
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA peptide-derived strategy for specifically targeting the mitochondria and ER of cancer cells: a new approach in fighting cancer†
Yang Sung
Sohn
 *a,
Anat
losub-Amir
*b,
Alfredo E.
Cardenas
*c,
Ola
Karmi
ad,
Merav Darash
Yahana
a,
Tal
Gruman
b,
Linda
Rowland
d,
Henri-Baptiste
Marjault
a,
Lauren J.
Webb
e,
Ron
Mittler
d,
Ron
Elber
c,
Assaf
Friedler
*b and
Rachel
Nechushtai
*a
*a,
Anat
losub-Amir
*b,
Alfredo E.
Cardenas
*c,
Ola
Karmi
ad,
Merav Darash
Yahana
a,
Tal
Gruman
b,
Linda
Rowland
d,
Henri-Baptiste
Marjault
a,
Lauren J.
Webb
e,
Ron
Mittler
d,
Ron
Elber
c,
Assaf
Friedler
*b and
Rachel
Nechushtai
*a
aThe Alexander Silberman Institute of Life Science, The Hebrew University of Jerusalem, Edmond J. Safra Campus at Givat Ram, Jerusalem 9190401, Israel. E-mail: rachel@mail.huji.ac.il
bInstitute of Chemistry, The Hebrew University of Jerusalem, Edmond J. Safra Campus at Givat Ram, Jerusalem 9190401, Israel. E-mail: assaf.friedler@mail.huji.ac.il
cInstitute for Computational Engineering and Science and Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, USA
dDepartment of Surgery, University of Missouri School of Medicine, Christopher S. Bond Life Sciences Center University of Missouri. 1201 Rollins St, Columbia, MO 65201, USA
eDepartment of Chemistry, The University of Texas at Austin, 2506 Speedway STOP A5300, Austin, TX 78712, USA
First published on 26th May 2022
Abstract
An effective anti-cancer therapy should exclusively target cancer cells and trigger in them a broad spectrum of cell death pathways that will prevent avoidance. Here, we present a new approach in cancer therapy that specifically targets the mitochondria and ER of cancer cells. We developed a peptide derived from the flexible and transmembrane domains of the human protein NAF-1/CISD2. This peptide (NAF-144-67) specifically permeates through the plasma membranes of human epithelial breast cancer cells, abolishes their mitochondria and ER, and triggers cell death with characteristics of apoptosis, ferroptosis and necroptosis. In vivo analysis revealed that the peptide significantly decreases tumor growth in mice carrying xenograft human tumors. Computational simulations of cancer vs. normal cell membranes reveal that the specificity of the peptide to cancer cells is due to its selective recognition of their membrane composition. NAF-144-67 represents a promising anti-cancer lead compound that acts via a unique mechanism.
Introduction
The mitochondrial-ER network plays a pivotal role in dictating life or death decisions in response to many different environmental and developmental stimuli. Recent studies identified a critical role for the ER, mitochondria and mitochondrial-associated membrane (MAM) network of cancer cells in promoting survival, tumor growth and metastasis.1,2 One of the proteins localized to the MAM, outer mitochondrial membranes (OMM) and ER is the iron-sulfur cluster [2Fe–2S] protein CISD2 (CDGSH iron-sulfur domain-containing protein 2; also known as nutrient autophagy factor 1; NAF-1).3,4 NAF-1 plays a key role in regulating calcium, reactive oxygen species (ROS), and iron homeostasis and signalling in cells.4–9 It also controls several different cell death and recycling pathways including apoptosis, ferroptosis and autophagy.4,10–13 Elevated levels of NAF-1 expression are associated with the progression of many human cancers and suppressing NAF-1 expression results in reduced cancer proliferation and tumor growth.5,14 Elevated levels of NAF-1 are also associated with poor prognosis of patients and NAF-1 is used as a diagnostic marker for several different cancers.5,14–20 NAF-1 is a homodimer, with each subunit containing a trans-membrane domain (residues 38–57), a disordered flexible loop (residues 58–67) and a CDGSH [2Fe–2S] soluble binding domain facing the cytosol (resides 68–135).3 Artificially enhancing NAF-1 expression in cancer cells promoted cellular proliferation, and this promotion was dependent on the degree of lability of the NAF-1 Fe–S clusters.14 NAF-1 is therefore a central regulator of many cellular processes important for cancer proliferation, and this function of NAF-1 is at least partially dependent on the function of its [2Fe–2S] clusters.3,18,21,22A major challenge in cancer-therapy is to develop molecules that induce cancer cell death but have no cytotoxic effect towards normal cells. Cell Penetrating Peptides (CPP) are promising candidates and well explored.23,24 CPP are peptides of about ≤30 amino acids able to penetrate the plasma membrane (PM) with an energy dependent (endocytosis) or energy independent (direct permeation) mechanism.25 They are frequently derived from the membrane translocation machinery, such as the TAT peptide that leads the permeation of the HIV Trans Activator Protein.26 Other sources of CPP are antimicrobial peptides (AMPs).27 For example, the peptide buforin IIb is an antimicrobial peptide derived from histone H2A that shows significant anticancer activity.28 The peptide discussed in the present manuscript is derived from a source not considered before. It is a fragment of a transmembrane protein, NAF-1 (NAF-144-67), containing a hydrophobic N terminal segment, which is embedded in the mitochondrial membrane, and a charged C-terminal part that is found in aqueous solution. Attempts of using CPP against cancer cells face the following challenges: (i) the CPP must be selective towards cancer cells; (ii) it must induce cell death; and (iii) it must survive degradation for a sufficient length of time to have an impact. Other desired properties are the ability of the peptide to induce cell death by itself without additional cargo and its ability to target a vital organelle(s) within cancer cells. To our knowledge, at present, there are no CPP satisfying all these