidad de Zaragoza and the “Servei de Microscòpia” of Universitat Autònoma de Barcelona. The authors additionally acknowledge Dr Olga Crespo for the fluorescence measurements. The authors are also thankful for the use of Servicios Científico Técnicos del CIBA (IACS-Universidad de Zaragoza).References
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS PubMed.
- R. Fenyves, M. Schmutz, I. J. Horner, F. V. Bright and J. Rzayev, J. Am. Chem. Soc., 2014, 136, 7762–7770 CrossRef CAS PubMed.
- J. Dupont, G. Liu, K. I. Niihara, R. Kimoto and H. Jinnai, Angew. Chem., Int. Ed., 2009, 48, 6144–6147 CrossRef CAS.
- A. Choucair, C. Lavigueur and A. Eisenberg, Langmuir, 2004, 20, 3894–3900 CrossRef CAS PubMed.
- S. Li, J. He, M. Zhang, H. Wang and P. Ni, Polym. Chem., 2016, 7, 1773–1781 RSC.
- M. Abad, G. Mendoza, L. Usón, M. Arruebo, M. Piñol, V. Sebastián and L. Oriol, Macromol. Biosci., 2022, 22, 2100528 CrossRef CAS.
- X. Wang and Z. An, Macromol. Rapid Commun., 2019, 40, 1–14 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS.
- S. Y. Khor, J. F. Quinn, M. R. Whittaker, N. P. Truong and T. P. Davis, Macromol. Rapid Commun., 2019, 40, 1–22 CrossRef.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef PubMed.
- F. L. Hatton, M. J. Derry and S. P. Armes, Polym. Chem., 2020, 11, 6343–6355 RSC.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS PubMed.
- Y. Kang, A. Pitto-Barry, H. Willcock, W. D. Quan, N. Kirby, A. M. Sanchez and R. K. O'Reilly, Polym. Chem., 2015, 6, 106–117 RSC.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- Y. Kang, A. Pitto-Barry, A. Maitland and R. K. O'Reilly, Polym. Chem., 2015, 6, 4984–4992 RSC.
- L. D. Blackman, K. E. B. Doncom, M. I. Gibson and R. K. O'Reilly, Polym. Chem., 2017, 8, 2860–2871 RSC.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- S. Chen, P. Shi and W. Zhang, Chin. J. Polym. Sci., 2017, 35, 455–479 CrossRef CAS.
- C. J. Mable, K. L. Thompson, M. J. Derry, O. O. Mykhaylyk, B. P. Binks and S. P. Armes, Macromolecules, 2016, 49, 7897–7907 CrossRef CAS PubMed.
- X. Shen, F. Huo, H. Kang, S. Zhang, J. Li and W. Zhang, Polym. Chem., 2015, 6, 3407–3414 RSC.
- F. Huo, C. Gao, M. Dan, X. Xiao, Y. Su and W. Zhang, Polym. Chem., 2014, 5, 2736–2746 RSC.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- M. Chen, J. W. Li, W. J. Zhang, C. Y. Hong and C. Y. Pan, Macromolecules, 2019, 52, 1140–1149 CrossRef.
- X. Dai, L. Yu, Y. Zhang, L. Zhang and J. Tan, Macromolecules, 2019, 52, 7468–7476 CrossRef CAS.
- S. Sugihara, S. P. Armes, A. Blanazs and A. L. Lewis, Soft Matter, 2011, 7, 10787–10793 RSC.
- K. L. Thompson, C. J. Mable, A. Cockram, N. J. Warren, V. J. Cunningham, E. R. Jones, R. Verber and S. P. Armes, Soft Matter, 2014, 10, 8615–8626 RSC.
- Q. Zhang, R. Wang, Y. Chen, L. Zhang and J. Tan, Langmuir, 2022, 38, 2699–2710 CrossRef CAS.
- J. R. Lovett, L. P. D. Ratcliffe, N. J. Warren, S. P. Armes, M. J. Smallridge, R. B. Cracknell and B. R. Saunders, Macromolecules, 2016, 49, 2928–2941 CrossRef CAS PubMed.
- S. Varlas, J. C. Foster, P. G. Georgiou, R. Keogh, J. T. Husband, D. S. Williams and R. K. O'Reilly, Nanoscale, 2019, 11, 12643–12654 RSC.
- L. P. D. Ratcliffe, K. J. Bentley, R. Wehr, N. J. Warren, B. R. Saunders and S. P. Armes, Polym. Chem., 2017, 8, 5962–5971 RSC.
