
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater as a monomer: synthesis of an aliphatic polyethersulfone from divinyl sulfone and water†
Karin
Ratzenböck
 ab,
Mir Mehraj
Ud Din
cd,
Susanne M.
Fischer
ab,
Ema
Žagar
e,
David
Pahovnik
e,
A. Daniel
Boese
f,
Daniel
Rettenwander
cd and
Christian
Slugovc
*ab
ab,
Mir Mehraj
Ud Din
cd,
Susanne M.
Fischer
ab,
Ema
Žagar
e,
David
Pahovnik
e,
A. Daniel
Boese
f,
Daniel
Rettenwander
cd and
Christian
Slugovc
*ab
aChristian Doppler Laboratory for Organocatalysis in Polymerization, Stremayrgasse 9, 8010 Graz, Austria. E-mail: slugovc@tugraz.at
bInstitute for Chemistry and Technology of Materials, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria
cDepartment of Material Science and Engineering, NTNU Norwegian University of Science and Technology, Sem Sælands vei 12, 7034 Trondheim, Norway
dInternational Christian Doppler Laboratory for Solid-State Batteries, NTNU Norwegian University of Science and Technology, Sem Sælands vei 12, 7034 Trondheim, Norway
eNational Institute of Chemistry, Department of Polymer Chemistry and Technology, Hajdrihova 19, 1000 Ljubljana, Slovenia
fPhysical and Theoretical Chemistry, Institute of Chemistry, University of Graz, Heinrichstrasse 28/IV, 8010 Graz, Austria
First published on 23rd May 2022
Abstract
Using water as a monomer in polymerization reactions presents a unique and exquisite strategy towards more sustainable chemistry. Herein, the feasibility thereof is demonstrated by the introduction of the oxa-Michael polyaddition of water and divinyl sulfone. Upon nucleophilic or base catalysis, the corresponding aliphatic polyethersulfone is obtained in an interfacial polymerization at room temperature in high yield (>97%) within an hour. The polyethersulfone is characterized by relatively high molar mass averages and a dispersity around 2.5. The polymer was tested as a solid polymer electrolyte with lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) as the salt. Free-standing amorphous membranes were prepared by a melt process in a solvent-free manner. The polymer electrolyte containing 15 wt% LiTFSI featured an oxidative stability of up to 5.5 V vs. Li/Li+ at 45 °C and a conductivity of 1.45 × 10−8 S cm−1 at room temperature.
Introduction
Water, covering around 71% of the earth's surface, is the world's most abundant natural resource.1 While the upcycling and polymerization of natural resources such as CO2 has been of high interest in polymer synthesis over the last decades,2 the use of water as a polymerizable substance has not yet been studied extensively. However, water is the epitome of a green monomer, being sustainable, environmentally friendly, non-toxic, abundant, and cheap. Water has only been employed in some multicomponent polymerization reactions in which three or more starting materials are combined in one step.3 Typical examples comprise the formation of polyamides by combining diisocyanides, bis(bromoalkyne)s and water,4 or by using benzoxazines, isocyanides and water.5 The synthesis of poly(N-acylsulfonamide)s with alkynes, sulfonyl azides, isatins and water has also been reported.6 However, in these studies, water is used among multiple components and does not cause a direct linkage in the backbone of the polymer. To the best of our knowledge, water functioning as one of two monomers has not been studied so far in any polymerization reaction. The concept thereof is intriguing.When selecting a reaction type for polymerizing water, the oxa-Michael addition is worth considering. Copolymerization of diols with difunctional Michael acceptors such as divinyl sulfone or diacrylates is a common process.7 However, water could also be regarded as the simplest oxygen based Michael donor which is able to form two bonds. Indeed, water is used as a Michael donor in Michael reactions even on an industrial scale as in the addition of water to acrolein yielding 3-hydroxypropanal.8 The addition of water to α,β-unsaturated ketones, for instance, catalyzed by trialkyl phosphines,9 amino acids,10 or indium(III) salts11 is known. Furthermore, a double Michael addition of water has been described in the synthesis of 2-cyanoethyl ether from acrylonitrile and aqueous NaOH.12 However, the oxa-Michael addition of water is rather an exception since water is more often used as an inert solvent in Michael reactions.13,14 Moreover, by using water in a polyaddition reaction one might anticipate at least two major challenges. Firstly, an equimolar amount of monomers is required to obtain reasonable molar masses (as the reaction presumably proceeds via a step-growth mechanism) and adjusting the stoichiometry is difficult. Secondly, reactive Michael donors are desired as water displays a lower nucleophilicity than alcohols.13,15 To obtain reasonable molar masses a high yield in each reaction step is necessary. Accordingly, the highly reactive difunctional Michael acceptor divinyl sulfone (DVS),16 which previously showed high reactivity with alcohols in oxa-Michael polymerization,17 was selected. The desired aliphatic polyethersulfone (PES) resulting from the potential polyaddition of water and DVS18 has recently been reported by Brendel and coworkers.19 They used a different but similar synthetic route performing the oxa-Michael polyaddition of 2-hydroxyethyl vinyl sulfone, priorly prepared by the oxidation of vinylmercaptoethanol. Herein, we present the use of water as a monomer in the oxa-Michael polyaddition with DVS. The obtained aliphatic polyethersulfone was further tested as a solid polymer electrolyte, which was prepared in a solvent-free manner.