criteria. We illustrate in this manuscript that NAF-144-67 does. NAF-144-67 selectively kills cancer cells (IC50 values of 12–18 μM), without affecting normal cells. It permeates through the PM of human epithelial breast cancer cells (MDA-MB-231), but not of healthy cells (MCF-10A), and targets their mitochondria and ER. Atomically detailed simulations of the interactions of NAF-144-67 with models of cancer and normal PM, explain the NAF-144-67 permeation specificity towards cancer cells. The negatively charged PM of cancer cells strongly attracts the positive charges of NAF-144-67, while the less negatively charged PM of healthy cells does not. The initial attraction and settling of the peptide at the membrane surface, demonstrated in this paper, is followed by efficient permeation of the transmembrane component of NAF-144-67 into the membrane core.29 Upon permeation, the peptide activates several cell death processes that display characteristics of ferroptosis, apoptosis and necroptosis. In vivo studies of NAF-144-67 using xenograft mice carrying human triple-negative breast cancer MDA-MB-231 tumors indicate a significant decrease in tumor growth. The peptide is selective towards cancer cells because it is derived from the NAF-1 sequence that resides in the mitochondrial and ER membranes. It is thus: (i) selective for cancer cells PMs, which resemble the mitochondrial membrane in their composition; and (ii) it specifically targets the mitochondria and ER of cancer cells where NAF-1 normally resides. Taken together, we are presenting a new approach in cancer therapy of targeting of the mitochondria and ER of cancer cells using peptides derived from native proteins of this network.
Results
NAF-144-67 is derived from the transmembrane and flexible loop regions of NAF-1 and forms a helix in a membrane-like environment
NAF-144-67 is derived from part of the NAF-1 transmembrane helix (residues 44–56) and its disordered loop (residues 57–67; Fig. 1a). The predicted structure of NAF-144-67 is shown in Fig. 1b. The CD spectrum of the peptide in saline (165 mM NaCl dissociated in water) shows a disordered nature. Whereas in the presence of 3 mM Dodecylphosphocholine (DPC) that form micelles, the CD spectrum of the peptide shifts to a helical structure combined with some disordered features (Fig. 1c).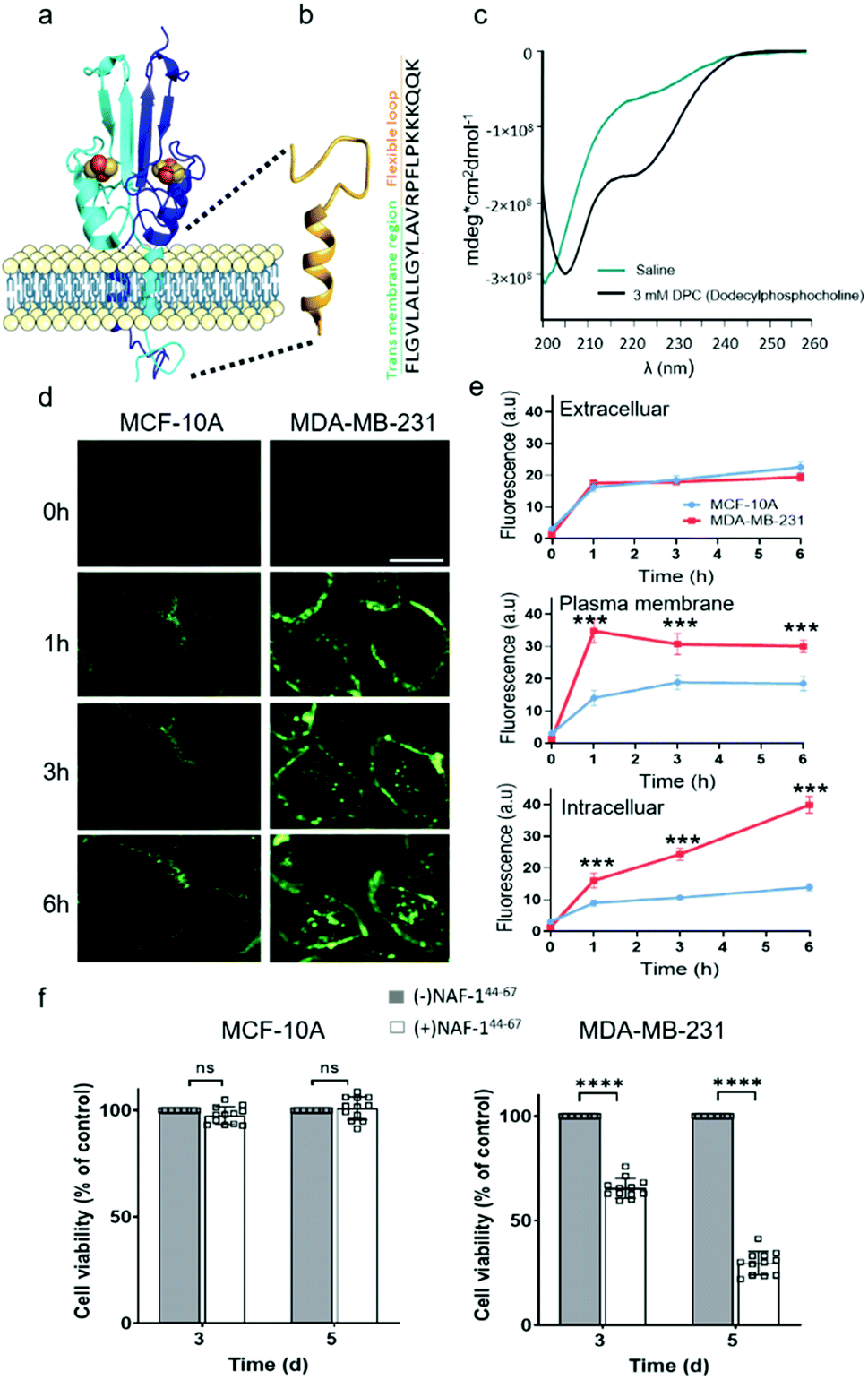 | ||
| Fig. 1 NAF-144-67, derived from the transmembrane and flexible loop regions of NAF-1, selectively kills cancer cells. (a) Schematic model of NAF-1 structure (PDB 3FNV). Dashed lines show the NAF-1 region from which the NAF-144-67 peptide is derived. (b) Predicted structure of NAF-144-67 showing the transmembrane and flexible regions. (c) The CD spectrum of NAF-144-67 in saline (turquoise) or 3 mM DPC (black). (d) Confocal fluorescence images of control MCF-10A (left) and MDA-MB-231 breast cancer cells (right) at different time points following treatment with Fl-NAF-144-67. The scale bar is 20 μm. (e) Quantitative analysis of Fl-NAF-144-67 penetration into MDA-MB-231 (red lines) or MCF-10A (blue lines) cells. Data is shown as mean ± SD of 25 cells per field calculated for each time-point obtained from n = 5 independent experiments. ***P < 0.001 by t-test. (f) Cytotoxicity of the NAF-144-67 peptide towards malignant MDA-MB-231 and control MCF-10A cells. ****P < 0.0001 by t-test. | ||
NAF-144-67 selectively permeates and kills cancer cells
To study how NAF-144-67 interacts with cells, we labelled it with fluorescein (Fl-NAF-144-67) and followed its translocation into triple negative malignant epithelial breast cancer (MDA-MB-231) and non-malignant control (MCF-10A) cells using confocal microscopy. Fl-NAF-144-67 fluorescence was primarily found outside of control MCF-10A cells, indicating that the peptide did not permeate normal cells. In contrast, a high Fl-NAF-144-67 fluorescence signal was found at the PM of MDA-MB-231 cancer cells at early stages, and inside MDA-MB-231 cancer cells at 3 and 6 hours of incubation, indicating that Fl-NAF-144-67 preferentially penetrated MDA-MB-231 cancer cells (Fig. 1d, e and S1†). To determine the effect of NAF-144-67 on cancer cells compared to normal healthy cells, we measured the viability of MDA-MB-231 and MCF-10A cells following treatment with 10 μM of NAF-144-67 for three and five days. The viability of MDA-MB-231 cancer cells treated with the peptide decreased by 40% at three days and by 80% at five days post treatment, whereas that of MCF-10A control cells remained unaffected (Fig. 1f). We also tested the penetration and cytotoxic effects of NAF-144-67 on other non-malignant immortalized cells, such as rat cardio myoblast H9C2 cells and human embryonic kidney 293 HEK cells. These cells showed no entry of Fl-NAF-144-67 and remained unaffected after 3 days of treatment (Fig. S2†).Molecular simulations of NAF-144-67 permeation into cancer cells
Molecular simulations were performed to determine the mechanisms governing the selective entry of NAF-144-67 into cancer cells. The PMs of control and cancer cells were modelled based on lipidomic analysis of control human mammary epithelial breast and MDA-MB-231 breast cancer cell lines.30 Studies of different cancer cells revealed that the outer layer of their PM is rich with negatively charged PS phospholipids, while the outer layer of a normal PM mostly contains zwitterionic phospholipids.31–33 Simulating the binding of NAF-144-67 to the two different membranes revealed that, compared to the PM of normal cells, the peptide forms many more electrostatic interactions with the PM of malignant cells (Fig. 2a–c). These interactions were primarily attributed to a higher number of hydrogen bonds formed between NAF-144-67 and the PM of cancer cells (Fig. 2c). Hydrogen bonds can be disrupted by high concentrations of charged ions, such as those present