
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNS3 helicase inhibitory potential of the marine sponge Spongia irregularis†
Enas Reda Abdelaleem‡
a,
Mamdouh Nabil Samy‡ a,
Taha F. S. Alib,
Muhamad Mustafac,
Mahmoud A. A. Ibrahimd,
Gerhard Bringmanne,
Safwat A. Ahmedf,
Usama Ramadan Abdelmohsen*ag and
Samar Yehia Desoukeya
a,
Taha F. S. Alib,
Muhamad Mustafac,
Mahmoud A. A. Ibrahimd,
Gerhard Bringmanne,
Safwat A. Ahmedf,
Usama Ramadan Abdelmohsen*ag and
Samar Yehia Desoukeya
aDepartment of Pharmacognosy, Faculty of Pharmacy, Minia University, 61519 Minia, Egypt. E-mail: usama.ramadan@mu.edu.eg
bDepartment of Medicinal Chemistry, Faculty of Pharmacy, Minia University, 61519 Minia, Egypt
cMedicinal Chemistry Department, Faculty of Pharmacy, Deraya University, New Minia 61111, Egypt
dComputational Chemistry Laboratory, Chemistry Department, Faculty of Science, Minia University, 61519 Minia, Egypt
eInstitute of Organic Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg, Germany
fDepartment of Pharmacognosy, Faculty of Pharmacy, Suez Canal University, Ismailia 41522, Egypt
gDepartment of Pharmacognosy, Faculty of Pharmacy, Deraya University, New Minia 61111, Egypt
First published on 21st January 2022
Abstract
In the current study, an investigation of the activity of the total extract of the marine sponge Spongia irregularis and its different fractions against the hepatitis C virus (HCV) was pursued. The results revealed that the ethyl acetate fraction exhibited the highest anti-HCV activity, with an IC50 value of 12.6 ± 0.05 μg ml−1. Chromatographic resolution of the ethyl acetate fraction resulted in the isolation of four known compounds, 1,3,7-trimethylguanine (1), 3,5-dihydroxyfuran-2(5H)-one (2), thymidine (3), and 1H-indazole (4). By using LC-HR-ESI-MS metabolic profiling, compounds 5–14 were also identified in the same fraction. Molecular docking calculations revealed the high binding affinity of compound 14 against the allosteric pocket of HCV NS3-NS4A and the active site of HCV NS3 helicase (−10.1 and −7.4 kcal mol−1, respectively). Molecular dynamics simulations, followed by molecular mechanics-generalized Born surface area energy calculations, demonstrated the structural and energetic stability of compound 14 in complex with HCV targets.
1. Introduction
Hepatitis C virus (HCV) is an RNA virus, a member of the flavivirus family. The HCV genome consists of approximately 9600 nucleotides and encodes a polyprotein consisting of 3011–3033 amino acid residues. The N-terminal HCV polyprotein region contains the structural proteins (core protein and two envelope glycoprotein proteins (E1 and E2)), while the non-structural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) are located in the remaining two thirds of the viral genome. The later proteins are responsible for different intracellular processes of the HCV life cycle.1 About 170 million people worldwide suffer from an HCV infection, which is thus considered as a viral pandemic. HCV naturally targets hepatocytes and, possibly, also B lymphocytes.2 HCV infection is a challenging public health problem in Egypt. Chronic HCV infection can develop several complications such as cirrhosis and hepatocellular carcinoma and it may lead to mortality. Many strategies have been developed to achieve the target of elimination of HCV infections in Egypt by 2030.3The marine environment is considered a treasure for discovering novel and new bioactive metabolites with varied chemical scaffolds and biological activities. Thus, it can play an important role as a source for the discovery of antiviral secondary metabolites from different marine organisms4 or even marine-associated microorganisms.5 Marine sponges which belong to genus Spongia, family Spongidae, order Dictyoceratida (according to “The World Porifera Database” and “The World Register of Marine Species”) were reported to have a valuable role in the discovery of marine lead compounds with wide varied biological activities.6,7 In these literatures, Spongia sponges were reported to exhibit antiviral activities towards different viruses. In 1987, three furanoditerpenes named spongiadiol, epispongiadiol, and isospongiadiol were isolated from a Caribbean marine sponge, Spongia sp. They were tested for their antiviral activities towards type 1 (HSV-1) Herpes simplex virus and showed antiviral activities against HSV-1 with IC50 values of 0.25, 12.5, and 2 μg ml−1, respectively, where IC50 of 50 and 0.5 μg ml−1 were detected for the standards ara-A and acyclovir, respectively.8
Moreover, two spongian diterpenes, spongiatriol and isospongiatriol, were isolated from Spongia officinalis.9 These diterpenes had previously been isolated from Hyattella aff. intestinalis and reported for their antiviral activities against adenovirus with IC50 values of 17.0 and 52.0 μM, respectively.10 This provoked us to evaluate the anti-HCV activity of the marine sponge Spongia irregularis and its different fractions with more-in-depth investigations of the most active fraction, including chromatographic separation and isolation of the secondary metabolites, LC-HR-ESI-MS metabolomic analysis, molecular docking, and molecular dynamics studies of both isolated and dereplicated metabolites. Despite the presence of various targets for HCV, the performed molecular docking calculations and molecular dynamics simulations will concentrate on the various critical viral proteins needed for HCV replication. These targets are protease (NS3), RNA-dependent RNA polymerase (NS5B), helicase (NS3), and the NS3–NS4A protein allosteric pocket located at the interface among the protease domain and helicase domain.11
2. Results and discussion
In the present study, investigations on the anti-HCV activity of the total extract and its different fractions were performed according to procedures described in the literature.12,13 The results, demonstrated in (Table 1 and Fig. S1†), showed that the ethyl acetate fraction exhibited the highest anti-HCV activity, with an IC50 value of 12.6 ± 0.05 μg ml−1. Also, the total extract revealed a moderate anti-HCV activity, with an IC50 of 17.9 ± 0.02 μg ml−1. Among the tested fractions, the petroleum ether fraction exhibited the lowest anti-HCV activity, with an IC50 value of 55.9 ± 0.1 μg ml−1, using cyclosporine as a positive control for the inhibition of replication of the hepatitis C virus.| Extract or fraction | IC50 (μg ml−1) |
|---|---|
| Total extract | 17.9 ± 0.02 |
| Petroleum ether fraction | 55.9 ± 0.1 |
| Ethyl acetate fraction | 12.6 ± 0.05 |
In addition, silica gel and sephadex LH 20 column chromatography and different spectroscopic techniques were followed for the chemical investigation of the ethyl acetate fraction. The chemical investigation afforded four known compounds (Fig. 1), a purine analogue, compound 1 (6 mg), which was identified as 1,3,7-trimethylguanine (Fig. S2–S7†), previously isolated from the marine sponge Latrunculia brevis14 and first discovered in the family Spongiidae, the γ-lactone 2 (10 mg), which was identified as 3,5-dihydroxyfuran-2(5H)-one (Fig. S8–S11†) and previously isolated from a soil-derived fungus, Clonostachys rosea,15 the nucleoside 3 (3 mg), which was evidenced to be thymidine (Fig. S12–S19†) and previously isolated from the marine sponge Haliclona sp.16 and 1H-indazole 4 (3 mg) (Fig. S20–S24†), which was earlier reported in the fungus Volvariella bombycina.17 Identification of the isolated compounds was performed using NMR spectroscopic techniques and confirmed by comparison with previously reported data. It is noteworthy that several nucleotide and nucleoside analogues were described as antiviral drugs, among them telbivudine, which was considered as the L-isomer of thymidine and exhibited anti-HBV activity.18 This could explain the high activity of the ethyl acetate fraction.
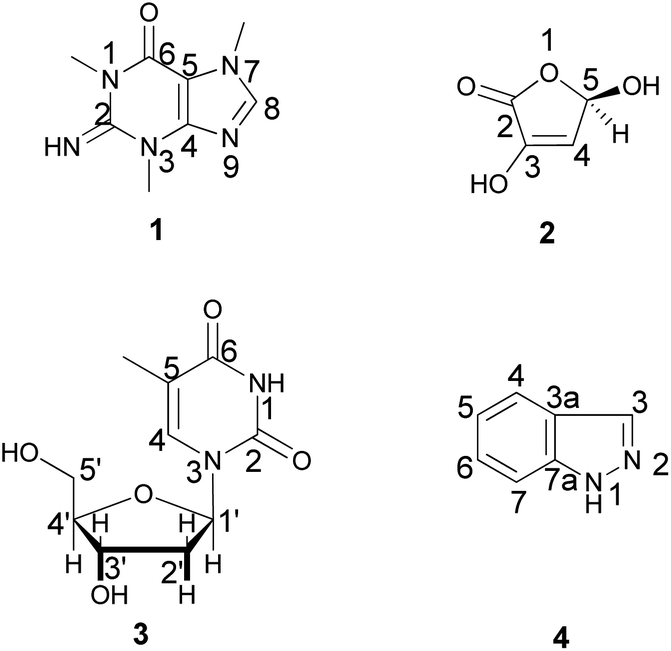 | ||
| Fig. 1 Compounds isolated from the ethyl acetate fraction of S. irregularis. | ||
Metabolomics was considered as a highly valuable tool aiming to rapidly identify natural products of an extract under defined conditions using gas or liquid chromatography combined with HRESIMS or NMR spectroscopic methods.19,20 In accordance with this, HRESIMS metabolomics analysis of the ethyl acetate fraction (Fig. S25†) resulted in the identification of ten compounds, 5–14 (Nakijiquinone F) (Table S1† and Fig. 2).