Results and discussion
Since base or nucleophilic catalysis is typically needed for oxa-Michael additions to proceed in a useful speed at ambient temperatures,7 the investigation started with a screening of different nucleophiles and bases. It is to note that DVS and water are hardly miscible and form a two phase system. Therefore, DVS and water were employed in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 and 5 mol% of the catalyst (in respect to DVS) was added to the stirred reaction mixture at room temperature. Only upon stirring and thus, increasing the interface between water and DVS, polymerization proceeds, finally resulting in the formation of a precipitate. The reaction was monitored via1H-NMR spectroscopy (by dissolving an aliquot of the reaction mixture in DMSO-d6 and assessing the conversion of the double bonds) after 1 h and 24 h. In all cases, the formation of the desired polymer (PES) together with an anticipated cyclic side product, 1,4-oxathiane 4,4-dioxide (1), was observed (Table 1, for details see ESI†). Results with nucleophiles such as 4-dimethylaminopyridine (DMAP) or quinuclidine (ABCO) showed above 90% double bond conversion after already one-hour reaction time (Table 1, entry 1, 2). By changing the bicyclic ABCO to a simple tertiary amine, such as triethylamine (TEA), the double bond conversion significantly decreased (74%; Table 1, entry 3). In case of N,N-diisopropylethylamine (DIPEA) hardly any conversion could be observed after 1 h (Table 1, entry 4). Because of its bicyclic structure, ABCO shows the highest nucleophilicity. TEA and DIPEA, however, are much weaker nucleophiles due to increased steric hindrance and thus, the reactivity decreases.20 Moreover, 1,1,3,3-tetramethylguanidine (TMG), often described as non-nucleophilic base, showed high efficiency (97% conversion after 1 h; Table 1, entry 5). In Michael additions, however, guanidines have also been reported to react as nucleophiles with the Michael acceptor.21 With triphenyl phosphine (PPh3) a purely nucleophilic catalyst was tested (Table 1, entry 6). The poor solubility of PPh3 in water might explain its weaker performance compared to other nucleophiles. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) proved to be a less suitable catalyst in this reaction, presumably due to quick hydrolysis under the employed conditions (Table 1, entry 7).22,23 With potassium tert-butoxide (KOtBu) or sodium hydroxide a much higher share of the cyclic side product 1 was observed. Moreover, a slightly lower double bond conversion compared to runs with nucleophiles like DMAP or TMG was achieved (Table 1, entry 8, 9). In the following, different reaction parameters (Table 2) were studied for DMAP and TMG catalyzed reactions. Moreover, KOtBu was investigated further to include a purely basic catalyst for comparison. Besides the double bond conversion, size exclusion chromatography (SEC) was used to assess the molar mass characteristics of the polymers. The reference PES synthesized with 5 mol% DMAP at room temperature (Table 2, entry 1) featured number and weight average molar masses (Mn, Mw) of 5.3 and 13.3 kg mol−1 and a dispersity of 2.5. In comparison, Brendel and coworkers reported number average molar masses between 2.2 and 3.7 kg mol−1 (Đ = 1.3–1.6) for the same polymer prepared from 2-hydroxyethyl vinyl sulfone (measured in LiCl/N,N-dimethylacetamide based on conventional calibration using poly(ethylene oxide) standards).19 Increasing the catalyst loading of DMAP to 10 mol%, led to a slight reduction in molar mass averages (Table 2, entry 2). Likewise, a reduction of the catalyst loading to 2 mol% DMAP gave even lower molar mass averages, which is in line with a lower double bond conversion in this case. With 5 mol% TMG, the molar mass averages are also slightly lower (Table 2, entry 4) than those of reference PES, which is the consequence of a more pronounced shoulder in the low molar mass range. However, results with 2 mol% TMG are competitive with those obtained for 5 mol% DMAP in terms of the molar mass averages and the double bond conversion. In all cases described so far, side product 1 only formed in small amounts. Performing the reaction with 5 mol% base (Table 2, entry 6) gave PES with distinctly lower molar mass averages compared to nucleophile catalyzed reactions. Furthermore, the influence of the amount of water was investigated. Increasing the amount of water from 10 to 20 equiv. (Table 2, entry 7) led only to a somewhat lower double bond conversion and slight tailing at the low molar mass peak end. However, using 100 equiv. water decreased the double bond conversion to only 76%, while SEC curve shift to a larger elution volume, which is reflected in reduced molar mass averages of the product (Table 2, entry 8). Additionally, the share of 1 was higher (7%). Using base catalysis and 100 equiv. water (Table 2, entry 9) low conversion of 81%, high share of 1 (13%), and low molar mass averages of the product were obtained. Finally, the influence of the temperature was assessed. At a low temperature of 4 °C the reaction is slower resulting in a double bond conversion of 72% after 1 h. Nevertheless, a polymer with high molar mass averages and somewhat broader molar mass distribution was obtained (Table 2, entry 10). Increasing the temperature to 40 °C resulted in the formation of shorter polymer strands in comparison to room temperature experiments (Table 2, entries 11 and 12). The share of 1 stayed unaltered under these reaction conditions. Doubling the reaction temperature to 80 °C led to a significant decrease in the molar mass averages. Predominantly OH-terminated oligomers (Fig. S3†) accompanied by high shares of side product 1 were obtained. In case of using KOtBu at 80 °C a mixture of 72% 1 and 28% bis(2-hydroxyethyl)sulfone (HES) formed after 30 min reaction time. Upon further heating HES is fully converted into 1. This transformation presumably proceeds via elimination of water at elevated temperatures and under basic conditions.24 In summary, PES is preferably formed at low temperature with nucleophiles as catalysts. At elevated temperatures and particularly if catalyzed by base, the formation of 1 and short hydroxy terminated oligomers is preferred.
:10 and 5 mol% of the catalyst (in respect to DVS) was added to the stirred reaction mixture at room temperature. Only upon stirring and thus, increasing the interface between water and DVS, polymerization proceeds, finally resulting in the formation of a precipitate. The reaction was monitored via1H-NMR spectroscopy (by dissolving an aliquot of the reaction mixture in DMSO-d6 and assessing the conversion of the double bonds) after 1 h and 24 h. In all cases, the formation of the desired polymer (PES) together with an anticipated cyclic side product, 1,4-oxathiane 4,4-dioxide (1), was observed (Table 1, for details see ESI†). Results with nucleophiles such as 4-dimethylaminopyridine (DMAP) or quinuclidine (ABCO) showed above 90% double bond conversion after already one-hour reaction time (Table 1, entry 1, 2). By changing the bicyclic ABCO to a simple tertiary amine, such as triethylamine (TEA), the double bond conversion significantly decreased (74%; Table 1, entry 3). In case of N,N-diisopropylethylamine (DIPEA) hardly any conversion could be observed after 1 h (Table 1, entry 4). Because of its bicyclic structure, ABCO shows the highest nucleophilicity. TEA and DIPEA, however, are much weaker nucleophiles due to increased steric hindrance and thus, the reactivity decreases.20 Moreover, 1,1,3,3-tetramethylguanidine (TMG), often described as non-nucleophilic base, showed high efficiency (97% conversion after 1 h; Table 1, entry 5). In Michael additions, however, guanidines have also been reported to react as nucleophiles with the Michael acceptor.21 With triphenyl phosphine (PPh3) a purely nucleophilic catalyst was tested (Table 1, entry 6). The poor solubility of PPh3 in water might explain its weaker performance compared to other nucleophiles. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) proved to be a less suitable catalyst in this reaction, presumably due to quick hydrolysis under the employed conditions (Table 1, entry 7).22,23 With potassium tert-butoxide (KOtBu) or sodium hydroxide a much higher share of the cyclic side product 1 was observed. Moreover, a slightly lower double bond conversion compared to runs with nucleophiles like DMAP or TMG was achieved (Table 1, entry 8, 9). In the following, different reaction parameters (Table 2) were studied for DMAP and TMG catalyzed reactions. Moreover, KOtBu was investigated further to include a purely basic catalyst for comparison. Besides the double bond conversion, size exclusion chromatography (SEC) was used to assess the molar mass characteristics of the polymers. The reference PES synthesized with 5 mol% DMAP at room temperature (Table 2, entry 1) featured number and weight average molar masses (Mn, Mw) of 5.3 and 13.3 kg mol−1 and a dispersity of 2.5. In comparison, Brendel and coworkers reported number average molar masses between 2.2 and 3.7 kg mol−1 (Đ = 1.3–1.6) for the same polymer prepared from 2-hydroxyethyl vinyl sulfone (measured in LiCl/N,N-dimethylacetamide based on conventional calibration using poly(ethylene oxide) standards).19 Increasing the catalyst loading of DMAP to 10 mol%, led to a slight reduction in molar mass averages (Table 2, entry 2). Likewise, a reduction of the catalyst loading to 2 mol% DMAP gave even lower molar mass averages, which is in line with a lower double bond conversion in this case. With 5 mol% TMG, the molar mass averages are also slightly lower (Table 2, entry 4) than those of reference PES, which is the consequence of a more pronounced shoulder in the low molar mass range. However, results with 2 mol% TMG are competitive with those obtained for 5 mol% DMAP in terms of the molar mass averages and the double bond conversion. In all cases described so far, side product 1 only formed in small amounts. Performing the reaction with 5 mol% base (Table 2, entry 6) gave PES with distinctly lower molar mass averages compared to nucleophile catalyzed reactions. Furthermore, the influence of the amount of water was investigated. Increasing the amount of water from 10 to 20 equiv. (Table 2, entry 7) led only to a somewhat lower double bond conversion and slight tailing at the low molar mass peak end. However, using 100 equiv. water decreased the double bond conversion to only 76%, while SEC curve shift to a larger elution volume, which is reflected in reduced molar mass averages of the product (Table 2, entry 8). Additionally, the share of 1 was higher (7%). Using base catalysis and 100 equiv. water (Table 2, entry 9) low conversion of 81%, high share of 1 (13%), and low molar mass averages of the product were obtained. Finally, the influence of the temperature was assessed. At a low temperature of 4 °C the reaction is slower resulting in a double bond conversion of 72% after 1 h. Nevertheless, a polymer with high molar mass averages and somewhat broader molar mass distribution was obtained (Table 2, entry 10). Increasing the temperature to 40 °C resulted in the formation of shorter polymer strands in comparison to room temperature experiments (Table 2, entries 11 and 12). The share of 1 stayed unaltered under these reaction conditions. Doubling the reaction temperature to 80 °C led to a significant decrease in the molar mass averages. Predominantly OH-terminated oligomers (Fig. S3†) accompanied by high shares of side product 1 were obtained. In case of using KOtBu at 80 °C a mixture of 72% 1 and 28% bis(2-hydroxyethyl)sulfone (HES) formed after 30 min reaction time. Upon further heating HES is fully converted into 1. This transformation presumably proceeds via elimination of water at elevated temperatures and under basic conditions.24 In summary, PES is preferably formed at low temperature with nucleophiles as catalysts. At elevated temperatures and particularly if catalyzed by base, the formation of 1 and short hydroxy terminated oligomers is preferred.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | DB conversion at 1 h [%]b | Side product 1 at 1 h [%]c | DB conversion at 24 h [%]b | Side product 1 at 24 h [%]c |
|
a Molar ratio of DVS:H2O:catalyst = 1:10:0.05; no extra solvent used; reaction stirred at room temperature.
b Double bond conversion is calculated from 1H-NMR spectra by setting signal intensities for protons from double bonds in relation to the intensities for emerging ethylene groups (see ESI for details).
c Percentage of 1 in relation to all ethylene groups.
|
|||||
| 1 | DMAP | 97 | 2 | 99 | 2 |
| 2 | ABCO | 91 | 2 | 98 | 4 |
| 3 | TEA | 74 | 4 | 74 | 6 |
| 4 | DIPEA | 6 | <1 | 57 | 3 |
| 5 | TMG | 97 | 4 | 98 | 4 |
| 6 | PPh3 | 47 | 2 | 92 | 3 |
| 7 | DBU | 48 | 4 | 58 | 4 |
| 8 | KOtBu | 89 | 26 | 94 | 27 |
| 9 | NaOH | 78 | 14 | 91 | 14 |
| Entry | Catalyst (loading [mol%]) | Water [equiv.] | T [°C] | DB conversion at 1 h [%]b | Side product 1 at 1 h [%]d | M w [kg mol−1]e | M n [kg mol−1]e | Đ |
|---|---|---|---|---|---|---|---|---|
| a No extra solvent used; reaction stirred at given temperature. b Double bond conversion is calculated from 1H-NMR spectra by setting signal intensities for protons from double bonds in relation to the intensities for emerging ethylene groups (see ESI for details). c Double bond conversion after 30 min; after 1 h full conversion was observed. d Percentage of 1 in relation to all ethylene groups. e Determined by size exclusion chromatography in LiBr/DMSO relative to PMMA standards after >24 h reaction time (double bond conversion was in all cases higher than 95%). | ||||||||
| 1 | DMAP (5) | 10 | 23 | 97 | 2 | 13.3 | 5.3 | 2.5 |
| 2 | DMAP (10) | 10 | 23 | 98 | 2 | 11.7 | 4.6 | 2.5 |
| 3 | DMAP (2) | 10 | 23 | 79 | 4 | 8.5 | 3.4 | 2.5 |
| 4 | TMG (5) | 10 | 23 | 97 | 4 | 11.5 | 4.4 | 2.6 |
| 5 | TMG (2) | 10 | 23 | 98 | 2 | 11.8 | 4.9 | 2.4 |
| 6 | KOtBu (5) | 10 | 23 | 89 | 26 | 2.7 | 1.3 | 2.1 |
| 7 | DMAP (5) | 20 | 23 | 91 | 2 | 13.3 | 4.6 | 2.9 |
| 8 | DMAP (5) | 100 | 23 | 76 | 7 | 7.5 | 3.2 | 2.3 |
| 9 | KOtBu (5) | 100 | 23 | 81 | 13 | 3.0 | 1.7 | 1.7 |
| 10 | DMAP (5) | 10 | 4 | 72 | 2 | 16.6 | 5.2 | 3.2 |
| 11 | DMAP (2) | 10 | 40 | 93c | 3 | 10.2 | 4.2 | 2.4 |
| 12 | DMAP (5) | 10 | 40 | 99c | 3 | 7.6 | 3.5 | 2.2 |
| 13 | DMAP (2) | 10 | 80 | 99c | 16 | 1.1 | 0.5 | 2.2 |
| 14 | DMAP (5) | 10 | 80 | >99c | 20 | n.d. | n.d. | n.d. |
| 15 | TMG (2) | 10 | 80 | >99c | 32 | 0.7 | 0.4 | 1.8 |
| 16 | KOtBu (5) | 10 | 80 | >99c | 72 | n.d. | n.d. | n.d. |
Detailed characterization of PES obtained from entry 1 in Table 2 by NMR spectroscopy and matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS) revealed very similar results as reported for PES obtained from 2-hydroxyethyl vinyl sulfone.19 In 1H-NMR spectroscopy, the oxa-Michael repeating unit gives rise to two triplets at 3.81 ppm and 3.42 ppm. As end groups 4-dimethylamino pyridinium (8.28, 7.04 ppm), alcohol units (3.78, 3.26 ppm) and vinyl groups (7.00–6.94, 6.25–6.19 ppm) were observed (Fig. S4†). MALDI-TOF MS confirmed the presence of all end groups identified by NMR spectroscopy (Fig. S12†) and 4-dimethylamino pyridinium end-capped species (m/z = 1329.26 Da, 1347.27 Da) were found as the most abundant series. In case of TMG catalysis, results of MALDI-TOF MS suggest that TMG is acting as nucleophile in the reaction. Additionally, PES species terminated by the hydrolysis products of TMG, dimethyl amine and 1,1-dimethyl urea,25 were observed (Fig. S15†). Thus, the lower molar mass obtained in the TMG catalyzed reaction (Table 2, entry 4) might be explained. PES originating from KOtBu catalyzed reactions (Table 2, entry 6) is terminated by either two vinyl or a vinyl and a hydroxyl end-group (Fig. S17†).