in a solution of MgCl2. We therefore tested the effect of MgCl2 on the binding of NAF-144-67 to membranes in silico (Fig. 2c) and in vivo (Fig. 2d). Addition of MgCl2 to the molecular simulation revealed that the number of hydrogen bonds formed between NAF-144-67 and the cancer cell PM decreased to that of NAF-144-67 interactions with normal cell PM (Fig. 2c). Addition of MgCl2 to MDA-MB-231 cells incubated with NAF-144-67 revealed that MgCl2 had a strong inhibitory effect on the cancer killing properties of NAF-144-67 (Fig. 2d). Taken together, our findings suggest that formation of hydrogen bonds between NAF-144-67 and the negatively charged PM of cancer cells plays a key role in determining its selectivity and anti-cancer activity. The entire permeation process of NAF-146-67 through a model DOPC membrane was calculated using the method of Milestoning,84 supporting the picture of strong electrostatic interactions with the membrane surface.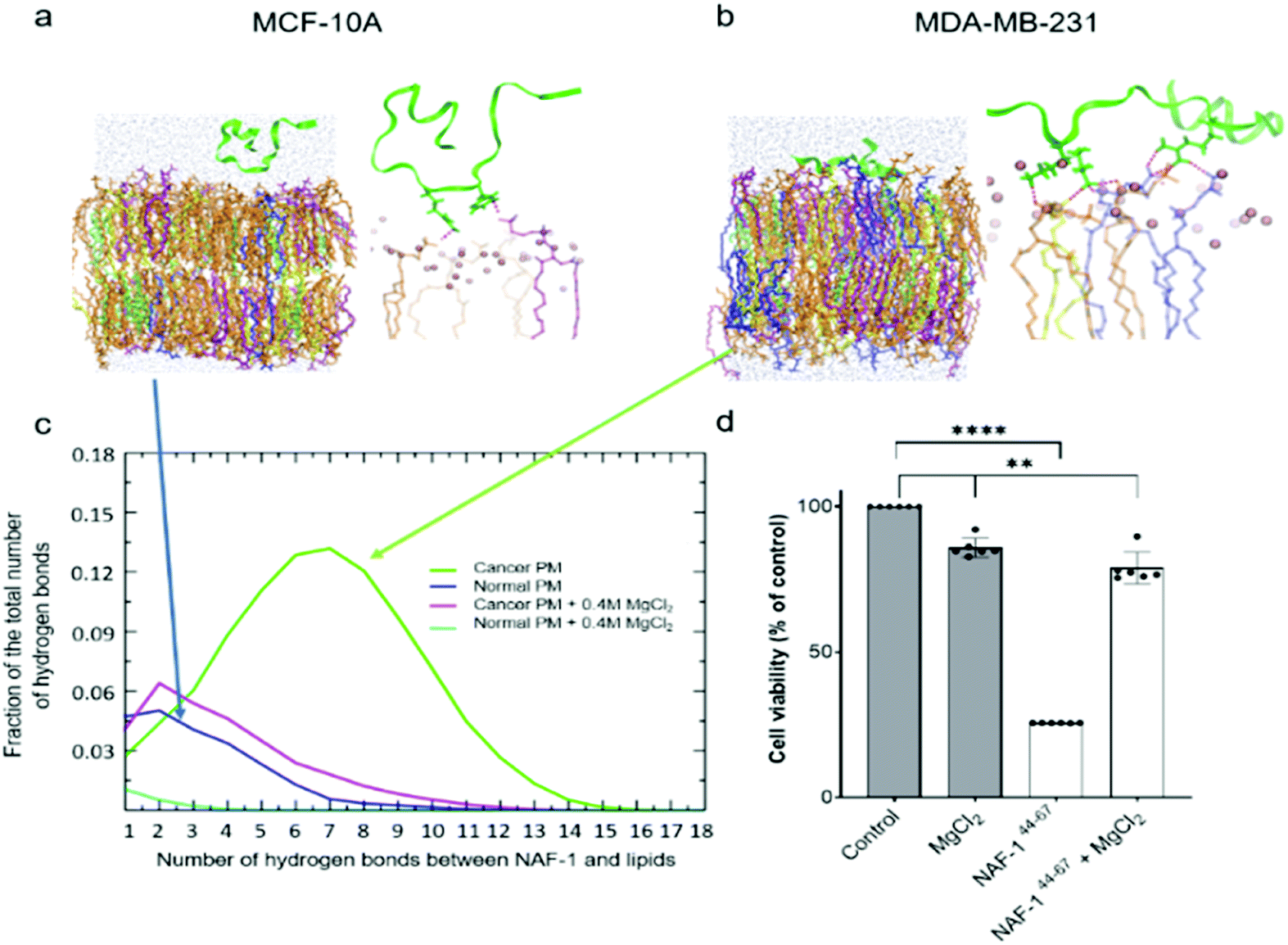 | ||
| Fig. 2 NAF-144-67 preferential permeation into epithelial breast cancer cells is dependent on peptide-lipids electrostatic interactions. (a) A molecular simulations snapshot of NAF-144-67 interaction with the surface of a normal cells' PM and detail view of the molecular snapshot shown for the peptide in the normal PM model. Phosphorus atoms at the membrane/water interface are shown as pink spheres. Side chains of the peptide are added to display the atoms forming hydrogen bonds with the lipids (shown in red). (b) A molecular simulations snapshot illustrating the stronger interaction and early insertion of NAF-144-67 through the cancer cells' membrane and detail view of the molecular snapshot for the peptide in the cancer PM model. (c) Normalized distribution of the number of hydrogen bonds formed between NAF-144-67 and the lipid molecules of models cancer and normal cell PMs obtained using MD simulations. Also shown are distributions of hydrogen bonding when MgCl2 is added to the aqueous solution of the normal and cancer plasma membranes. (d) Cell viability measurement of cancer cells treated with NAF-144-67 for 6 hours in the presence or absence of magnesium chloride (MgCl2). ****P < 0.0001, **P < 0.01 by t-test. In (a) and (b) the NAF-1 peptide is represented as green ribbons. The lipids are shown using bond representations: PS in blue, PC in orange, PE in yellow, PSM in purple and cholesterol in lime. The oxygen atoms of the water molecules are represented with van der Waals spheres. | ||
NAF-144-67 targets the mitochondria and ER of cancer cells
Because NAF-1 resides on the cytosolic-facing side of the ER, mitochondrial and MAM membranes, we studied the effects of NAF-144-67 on these organelles within cancer cells. NAF-144-67 caused a loss of mitochondria membrane potential (MMP) and degradation of ER in cancer cells following six hours of incubation. In contrast, the nuclei and PM remained relatively intact, highlighting the specificity of NAF-144-67 to the mitochondria and ER (Fig. 3a and b). In addition, the Fl-NAF-144-67 peptide colocalized to the mitochondria and ER (Fig. S3 and S6†). Transmission electron microscopy (TEM) analysis of MDA-MB-231 cancer cells treated with NAF-144-67 further revealed that NAF-144-67 induced various abnormalities in mitochondria size, cristae integrity and ER structures (Fig. 3c and d); without any apparent effects on the PM and nuclei (Fig. S4†). Mitochondrial functional studies (Fig. S5†) agree with these TEM results. Thus, mitochondrial respiration was significantly decreased, while glycolysis was affected to a much less extent (Fig. S5†).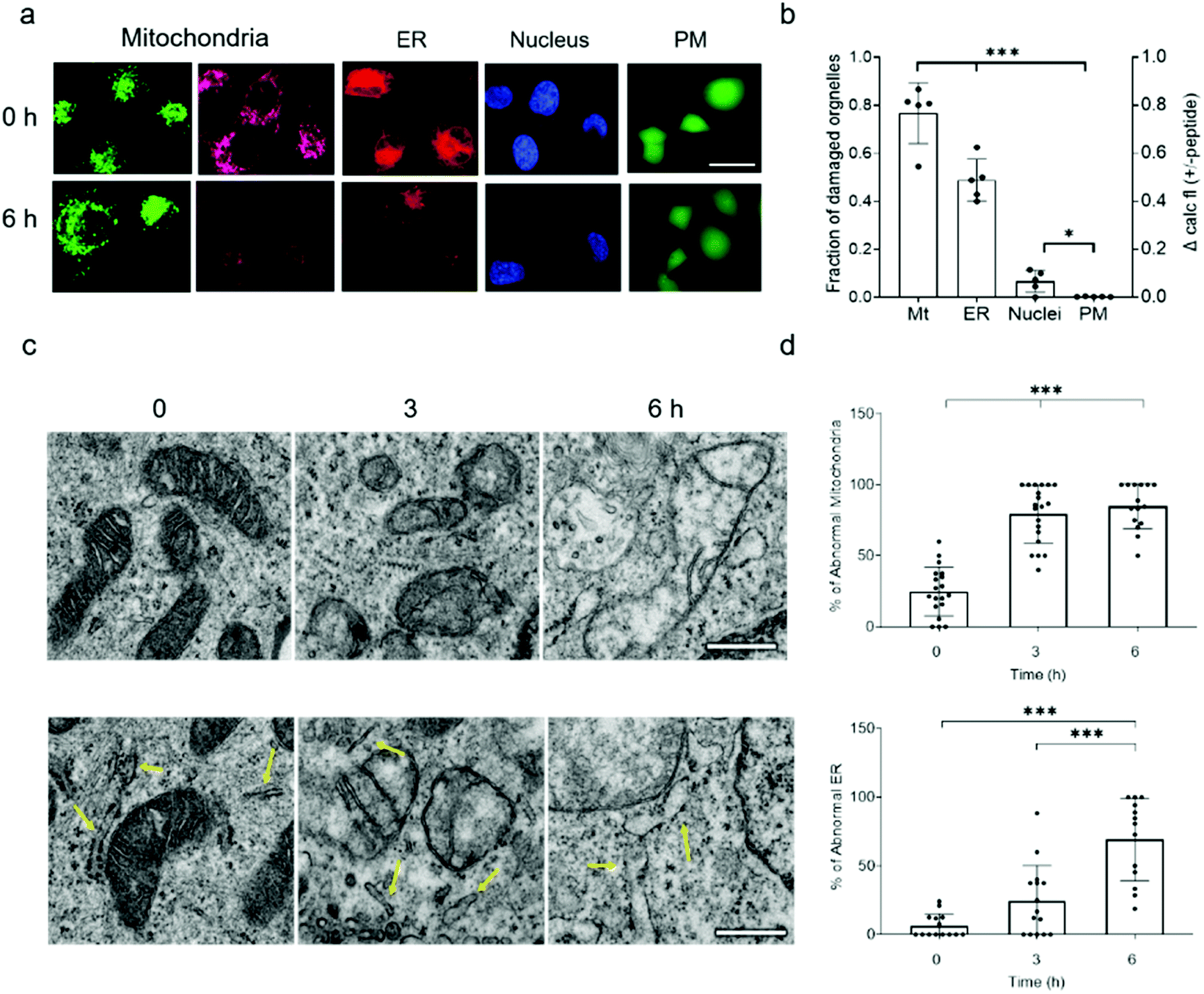 | ||
| Fig. 3 NAF-144-67 targets the mitochondria and ER of cancer cells without causing any apparent damage to the PM and nuclei. (a) Representative confocal images of MDA-MB-231 cells incubated with NAF-144-67. The images were taken at time 0 and after six hours of incubation with the peptide. Cells are stained with MMP-independent mitotracker green or MMP-dependent Rhodamine 800 (mitochondria), DsRed (ER), Hoechst 342 (nuclei), or Calcein AM for testing PM integrity. The scale bar is 20 μm. (b) Degree of damage to the different intracellular structures and PM following six hours of incubation with 10 μM NAF-144-67. Data is shown as mean ± SD of 75 cells from 15 different fields per time-point, from 5 independent experiments. ***P < 0.001 by t-test. (c) Representative TEM images of structural alterations in mitochondria (top) and ER (bottom) at different time points following incubation with NAF-144-67. The scale bar is 500 nm. (d) Statistical analysis of structural alterations in mitochondrial (top) and ER (bottom) at different time points following incubation with NAF-144-67. ***P < 0.001 by t-test. No changes were observed in PM and nuclei structures by TEM (Fig. S4†). | ||
Taken together, our findings reveal that while NAF-144-67 penetrated the PM of cancer cells (Fig. 1), it did not cause any apparent damage to it, or to the nuclei (Fig. 3 and S6†). Instead, NAF-144-67 primarily damaged the ER and mitochondria of cancer cells (Fig. 3 and S6†). In contrast, NAF-144-67 did not penetrate the PM of normal cells (Fig. 1) and therefore did not damage their content (Fig. 3).
NAF-144-67 induces in cancer cells a cell death process that primarily resembles apoptosis