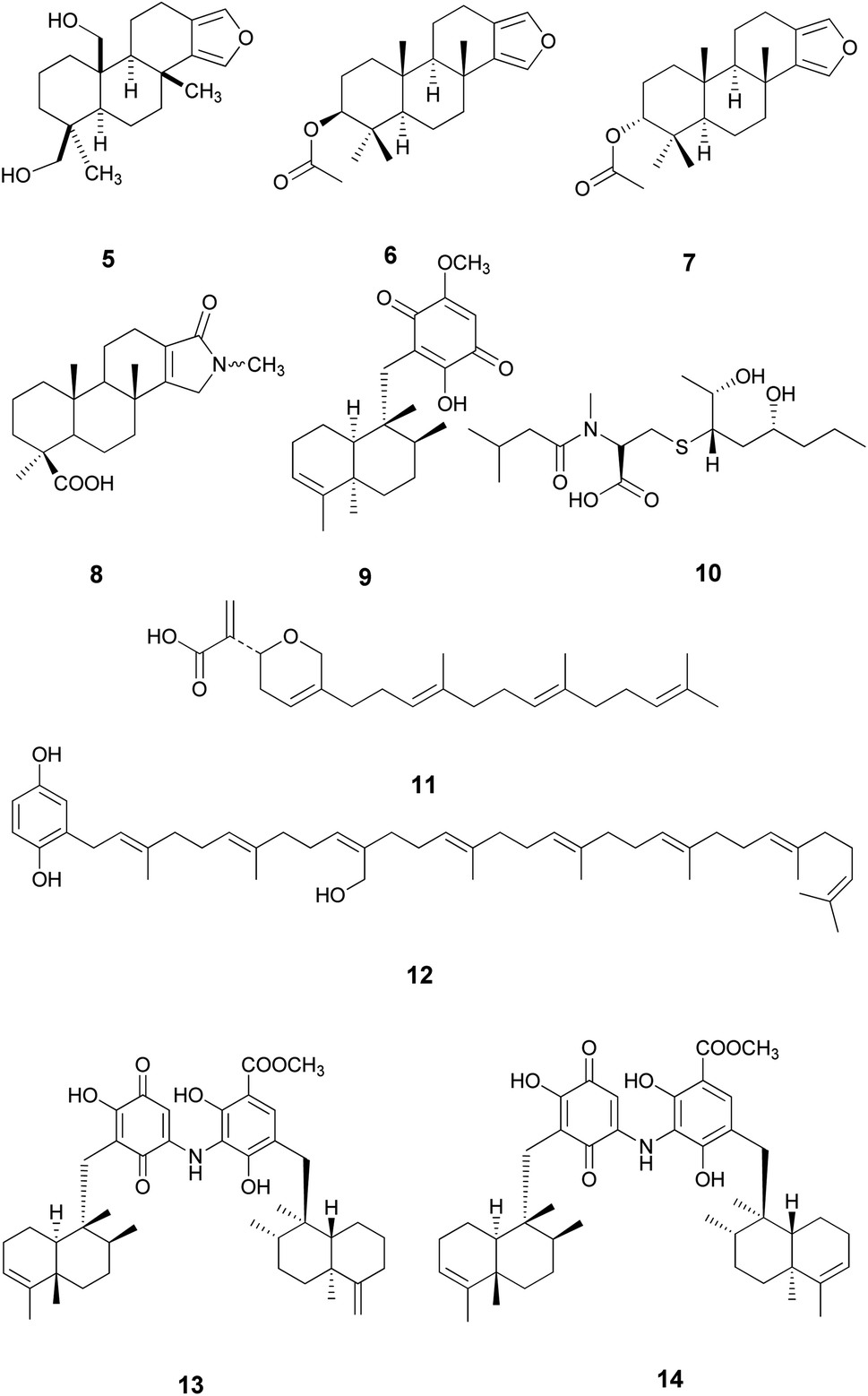 | ||
| Fig. 2 Annotated compounds from the ethyl acetate fraction of S. irregularis. | ||
Additionally, docking studies of both isolated and dereplicated metabolites were carried out to evaluate their binding affinity to different targets for HCV. Prior to the molecular docking analysis for the isolated and annotated compounds, the co-crystallized ligands were first re-docked to validate the performance of the employed technique. Molecular docking calculations were performed against the binding sites of protease (NS3, PDB ID: 6NZT), HCV RNA-dependent RNA polymerase (NS5B, PDB ID: 3H2L), and helicase (NS3, PDB ID: 4OKS), as well as the allosteric pocket in the HCV NS3–NS4A protein (a newly discovered allosteric pocket located at the interface between the protease domain and the helicase domain, PDB ID: 4B73), see Fig. S26(A).† The predicted binding modes and scores of the native ligands are given in (Table 2). According to the predicted binding modes, the employed molecular docking technique predicts the correct binding mode of the native ligands, with RMSD values of 0.7424, 0.4939, 0.3114, and 0.9157 Å, respectively.
| Ligand | Binding affinity ΔG (kcal mol−1) | |||
|---|---|---|---|---|
| Protease (6NZT) | Polymerase (3H2I) | Helicase (4OKS) | Protease–helicase allosteric (4B73) | |
| 1 | −5.1 | −5.50 | −4.9 | −5.44 |
| 2 | −4.1 | −4.5 | −4.1 | −4.28 |
| 3 | −6.0 | −6.3 | −5.5 | −6.3 |
| 4 | −4.2 | −4.3 | −4.2 | −4.8 |
| 5 | −6.1 | −6.2 | −5.6 | −6.0 |
| 6 | −5.8 | −6.6 | −5.6 | −6.7 |
| 7 | −5.7 | −6.2 | −5.6 | −6.1 |
| 8 | −6.0 | −6.1 | −5.7 | −6.0 |
| 9 | −5.5 | −6.5 | −5.8 | −6.4 |
| 10 | −6.4 | −6.9 | −6.5 | −8.2 |
| 11 | −6.4 | −7.5 | −6.4 | −7.8 |
| 12 | −5.6 | −7.3 | −7.1 | −7.2 |
| 13 | −7.0 | −9.5 | −7.3 | −7.8 |
| 14 | −5.8 | −9.3 | −7.4 | −10.1 |
| Co-crystallized ligand | −11.6 (0.7) | −9.6 (0.5) | −7.5 (0.3) | −9.0 (0.9) |
Compound 13, previously isolated from Spongia sp.,21 showed the best binding affinity (−7.0 kcal mol−1), among the docked compounds, to the NS3 protease active site, whereas the native ligand showed a binding affinity of −11.6 kcal mol−1 (Table S2†). Unfortunately, compound 13 did not show any interactions within the protease active site, indicating that our screened compounds are less likely to target NS3 protease active site Fig. 3 and S26(B)†.