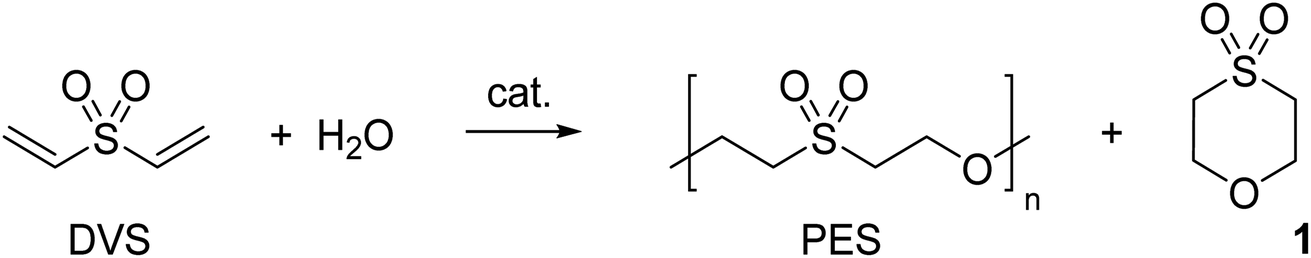
In a next step, the reaction progress was investigated with 10 equiv. water, room temperature and 5 mol% DMAP as reaction conditions. As shown in Fig. 1a and b, DVS is quickly consumed. The first species which can be observed by NMR spectroscopy (after 25 s reaction time) comprise the DMAP adduct with DVS, very small amounts of 1 and as major product the trimer made of two equivalents of DVS and one equivalent of water (Fig. 1a). Interestingly, 2-hydroxyethyl vinyl sulfone (or the according alkoxide) could not be observed. Between 3 to 6 min the initially heterogeneous reaction mixture becomes homogeneous. At this stage, mainly vinyl sulfone terminated oligomers are present. After 10–15 min reaction time, the formation of an oily precipitate was noted. Only about 2–3% of DVS is not consumed and oligomerization has proceeded (Fig. 1a and b). As time goes on, the oligomers grow into a polymer indicated by a downfield shift of the oxa-Michael repeating unit towards 3.81 ppm (Fig. 1a and S20†). After about 45 min the initially oily precipitate has turned into an off-white, brittle solid. Cycle 1 can be detected from the very beginning and its share is only marginally increasing over time (Fig. 1b). In case of base catalysis with 5 mol% KOtBu a slower double bond conversion and a higher share of 1 evolving over time was detected (Fig. S23†). In this case, no interim homogenization of the reaction mixture was observed and precipitation occurred only after approx. 2.5 h reaction time. Monitoring the evolution of the molar masses over time revealed a distinctly faster increase of the molar masses in case of DMAP catalyzed reactions compared to KOtBu catalyzed ones (Fig. 1c). Moreover, the reaction was performed as a 1 M solution of DVS in dimethyl sulfoxide in the presence of 10 equiv. water and 5 mol% DMAP at room temperature. After 1 h the same products, detected already after 25 s in the solvent-free reaction, were observed (Fig. S28†). This illustrates the importance of undiluted reaction conditions for obtaining a fast polymerization.
 | ||
| Fig. 1 (a) 1H-NMR spectra and photographs of the reaction vessel at indicated time; reaction conditions in every case: molar ratio of DVS:H2O:catalyst = 1:10:0.05; no extra solvent used; reaction stirred at room temperature; (b) reaction progress showing the development of the relative amount of different chemical entities over time (DMAP catalyzed); (c) normalized refractive index responses in SEC chromatograms show different profiles of peak shifting towards lower elution volume (increase in molar mass) with reaction time for DMAP and KOtBu catalyzed reactions. | ||
Mechanistically, for the polymerization of DVS and water in the first step hydroxide ions need to be formed either by a base (Scheme 1, A) or by a nucleophile (B). In the latter case, hydroxide is generated upon the addition of the nucleophile to the Michael acceptor, forming a basic zwitter-ionic species that subsequently is protonated by water.9,26 In other words, both base and nucleophilic catalysis generate hydroxide ions but different cations are present, e.g. K+ (in case of KOtBu) or the presumably much softer nucleophile derived cation shown in Scheme 1 as B. In any case, the hydroxide is reacting with DVS to form the alkoxide C. The alkoxide C immediately undergoes oxa-Michael reaction with another DVS generating, together with another equivalent of water, the polymeric unit D and hydroxide. Alternatively, C might react by an intramolecular oxa-Michael reaction resulting in 1 and hydroxide upon protonation with water. However, in case of nucleophilic catalysis 1 might also be obtained by an intramolecular nucleophilic substitution reaction of the zwitter-ion E. E originates from the oxa-Michael addition of the hydroxide to the vinyl group in ion-pair B. Subsequently, upon addition of DVS and protonation by another water molecule, the first member of the homologous series of ion-pairs F is formed. However, this propagation pathway can be considered as less important compared to the main propagation pathway (Scheme 1).