Proteomics analysis of NAF-144-67-treated MCF-10A and MDA-MB-231 cells revealed that the expression of several different proteins associated with apoptosis, ferroptosis, necrosis and autophagy was significantly and specifically altered in MDA-MB-231 cells, with the highest proportion of these belonging to apoptotic-like pathways (Fig. 4a. S8, S9 and S10†). Supporting these findings, vesicles resembling apoptotic bodies were also observed in NAF-144-67-treated MDA-MB-231 cells (Fig. 4b and S7†), and cyclosporine A (CsA), an inhibitor of apoptosis, inhibited NAF-144-67-induced cell death of MDA-MB-231 spheroids by about 50% (Fig. 4c). In addition, release of cytochrome c from isolated mitochondria (a major hallmark of apoptosis) was observed in isolated mitochondria treated with the NAF-144-67 peptide (Fig. 4d and e). These results suggest that apoptosis, and perhaps other cell death pathways are activated in cancer cells by treatment with NAF-144-67.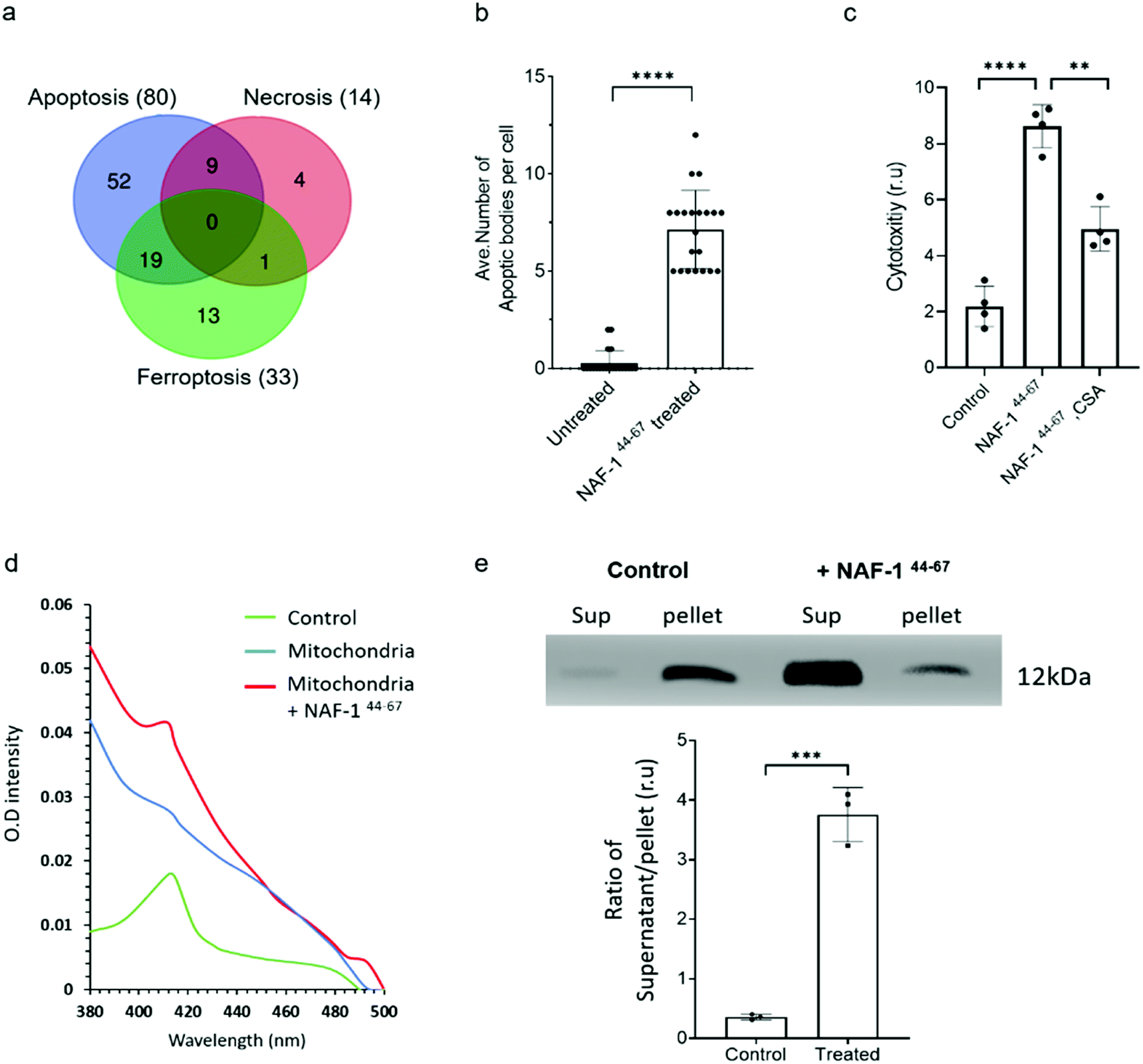 | ||
| Fig. 4 NAF-144-67 primarily activates apoptosis in cancer cells. (a) Venn diagram showing the overlap between different cell death pathways associated proteins specifically altered in their expression in cancer cells following treatment with NAF-144-67. Most of the proteins (80) affected by the NAF-144-67 peptide belong to the apoptosis cell death pathway. (b) Formation of apoptotic-like bodies (Fig. S7†) in cancer cells following treatment with NAF-144-67. (c) The effect of cyclosporine A, CsA, an inhibitor of apoptosis on the anti-cancer activity of NAF-144-67. MDA-MB-231 spheroids were incubated with NAF-144-67 in the presence or absence of CsA and assayed for cell viability using an IncuCyte apparatus. (d) Isolated mitochondria from MDA-MB-231 breast cancer cells were treated or untreated with NAF-144-67. Mitochondria were precipitated by centrifugation and pellet and soluble fractions collected. The adsorption spectrum with characteristic 420 nm absorption peak of cytochrome C (control) was measured in the supernatant of treated and untreated mitochondria. The absorption of purified cytochrome C (green trace), supernatant of untreated mitochondria (blue trace), and supernatant of mitochondria treated with the NAF-1144-67 peptide (red trace). (e) Control and the peptide treated mitochondrial fractions (supernatant, Sup) and pellet, were analysed by western blots with antibodies against cytochrome C [top panel is showing cytochrome C levels in supernatant and pallet from MDA-MB-231 cells treated or nontreated with NAF-144-67; (bottom panel is showing the ratio of Cytochrome C protein levels in supernatant and pellet of control (non-treated) and peptide treated mitochondrial fractions]. The results include all data points measured in three different experiments. **P < 0.01, ***P < 0.001,****P < 0.0001 compared to control; Student's t-test. | ||
Sequence analysis and development of stable variants of NAF-144-67
To reveal the minimal sequence required for its activity, we designed four shorter peptides derived from different regions of NAF-144-67: the transmembrane region (NAF-144-58; M0; Fig. 5a), part of the transmembrane region and flexible loop (NAF-153-67; M1; Fig. 5a), the flexible loop (NAF-157-67; M2; Fig. 5a), and both termini (NAF-144-52+62-67; M3; Fig. 5a). CD spectroscopy revealed that NAF-153-67 and NAF-157-67, which were mostly derived from the flexible loop, are disordered, whereas NAF-144-52+62-67, derived from both the transmembrane helix and the disordered loop, displayed helical and disordered features (Fig. S11a†). NAF-144-58, the transmembrane derived peptide, was not soluble. Compared to all its derivatives, the parent NAF-144-67 peptide exhibited the most potent killing effect towards MDA-MB-231 cells (by more than 50%; Fig. 5b), indicating that the full-length sequence of the peptide is required for its activity. Compared to its different variants, the full-length sequence of NAF-144-67 also contained more amino acids that could form hydrogen bonds with the PM of cancer cells (Fig. 2b), potentially allowing it to bind better to the cancer cell PM that contains a larger number of negatively charged phospholipids than the PM of a normal cell.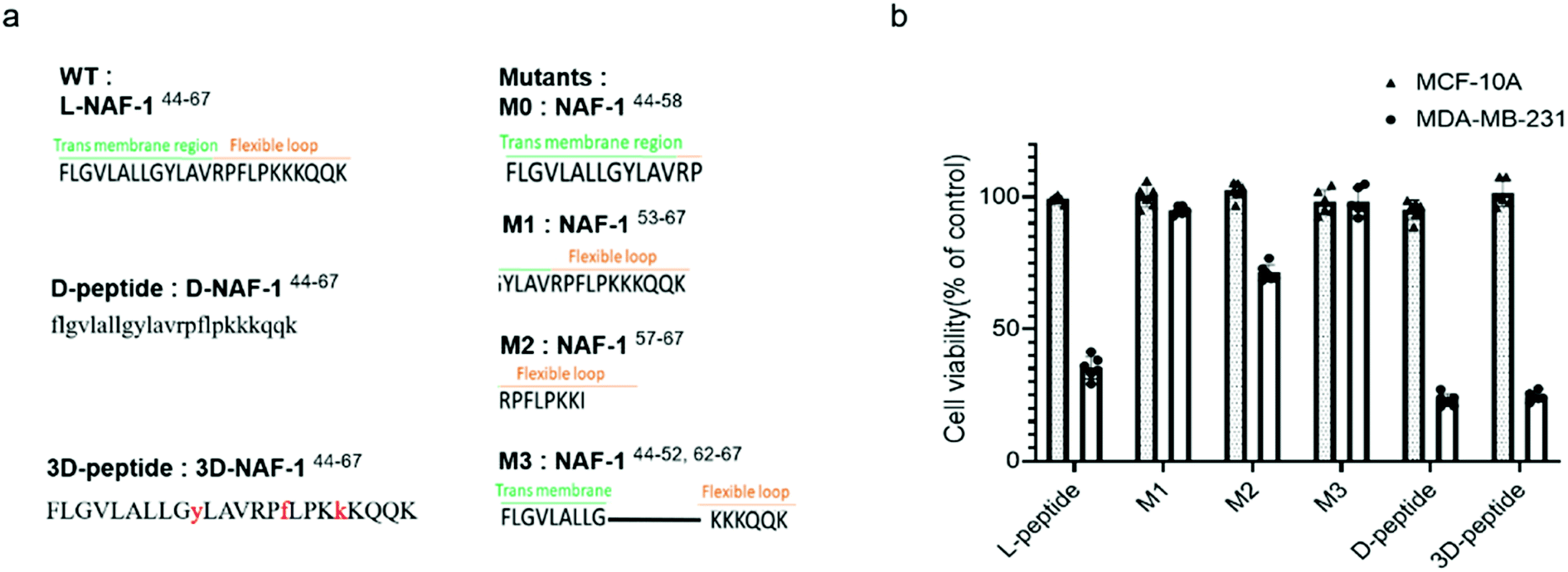 | ||
| Fig. 5 The full sequence of NAF-144-67 is required for its anti-cancer activity, and NAF-144-67 can be stabilized by incorporating D amino acids. (a) The full sequence of NAF-144-67 and its four mutations, D and 3D peptide sequence. (b) Effect of the NAF-1 derived peptides (10 μM) on the viability of cancer cells (blank, MDA-MB-231) and normal cells (dotted, MCF-10A) after 5 days. L-peptide refers to the original full-length NAF-144-67 peptide. | ||
To improve the stability of NAF-144-67 in biological systems, we incorporated D amino acid residues into its sequence. We synthesized a peptide in which all residues were replaced by D amino acids (D-NAF-144-67; Fig. 5a and S11b†). In addition, we designed a peptide in which only specific residues were replaced by D amino acids (3D-NAF-144-67; Fig. 5a and S11b†). For the latter, we used the Prosper server,34 which predicted that the most probable protease cleavage sites within NAF-144-67 are Y53, F59, and K63, and replaced these residues with D-amino acids to form the peptide termed 3D-NAF-144-67 (Fig S11b and c†). In the presences of Trypsin, the L peptide was fully cleaved after 400 min, while the D and 3D peptides showed no cleavage after 24 hours (Fig. S11c†). In the presence of Chymotrypsin, the L peptide was fully cleaved after 100 min, whereas after 540 min only 65% of the 3D peptide and 10% of the D peptide were cleaved (Fig. S11c†). As expected, the CD spectrum of D-NAF-144-67 was a mirror image of L-NAF-144-67 and showed a combination of helical features with some disordered domains (Fig. S11b†). The CD spectrum of the 3D-NAF-144-67 revealed that while the peptide had a larger proportion of disordered features, it kept some of its helical nature (Fig. S11b†). The peptides derived from NAF-144-67 induced cell death with an IC50 value of 18.3 ± 0.4 μM for the L peptide, 12.8 ± 0.2 μM for the D peptide, and 12.5 ± 0.6 μM for the 3D peptide (Fig. S11d†), indicating that the improved stability by the D amino acid residues did not interfere with the cancer killing activity of the peptide.