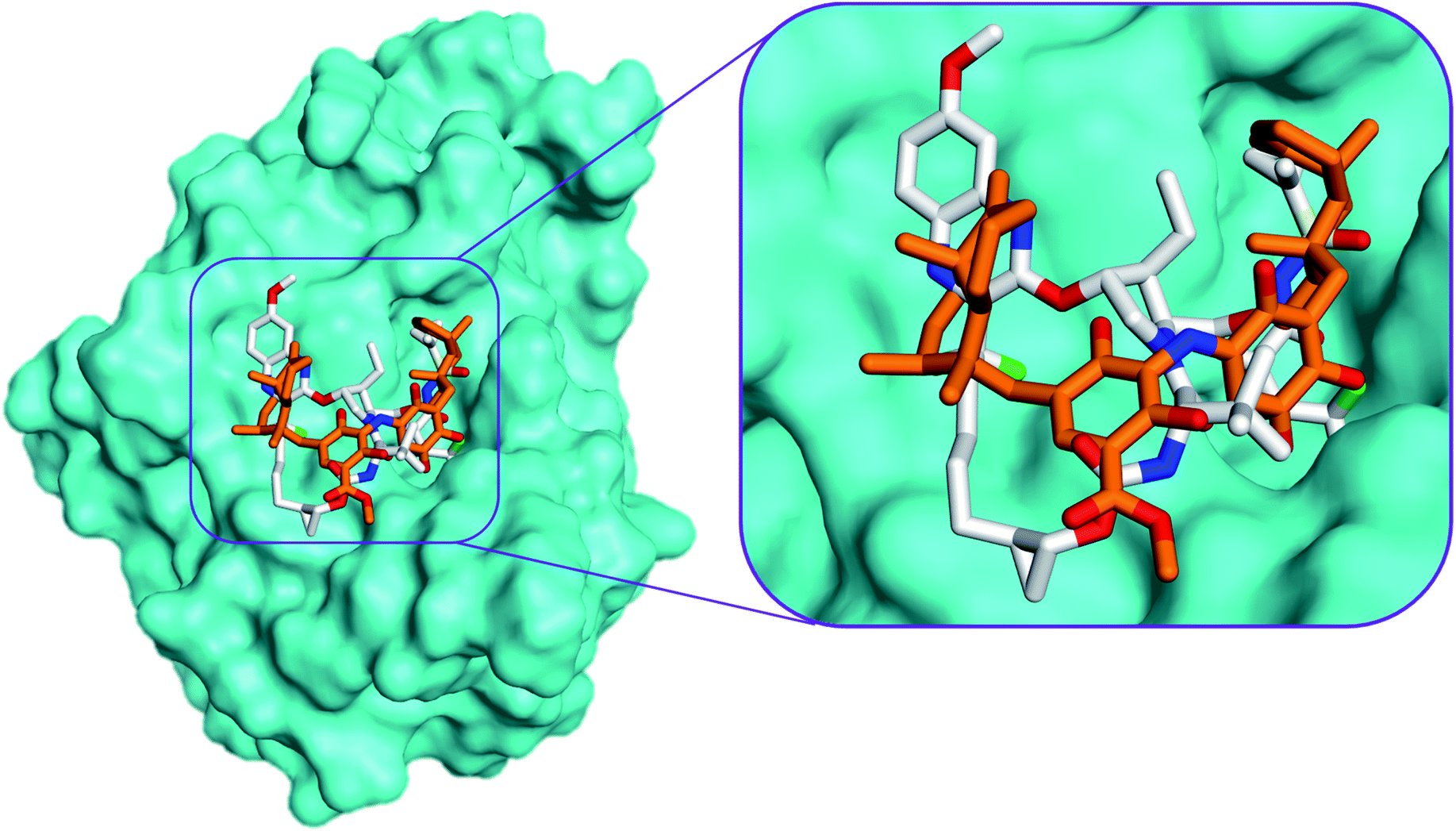 | ||
| Fig. 3 Comparison of the modelled binding mode of the co-crystallized ligand voxilaprevir (white stick model) and the top-scoring docked pose of compound 13 (orange stick model) to the HCV NS3 protease active site (PDB code 6NZT) as predicted by MOE 2019.01. | ||
Compound 13 showed the highest binding affinity to the active site of NS5B HCV RNA-dependent RNA polymerase (−9.5 kcal mol−1) compared to the screened compounds and was comparable to the native ligand (9.6 kcal mol−1), (Table S3†). In addition, 13 exhibited a substantial ability to bind within the palm I region, showing a hydrogen bond between the hydroxy group and Cys 366 amino acid, with a distance of 2.61 Å, whereas the quinone oxygen formed a hydrogen bond with the key amino acid Ser 367, with a distance of 2.85 Å (Fig. 4 and S27†). These essential interactions may account for the influential binding affinity of 13 to the RNA polymerase active site.
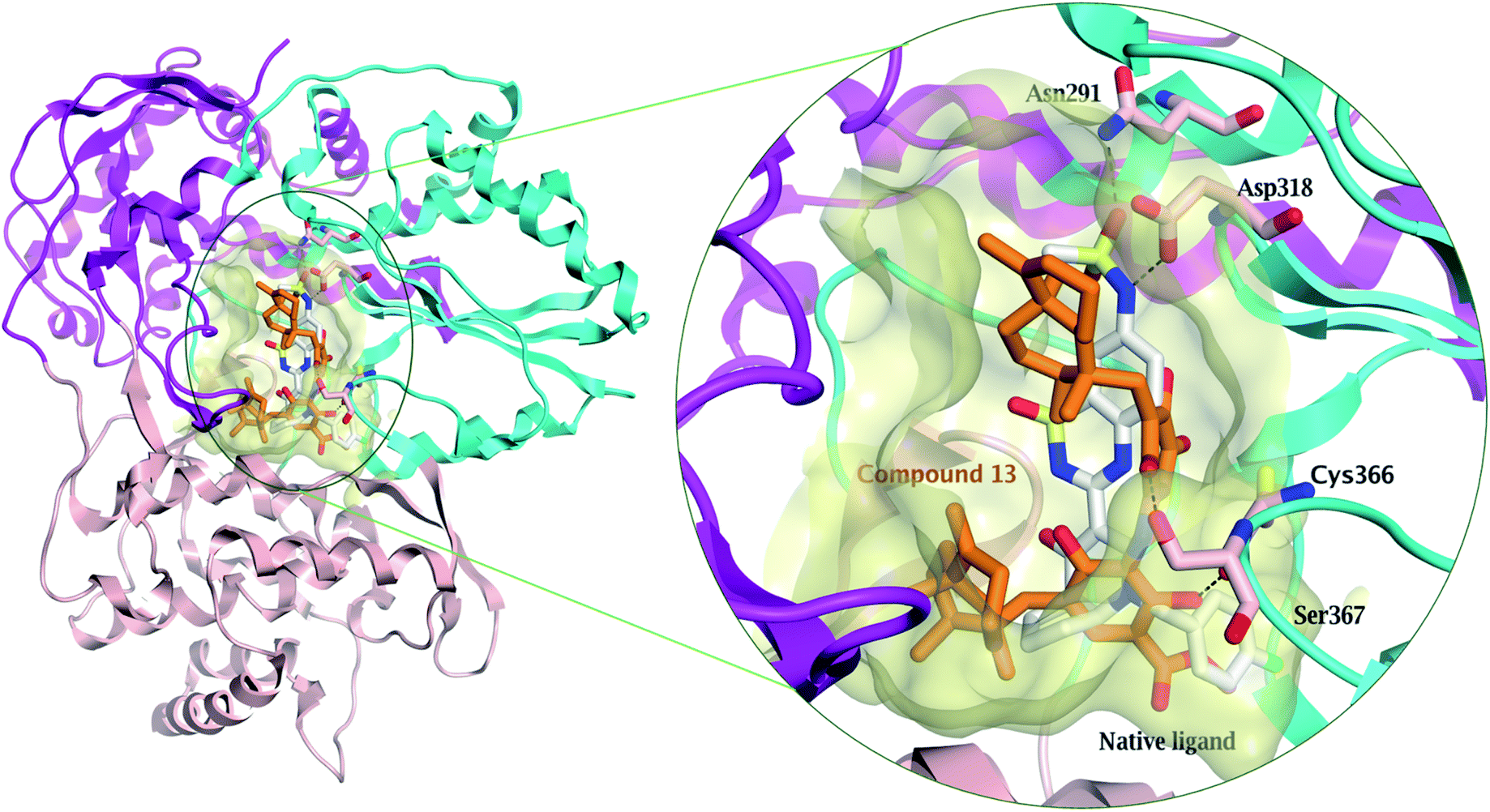 | ||
| Fig. 4 Comparison of the modelled binding mode of the native ligand (a novel bicyclic dihydropyridinone inhibitor) (white stick model) and the top-scoring docked pose of compound 13 (orange stick model) to the HCV NS5B polymerase active site (PDB code: 3H2L) as predicted by MOE 2019.01. Hydrogen bonds are shown as black dashed lines. | ||
Compound 14, previously isolated from Spongia sp.,21 showed a good binding to the NS3 helicase active site and showed a comparable binding affinity to that of the native ligand (−7.4 and −7.5 kcal mol−1, respectively) (Table S4†). Interestingly, the key amino acid Trp 501 formed one hydrogen bond with the carboxylate oxygen of 14 and three H–π interactions (Fig. 5 and S28†). Moreover, the methyl protons in the ester moiety of 14 showed a hydrogen bond with the carbonyl oxygen of Asn 556. Preferentially, the hydrophobic octahydronaphthalenyl methyl moiety penetrated deeply within the lipophilic pocket's active site, which might contribute to the strong affinity of compound 14 to the NS3 helicase active site.