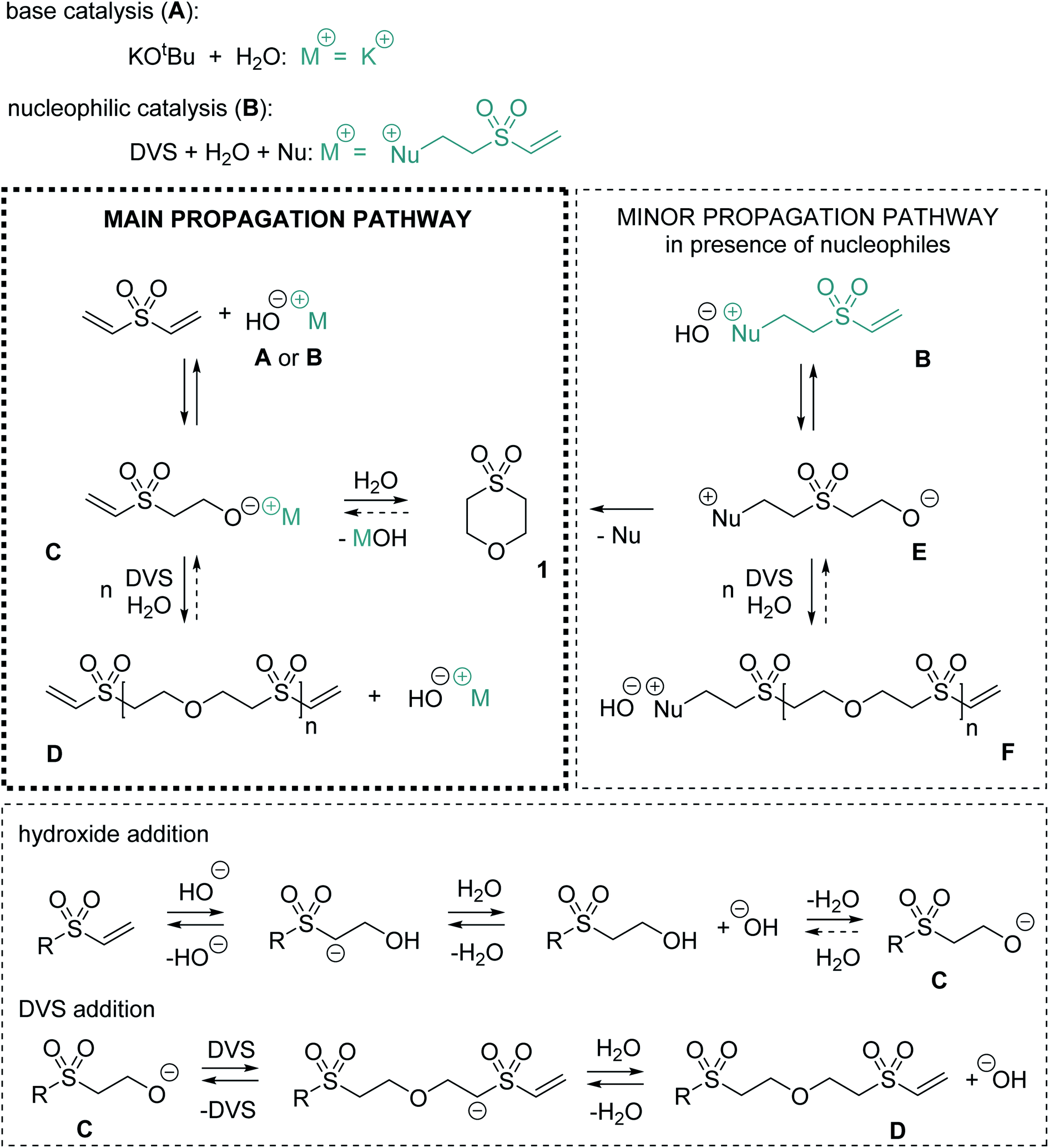 | ||
| Scheme 1 Mechanistic understanding of the reaction. | ||
Based on this mechanistic sketch several peculiarities of the reaction need to be discussed. The first question is why excess of water allows for the formation of a polymer instead of causing hydroxy terminated oligomers as one would expect. An explanation could be the immiscibility of DVS and water. Thus, most likely an interfacial polyaddition reaction takes place in which stoichiometry is adjusted at the interface and excess of water does not interfere with the polymerization.27 For a deeper insight, the reaction of methyl vinyl sulfone and excess of water, two miscible reagents, was studied. Upon addition of 5 mol% DMAP (or KOtBu) bis-(2-methanesulfonyl-ethyl) ether is obtained as the only product (Fig. S29–S34†). Similarly, as aforementioned, the reaction of acrylonitrile in alkaline water yields the corresponding ether derivative.12 Accordingly, the poor miscibility of DVS and water as explanation is not sufficient. Studying the reaction of DVS and D2O (DMAP or KOtBu catalyzed) via NMR spectroscopy revealed in the first step partial deuteration of the internal carbon atom of the vinyl group (Scheme S1†). In the end, PES completely deuterated in α-position to the sulfone-group formed (Fig. S38–S41†). Without the addition of a catalyst, no deuterium is incorporated into the vinyl group. These findings show that the attack and the elimination of the hydroxide are fast, reversible processes, i.e.C is in equilibrium with the vinyl species. Upon the reaction of the alkoxide C with another DVS towards D, C is withdrawn from this equilibrium. The higher reactivity of alkoxide C in oxa-Michael reactions compared to hydroxide is plausible. Alkoxides are believed to be better nucleophiles than hydroxide15 and the corresponding alcohol of C is at least about 3.8 orders of magnitude more acidic than water (Table S2†).28 Accordingly, a hydroxide will most likely undergo a fast acid base reaction with alcohol-terminated species, forming the corresponding alkoxide and water. The alkoxide will then react further with DVS.29 Moreover, Gibbs' free energies of model substrates were calculated using highly accurate Coupled-Cluster (CCSD(T)) methods at the basis set limit. Results suggest that oxa-Michael products of DVS and alcohols are thermodynamically more stable than 2-hydroxyethyl vinyl sulfone or HES formed by the addition of water to DVS (Fig. S42†).