NAF-144-67 reduces tumor mass in xenograft mice injected with MDA-MB-231 triple-negative breast cancer cells
In vivo tumor growth assays were performed in compliance with the Hebrew University Authority for biological and biomedical models (NS-17-15262-4). MDA-MB-231 triple-negative human breast cancer cells (3 × 106) were injected subcutaneously to 7 to 8 week-old athymic nude (FOXN1NU) mice. Twenty-eight days after tumor inoculation mice were treated for 3 weeks with either 6 doses of 3D-NAF-144-67 50 μM (0.875 mg kg−1) or, as control, with 9 doses of doxorubicin (5 mg kg−1). Treatment of mice with 3D-NAF-144-67 dramatically reduced tumor growth relative to control untreated mice (Fig. 6a and b). A similar effect of reduction in tumor growth was also found for the potent chemotherapy agent doxorubicin (Fig. 6a and b). However, while doxorubicin treatment caused a severe decrease in body weight during treatment, mice treated with 3D-NAF-144-67 showed no change in body weight, demonstrating the important non-toxic effect of NAF-144-67 on mice (Fig. 6c). At the termination of the experiment, tumor weight was measured revealing a significant effect of 3D-NAF-144-67 on decreasing tumor weight in mice (Fig. 6b). Survival curve assays were performed on younger athymic nude (FOXN1NU) mice, aged 5–6 weeks, that were inoculated subcutaneously with a larger amount (5 × 106) of MDA-MB-231 cells. Rapid tumor growth was established due to the younger age of mice and the higher number of cells inoculated. Mice were treated one week after tumor inoculation for 3 weeks with 6 doses of 3D-NAF-144-67 peptide 50 μM (0.875 mg kg−1) and a survival curve was established showing a clear mice survival effect on mice treated with the 3D-NAF-144-67 peptide compared to control saline (Fig. S12†).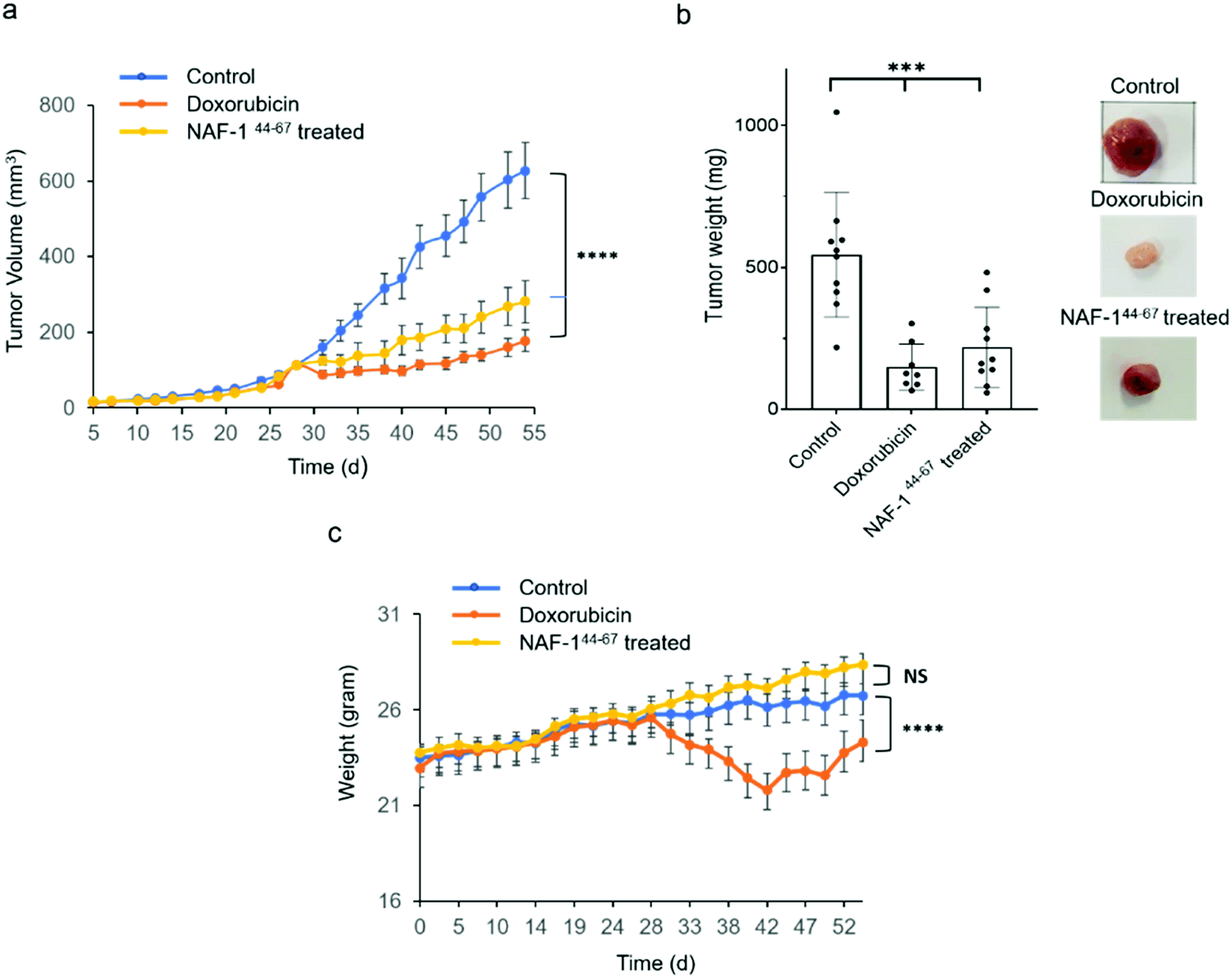 | ||
| Fig. 6 Treatment with NAF-144-67 reduces tumor size in xenograft mice injected with MDA-MB-231 triple-negative breast cancer cells. (a) The effect of NAF-144-67 or doxorubicin on the volume of MDA-MB-231 tumors developing in xenograft mice. (b) Bar graph (left) and representative images (right) showing the weight and appearance of tumors obtained at termination (week 55) from treated and untreated mice. (c) Changes in body weight of treated and untreated mice. ***P < 0.001, ****P < 0.0001 by t-test. | ||
Discussion
We describe a peptide, derived from the human NAF-1/CISD2 protein (NAF-144-67; Fig. 1), which selectively penetrates cancer cells and causes their death without affecting normal cells (Fig. 2), using a unique mechanism. NAF-144-67 is derived from parts of the transmembrane region and flexible loop of NAF-1 and is mainly helical in DPC. In contrast to many other anti-cancer peptides that target the PM of cancer cells,35–39 NAF-144-67 primarily targets their mitochondria and ER (Fig. 4). Targeting both the mitochondria and ER could be a promising anti-cancer strategy, since the mitochondrial-ER network of cancer cells significantly contributes to the chemoresistance and survival of cancer cells by helping the cancers cells adopt to stress.40–45Peptides with anti-cancer activity targeting mitochondrial proteins and membrane were previously described.23,29,30 Many of them are chimeric peptides that include a functional sequence that induces apoptosis and a targeting sequence that targets the peptide to the mitochondria.40,46,47 To the best of our knowledge, NAF-144-67 is the first peptide that selectively penetrates the PM of cancer cells and targets both the ER and the mitochondria (Fig. 4). This behavior of NAF-144-67 is likely a result of the dual targeting of NAF-1 to the ER and mitochondrial membranes in almost all cells studied to date,10,11 and the similarities observed between the PM of cancer cells and the ER and mitochondrial membranes of healthy cells48 [Table S1†]. NAF-144-67 could therefore interact with the PM of cancer cells as it would interact with the ER or mitochondrial membranes of normal cells, penetrate them without any impact on their integrity, since they do not serve as the target of NAF-1 localization, and continue to interact with and disrupt the cancer cell ER and mitochondrial membranes that are the target of NAF-1 localization. In contrast, since NAF-144-67 does not penetrate the PM of normal cells, that do not resemble ER or mitochondrial membranes of normal cells and do not serve as the target of NAF-1 localization, it does not reach the ER and mitochondrial membranes of normal cells and does not disrupt them.
Trans-membrane regions of proteins, with or without positively charged amino acids proceeding them, were shown to be responsible for the localization of proteins to their proper organelles, and changing the charge, length or hydrophobicity of this trans-membrane region may change protein localization.49–52 Our simulations show that the trans-membrane sequence of NAF-144-67 is ideal for localizing it to the ER and mitochondrial membranes. The targeting strategy, pursued here, could therefore result in the targeting of the mitochondria and ER of a wide range of cancer cells with very high specificity.
In contrast to other CPP, NAF-144-67 appears to induce a combination of cell death pathways in cancer cells that primarily includes apoptosis, but also necrosis and ferroptosis, suggesting that NAF-1 could represent a crosstalk point between these different cell death pathways.53,54 Ferroptosis is a cell death mechanism that is dependent on iron and ROS levels in the cell,55–58 and NAF-1 regulates the accumulation of ROS and iron in cells.4 Suppressing NAF-1 in human epithelial breast cancer cells resulted in increased accumulation of Fe and ROS in the mitochondria.5,14 In addition, suppressing NAF-1 levels resulted in apoptosis and ferroptosis activation in cancer cells.59,60 The activation of ferroptosis and apoptosis by NAF-144-67 suggests therefore that this peptide could inhibit the function of the endogenous NAF-1 protein of cancer cells inducing ferroptosis and apoptosis, and that this process may have induced other cell death pathways such as necroptosis.
Taken together, our findings suggest that NAF-144-67 may induce cancer cell death by two possible mechanisms. The peptide could insert itself into the membranes of the ER and mitochondria, compromising their integrity (that would cause for example leakage of cytochrome c) and activate apoptosis and/or other cell death pathways. Alternatively, or at the same time, the peptide may interfere with the dimerization process of NAF-1, inactivating it and causing activation of different cell death pathways that NAF-1 is protecting cancer cells from (inhibiting the dimerization of a transmembrane region of a protein was shown for example for the receptors ErbB2 and NRP161–63). Because a version of the peptide that contained only D amino acids had a similar killing activity to that of the L amino acid peptide (Fig. 5b), we favour the first mechanism over interfering with dimerization. Regardless of the exact mechanism of cell death initiation, the NAF-144-67 peptide is unique in the fact that it selectively penetrates through the PM of cancer cells, targeting their mitochondria and ER, and causing cytochrome c release and apoptosis/cell death activation, specifically in cancer cells.