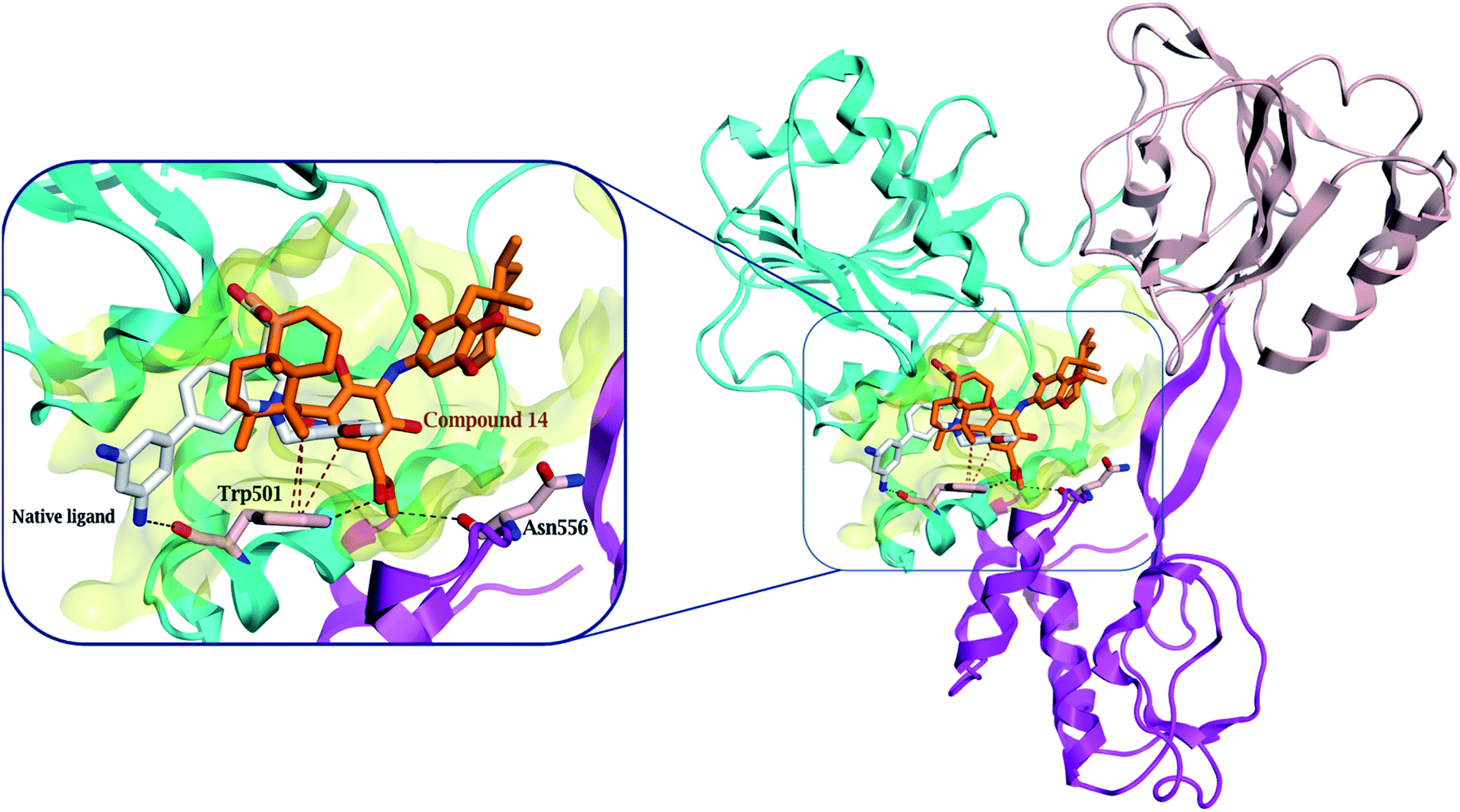 | ||
| Fig. 5 Comparison of the modelled binding mode of the native ligand (white stick model) and the top-scoring docked pose of compound 14 (orange stick model) to the HCV NS3 helicase active site (PDB code 4OKS), as predicted by MOE 2019.01. Hydrogen bonds and H–π interactions are shown as black and red dashed lines, respectively. | ||
Surprisingly, compound 14 displayed a better binding affinity than the native ligand and the screened compounds to HCV NS3–NS4A protein allosteric site (−10.1 kcal mol−1) (Table S5†). Compound 14 fitted perfectly within the allosteric active site and formed two essential hydrogen bonds with Asp 79 and Glu 628 amino acids residues (Fig. 6 and S29†). Besides, the quinone six-membered ring showed an H–π interaction with Val 524 amino acid. Consequently, the HCV NS3-NS4A allosteric site could be claimed as a plausible target for compound 14.
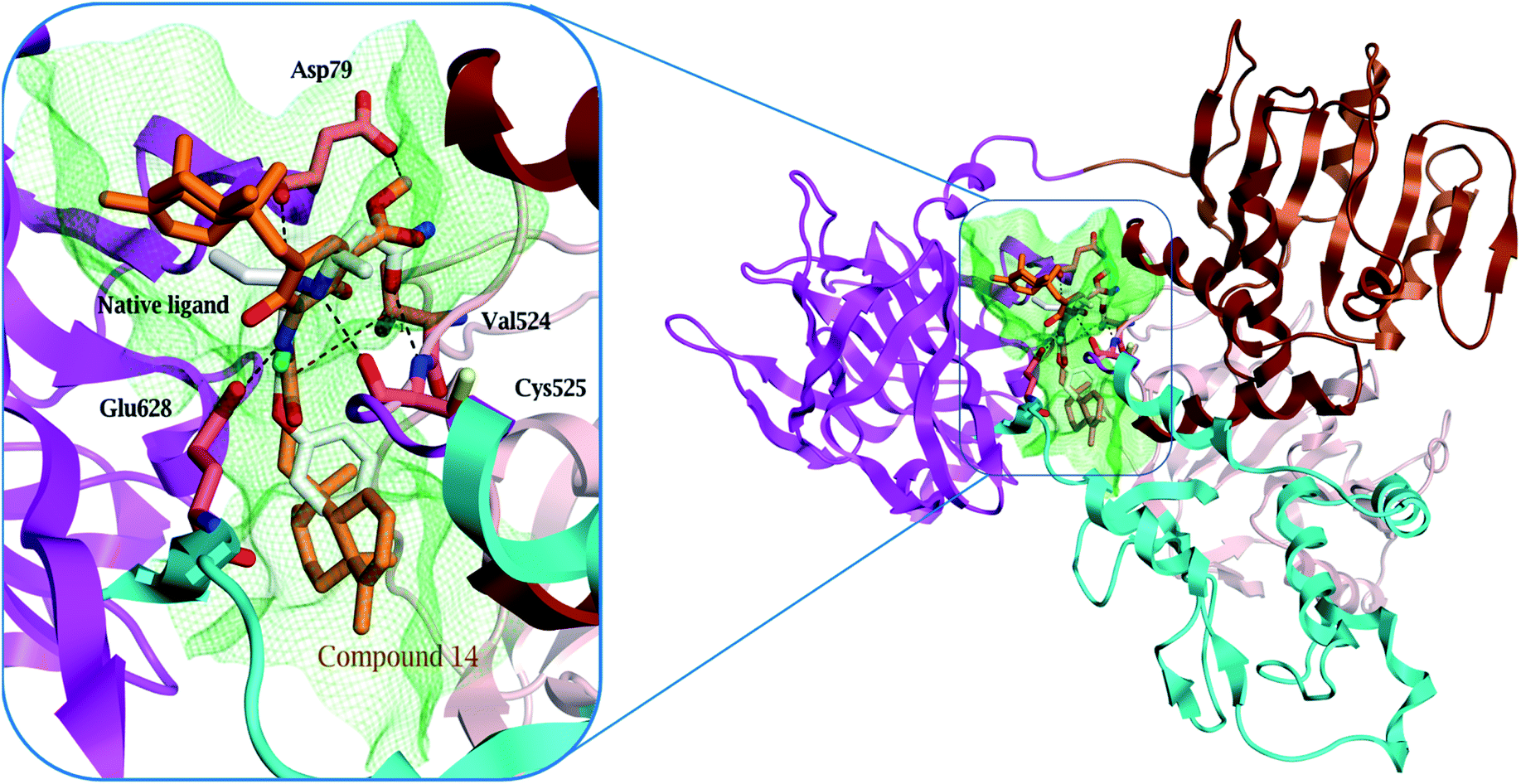 | ||
| Fig. 6 Comparison of modelled binding mode of the native ligand (white stick model) and the top-scoring docked pose of compound 14 (orange stick model) to the allosteric site on the HCV NS3–NS4A protein, located between the protease and helicase domains of the HCV NS3 protein (PDB code: 4B73) as predicted by MOE 2019.01. Hydrogen bonds and H–π interactions are shown as black and red dashed lines, respectively. | ||
According to the above-mentioned docking analyses, compound 14 showed excellent binding affinities and interactions within NS3 helicase active site and the allosteric site in the HCV NS3–NS4 protein. Because of this reason, compound 14 is the most likely anti-HCV candidate among the screened compounds isolated from S. irregularis.