The second question is why the polymerization catalyzed by base is slower compared to its nucleophilic catalyzed version. A first hypothesis that zwitter-ion terminated species (e.g.E) are preferably growing was discarded, because of the findings from monitoring the early steps of the reaction (Fig. 1a). Instead, it is likely that ion-pairs B or the higher homologues F act as phase transfer catalysts.30 As expected, without stirring, hardly any reaction takes place in either phase of the biphasic reaction mixture (Fig. S45†). Investigating the two phases by 1H-NMR spectroscopy (after shaking of the NMR tube) revealed that reaction products are mainly observed in the water phase, which is not surprising since the active species are of ionic nature. Additionally, a cloudy colorless precipitate, most likely PES, forms in the aqueous phase over time. Based on these findings, it is hypothesized that ion-pairs B and F facilitate the transfer of DVS (and later of vinyl species D) into the aqueous phase in which those species immediately trap alkoxide C (or higher homologues of C). The formed neutral species are then either soluble in the DVS phase or precipitate from the water phase. Thus, a back reaction is impeded. With this assumption, also an explanation for the preferential formation of side product 1 in the basic regime at low temperature (Table 2, entry 6) and the higher share of 1 in DMAP catalyzed reactions with more water (Table 2, entry 8) becomes available. Formation of 1 is competing with the formation of the polymer and becomes more important the lower the DVS (or D) concentration is in the water phase. If a low amount of DVS (or D) is present, an intramolecular reaction is favored. However, at a later stage of the reaction, intramolecular cyclisation leading to larger cyclic oligomers is less probable but still conceivable. At a higher temperature of 80 °C (Table 2, entries 13–16) DVS and water are miscible and the outcome of the reaction is drastically changed due to an imbalanced stoichiometry. The excess of water favors the formation of cycle 1 and hydroxy terminated oligomers, most probably because the equilibrium under homogenous conditions is rather on the side of well water soluble species like HES, 1, B or F (Fig. S49†). Furthermore, 1 can be prepared by reacting HES at 80 °C with catalytic amounts of base within 24 h in virtually quantitative yield (Fig. S50†), illustrating the reversibility of the oxa-Michael reaction of water with DVS. Similarly, base catalyzed depolymerization of related polymers has been described in literature, supporting the claims on reversibility made here.31
Thermal properties and use in a polymer electrolyte
PES prepared from water and DVS exhibits very similar thermal properties compared to the recently disclosed PES made from 2-hydroxyethyl vinyl sulfone.19 Differential scanning calorimetry (DSC) revealed a glass transition temperature (Tg) around room temperature (determined from the second heat run with 20 °C min−1, Fig. S51†). In the first heat run a broad endothermic signal around 80 °C is observed indicating melting, which is no longer observed in the following heating and cooling runs. X-ray powder diffraction (XRD) measurements confirmed that in the original, brittle state the polymer is semi-crystalline and after melting the material is amorphous (Fig. S52†). From thermogravimetric analysis (TGA) of the semi-crystalline material a mass loss of 2–3% until 150 °C was observed and 5% mass loss occurred at 184 °C. After the exposure of PES to 120 °C for 1 h to obtain an amorphous sample, the material revealed a higher thermal stability. Negligible mass loss until 150 °C and 5% mass loss at 210 °C were observed (Fig. S53†). Thus, melting of the initially obtained, semi-crystalline material goes in hand with some mass loss.The polymer's low melting point and amorphous character of the solidified melt motivated us to prepare solid polymer electrolytes (SPE) with lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) as salt.32 Interestingly, despite the appealing properties of sulfone based liquid electrolytes, in particular their wide electrochemical window, the application of polyethersulfones as SPEs is rare.33 A recent study on poly(ether-thioethers) including poly(ether-sulfoxide) and poly(ether-sulfone) with varying methylene spacers shows advances in this field.34 Further, a theoretical study showed that a copolymer with alternating sulfonyl and ethylene oxide repeating units with LiTFSI as salt allows for a higher cation transference number than the standard SPE poly(ethylene oxide) (PEO), as the affinity of sulfonyl group for TFSI− is increased.35 Free-standing membranes of the SPE (Fig. 2) were prepared under solvent-free conditions by simply melting crude (semi-crystalline) PES together with LiTFSI at 120 °C. The finely grounded, dried polymer (without work-up) was mixed thoroughly with varying amounts of LiTFSI salt (0–20 wt%) inside a glovebox. In the following, the mixture was melted on a Teflon plate in an oven at 120 °C overnight.
 | ||
| Fig. 2 Photograph of dried, crude, semi-crystalline PES (left) which is blended with 15 wt% LiTFSI and subsequently heated at 120 °C forming a homogenous melt (middle). From the melt, free-standing amorphous SPE membranes are punched (right). | ||
Thereby, a soft, slightly sticky, formable material was obtained. After pressing, membranes of the SPE, which could easily be peeled off the Teflon plate, were cut (12 mm diameter, 0.9–1.0 mm thickness). XRD spectra of both, the melted polymer and the SPE membrane, look alike (Fig. S56†). The absence of reflections related to LiTFSI crystals indicates a uniform distribution of Li salt in the polymer. Thermal stability was studied by TGA and DSC (Fig. S57 and S58†). Again, the SPE shows similar properties to the melted polymer (without salt). The glass transition temperature of the SPE determined by DSC is slightly lower than of melted PES (7.6 °C against 13.3 °C). Thermal stability is identical as the material with LiTFSI is also stable up to 210 °C (mass loss 5%).
The Li-ion conductivity of such membranes with and without added LiTFSI as conductive salt was evaluated by electrochemical impedance spectroscopy. PES without the addition of LiTFSI can be regarded as an insulator at room temperature (Fig. S59a†). Even after the addition of 5 wt% LiTFSI neither a DC plateau nor an electrode polarization is observable. Only with 10 wt% LiTFSI a considerable Li-ion conductivity of 7.14 × 10−9 S cm−1 is observed. The highest Li-ion conductivity of 1.45 × 10−8 S cm−1 at room temperature was achieved by adding 15 wt% LiTFSI to PES. This conductivity is close to values of PEO (without any additives such as plasticizers), which are typically in the range of 10−7 to 10−8 S cm−1 at room temperature.36 Any further increase in the salt concentration does not lead to a significant improvement in the Li-ion conductivity. In a second step, the temperature dependency of the conductivity of the sample containing 15 wt% LiTFSI was studied in the range from 25 to 100 °C (Fig. S59b†). The determined activation energy turned out to be 1.02 eV. At 70 °C a conductivity of 1.6 × 10−6 S cm−1 is obtained. The temperature dependent Li-ion conductivity follows an Arrhenius behavior and no indication of an abrupt change in the Li-ion conductivity is observed by increasing the temperature (Fig. S59c†). Further, the electrochemical stability of polymer electrolytes is a key parameter in terms of their application in solid-state lithium metal batteries. Accordingly, the stability of PES containing 15 wt% LiTFSI was investigated by cyclic voltammetry using a stainless-steel electrode and lithium metal as the reference electrode. Oxidative stability up to 5.5 V vs. Li/Li+ was found at 45 °C (Fig. S61†), which exceeds the oxidative stability of conventionally used PEO polymer electrolytes (about 4 V).37 The combination of high electrochemical stability and appealing conductivity as well as the solvent-free membrane preparation makes a potential application of PES as polymer electrolyte in high voltage lithium metal batteries interesting. However, further studies are needed to optimize its properties and to study the lithium dendrite suppression capabilities of this polymer electrolyte or its ionic dynamics and transference number.