Materials and methods
Peptides synthesis
Peptides were synthesized on a Liberty Blue Microwave Assisted Peptide Synthesizer (CEM) using standard Fmoc chemistry and DIC/Oxyma as coupling reagents. The peptides were labeled with 5(6)-carboxyfluorescein at their N termini as described64 and cleaved from the resin as described.65 The peptides were purified on a MERCK-Hitachi HPLC using a reverse-phase C18 semi-preparative column with a gradient of ACN/TDW. ESI mass spectrometry and analytical HPLC were used to verify the identity and purity of the peptides.Prediction of proteases cleavage sites in the peptides
Prediction of the protease cleavage sites in NAF-144-67 was performed using PROSPER: PROtease Specificity Prediction servER34Circular dichroism (CD)
CD spectra of different NAF-144-67 derived peptides were recorded as described.66 100 μM peptides were dissolved in 165 mM NaCl (saline) or 3 mM n-dodecylphosphocholine (DPC).Peptide stability against protease cleavage
Peptide stocks of 500 μl in concentrations of 450 μM were added 0.001 mg ml−1 protease (trypsin/chymotrypsin) and incubated at 22 °C. Samples of 40 μl were taken before adding the protease and after 15 min, 30 min, 45 min, 1 h, 2 h, 3 h, 4 h, 5 h, 7 h, 9 h and 24 h. 7 μl of 2% TFA in TDW were added to stop the proteolysis. 60 μl TDW were then added and the samples were analyzed using analytical HPLC.Determination of IC50
MDA-MB-231 cells were planted at a density of 10 × 103 in a 96 well plate and treated with NAF-144-67 peptides at the series of concentrations (1.25 μM, 2.5 μM, 5 μM, 10 μM, 20 μM, 40 μM, 80 μM, 120 μM, 140 μM, 160 μM) and presto-blue probes (Invitrogen) used for accessing cell death. % of cell death was calculated in reference to cells without the peptide. The IC50 values were calculated by fitting the plot to the Hill equation: Y = B + ((T–B)/(1 + 10(log(EC50)-X) × Hill slope)) T and B: top and bottom plateaus in the Y axis; EC50: the X value that gives the half Y value between top and bottom Y plateaus. Hill slop is the curve slope.Cell culture
Malignant epithelial breast cells (MDA-MB-231) were grown in 5% CO2 RPMI medium 1640 supplemented with 10% FCS, L-glutamine, and antibiotics (Biological Industries). Control epithelial breast cells (MCF-10A) were maintained in complete growth medium consisting of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Dulbecco's modified Eagle's medium and Ham's F12 medium supplemented with horse serum (5%), epidermal growth factor (20 ng ml−1), cholera toxin (CT, 0.1 μg mg−1), insulin (10 μg ml−1), hydrocortisone (500 ng ml−1), and penicillin/streptomycin (1 unit per ml). Cells were plated one day prior to the experiments on 96-well plates or 4-Chamber Glass Bottom Dish for cell viability or microscopic measurement.59
:1 mixture of Dulbecco's modified Eagle's medium and Ham's F12 medium supplemented with horse serum (5%), epidermal growth factor (20 ng ml−1), cholera toxin (CT, 0.1 μg mg−1), insulin (10 μg ml−1), hydrocortisone (500 ng ml−1), and penicillin/streptomycin (1 unit per ml). Cells were plated one day prior to the experiments on 96-well plates or 4-Chamber Glass Bottom Dish for cell viability or microscopic measurement.59
Uptake of fluorescein labelled peptide into cells
Control epithelial breast cells (MCF-10A) and malignant epithelial breast cells (MDA-MB-231) were plated at a density of 2 × 105 cells in 4-Chamber Glass Bottom Dish with 1 ml growth medium and incubated with the Fl-NAF-144-67 peptide (10 μM) for 24 h.59 Nuclei were stained with Hoechst 33342 and mitochondria with Rhodamine 800. Fluorescein fluorescence was observed with a Nikon A1R confocal microscope with GaAsP detectors. Image analysis was conducted using NIS-Elements confocal microscope imaging software.59Cell viability experiments
Cell viability was assayed following incubation of the NAF-144-67 peptide (10 μM) with MCF-10A or MDA-MB-231 cells plated at a density of 1 × 104 cells per well in 96-well plates. Cells were incubated with the peptide for 3 and 5 days. For the measurement of divalent cations effect, cells were incubated with the peptide in the presence or absence of MgCl2 (0.1 M) following 3 washes (1 ml each) with DMEM-Hepes the cells' viability was determined with the fluorescent redox probe, Presto-Blue.67 The fluorescence of Presto-Blue was recorded using a plate-reader (Tecan Safire) after 1 h of incubation at 37 °C (λex = 560 nm; λem = 590 nm).Peptide-induced intracellular structure damage assessment
Epithelial breast cells (MCF-10A) and malignant epithelial breast cells (MDA-MB-231) were pre-stained with Rhodamine 800 and Rhodamine B-[(1,10-phenanthrolin-5-yl)-aminocarbonyl] benzyl ester (RPA), for mitochondria, Hoechst 33342 for nuclei, Calcein-AM for plasma membrane integrity, or modified to stably express pDsRed for ER, and incubated with NAF-144-67 (10 μM). Cell images were captured with confocal microscope at 0, 1, 3 and 6 h post incubation. Intracellular structure damage was assessed by quantifying the decrease in fluorescence change of each intracellular compartments due to the addition of the peptide. Image analysis was conducted using ImageJ.14,59
Evaluation of the cytotoxicity of the spheroids of breast cancer cells
The MDA-MB-231 cells were seeded in a 96 well ULA plate (Corning 7007) at a density of 2000 and allowed to culture for an additional 3 days to form the spheroids. The resulting spheroids were then treated with 10 μM 3D-NAF-144-67 and cell death inhibitors (2 μM ferrostatin, 1 μM cyclosporine A, 50 μM necrostatin) and the IncuCyte® Red cytotoxicity reagent (Essen Bioscience Cat #4632) to probe the cell death. Cell images were recorded every two hours, and the result in red color, reflecting cell death, was analyzed with the IncuCyte Zoom system (Essen Bioscience).Transmission electron microscopy (TEM)
Unless otherwise stated, all reagents were purchase from Electron Microscopy Sciences and all specimen preparation was performed at the Electron Microscopy Core Facility, University of Missouri (https://emc.missouri.edu/). Cells were fixed in 2% paraformaldehyde, 2% glutaraldehyde in 100 mM sodium cacodylate buffer pH 7.35. Cells were then rinsed with 100 mM sodium cacodylate buffer, pH 7.35 containing 130 mM sucrose. Secondary fixation was performed using 1% osmium tetroxide (Ted Pella, Inc. Redding, California). Specimens were then incubated at 4 °C for 1 hour, rinsed with cacodylate buffer, followed by rinsing in with distilled water. En bloc staining was performed using 1% aqueous uranyl acetate (4 °C overnight). A graded dehydration series was performed using ethanol at 4 °C, transitioned into acetone, and dehydrated tissues were then infiltrated with Epon resin for 24 hours at room temperature and polymerized at 60 °C overnight. Sections were cut to a thickness of 75 nm using an ultramicrotome (Ultracut UCT, Leica Microsystems, Germany) and a diamond knife (Diatome, Hatfield PA). Images were acquired with a JEOL JEM 1400 transmission electron microscope (JEOL, Peabody, MA) at 80 kV on a Gatan Ultrascan 1000 CCD (Gatan, Inc, Pleasanton, CA) as previously described.68,69 Results were evaluated from 5–10 different cells randomly selected, every cell was evaluated for their nuclear and general morphology in low magnification. In higher magnification averaged over 20 fields per cell; at least 100–200 mitochondria and 200–300 ER were counted. Mitochondrial and ER damage were expressed as the ratio of damaged organelle to total number of the organelle, in three independent experiments.9Proteomic analysis
Cells (MDA-MB-231 breast cancer epithelial cells and MCF-10A control epithelial cells) were grown to 80% confluency and treated with 10 μM 3D-NAF-144-67 peptide for (0, 1, 3 and 6 hours). At the different time points cells were sampled and subjected to protein extraction using 0.175 M Tris–HCl, pH 8.8, 5% SDS, 15% glycerol, 0.3 M DTT, as described in.70 Protein samples were precipitated with 100% of Acetone, washed, resuspended in urea/thiourea buffer (6 M urea, 2 M thiourea, 100 mM ammonium bicarbonate, pH 8.0) and subjected to protein quantification using 660 nm protein assay. An equal amount of protein (40 μg) from each sample was reduced by DTT, alkylated by IAA and digested by LysC/trypsin overnight. Digested peptides were purified by C18 ziptips. All resuspended samples were analyzed by Bruker timsTOF pro DIA-PASEF acquisition mode with a 90 min LC gradient. The spectral library generation and DIA-PASEF data analysis were completed by Spectronaut (v14).For spectral library generation, 4 μg of purified peptides from each sample were combined and fractionated by high pH reversed-phase peptide fractionation kit according to the manufacturer's protocol (Thermofisher, Waltham, MA). Each of 8 high pH fractions was acquired by 60 min DDA-PASEF70 and searched against the Uniprot-Human database (UP000005640, 20381 entries) with the Pulsar algorithm in Spectronaut using the following criteria: spectrum, peptide and protein FDR 1%, min 3 and max 6 best fragments per peptide. A total 97495 precursors, 78084 peptides and 8246 protein groups were included in the library. For dia-pasef data analysis: precursor Qvalue (precursor FDR), protein Qvalue (protein FDR) 1%. MS2 area quantification were filtered based on Qvalue and final PG. quantity was cut-off by 30 (1% quantile). A protein was identified in at least 2 samples per group. On average, >6400 proteins were identified in each sample. A total 7431 proteins were identified in all samples. The global sum normalization method was applied.
The statistical analysis included 1- Likelihood Ratio Test (LRT) for MDA-MB-231 cell type to test the effect of treatment in a series of time course; and 2- Likelihood Ratio Test (LRT) for MCF-10A cell type to test the effect of treatment in a series of time course. Significant changes: p value < 0.01. Proteomics data was deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD030509.
Animal studies
This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication no. 85-23 Rev. 1985) and was approved by the Hebrew University Animal Care and Use; The Authority for biological and biomedical models (NS-17-15262-4).In our protocol we used a known human tumor xenograft model for human breast cancer. We injected MDA-MB-231 human breast cancer cells (3 × 106 cells or 5 × 106) subcutaneously to athymic nude (FOXN1NU) at two different age groups 7–8- or 5–6 weeks old mice. Mice weight and tumor size was measured through the experiment. Age groups 5–6 weeks mice were treated one week after tumor inoculation for 3 weeks with 6 doses of NAF-1 peptide 50 μM (17.5 microgram/mouse) and survival curve was established. Age groups 7–8 weeks mice were treated 28 days after tumor inoculation for 3 weeks with either 6 doses of NAF-1 peptide 50 μM (17.5 microgram/mouse) or 9 doses of Doxorubicin (5 mg kg−1). Tumor areas were calculated according to the formula V = ½ (Length × Width2). At the end of each experiment the animals were euthanized the tumor was remove, weighed, photographed, and preserved in 10% PFA (paraformaldehyde).
Mitochondrial bioenergetics, oxygen consumption rate (OCR), cellular glycolysis and extracellular acidification rate (ECAR)
OCR and ECAR were measured using a Seahorse XFp, Agilent, cell mito-stress analyzer (Agilent Technologies, Inc., Santa Clara, CA, USA) with the XF Cell Mito Stress Kit and XF Glycolysis Stress Kit (Agilent Technologies, Inc., Santa Clara, CA, USA), according to the manufacturer's instructions. MDA-MB-231 cells (40000) were grown to approximately 80% confluence in complete medium overnight before the experiment. The initial medium was then exchanged with a seahorse-running medium consists of Dulbecco's modified Eagle's medium (DMEM) base without glucose, L-Glutamine 2 mM, sodium pyruvate 1 mM, glucose added to a final concentration of 25 mM, pH adjusted to be 7.3–7.4. Then microplates containing cells were incubated at 37 °C without CO2 for 1 h before the assay. Plates were then placed into the XFp analyzer. The OCR was calculated after the sequential additions of oligomycin A 1.5 μM, FCCP 0.75 μM, antimycin A/rotenone 1 μM, using an XF Cell Mito Stress Test kit. The ECAR medium initially did not contain glucose, and then measured after the addition of glucose 10 mM, oligomycin A 2 μM and 2-DG 50 mM. Results were expressed as mean ± SD of three independent experiments. All measurements were recorded at set-interval time points. All materials and compounds obtained from Seahorse Bioscience. Calculations of the OCR and the ECAR tests parameters performed according to the manufacturer recommendations, using the equations described in.71
The cells were or were not treated with NAF-144-67 peptide of 20 μM incubated for 3 h before the experiment is taking place of OCR and ECAR.