To evaluate the binding affinity and stability of compounds 13 and 14 in complex with HCV targets, molecular dynamics (MD) simulations were executed throughout 25 ns. The binding energies (ΔGbinding) were estimated using the MM-GBSA approach and tabulated in Table 3. According to the estimated MM-GBSA binding energies (Table 3), compound 14 revealed superior binding affinity towards NS3-NS4A allosteric site with a ΔGbinding value of −66.4 kcal mol−1, compared to the native ligand 4AV (−43.2 kcal mol−1). Compound 14 also demonstrated a good binding affinity against HCV helicase with ΔGbinding similar to the co-crystallized 2T9 ligand (−41.6 and −44.1 kcal mol−1, respectively). Towards HCV polymerase, compound 13, compound 14, and the native ligand YAK showed robust binding affinities (ΔGbinding) with values of −45.4, −42.5, and −40.6 kcal mol−1, respectively (Table 3).
| Compound name | Docking score (kcal mol−1) | MM-GBSA binding energy (ΔGbinding) (kcal mol−1) |
|---|---|---|
| Protease | ||
| 13 | −7.0 | −30.0 |
| 14 | −5.8 | −7.4 |
| LGP | −11.6 | −44.1 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Polymerase | ||
| 13 | −9.5 | −45.4 |
| 14 | −9.3 | −42.5 |
| YAK | −9.6 | −40.6 |
|
||
| Helicase | ||
| 13 | −7.3 | −23.7 |
| 14 | −7.4 | −41.6 |
| 2T9 | −7.5 | −30.9 |
|
||
| Allosteric | ||
| 13 | −7.8 | −32.1 |
| 14 | −10.1 | −66.4 |
| 4VA | −9.0 | −43.2 |
Decomposition of the binding energy was performed to reveal the nature of the dominant ligand–target interactions (Fig. 7). According to data presented in Fig. 7, Evdw interactions were dominant forces in the binding of compound 13, compound 14, and co-crystallized inhibitor with the HCV targets, with values up to −74.7 kcal mol−1. Besides, electrostatic interactions (Eele) were a convenient participator for the binding affinities of compound 13, compound 14, and co-crystallized inhibitor with HCV targets, with values up to −56.1 kcal mol−1. Together these results provide quantitative data of the binding affinities of compounds 13 and 14 as potential anti-HCV drug candidates.
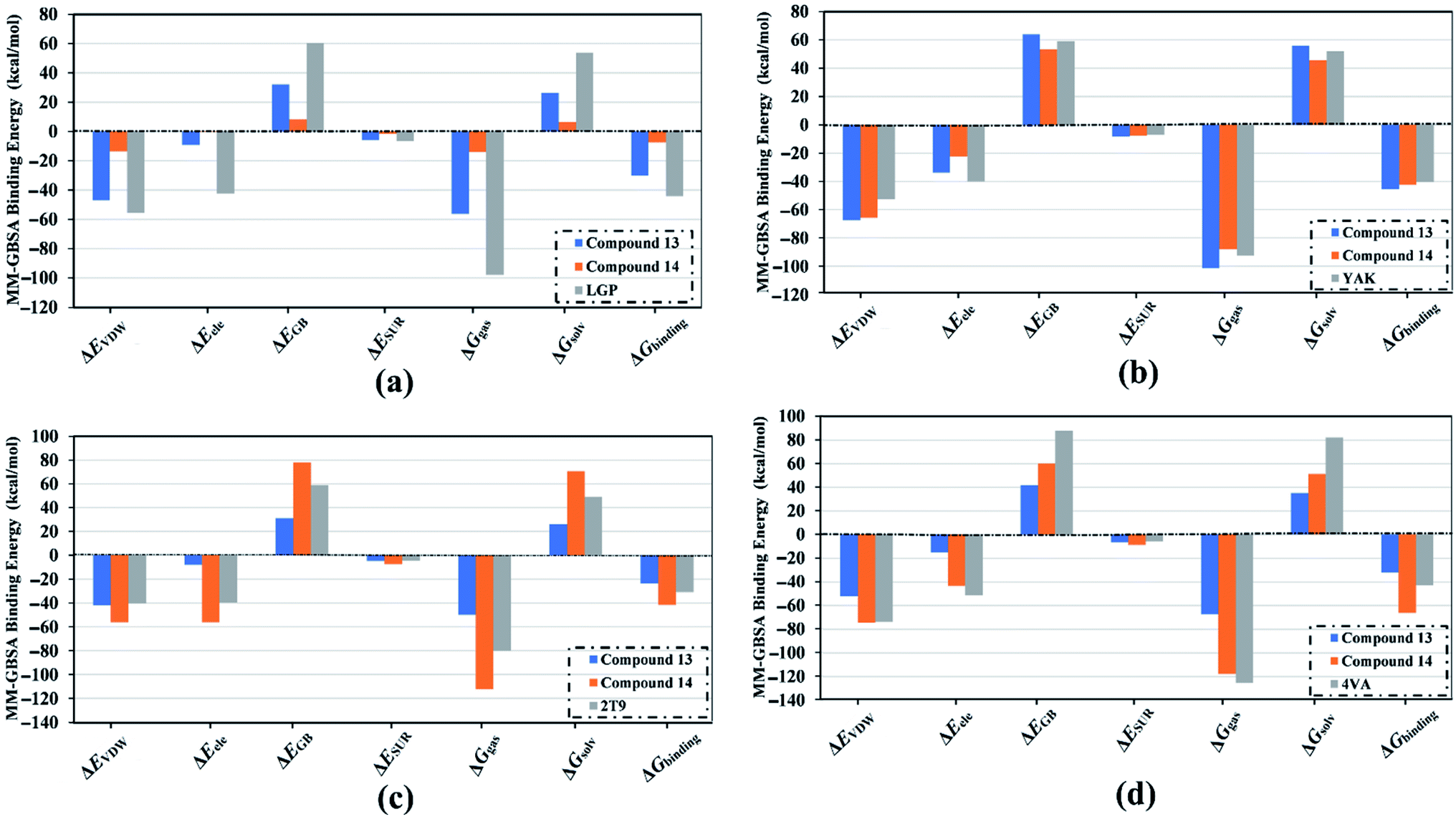 | ||
| Fig. 7 Components of the MM-GBSA binding energies for the investigated inhibitors in complex with (a) HCV NS3 protease, (b) HCV NS5B polymerase, (c) HCV NS3 helicase, and (d) allosteric site on the HCV NS3–NS4A protein throughout 25 ns MD simulations. | ||
Post-MD analyses were also carried out to demonstrate the energetical and structural stabilities for the explored compounds complexed with HCV targets. The structural and energetical analyses included root-mean-square deviation (RMSD) and binding energy per-frame.
MM-GBSA binding energy per-frame for compound 13, compound 14, and co-crystallized ligands with HCV targets were inspected over 25 ns MD simulations (Fig. 8). What stands out in Fig. 8 is the overall stabilities for the investigated compounds complexed with HCV targets throughout the MD simulation. These findings disclose good sufficient stabilities of the inhibitor–receptor complexes. The RMSD for the entire complex backbone atoms showed that all inspected complexes fulfilled steadiness in a short time (Fig. 9). The RMSD values of all complexes oscillated about 0.25 nm. These findings emphasize that all scrutinized inhibitors are tightly bonded in the HCV targets and do not impact the comprehensive topology of the HCV targets. Ultimately, these post-MD energetic and structural analyses evinced the great constancy of the studied inhibitor–receptor complexes throughout 25ns MD simulations.