- W. Zhou, Q. Qu, W. Yu and Z. An, ACS Macro Lett., 2014, 3, 1220–1224 CrossRef CAS.
- S. Xu, J. Yeow and C. Boyer, ACS Macro Lett., 2018, 7, 1376–1382 CrossRef CAS PubMed.
- M. Nardi, T. Scherer, L. Yang, C. Kübel, C. Barner-Kowollik and E. Blasco, Polym. Chem., 2021, 12, 1627–1634 RSC.
- Q. Zhao, Q. Liu, C. Li, L. Cao, L. Ma, X. Wang and Y. Cai, Chem. Commun., 2020, 56, 4954–4957 RSC.
- H. Yao, Y. Ning, C. P. Jesson, J. He, R. Deng, W. Tian and S. P. Armes, ACS Macro Lett., 2017, 6, 1379–1385 CrossRef CAS.
- X. Chen, L. Liu, M. Huo, M. Zeng, L. Peng, A. Feng, X. Wang and J. Yuan, Angew. Chem., Int. Ed., 2017, 56, 16541–16545 CrossRef CAS PubMed.
- H. Khan, S. Chen, H. Zhou, S. Wang and W. Zhang, Macromolecules, 2017, 50, 2794–2802 CrossRef CAS.
- E. Raphael, M. J. Derry, M. Hippler and S. P. Armes, Chem. Sci., 2021, 12, 12082–12091 RSC.
- G. Mellot, J. M. Guigner, L. Bouteiller, F. Stoffelbach and J. Rieger, Angew. Chem., Int. Ed., 2019, 58, 3173–3177 CrossRef CAS PubMed.
- P. Gao, H. Cao, Y. Ding, M. Cai, Z. Cui, X. Lu and Y. Cai, ACS Macro Lett., 2016, 5, 1327–1331 CrossRef CAS PubMed.
- T. N. Tran, S. Piogé, L. Fontaine and S. Pascual, Macromol. Rapid Commun., 2020, 41, 1–5 CrossRef.
- L. Romero-Azogil, N. J. W. Penfold and S. P. Armes, Polym. Chem., 2020, 11, 5040–5050 RSC.
- J. Tan, D. Liu, Y. Bai, C. Huang, X. Li, J. He, Q. Xu, X. Zhang and L. Zhang, Polym. Chem., 2017, 8, 1315–1327 RSC.
- N. Zaquen, J. Yeow, T. Junkers, C. Boyer and P. B. Zetterlund, Macromolecules, 2018, 51, 5165–5172 CrossRef CAS.
- J. Tan, H. Sun, M. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 CrossRef CAS PubMed.
- J. Tan, Q. Xu, Y. Zhang, C. Huang, X. Li, J. He and L. Zhang, Macromolecules, 2018, 51, 7396–7406 CrossRef CAS.
- N. J. W. Penfold, J. R. Whatley and S. P. Armes, Macromolecules, 2019, 52, 1653–1662 CrossRef CAS.
- A. Concellón, R. Clavería-Gimeno, A. Velázquez-Campoy, O. Abian, M. Piñol and L. Oriol, RSC Adv., 2016, 6, 24066–24075 RSC.
- A. Concellón, E. Blasco, A. Martínez-Felipe, J. C. Martínez, I. Šics, T. A. Ezquerra, A. Nogales, M. Piñol and L. Oriol, Macromolecules, 2016, 49, 7825–7836 CrossRef.
- M. Abad, A. Martínez-Bueno, G. Mendoza, M. Arruebo, L. Oriol, V. Sebastián and M. Piñol, Polymers, 2021, 13, 684 CrossRef CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS.
- T. Janoschka, A. Teichler, A. Krieg, M. D. Hager and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1394–1407 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, in Polymer Science: A Comprehensive Reference, Elsevier, 2012, vol. 3, pp. 181–226 Search PubMed.
- A. Concellón, E. Blasco, M. Piñol, L. Oriol, I. Díez, C. Berges, C. Sánchez-Somolinos and R. Alcalá, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3173–3184 CrossRef.
- T. J. Neal, N. J. W. Penfold and S. P. Armes, Angew. Chem., Int. Ed., 2022, 61, e202207376 CrossRef CAS PubMed.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: mDAP solubility tests; Fig. S1–S16 and Table S1. See DOI: https://doi.org/10.1039/d2py01250b |
| This journal is © The Royal Society of Chemistry 2023 |