Mitochondrial fraction isolation
Mitochondria were isolated following the protocol published by Lampl et al.72 or by using the mitochondria isolation kit for cultured cells from abcam (ab110170). Briefly, 30 × 105 cells were collected and washed with pre-cooled PBS 1× twice and centrifuge at 600×g for 5 min at 4 °C. The cell pellet was resuspended into 1 ml of mitochondrial isolation buffer (200 mM sucrose, 10 mM Tris/MOPS, pH 7.4, and 1 mM EGTA/Tris). Then, the cells were broken using a syringe and a 27-gauge ½ inch needle. The cell debris were centrifuged and the supernatant containing the mitochondria crude were collected. The mitochondria crude extract was centrifugated at 10,000×g for 10 min at 4 °C and the pellet containing the enriched mitochondrial fraction was resuspended in mitochondrial isolation buffer at 4 °C.Measurements of cytochrome C release
5 mg ml−1 of isolated mitochondria were incubated with or without 20 μM of NAF-144-67 peptide for 30 min in isolation buffer. Mitochondrial fractions were then centrifuged for 10 minutes at 10,000×g at 4 °C. The supernatant was then delicately separated from the pellet. 200 μl of the supernatants of the mitochondrial fraction (with and without treatment with NAF-144-58) were loaded in a quartz cuvette to measure the absorption spectrum. The cytochrome c spectrum was recorded with the Varian Cary 300 Bio UV-visible Spectrophotometer. The optical density of supernatants was recorded against the isolation buffer as a reference from 380 to 500 nm. 0.25 μM of cytochrome C spectrum was also recorded as positive control. The release of cytochrome C from the enriched mitochondrial fraction was also measured by western blot. Similar volume of pellet and supernatant were prepared for western blot using denaturation buffer and the equal volume of solution was loaded and run into SDS-PAGE gel (15% acrylamide). The proteins were transferred to nitrocellulose blots and incubated with antibody against cytochrome C (1/500; ab13575-abcam). Anti-mouse IgG from Jackson ImmunoResearch Laboratories (West Grove, PA) were used as secondary antibodies.Computer simulations
942 and 14739 water molecules to the mitochondrial and ER membrane models, respectively to fully hydrate the bilayer systems. We also included K+ ions to neutralize the negative charged membranes, and an additional 150 mM KCl ions. To prepare the models for the normal and cancer plasma membranes a similar procedure was followed. In this case, the number of lipids in the membrane patches was 300 and the number of water molecules was 15170 (for the cancer plasma membrane) and 15017 (for the normal plasma membrane).
Conclusions
In summary, we present a new approach for anticancer therapy using a unique CPP derived from a protein localized to the mitochondria-ER network (NAF-144-67). This CPP interacts with and transverse the PM of cancer cells that have a somewhat similar composition to that of mitochondria and ER membranes and triggers a combination of different cell death pathways, including ferroptosis and apoptosis, regulated by the native protein it is derived from. In contrast, the peptide does not penetrate the PM of healthy cells that do not resemble mitochondria and ER membranes.Data availability
Proteomics data was deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD030509”). We added representative mass spectra to the ESI.† Please find below a special account access info for viewing the data online: Reviewer account details: Username: reviewer_pxd030509@ebi.ac.uk. Password: pLMCoJad.Author contributions
Conceptualization: RN, AF, RE and RM. Experimental design: RN, AF, RM and RE. Experimental data collection and analysis: Y-SS, A-IA, OK, AEC, MDY, LR, LJW. Computational studies and analysis: AEC, and RE. LJW contributed to experiments on simplified systems. Funding acquisition: RN, AF, RM, LJW and RE. Wrote the manuscript: Y-SS, A-IA, OK, AEC, RN, RE, AF and RM.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by the MINERVA Center for Bio-hybrid Complex Systems at the Hebrew University and by the support of the BSF grant number 2020094 to R. N, A. F, L. J. W., and R. E. and the NIH, Grant no. GM 59796 (to R. E.) and GM 111364 (to R. E., L. J. W., R. M.); the Welch Foundation, Grant no. F-1896 (to R. E) and F-1722 (to L. J. W.); and Part of the computations were done using HPC resources from the Texas Advanced Computing Center (TACC) at the University of Texas at Austin. We gratefully acknowledge the use of facilities at the Texas Materials Institute at the University of Texas at Austin.References
- H. Kato and H. Nishitoh, Front. Oncol., 2015, 5, 93 CrossRef PubMed.
- G. Morciano, S. Marchi, C. Morganti, L. Sbano, M. Bittremieux, M. Kerkhofs, M. Corricelli, A. Danese, A. Karkucinska-Wieckowska and M. R. Wieckowski, Neoplasia, 2018, 20, 510–523 CrossRef CAS PubMed.
- A. R. Conlan, H. L. Axelrod, A. E. Cohen, E. C. Abresch, J. Zuris, D. Yee, R. Nechushtai, P. A. Jennings and M. L. Paddock, J. Mol. Biol., 2009, 392, 143–153 CrossRef CAS PubMed.
- S. Tamir, M. L. Paddock, M. Darash-Yahana-Baram, S. H. Holt, Y. S. Sohn, L. Agranat, D. Michaeli, J. T. Stofleth, C. H. Lipper and F. Morcos, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 1294–1315 CrossRef CAS PubMed.
- Y.-S. Sohn, S. Tamir, L. Song, D. Michaeli, I. Matouk, A. R. Conlan, Y. Harir, S. H. Holt, V. Shulaev and M. L. Paddock, Proc. Natl. Acad. Sci., 2013, 110, 14676–14681 CrossRef CAS PubMed.
- S. Tamir, J. A. Zuris, L. Agranat, C. H. Lipper, A. R. Conlan, D. Michaeli, Y. Harir, M. L. Paddock, R. Mittler and Z. I. Cabantchik, PLoS One, 2013, 8, e61202 CrossRef PubMed.
- C. H. Lipper, M. L. Paddock, J. N. Onuchic, R. Mittler, R. Nechushtai and P. A. Jennings, PLoS One, 2015, 10, e0139699 CrossRef PubMed.
- N. C. Chang, M. Nguyen and G. C. Shore, Autophagy, 2012, 8, 856–857 CrossRef CAS PubMed.
- O. Karmi, Y. S. Sohn, H. B. Marjault, T. Israeli, G. Leibowitz, K. Ioannidis, Y. Nahmias, R. Mittler, I. Z. Cabantchik and R. Nechushtai, Antioxidants, 2021, 10(8), 1160 CrossRef CAS PubMed.
- S. Amr, C. Heisey, M. Zhang, X.-J. Xia, K. H. Shows, K. Ajlouni, A. Pandya, L. S. Satin, H. El-Shanti and R. Shiang, Am. J. Hum. Genet., 2007, 81, 673–683 CrossRef CAS PubMed.
- Y.-F. Chen, C.-H. Kao, Y.-T. Chen, C.-H. Wang, C.-Y. Wu, C.-Y. Tsai, F.-C. Liu, C.-W. Yang, Y.-H. Wei and M.-T. Hsu, Genes Dev., 2009, 23, 1183–1194 CrossRef CAS PubMed.
- C.-C. Lin, T.-H. Chiang, W.-J. Chen, Y.-Y. Sun, Y.-H. Lee and M.-S. Lin, Injury, 2015, 46, 2341–2350 CrossRef PubMed.
- N. C. Chang, M. Nguyen, M. Germain and G. C. Shore, EMBO J., 2010, 29, 606–618 CrossRef CAS PubMed.
- M. Darash-Yahana, Y. Pozniak, M. Lu, Y.-S. Sohn, O. Karmi, S. Tamir, F. Bai, L. Song, P. A. Jennings and E. Pikarsky, Proc. Natl. Acad. Sci., 2016, 113, 10890–10895 CrossRef CAS PubMed.
- B. Chen, S. Shen, J. Wu, Y. Hua, M. Kuang, S. Li and B. Peng, Int. J. Clin. Exp. Path., 2015, 8, 13725 CAS.
- L. Wang, F. Ouyang, X. Liu, S. Wu, H.-m. Wu, Y. Xu, B. Wang, J. Zhu, X. Xu and L. Zhang, Oncotarget, 2016, 7, 3791 CrossRef PubMed.
- L. Yang, S. Hong, Y. Wang, Z. He, S. Liang, H. Chen, S. He, S. Wu, L. Song and Y. Chen, Oncotarget, 2016, 7, 22720 CrossRef PubMed.
- Y. Yang, Y.-s. Bai and Q. Wang, Oncol. Res., 2017, 25, 605 CrossRef PubMed.
- S.-M. Li, C.-H. Chen, Y.-W. Chen, Y.-C. Yen, W.-T. Fang, F.-Y. Tsai, J.-L. Chang, Y.-Y. Shen, S.-F. Huang and C.-P. Chuu, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- L. Liu, M. Xia, J. Wang, W. Zhang, Y. Zhang and M. He, Med. Oncol., 2014, 31, 183 CrossRef PubMed.
- W. D. Foulkes, I. E. Smith and J. S. Reis-Filho, N. Engl. J. Med., 2010, 363, 1938–1948 CrossRef CAS PubMed.
- O. Karmi, Y. S. Sohn, S. I. Zandalinas, L. Rowland, S. King, R. Nechushtai and R. Mittler, Free Radical Biol. Med., 2021, 176, 92–104 CrossRef CAS PubMed.
- S. Jones, T. Holm, I. Mäger, Ü. Langel and J. Howl, Chem. Biol., 2010, 17, 735–744 CrossRef CAS PubMed.
- M. Lukanowska, J. Howl and S. Jones, Biotechnol. J., 2013, 8, 918–930 CrossRef CAS PubMed.
- G. C. Kim, D. H. Cheon and Y. Lee, Biochim. Biophys. Acta – Proteins Proteom., 2021, 1869(4), 140604 CrossRef CAS PubMed.
- A. D. Frankel and C. O. Pabo, Cell, 1988, 55, 1189–1193 CrossRef CAS PubMed.
- J. K. Boparai and P. K. Sharma, Protein Pept. Lett., 2020, 27, 4–16 CrossRef CAS PubMed.
- H. S. Lee, C. B. Park, J. M. Kim, S. A. Jang, I. Y. Park, M. S. Kim, J. H. Cho and S. C. Kim, Cancer Lett., 2008, 271, 47–55 CrossRef CAS PubMed.
- R. Worch, A. Dudek, P. Borkowska and P. Setny, Int. J. Mol. Sci., 2021, 22, 5301 CrossRef CAS PubMed.