Conclusion
For the first time water was used as a monomer in a polyaddition reaction with DVS. Initially expected challenges concerning the need for equimolar amounts of monomers and issues in terms of low reactivity of water were overcome. Excess of water is tolerated due to an interfacial polymerization and most likely the favorable behavior of zwitterions as phase transfer catalysts in case of nucleophilic catalysis. Addition of hydroxide to DVS, a highly reactive Michael acceptor, is directed towards the formation of PES as formed alkoxides can immediately react with DVS in the presence of water. Moreover, due to precipitation of the polymer the equilibrium is shifted. Thus, we were able to synthesize aliphatic polyethersulfones (PES) with number molar mass averages of around 5 kg mol−1 and a dispersity of about 2.5 at room temperature using base or nucleophilic catalysis. Additionally, the formed PES was studied as solid polymer electrolyte. Free-standing polymer electrolyte membranes were obtained in a solvent-free manner by dissolving lithium bis(trifluoromethanesulfonyl)imide in the melt of PES. The polymer electrolyte revealed Li-ion conductivities of 10−8 and 10−6 S cm−1 at room temperature and 70 °C, respectively, and is stable up to 5.5 V versus Li/Li+ at 45 °C.Data availability
All the data is provided in the ESI.†Author contributions
KR, MMUD, SMF, EŽ and DP conducted the studies and data analysis. ADB, DR, CS, EŽ and DP supervised the research and provided resources. DR and CS made funding acquisition. CS conceptualized the research. All authors contributed to the manuscript writing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Austrian Federal Ministry for Digital and Economic Affairs, the National Foundation for Research, Technology and Development, and the Christian Doppler Research Association (Christian Doppler Laboratory for Organocatalysis in Polymerization and International Christian Doppler Laboratory for Solid-State Batteries) is gratefully acknowledged. We are thankful for assistance and support from Petra Kaschnitz, Karin Bartl, Lukas Ladenstein and Jasmina Turnšek.References
- W. Han in International Encyclopedia of Geography: People, the Earth, Environment and Technology, ed. D. Richardson, N. Castree, M. F. Goodchild, A. Kobayashi, W. Liu and R. A. Marston, John Wiley & Sons, Ltd, Oxford, UK, 2016, pp. 1–10, DOI:10.1002/9781118786352.wbieg0666.
- (a) B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Advances in the use of CO2 as a renewable feedstock for the synthesis of polymers, Chem. Soc. Rev., 2019, 48, 4466–4514, 10.1039/c9cs00047j; (b) Y. Zhu, C. Romain and C. K. Williams, Sustainable polymers from renewable resources, Nature, 2016, 540, 354–362, DOI:10.1038/nature21001.
- R. Kakuchi, Multicomponent Reactions in Polymer Synthesis, Angew. Chem., Int. Ed., 2014, 53, 46–48, DOI:10.1002/anie.201305538.
- J. Zhang, W. Wang, Y. Liu, J. Z. Sun, A. Qin and B. Z. Tang, Facile Polymerization of Water and Triple-Bond Based Monomers toward Functional Polyamides, Macromolecules, 2017, 50, 8554–8561, DOI:10.1021/acs.macromol.7b01592.
- J. Zhang, W. Shi, Q. Liu, T. Chen, X. Zhou, C. Yang, K. Zhang and Z. Xie, Atom-economical, room-temperature, and high-efficiency synthesis of polyamides via a three-component polymerization involving benzoxazines, odorless isocyanides, and water, Polym. Chem., 2018, 9, 5566–5571, 10.1039/C8PY01256C.
- L. Xu, F. Zhou, M. Liao, R. Hu and B. Z. Tang, Room temperature multicomponent polymerizations of alkynes, sulfonyl azides, and N-protected isatins toward oxindole-containing poly(N-acylsulfonamide)s, Polym. Chem., 2018, 9, 1674–1683, 10.1039/C7PY01983A.
- (a) S. Strasser and C. Slugovc, Nucleophile-mediated oxa-Michael addition reactions of divinyl sulfone – a thiol-free option for step-growth polymerisations, Catal. Sci. Technol., 2015, 5, 5091–5094, 10.1039/C5CY01527H; (b) H. Yang, Y. Zuo, J. Zhang, Y. Song, W. Huang, X. Xue, Q. Jiang, A. Sun and B. Jiang, Phosphazene-catalyzed oxa-Michael addition click polymerization, Polym. Chem., 2018, 9, 4716–4723, 10.1039/C8PY01089G.
- D. Arntz, A. Fischer, M. Höpp, S. Jacobi, J. Sauer, T. Ohara, T. Sato, N. Shimizu and H. Schwind, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2007, DOI:10.1002/14356007.a01_149.pub2.
- I. C. Stewart, R. G. Bergman and F. D. Toste, Phosphine-Catalyzed Hydration and Hydroalkoxylation of Activated Olefins: Use of a Strong Nucleophile to Generate a Strong Base, J. Am. Chem. Soc., 2003, 125, 8696–8697, DOI:10.1021/ja035232n.
- V. Resch, C. Seidler, B.-S. Chen, I. Degeling and U. Hanefeld, On the Michael Addition of Water to α,β-Unsaturated Ketones Using Amino Acids, Eur. J. Org. Chem., 2013, 7697–7704, DOI:10.1002/ejoc.201301230.
- J.-J. Yun, M.-L. Zhi, W.-X. Shi, X.-Q. Chu, Z.-L. Shen and T.-P. Loh, Indium(III)-Catalyzed Hydration and Hydroalkoxylation of α,β-Unsaturated Ketones in Aqueous Media, Adv. Synth. Catal., 2018, 360, 2632–2637, DOI:10.1002/adsc.201800301.
- T. R. Krishna and N. Jayaraman, Synthesis of Poly(propyl ether imine) Dendrimers and Evaluation of Their Cytotoxic Properties, J. Org. Chem., 2003, 68, 9694–9704, DOI:10.1021/jo035072y.
- V. Resch and U. Hanefeld, The selective addition of water, Catal. Sci. Technol., 2015, 5, 1385–1399, 10.1039/C4CY00692E.
- R. N. Butler and A. G. Coyne, Water: Nature's Reaction Enforcers - Comparative Effects for Organic Synthesis “In-Water” and “On-Water”, Chem. Rev., 2010, 110, 6302–6337, DOI:10.1021/cr100162c.
- T. B. Phan and H. Mayr, Comparison of the nucleophilicities of alcohols and alkoxides, Can. J. Chem., 2005, 83, 1554–1560, DOI:10.1139/v05-170.
- D. S. Allgäuer, H. Jangra, H. Asahara, Z. Li, Q. Chen, H. Zipse, A. R. Ofial and H. Mayr, Quantification and Theoretical Analysis of the Electrophilicities of Michael Acceptors, J. Am. Chem. Soc., 2017, 139, 13318–13329, DOI:10.1021/jacs.7b05106.
- S. Strasser, C. Wappl and C. Slugovc, Solvent-free macrocyclisation by nucleophile-mediated oxa-Michael addition polymerisation of divinyl sulfone and alcohols, Polym. Chem., 2017, 8, 1797–1804, 10.1039/C7PY00152E.