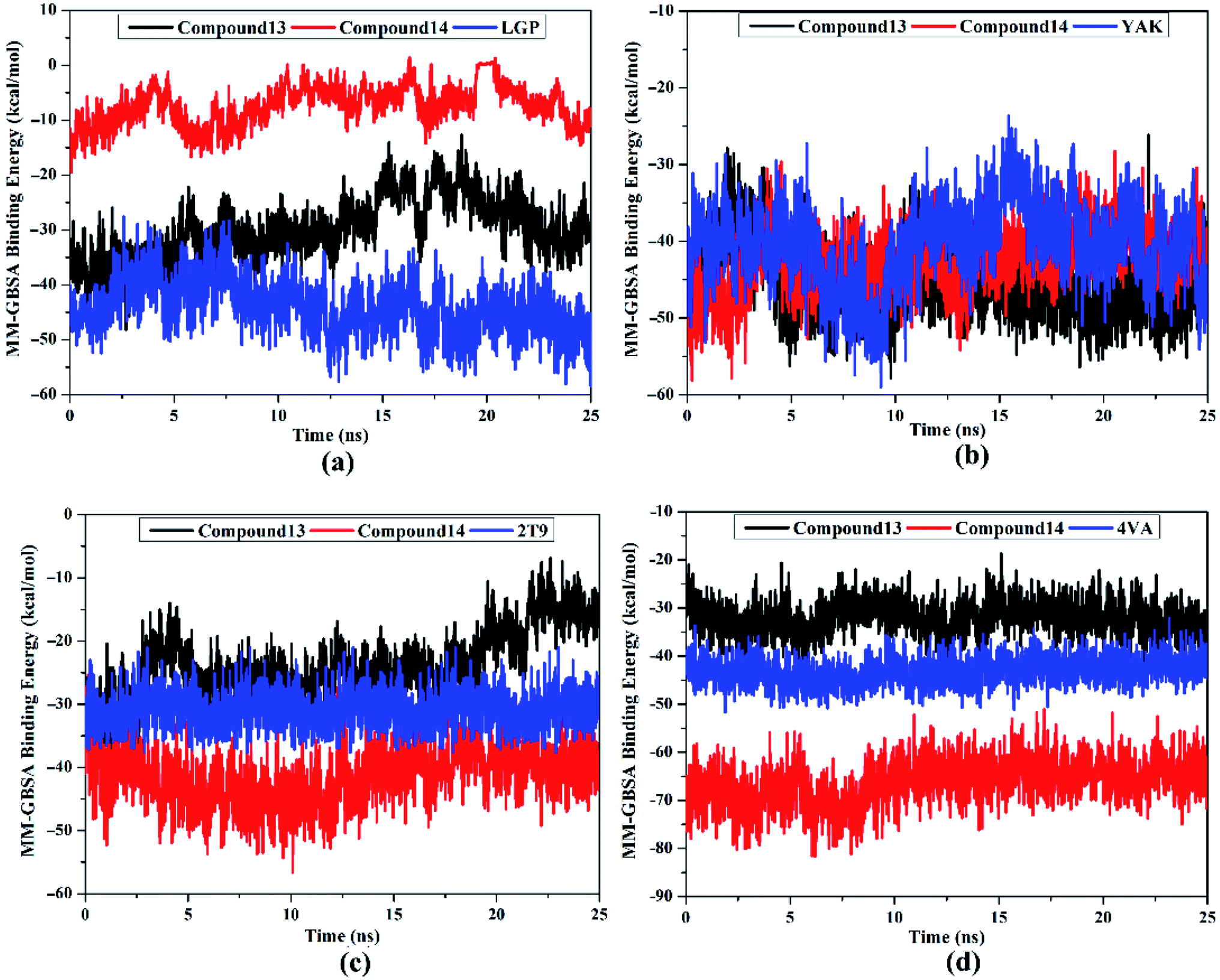 | ||
| Fig. 8 Estimated MM-GBSA binding energy per frame for compounds 13 (in black), 14 (in red), and the native ligand (in blue) in complex with (a) HCV NS3 protease, (b) HCV NS5B polymerase, (c) HCV NS3 helicase, and (d) allosteric site on the HCV NS3–NS4A protein throughout 25 ns MD simulations. | ||
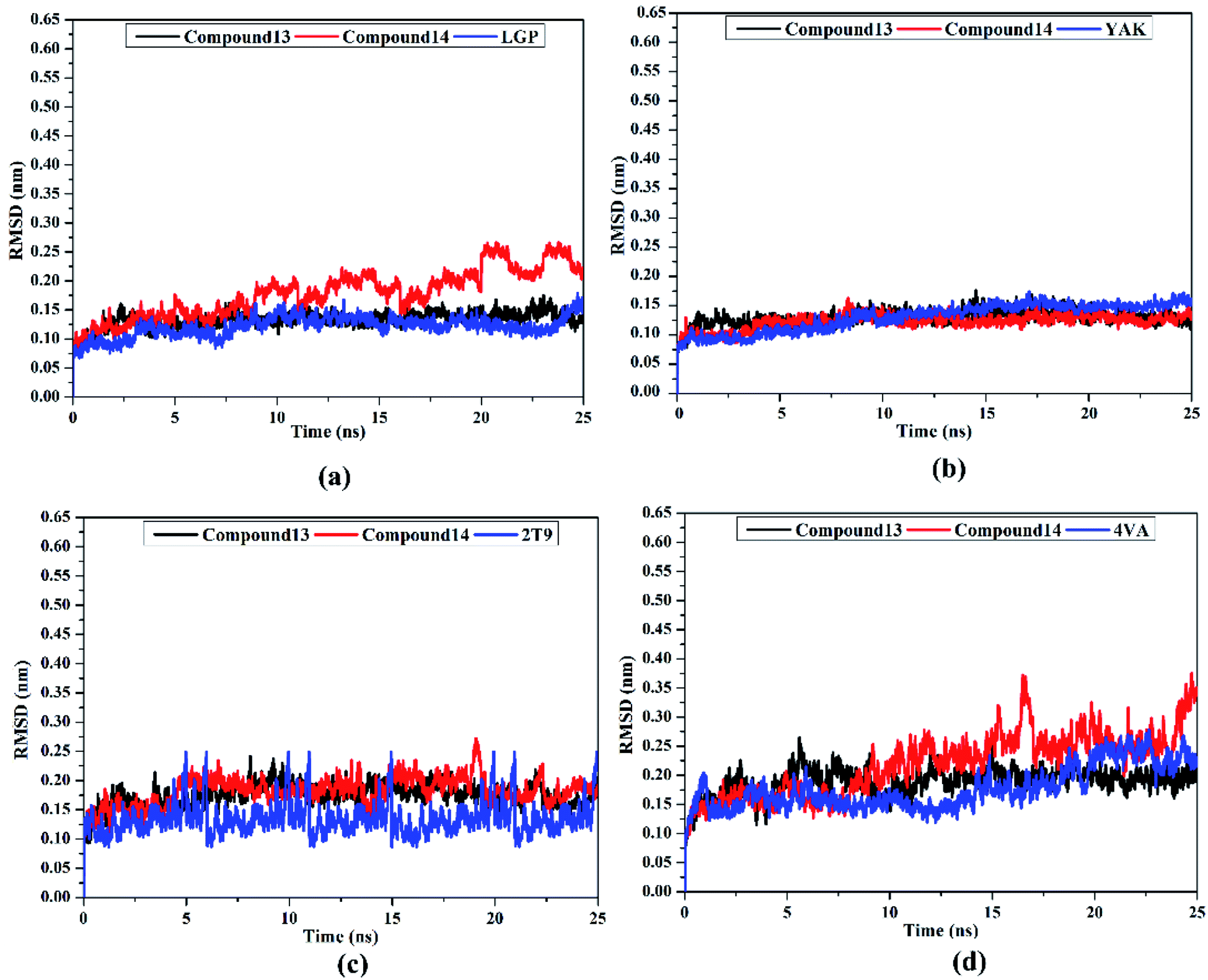 | ||
| Fig. 9 Root-mean-square deviation (RMSD) of the backbone atoms from the initial structure of compounds 13 (in black), 14 (in red), and the native ligand (in blue) in complex with (a) HCV NS3 protease, (b) HCV NS5B polymerase, (c) HCV NS3 helicase, and (d) allosteric site on the HCV NS3–NS4A protein throughout 25 ns MD simulations. | ||
2.1. In silico ADMET and drug-likeness profiling
In the ADME (Absorption, distribution, metabolism and excretion) analyses, no violations toward Lipinski's rule were observed for the investigated compounds, which, therefore possessed agreeable drug-likeness and pharmacokinetic features with no possible toxicity, except for compounds 13 and 14, which showed two violations in the molecular weight and LogP and the low predicted GIT absorption (Tables S6 and S7†). These violations would prompt further optimization of compound 14 to be better absorbed in future in vivo models.