- M. L. Dória, Z. Cotrim, B. Macedo, C. Simões, P. Domingues, L. Helguero and M. R. Domingues, Breast Cancer Res. Treat., 2012, 133, 635–648 CrossRef PubMed.
- A. C. Alves, D. Ribeiro, C. Nunes and S. Reis, Biochim. Biophys. Acta - Biomembr., 2016, 1858, 2231–2244 CrossRef CAS PubMed.
- R. Zwaal, P. Comfurius and E. Bevers, Cell. Mol. Life Sci., 2005, 62, 971–988 CrossRef CAS PubMed.
- S. Riedl, B. Rinner, M. Asslaber, H. Schaider, S. Walzer, A. Novak, K. Lohner and D. Zweytick, Biochim. Biophys. Acta – Biomembr., 2011, 1808, 2638–2645 CrossRef CAS PubMed.
- J. Song, H. Tan, A. J. Perry, T. Akutsu, G. I. Webb, J. C. Whisstock and R. N. Pike, PLoS One, 2012, 7, e50300 CrossRef CAS PubMed.
- C. Borghouts, C. Kunz and B. Groner, J. Pept. Sci., 2005, 11, 713–726 CrossRef CAS PubMed.
- J. S. Mader and D. W. Hoskin, Expert Opin. Invest. Drugs, 2006, 15, 933–946 CrossRef CAS PubMed.
- D. W. Hoskin and A. Ramamoorthy, Biochim. Biophys. Acta – Biomembr., 2008, 1778, 357–375 CrossRef CAS PubMed.
- R. J Boohaker, M. W Lee, P. Vishnubhotla, J. LM Perez and A. R Khaled, Curr. Med. Chem., 2012, 19, 3794–3804 CrossRef PubMed.
- M. R. Felício, O. N. Silva, S. Gonçalves, N. C. Santos and O. L. Franco, Front. Chem., 2017, 5, 5 Search PubMed.
- J. E. Constance and C. S. Lim, Ther. Delivery, 2012, 3, 961–979 CrossRef CAS PubMed.
- S. Wen, D. Zhu and P. Huang, Future Med. Chem., 2013, 5, 53–67 CrossRef CAS PubMed.
- V. Panda, P. Khambat and S. Patil, Int. J. Clin. Med., 2011, 2(4), 515–529 CrossRef CAS.
- F. Martinon, Acta Oncol., 2012, 51, 822–830 CrossRef CAS PubMed.
- T. Avril, E. Vauleon and E. Chevet, Oncogenesis, 2017, 6, e373 CrossRef CAS PubMed.
- H. Urra, E. Dufey, T. Avril, E. Chevet and C. Hetz, Trends Cancer, 2016, 2, 252–262 CrossRef PubMed.
- H. Ellerby, W. Arap, L. Ellerby, R. Kain, R. Andrusiak, G. Rio, S. Krajewski, C. Lombardo, R. Rao and E. Ruoslahti, Nat. Med., 1999, 5, 1032–1038 CrossRef CAS PubMed.
- S. Farsinejad, Z. Gheisary, S. E. Samani and A. M. Alizadeh, Tumor Biol., 2015, 36, 5715–5725 CrossRef CAS PubMed.
- D. Casares, P. V. Escribá and C. A. Rosselló, Int. J. Mol. Sci., 2019, 20, 2167 CrossRef CAS PubMed.
- M. Nguyen, D. Millar, V. W. Yong, S. Korsmeyer and G. Shore, J. Biol. Chem., 1993, 268, 25265–25268 CrossRef CAS PubMed.
- I. Arnold, H. Fölsch, W. Neupert and R. A. Stuart, J. Biol. Chem., 1998, 273, 1469–1476 CrossRef CAS PubMed.
- N. Borgese, S. Colombo and E. Pedrazzini, Int. J. Cell Biol., 2003, 161, 1013–1019 CrossRef CAS PubMed.
- D. M. Walther and D. Rapaport, Biochimica et Biophysica Acta – Molecular Cell Research is Biochim. Biophys. Acta, 2009, 1793, 42–51 CrossRef CAS PubMed.
- T. Vanden Berghe, W. J. Kaiser, M. J. Bertrand and P. Vandenabeele, Mol. Cell. Oncol., 2015, 2, e975093 CrossRef PubMed.
- S. H. Hong, D.-H. Lee, Y.-S. Lee, M. J. Jo, Y. A. Jeong, W. T. Kwon, H. A. Choudry, D. L. Bartlett and Y. J. Lee, Oncotarget, 2017, 8, 115164 CrossRef PubMed.
- Y. Xie, W. Hou, X. Song, Y. Yu, J. Huang, X. Sun, R. Kang and D. Tang, Cell Death Differ., 2016, 23, 369–379 CrossRef CAS PubMed.
- H. Yu, P. Guo, X. Xie, Y. Wang and G. Chen, J. Cell. Mol. Med., 2017, 21, 648–657 CrossRef CAS PubMed.
- A. R. Bogdan, M. Miyazawa, K. Hashimoto and Y. Tsuji, Trends Biochem. Sci., 2016, 41, 274–286 CrossRef CAS PubMed.
- W. S. Yang and B. R. Stockwell, Trends Cell Biol., 2016, 26, 165–176 CrossRef CAS PubMed.
- S. H. Holt, M. Darash-Yahana, Y. S. Sohn, L. Song, O. Karmi, S. Tamir, D. Michaeli, Y. Luo, M. L. Paddock and P. A. Jennings, J. Cell Sci., 2016, 129, 155–165 CAS.
- S. D. King, C. F. Gray, L. Song, R. Mittler and P. A. Padilla, PLoS One, 2021, 16, e0245174 CrossRef CAS PubMed.
- A. Arpel, P. Sawma, C. Spenlé, J. Fritz, L. Meyer, N. Garnier, I. Velázquez-Quesada, T. Hussenet, S. Aci-Sèche and N. Baumlin, Cell Rep., 2014, 8, 1714–1721 CrossRef CAS PubMed.
- A. Bennasroune, M. Fickova, A. Gardin, S. Dirrig-Grosch, D. Aunis, G. Crémel and P. Hubert, Mol. Biol. Cell, 2004, 15, 3464–3474 CrossRef CAS PubMed.
- C. Nasarre, M. Roth, L. Jacob, L. Roth, E. Koncina, A. Thien, G. Labourdette, P. Poulet, P. Hubert and G. Crémel, Oncogene, 2010, 29, 2381–2392 CrossRef CAS PubMed.
- P. J. Weber, J. E. Bader, G. Folkers and A. G. Beck-Sickinger, Bioorg. Med. Chem. Lett., 1998, 8, 597–600 CrossRef CAS PubMed.
- R. Gabizon, M. Mor, M. M. Rosenberg, L. Britan, Z. Hayouka, M. Kotler, D. E. Shalev and A. Friedler, Pept. Sci., 2008, 90, 105–116 CrossRef CAS PubMed.
- A. I. Amir, M. van Rosmalen, G. Mayer, M. Lebendiker, T. Danieli and A. Friedler, Sci. Rep., 2015, 5, 1–11 Search PubMed.
- M. Sonnaert, I. Papantoniou, F. P. Luyten and J. Schrooten, Tissue Eng. – C: Methods, 2015, 21, 519–529 CrossRef CAS PubMed.
- X. Pan, S. A. Sands, Y. Yue, K. Zhang, S. M. LeVine and D. Duan, Hum. Gene Ther., 2019, 30, 1039–1051 CrossRef CAS PubMed.
- S. I. Zandalinas, L. Song, S. Sengupta, S. A. McInturf, D. G. Grant, H. B. Marjault, N. A. Castro-Guerrero, D. Burks, R. K. Azad and D. G. Mendoza-Cozatl, Plant J., 2020, 101, 1152–1169 CrossRef CAS PubMed.
- B. Yang, R. Li, P. N. Liu, X. Geng, B. P. Mooney, C. Chen, J. Cheng, K. L. Fritsche, D. Q. Beversdorf and J. C. Lee, J. Proteome Res., 2020, 19, 2236–2246 CrossRef CAS PubMed.
- A. S. Divakaruni, A. Paradyse, D. A. Ferrick, A. N. Murphy and M. Jastroch, Methods Enzymol., 2014, 547, 309–354 CAS.
- S. E. Horvath and G. Daum, Prog. Lipid Res., 2013, 52, 590–614 CrossRef CAS PubMed.
- L. Brignac-Huber, J. R. Reed and W. L. Backes, Mol. Pharmacol., 2011, 79, 549–557 CrossRef CAS PubMed.
- F. Zambrano, S. Fleischer and B. Fleischer, Biochim Biophys Acta Mol Cell Biol Lipids., 1975, 380, 357–369 CAS.
- S. S. Bird, V. R. Marur, I. G. Stavrovskaya and B. S. Kristal, Metabolomics, 2013, 9, 67–83 CrossRef CAS PubMed.
- A. Y. Andreyev, E. Fahy, Z. Guan, S. Kelly, X. Li, J. G. McDonald, S. Milne, D. Myers, H. Park and A. Ryan, J. Lipid Res., 2010, 51, 2785–2797 CrossRef CAS PubMed.
- S. Hofmann, M. Krajewski, C. Scherer, V. Scholz, V. Mordhorst, P. Truschow, A. Schöbel, R. Reimer, D. Schwudke and E. Herker, Biochim. Biophys. Acta – Mol. Cell Biol. Lipids, 2018, 1863, 1041–1056 CrossRef CAS PubMed.
- R. Hovius, J. Thijssen, P. van der Linden, K. Nicolay and B. de Kruijff, FEBS Lett., 1993, 330, 71–76 CrossRef CAS PubMed.
- B. Antonny, S. Vanni, H. Shindou and T. Ferreira, Trends Cell Biol., 2015, 25, 427–436 CrossRef CAS PubMed.
- M. L. Valentine, A. E. Cardenas, R. Elber and C. R. Baiz, Biophys. J., 2018, 115, 1541–1551 CrossRef CAS PubMed.
- E. L. Wu, X. Cheng, S. Jo, H. Rui, K. C. Song, E. M. Dávila-Contreras, Y. Qi, J. Lee, V. Monje-Galvan, R. M. Venable, J. B Klauda and W. Im, J. Comput. Chem., 2014, 35(27), 1997–2004 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19–25 CrossRef.
- Y. Shen, J. Maupetit, P. Derreumaux and P. Tufféry, J. Chem. Theory Comput., 2014, 10, 4745–4758 CrossRef CAS PubMed.
- A. E. Cardenas, C. I. Drexler, R. Nechushtai, R. Mittler, A. Friedler, L. J. Webb and R. Elber, J. Phys. Chem. B, 2022, 126, 2834–2849 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01934e |
| This journal is © The Royal Society of Chemistry 2022 |