- (a) C. M. Welch, Polymeric adducts of divinyl sulfone with water as crosslinking agents for cellulose, US Pat., 3281204A, 1966 Search PubMed; (b) C. M. Welch, Polymeric adducts of divinyl sulfone with water and preparation thereof, US Pat., 3316308A, 1967 Search PubMed.
- N. Ziegenbalg, R. Lohwasser, G. D'Andola, T. Adermann and J. C. Brendel, Oxa-Michael polyaddition of vinylsulfonylethanol for aliphatic polyethersulfones, Polym. Chem., 2021, 12, 4337–4346, 10.1039/D1PY00256B.
- J. Ammer, M. Baidya, S. Kobayashi and H. Mayr, Nucleophilic reactivities of tertiary alkylamines, J. Phys. Org. Chem., 2010, 23, 1029–1035, DOI:10.1002/poc.1707.
- B. Maji, D. S. Stephenson and H. Mayr, Guanidines: Highly Nucleophilic Organocatalysts, ChemCatChem, 2012, 4, 993–999, DOI:10.1002/cctc.201200143.
- A. M. Hyde, R. Calabria, R. Arvary, X. Wang and A. Klapars, Investigating the Underappreciated Hydrolytic Instability of 1,8-Diazabicyclo[5.4.0]undec-7-ene and Related Unsaturated Nitrogenous Bases, Org. Process Res. Dev., 2019, 23, 1860–1871, DOI:10.1021/acs.oprd.9b00187.
- D. Edinger, H. Weber, E. Žagar, D. Pahovnik and C. Slugovc, Melt Polymerization of Acrylamide Initiated by Nucleophiles: A Route toward Highly Branched and Amorphous Polyamide 3, ACS Appl. Polym. Mater., 2021, 3, 2018–2026, DOI:10.1021/acsapm.1c00084.
- J. Kaur, R. Krishnan, B. Ramalingam and S. Jana, Hydroxyethyl sulfone based reactive coalescing agents for low-VOC waterborne coatings, RSC Adv., 2020, 10, 17171–17179, 10.1039/D0RA00753F.
- C. A. Lewis and R. Wolfenden, The Nonenzymatic Decomposition of Guanidines and Amidines, J. Am. Chem. Soc., 2014, 136, 130–136, DOI:10.1021/ja411927k.
- S. Chatani, R. J. Sheridan, M. Podgórski, D. P. Nair and C. N. Bowman, Temporal Control of Thiol-Click Chemistry, Chem. Mater., 2013, 25, 3897–3901, DOI:10.1021/cm402229j.
- F. Zhang, J.-b. Fan and S. Wang, Interfacial Polymerization: From Chemistry to Functional Materials, Angew. Chem., Int. Ed., 2020, 59, 21840–21856, DOI:10.1002/anie.201916473.
- Calculated using the pKa prediction platform available at pka.luo-group.com see: Q. Yang, Y. Li, J.-D. Yang, Y. Liu, L. Zhang, S. Luo and J.-P. Cheng, Holistic Prediction of the pKa in Diverse Solvents Based on a Machine-Learning Approach, Angew. Chem., Int. Ed., 2020, 59, 19282–19291, DOI:10.1002/anie.202008528.
- (a) S. M. Fischer, S. Renner, A. D. Boese and C. Slugovc, Electron-rich triarylphosphines as nucleophilic catalysts for oxa-Michael reactions, Beilstein J. Org. Chem., 2021, 17, 1689–1697, DOI:10.3762/bjoc.17.117; (b) K. Ratzenböck, D. Pahovnik and C. Slugovc, Step-growth polymerisation of alkyl acrylates via concomitant oxa-Michael and transesterification reactions, Polym. Chem., 2020, 11, 7476–7480, 10.1039/D0PY01271H.
- K. Piradashvili, E. M. Alexandrino, F. R. Wurm and K. Landfester, Reactions and Polymerizations at the Liquid–Liquid Interface, Chem. Rev., 2016, 116, 2141–2169, DOI:10.1021/acs.chemrev.5b00567.
- (a) T. Sasaki, T. Kondo, M. Noro, K. Saida, H. Yaguchi and Y. Naka, Photoinduced depolymerization in poly(olefin sulfone) films comprised of volatile monomers doped with a photobase generator, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1462–1468, DOI:10.1002/pola.25898; (b) C. M. Possanza Casey and J. S. Moore, Base-Triggered Degradation of Poly(vinyl ester sulfone)s with Tunable Sensitivity, ACS Macro Lett., 2016, 5, 1257–1260, DOI:10.1021/acsmacrolett.6b00698; (c) O. P. Lee, H. Lopez Hernandez and J. S. Moore, Tunable Thermal Degradation of Poly(vinyl butyl carbonate sulfone)s via Side-Chain Branching, ACS Macro Lett., 2015, 4, 665–668, DOI:10.1021/acsmacrolett.5b00234.
- A. Arya and A. L. Sharma, Polymer electrolytes for lithium ion batteries: a critical study, Ionics, 2017, 23, 497–540, DOI:10.1007/s11581-016-1908-6.
- W. Wu, Y. Bai, X. Wang and C. Wu, Sulfone-based high-voltage electrolytes for high energy density, Chin. Chem. Lett., 2021, 32, 1309–1315, DOI:10.1016/j.cclet.2020.10.009.
- J. M. Sarapas and G. N. Tew, Poly(ether–thioethers) by Thiol–Ene Click and Their Oxidized Analogues as Lithium Polymer Electrolytes, Macromolecules, 2016, 49, 1154–1162, DOI:10.1021/acs.macromol.5b02513.
- A. France-Lanord, Y. Wang, T. Xie, J. A. Johnson, Y. Shao-Horn and J. C. Grossman, Effect of Chemical Variations in the Structure of Poly(ethylene oxide)-Based Polymers on Lithium Transport in Concentrated Electrolytes, Chem. Mater., 2020, 32, 121–126, DOI:10.1021/acs.chemmater.9b02645.
- D. Zhou, D. Shanmukaraj, A. Tkacheva, M. Armand and G. Wang, Polymer Electrolytes for Lithium-Based Batteries: Advances and Prospects, Chem, 2019, 5, 2326–2352, DOI:10.1016/j.chempr.2019.05.009.
- (a) Y. Xia, T. Fujieda, K. Tatsumi, P. P. Prosini and T. Sakai, Thermal and electrochemical stability of cathode materials in solid polymer electrolyte, J. Power Sources, 2001, 92, 234–243, DOI:10.1016/S0378-7753(00)00533-4; (b) K. Nie, X. Wang, J. Qiu, Y. Wang, Q. Yang, J. Xu, X. Yu, H. Li, X. Huang and L. Chen, ACS Energy Lett, 2020, 5, 826–832, DOI:10.1021/acsenergylett.9b02739.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02124b |
| This journal is © The Royal Society of Chemistry 2022 |