3. Experimental
3.1. Biological material
A sample of the marine sponge Spongia sp. was obtained from the Red Sea (Ras Mohamed, Sinai, Egypt) at a depth of 10 m in august 2008. The sponge sample was identified by R. W. M. van Soest (University of Amsterdam, Netherlands). The sponge biomass was freezed and transported to the laboratory in a plastic bag containing seawater. The sponge body was cut into small pieces and let to dry. A voucher specimen (SAA 5) was deposited in the Department of Pharmacognosy, Faculty of Pharmacy, Suez Canal University, Ismailia, Egypt.3.2. Extraction, fractionation, and isolation
The sponge body of S. irregularis was extracted using 95% ethanol, then the dried ethanolic extract (15.3 g) was further mixed with the smallest-possible amount of distilled H2O to produce a suspension and transferred to a glass separating funnel to be fractionated. Different organic solvents, such as petroleum ether and ethyl acetate (EtOAc), were used for the liquid–liquid fractionation, affording three successive fractions: a petroleum ether fraction (3.8 g), an EtOAc fraction (1 g) and the mother liquor.The ethyl acetate fraction was chromatographed on Sephadex LH-20 (Merck, Bremen, Germany) as a stationary phase, using methanol as the eluting solvent resulting in three subfractions (I, II, and III). Subfraction I mainly contained coloring matter and did not show interesting spots in thin layer chromatography. Subfraction II was further chromatographed on a silica-gel column (gradient elution using dichloromethane-methanol) resulting in the isolation of three compounds 1, 2, and 3. Subfraction III was further chromatographed on Sephadex LH-20, again using methanol as the eluting solvent, resulting in the isolation of compound 4. The isolated compounds were identified using different spectroscopic techniques (Bruker Avance HD 400 MHz and 500 MHz NMR spectrometer (Germany)).
3.3. Evaluation of the antiviral activity
HCV replicon cells (Vero E6 cells) were inoculated at 26 × 104 cells (per well in a 48-well plate) 24 h before the experiment (Reblikon, Mainz, Germany). The total extract of S. irregularis and its fractions were added at different concentrations (1–200 μg ml−1) to the culture broth. After 72 h, cell culture lysis reagent was utilized for the harvesting and lysis of the treated cells. A luciferase assay system was evaluated to measure luciferase activity and the resulting luminescence was detected by a plate reader (PerkinElmer). The result was complementary to the expression level of the HCV replicon.12 The MTS assay was utilized to monitor the cytotoxicity, which could be ascribed to some compounds in the samples13 using cyclosporin as a positive control for the inhibition of replication of the hepatitis C virus. The IC50 values were determined using the software (GraphPad Prism).3.4. LC-HR-MS metabolic analysis
The ethyl acetate fraction of S. irregularis was further subjected to LC-HR-ESI-MS metabolomics analysis using Accela HPLC (Thermo Fisher Scientific, Bremen, Germany) according to a procedure described earlier.19,20,22 The injected volume was 10 μl and the temperature of column was adjusted at 20 °C. The sample was exposed to a gradient chromatographic separation technique at a rate of 300 μl min−1 using purified water [total organic carbon (TOC) was 20 ppb] and acetonitrile, each containing 0.1% formic acid. After that, the elution was carried out with 10% acetonitrile, and then the polarity was gradually increased to 100% acetonitrile within 30 min. This step was followed by an isocratic elution for 5 min preceding a gradient decrease to 10% acetonitrile for 1 min. The HR-ESI-MS results were supplied in both negative and positive ionization modes. The raw data were analyzed using the MZmine 2.12 software to achieve better identification of compounds.23 The detailed steps are illustrated in the ESI Data.†3.5. Molecular docking calculation
Four X-ray crystal structures (PDB codes: 6NZT, 3H2L, 4OKS, 4B73) were retrieved from the Protein Data Bank using the Molecular Operating Environment software (MOE 2019.01, Chemical Computing Group, Montreal, QC, Canada),24–27. These co-crystal protein structures represent the most crucial four non-structural HCV protein targets with their native ligands binding within the active site. The QuickPrep module in the MOE software was used to prepare all the protein structures used in this docking study28,29. Using the MOE build suite, 14 dereplicated chemical structures were drawn, and then energy-minimized using the MOE default force field. Non-essential ligands and water were removed before docking. The native ligand in each co-crystal protein was taken as a center for the docking calculation grid, where the MOE induced-fit protocol was used.30 The generated docking poses were ranked according to their binding free energy and visualized using the MOE software.3.6. Molecular dynamics simulations
Molecular dynamics (MD) simulations were conducted for the most potent recognized compounds and the co-crystallized inhibitors complexed with the most crucial four non-structural HCV protein targets using AMBER16 software.31 The details of the utilized MD simulations are elucidated in ref. 32–35. In the MD simulations, AMBER force field 14SB and general AMBER force field (GAFF2) were employed to characterize the HCV targets and the investigated inhibitors, respectively.36,37 The docked structures of the investigated inhibitors in complex with the HCV targets were neutralized by inserting appropriate counterions. Additionally, all investigated systems were solvated in a truncated octahedron box of TIP3P water molecules with an average distance of 12 Å. Solvated complexes were minimized using 5000 steps and thereafter gently heated from 0 K to 300 K during a brief interval (50 ps). All investigated complexes were simulated for 1 ns equilibration and 25 ns of production with the assistance of periodic boundary conditions and NPT ensemble. Molecular mechanic generalized Born surface area (MM-GBSA) approach was applied to compute binding free energies of the investigated inhibitors complexed with HCV targets. The MM-GBSA (ΔGbinding) binding energies were calculated based on the uncorrelated snapshots collected throughout the MD simulations. All MD simulations were executed using a GPU version of pmemd (pmemd.cuda) implemented inside AMBER16 software on the CompChem hybrid GPU/CPU cluster (http://hpc.compchem.net).3.7. In silico ADMET and drug-likeness profiling
The online SwissADME database was used to predict the pharmacokinetic properties and medicinal-chemistry parameters.384. Conclusion
Marine organisms constitute a rich reservoir of novel and new potent bioactive metabolites with a broad variety of chemical structures and promising biological activities. Bioassay guided study of the marine sponge Spongia irregularis demonstrated a significant anti-HCV potential of the ethyl acetate fraction, with an IC50 value of 12.6 ± 0.05 μg ml−1. Additionally, chemical investigation of this fraction afforded four isolated compounds, which were identified using different spectroscopic methods. Another ten compounds were identified by metabolomic analysis using HRESIMS. Molecular docking calculations revealed that compound 14 (Nakijiquinone F)21 showed the best binding affinity to the active site of the NS3 helicase (−7.4 kcal mol−1), approximately the same as the native ligand (−7.5 kcal mol−1). In addition, it exhibited better affinity than the native ligand in the allosteric pocket of the HCV NS3–NS4A protein (−10.1 and −9.0 kcal mol−1, respectively). Thus, compound 14 (Nakijiquinone F) is the most likely anti-HCV candidate among the screened compounds isolated from S. irregularis. The energetic and structural analyses throughout 25 ns MD simulations pointed up high stability for the investigated inhibitors complexed with HCV multitargets. Clinical investigations are planned to further explicate the role of these compounds as putative drug candidates against HCV multitargets.Author contributions
U. R. A. and S. Y. D. planned the outlines of our work. S. A. A collected the sponge material and carried out the extraction step. E. R. A. carried out the fractionation, the chromatographic isolation of compounds and the anti HCV investigations. E. R. A. and G. B. carried out the spectroscopic analysis. E. R. A., M. N. S. and U. R. A. identified both isolation and dereplicated compounds. T. F. A., M. A. A. I. and M. M. carried out the molecular docking and molecular dynamics calculations. E. R. A., T. F. A. and M. N. S. wrote the manuscript. U. R. A. and S. Y. D. discussed the results scientifically and contributed to the editing of paper.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank Minia University for supporting this work.References
- O. V. Kalinina and A. V. Dmitriev, Mol. Genet. Microbiol. Virol., 2015, 30, 64–70 CrossRef.
- G. Lauer and B. Walker, N. Engl. J. Med., 2001, 345, 41–52 CrossRef CAS PubMed.
- E. M. El-Ghitany, j. high inst. public health., 2019, 49, 1–9 CrossRef.
- V. Gogineni, R. F. Schinazi and M. T. Hamann, Chem. Rev., 2015, 115, 9655–9706 CrossRef CAS PubMed.
- S. Z. Moghadamtousi, S. Nikzad, H. A. Kadir, S. Abubakar and K. Zandi, Mar. Drugs, 2015, 13, 4520–4538 CrossRef CAS PubMed.
- P. Máximo, L. M. Ferreira, P. Branco, P. Lima and A. Lourenço, Mar. Drugs, 2016, 14, 139–209 CrossRef PubMed.
- E. R. Abdelaleem, M. N. Samy, S. Y. Desoukey, M. Liu, R. J. Quinn and U. R. Abdelmohsen, RSC Adv., 2020, 10, 34959–34976 RSC.
- S. Kohmoto, O. J. McConnell, A. Wright and S. Cross, Chem. Lett., 1987, 16, 1687–1690 CrossRef.
- G.-Y. Han, D.-Y. Sun, L.-F. Liang, L.-G. Yao, K.-X. Chen and Y.-W. Guo, Fitoterapia, 2018, 127, 159–165 CrossRef CAS PubMed.
- P. Ahmadi, T. Haruyama, N. Kobayashi, N. J. de Voogd and J. Tanaka, Chem. Pharm. Bull., 2017, 65, 874–877 CrossRef CAS PubMed.
- A. A. Elgoud Said, A. H. Afifi, T. F. S. Ali, M. N. Samy, U. R. Abdelmohsen, M. A. Fouad and E. Z. Attia, RSC Adv., 2021, 11, 32740–32749 RSC.
- D. C. Montefiori, Curr. Protoc. Immunol., 2004, 64, 1–17 CrossRef PubMed.
- D.-R. Hwang, Y.-C. Tsai, J.-C. Lee, K.-K. Huang, R.-K. Lin, C.-H. Ho, J.-M. Chiou, Y.-T. Lin, J. T. A. Hsu and C.-T. Yeh, Antimicrob. Agents Chemother., 2004, 48, 2876–2882 CrossRef CAS PubMed.
- N. B. Perry, J. W. Blunt and M. H. G. Munro, J. Nat. Prod., 1987, 50, 307–308 CrossRef CAS.
- N. M. Abdel-Wahab, H. Harwoko, W. E. G. Müller, A. Hamacher, M. U. Kassack, M. A. Fouad, M. S. Kamel, W. Lin, W. Ebrahim, Z. Liu and P. Proksch, Bioorg. Med. Chem., 2019, 27, 3954–3959 CrossRef CAS PubMed.
- B. Wang, J. Dong, X. Zhou, K. J. Lee, R. Huang, S. Zhang and Y. Liu, Z. Naturforsch., C: J. Biosci., 2009, 64, 143–148 CrossRef CAS PubMed.
- G. H. Xu, S. J. Choo, Y. H. Kim, I. J. Ryoo, S. J. Seok, J. S. Ahn and I. D. Yoo, J. Microbiol. Biotechnol., 2010, 20, 78–81 CrossRef CAS PubMed.
- J. Fung, C. L. Lai, W. K. Seto and M. F. Yuen, J. Antimicrob. Chemother., 2011, 66, 2715–2725 CrossRef CAS PubMed.
- L. Macintyre, T. Zhang, C. Viegelmann, I. J. Martinez, C. Cheng, C. Dowdells, U. R. Abdelmohsen, C. Gernert, U. Hentschel and R. Edrada-Ebel, Mar. Drugs, 2014, 12, 3416–3448 CrossRef PubMed.
- K. A. Youssif, A. M. Elshamy, M. A. Rabeh, N. Gabr, W. M. Afifi, M. A. Salem, A. Albohy, U. R. Abdelmohsen and E. G. Haggag, ChemistrySelect, 2020, 5, 12278–12286 CrossRef CAS.
- Y. Takahashi, T. Kubota and J. i. Kobayashi, Bioorg. Med. Chem., 2009, 17, 2185–2188 CrossRef CAS PubMed.
- Y. Elsayed, J. Refaat, U. R. Abdelmohsen, E. M. Othman, H. Stopper and M. A. Fouad, Phytochem. Anal., 2018, 29, 543–548 CrossRef CAS PubMed.
- K. A. Youssif, E. G. Haggag, A. M. Elshamy, M. A. Rabeh, N. M. Gabr, A. Seleem, M. A. Salem, A. S. Hussein, M. Krischke, M. J. Mueller and U. R. Abdelmohsen, PLoS One, 2019, 14, 1–19 CrossRef PubMed.
- J. G. Taylor, S. Zipfel, K. Ramey, R. Vivian, A. Schrier, K. K. Karki, A. Katana, D. Kato, T. Kobayashi, R. Martinez, M. Sangi, D. Siegel, C. V. Tran, Z. Y. Yang, J. Zablocki, C. Y. Yang, Y. Wang, K. Wang, K. Chan, O. Barauskas, G. Cheng, D. Jin, B. E. Schultz, T. Appleby, A. G. Villasenor and J. O. Link, Bioorg. Med. Chem. Lett., 2019, 29, 2428–2436 CrossRef CAS PubMed.
- F. Ruebsam, D. E. Murphy, C. V. Tran, L. S. Li, J. Zhao, P. S. Dragovich, H. M. McGuire, A. X. Xiang, Z. Sun, B. K. Ayida, J. K. Blazel, S. H. Kim, Y. Zhou, Q. Han, C. R. Kissinger, S. E. Webber, R. E. Showalter, A. M. Shah, M. Tsan, R. A. Patel, P. A. Thompson, L. A. Lebrun, H. J. Hou, R. Kamran, M. V. Sergeeva, D. M. Bartkowski, T. G. Nolan, D. A. Norris, J. Khandurina, J. Brooks, E. Okamoto and L. Kirkovsky, Bioorg. Med. Chem. Lett., 2009, 19, 6404–6412 CrossRef CAS PubMed.
- S. R. LaPlante, A. K. Padyana, A. Abeywardane, P. Bonneau, M. Cartier, R. Coulombe, A. Jakalian, J. Wildeson-Jones, X. Li, S. Liang, G. McKercher, P. White, Q. Zhang and S. J. Taylor, J. Med. Chem., 2014, 57, 2074–2090 CrossRef CAS PubMed.
- S. M. Saalau-Bethell, A. J. Woodhead, G. Chessari, M. G. Carr, J. Coyle, B. Graham, S. D. Hiscock, C. W. Murray, P. Pathuri, S. J. Rich, C. J. Richardson, P. A. Williams and H. Jhoti, Nat. Chem. Biol., 2012, 8, 920–925 CrossRef CAS PubMed.
- H. I. Ciftci, M. O. Radwan, S. E. Ozturk, N. G. Ulusoy, E. Sozer, D. E. Ellakwa, Z. Ocak, M. Can, T. F. S. Ali, H. I. Abd-Alla, N. Yayli, H. Tateishi, M. Otsuka and M. Fujita, Molecules, 2019, 24, 1–15 CrossRef PubMed.
- M. O. Radwan, H. I. Ciftci, T. F. S. Ali, D. E. Ellakwa, R. Koga, H. Tateishi, A. Nakata, A. Ito, M. Yoshida, Y. Okamoto, M. Fujita and M. Otsuka, Molecules, 2019, 24, 1–17 CrossRef PubMed.
- M. O. Radwan, D. Takaya, R. Koga, K. Iwamaru, H. Tateishi, T. F. S. Ali, A. Takaori-Kondo, M. Otsuka, T. Honma and M. Fujita, Bioorg. Med. Chem., 2020, 28, 115409 CrossRef CAS PubMed.
- D. A. Case, R. M. Betz, D. S. Cerutti, T. E. Cheatham, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, T. Luchko, R. Luo, B. Madej, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, C. Sagui, C. L. Simmerling, W. M. Botello-Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao, P. A. Kollman, AMBER, 2016 Search PubMed.
- M. A. A. Ibrahim, K. A. A. Abdeljawaad, A. H. M. Abdelrahman, O. R. Alzahrani, F. M. Alshabrmi, E. Khalaf, M. F. Moustafa, F. Alrumaihi, K. S. Allemailem, M. E. S. Soliman, P. W. Pare, M. F. Hegazy and M. A. M. Atia, Antibiotics, 2021, 10, 934 CrossRef CAS PubMed.
- M. A. A. Ibrahim, A. H. M. Abdelrahman, M. A. M. Atia, T. A. Mohamed, M. F. Moustafa, A. R. Hakami, S. A. M. Khalifa, F. A. Alhumaydhi, F. Alrumaihi, S. H. Abidi, K. S. Allemailem, T. Efferth, M. E. Soliman, P. W. Paré, H. R. El-Seedi and M.-E. F. Hegazy, Mar. Drugs, 2021, 19, 391 CrossRef CAS PubMed.
- M. A. A. Ibrahim, E. A. A. Badr, A. H. M. Abdelrahman, N. M. Almansour, G. A. H. Mekhemer, A. M. Shawky, M. F. Moustafa and M. A. M. Atia, Mol. Inf., 2021, 40, 2060039 CrossRef.
- M. A. A. Ibrahim, E. A. A. Badr, A. H. M. Abdelrahman, N. M. Almansour, A. M. Shawky, G. A. H. Mekhemer, F. Alrumaihi, M. F. Moustafa and M. A. M. Atia, Cell Biochem. Biophys., 2021, 79, 189–200 CrossRef CAS.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08321j |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |