
Conformation-dependent antioxidant properties of β-carotene†
L.
Sandhiya‡
 * and
H.
Zipse
* and
H.
Zipse
Department of Chemistry, LMU München, Germany. E-mail: sandhiya@nistads.res.in
First published on 26th November 2021
Abstract
The antioxidant capacity of β-carotene has been studied in terms of H-atom abstraction reactions using quantum chemical methods. These oxidation reactions are studied for the all-trans as well as 15,15′-cis isomers (15Z) of β-carotene, as the latter is only ∼10 kJ mol−1 less stable than the all-trans isomer in the gas phase and about 9 kJ mol−1 less stable in aqueous solution. Hydrogen abstraction from the rotamers obtained through C–C single and double bond rotations has been shown to play an important role in determining the antioxidant capacity of β-carotene. Hydrogen abstraction from the C4 and C5-CH3 positions of the β-ionone rings and the C7 and C9 positions along the polyene chain of β-carotene by the hydroxyl radical have been studied. In the all-trans form the most favorable H-atom abstraction reaction occurs at the C4 position of the terminal regions of the polyene π-system of β-carotene, closely followed by hydrogen abstraction from the C5 methyl position. The H-atom abstraction reactions are more exothermic in water than in the gas phase due to solvation energies for the water product.
I. Introduction
Carotenoids are omnipresent accessory pigments which play an important role in photosynthesis and protect living organisms from photodynamic damage.1–4 β-Carotene is known well for its antioxidant activity and its effectiveness as a quencher of singlet oxygen and an array of reactive free radicals.5–8 Thus, the antioxidant activity of β-carotene is tightly linked to its reaction with free radicals. The most important oxidation mechanisms for carotenoids are H-atom abstraction by free radicals, radical addition to the carotenoid π-system, and electron transfer reactions.9 The significance of these oxidation mechanisms depends on the structure of the carotenoid and the nature of the reacting radical.10 The oxidation of β-carotene by the nitrogen dioxide radical has recently been studied both experimentally10–12 and theoretically.13 The experimental study by Baker et al.11 reported the formation of 4-nitro-β-carotene as the main product. A density functional theory (DFT) study on the solution phase oxidation mechanisms of β-carotene with NO2 supported the experimental study on theoretical grounds by pointing out that the formation of 4-nitro-β-carotene is due to initial H-atom abstraction from the C4 position in β-carotene.13 Another DFT study by Gao et al. reported the deprotonation of the β-carotene radical cation at different C-radical sites of the polyene chain.14The antioxidative reactivity of β-carotene was studied in terms of hydrogen atom transfer reactions between β-carotene and free radicals using various DFT methods.15 This study concluded that the C4 and C4′ positions of β-carotene (Scheme 1) are most reactive. The same group has performed DFT studies on the donor–acceptor capability of carotenoids and described donor–acceptor maps for carotenoids.16 Galano et al. studied the reaction between carotenoids and peroxyl radical in polar and non-polar environments, and concluded that the oxidation reaction proceeds mainly by adduct formation in both environments.17 A theoretical study on the radical chain propagation of carotenoids with a variety of free radicals reported that the oxidation reactions are controlled to a large extent by the nature of the free radical.18 The electron transfer reaction between carotenoids and superoxide anion was studied in lipid media and it was established that the antioxidant capability of carotenoids depends in this case on its ability to prevent the formation of reactive oxygen species (ROS).19 Another theoretical study on electron-transfer reactions between carotenoids and free radicals emphasized the importance of the electron affinity of the free radicals in these processes.20 Earlier experimental studies on β-carotene reveal that, in addition to the all-trans isomer, many stereoisomers of β-carotene exist in biological systems which possess different antioxidant properties.21–23 A recent theoretical study showed that cis carotenoids display largely similar antioxidative properties as compared to the trans isomers.24 Also in photosynthetic systems, the conformation of the carotenoids plays a major role.25 The conformational dependence of the electronic absorption spectra of carotenoids is well investigated.26,27
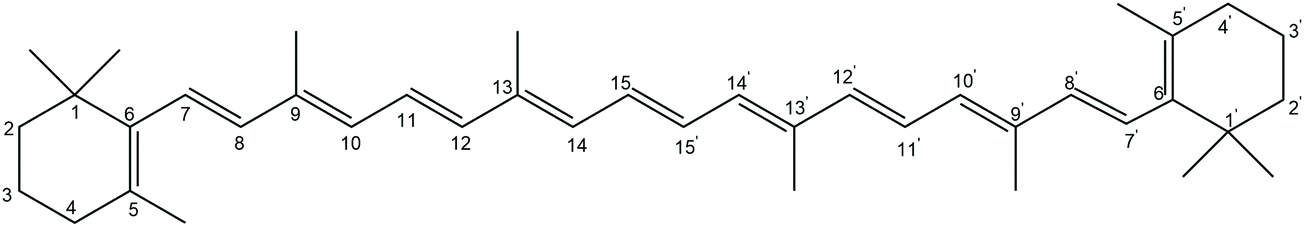 | ||
| Scheme 1 Molecular structure of β-carotene. | ||
Even though a larger number of studies have already analyzed the structures and stabilities of carotenoids and their radicals, some fundamental questions remain unanswered with respect to the reactivity of carotenoids towards free radicals as a measure of their antioxidant capability. In particular, the reactivity of β-carotene in polar media such as water has considerable practical importance. Since different rotamers of β-carotene may have different antioxidative properties, it is furthermore important to properly describe radical reactions at various atomic sites of β-carotene conformational isomers. This is quite essential as only very limited literature information exists with respect to the favourability of oxidation from different sites. The above mentioned exposures prompted us to study the conformational dependent oxidation of β-carotene by hydroxyl radical. The isomers and conformers resulting from C–C and the central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C rotations along the polyene chain of β-carotene are studied and the thermochemistry of the H-atom abstraction from these isomers will be elucidated in detail. Since β-carotene has several potentially reactive sites, understanding the energetics of the oxidation reactions at all of these sites may be required to fully describe the antioxidant properties of β-carotene. The paper is organized as follows: section II describes the computational methodology employed in the work. This is followed by model studies for the evaluation of computational methodology, results and discussion of the structure and energetics of the β-carotene isomers and the H-atom abstraction reactions in section III. The main conclusions arrived from the present work are summarized in section IV.
C rotations along the polyene chain of β-carotene are studied and the thermochemistry of the H-atom abstraction from these isomers will be elucidated in detail. Since β-carotene has several potentially reactive sites, understanding the energetics of the oxidation reactions at all of these sites may be required to fully describe the antioxidant properties of β-carotene. The paper is organized as follows: section II describes the computational methodology employed in the work. This is followed by model studies for the evaluation of computational methodology, results and discussion of the structure and energetics of the β-carotene isomers and the H-atom abstraction reactions in section III. The main conclusions arrived from the present work are summarized in section IV.
II. Computational methodology
A hierarchy of computational methods based on previous studies28,29 and benchmark calculations on model systems are employed to study the structure and energetics of β-carotene isomers and their radicals. The geometries of all systems involved in the oxidation process have been optimized at the (U)B3LYP/6-31G(d) level of theory.30,31 The frequency calculations were performed at the same level of theory and all minima were confirmed with all positive frequencies. Single point energy calculations were then performed at (U)B3LYP level with larger basis sets such as 6-311++G(d,p) and 6-311++G(3df,2p)32,33 in order to assess the magnitude of basis set effects. The thermochemistry has also been studied using the recently developed double hybrid ROB2-PLYP DFT method.34 This method has been shown to predict thermochemical data with a mean absolute deviation from experiment of 9.1 kJ mol−1 when tested against the G3/05 data set. To understand the effect of basis set size on the energetics calculated at ROB2-PLYP level, three different basis sets such as 6-31G(d), 6-311++G(d,p) and 6-311+G(3df,2p) are employed. The energies are calculated for a temperature of 298.15 K in the gas phase and the thermal corrections to the enthalpy and Gibb's free energy are obtained at the (U)B3LYP/6-31G(d) level of theory. For comparison, thermochemical data for the model systems has also been calculated using the G3B3,35 G3(MP2)-RAD,36 and CBS-QB337 composite methods. In order to quantify the effect of solvent on the thermochemistry of the reactions of β-carotene with hydroxyl radical, the polarizable continuum model (PCM)38,39 is employed by performing single point energy calculations at B3LYP/6-311++G(d,p) level of theory on (U)B3LYP/6-31G(d) optimized geometries. The required URCCSD(T) calculations have been performed using MOLPRO 2012.1,40 and all other calculations employed Gaussian09, Rev. D.01.41III. Results and discussion
A. C–H bond activation in unsaturated hydrocarbons
In order to identify suitable quantum chemical methods for studying the oxidation of β-carotene by OH radical, the oxidation of structurally similar polyenes such as propene (2), penta-1,3-diene (5), hepta-1,3,5-triene (7) and toluene (9) has been studied first.The H-atom abstraction by OH radical from the methyl group of the reference systems shown in Scheme 2 generate the respective alkyl radicals along with a water molecule. The thermochemistry of the reactions calculated at different levels of theory have been summarized in Table 1. The thermochemistry of H-atom abstraction reactions from other C-atom sites of the reference molecules are given in ESI.† As can be observed from Table 1, all these H-atom abstraction reactions are thermodynamically favourable and the exothermicity and exergonicity increases with larger polyene chains. In terms of methodology, it is to be noted that reaction energies obtained using the compound methods are in close agreement with the available experimental data. The (U)B3LYP and ROB2-PLYP methods are in line with the experimental results and with those from the compound methods provided that sufficiently large basis sets are used. This is clearly not the case for the 6-31G(d) basis set, which largely underestimates the reaction energies. These findings are also in line with our previous study on the oxidation of hydrocarbons.42 Because of the size of carotenoids, the calculations will in the following be limited to the (U)B3LYP/6-31G(d), (U)B3LYP/6-311++G(d,p) and ROB2-PLYP/6-311+G(d,p) levels of theory. The results obtained at (U)B3LYP/6-311++G(d,p) level of theory are discussed in the following sections.
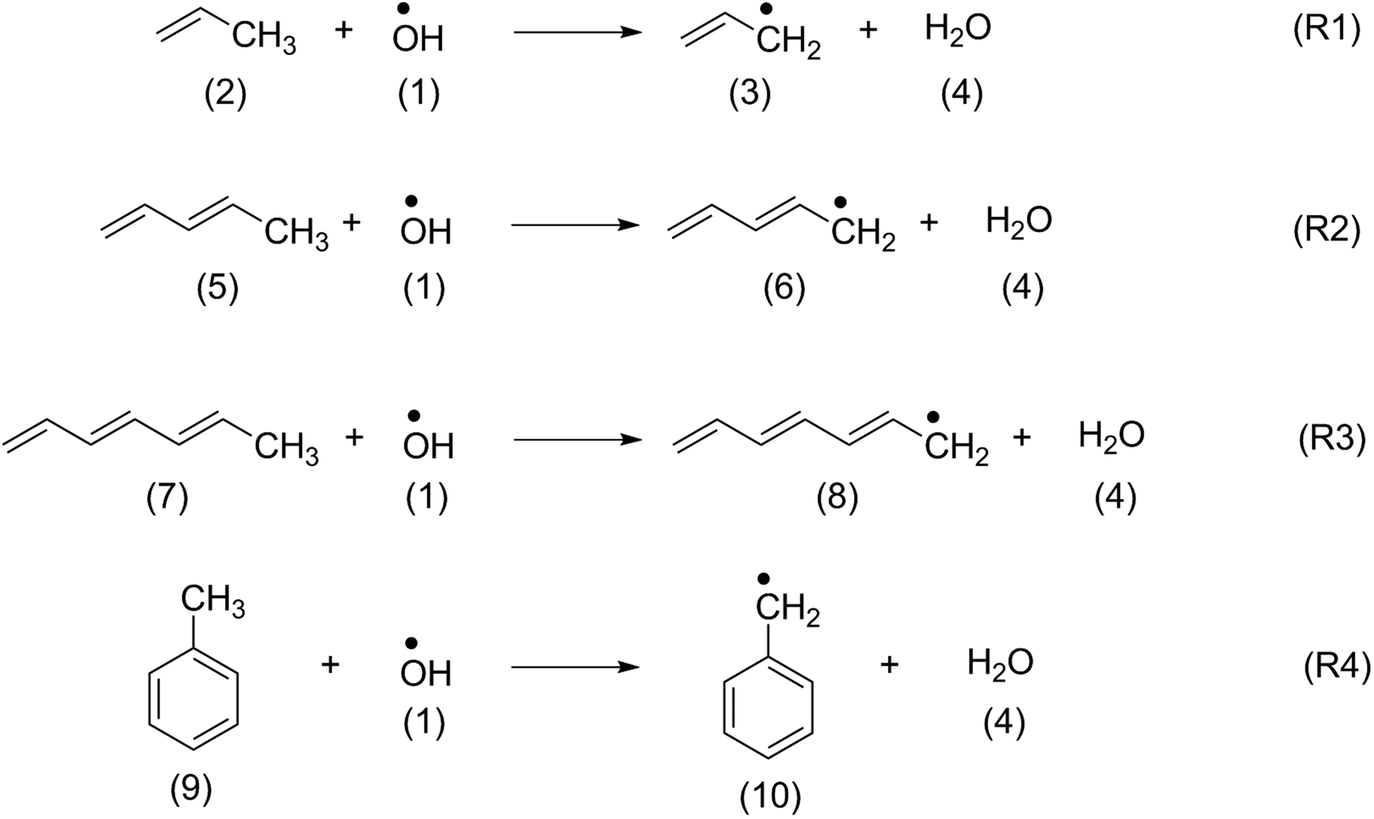 | ||
| Scheme 2 H-Atom abstraction reactions from the methyl group of reference systems. | ||
| Level of theory | Propene (R1) | Penta-1,3-diene (R2) | Hepta-1,3,5-triene (R3) | Toluene (R4) | |||||
|---|---|---|---|---|---|---|---|---|---|
| ΔH298 [kJ mol−1] | ΔG298 [kJ mol−1] | ΔH298 [kJ mol−1] | ΔG298 [kJ mol−1] | ΔH298 [kJ mol−1] | ΔG298 [kJ mol−1] | ΔH298 [kJ mol−1] | ΔG298 [kJ mol−1] | ||
| a Ref. 43. b Ref. 44. c Ref. 50. | |||||||||
| Exp. | −125.6 ± 1.7a | −149.8 ± 12a | −121 ± 5a,b | ||||||
| −128.0 ± 3.0b | |||||||||
| −131.5 ± 0.6c | |||||||||
| Compound methods | CBS-QB3 | −133.5 | −136.3 | −160.1 | −162.5 | −175.4 | −177.5 | −119.3 | −118.2 |
| G3B3 | −128.2 | −131.0 | −149.0 | −151.5 | −157.9 | −160.2 | −112.8 | −111.5 | |
| G3(MP2)-RAD | −130.9 | −133.7 | −152.7 | −155.1 | −165.2 | −167.4 | −120.0 | −118.7 | |
| Double hybrid DFT | ROB2-PLYP/6-311+G(3df,2p) | −129.8 | −132.7 | −152.6 | −155.1 | −165.3 | −167.6 | −118.7 | −117.4 |
| ROB2-PLYP/6-311+G(d,p) | −122.6 | −125.5 | −144.9 | −147.4 | −157.4 | −159.7 | −110.9 | −109.5 | |
| ROB2-PLYP/6-31G(d) | −92.3 | −95.1 | −114.4 | −126.9 | −126.9 | −129.2 | −80.6 | −79.3 | |
| Hybrid-DFT | (U)B3LYP/6-311++G(3df,2p) | −135.2 | −137.0 | −159.4 | −161.9 | −172.6 | −174.9 | −123.0 | −125.7 |
| (U)B3LYP/6-311++G(d,p) | −129.6 | −132.5 | −154.7 | −157.16 | −167.9 | −170.2 | −118.3 | −116.9 | |
| (U)B3LYP/6-31G(d) | −102.3 | −105.2 | −127.4 | −129.85 | −140.6 | −142.9 | −90.8 | −89.4 | |
| (U)MPW1K/6-31G(d,p) | −95.5 | −98.3 | −120.8 | −123.2 | −135.1 | −137.3 | −81.5 | −82.0 | |
B. Energetics and structures of the isomers of β-carotene
In order to understand the relationship between structural and antioxidant properties of β-carotene, the stabilities of different conformational isomers of β-carotene has been identified first. This includes rotations around the formal single and double bonds and the centrosymmetic C15C15′ double bond. As can be seen in the optimized structures of the all-trans (15E) and 15,15′-cis (15Z) isomers of β-carotene in Fig. 1, the end groups of the polyene chain are twisted out of the plane and the methyl groups also lie out of the plane in order to minimize steric crowding of the methyl groups. The structure of the all-trans isomer of β-carotene is in good agreement with the X-ray crystal structure.45 Higher energy di-cis and tri-cis structures resulting from rotations around selected C–C single bonds are also included in the current study. The conformational space along the polyene chain is largely dependent on the twisting of the end groups of β-carotene in addition to rotation around the C–C single and CC double bonds. Accordingly, three different conformations with respect to the end groups are observed. In one of the conformations, the end groups are twisted by about 45–49° out of the plane of the polyene chain and in the second conformation, one of the end rings is out of plane by 45–49° and the other end ring is in plane with the polyene chain with d(5–6–7–8) = ∼170°. In the third conformational space, both the end rings are in plane with the polyene chain. From Fig. 1, it can be seen that, the conformation where the terminal cyclohexenyl groups are twisted by about 45–49° out of the plane of the polyene chain is the most favourable conformation for all the rotamers studied. This compares quite well with the trend in twist angles calculated for similar types of polyenes.46–49
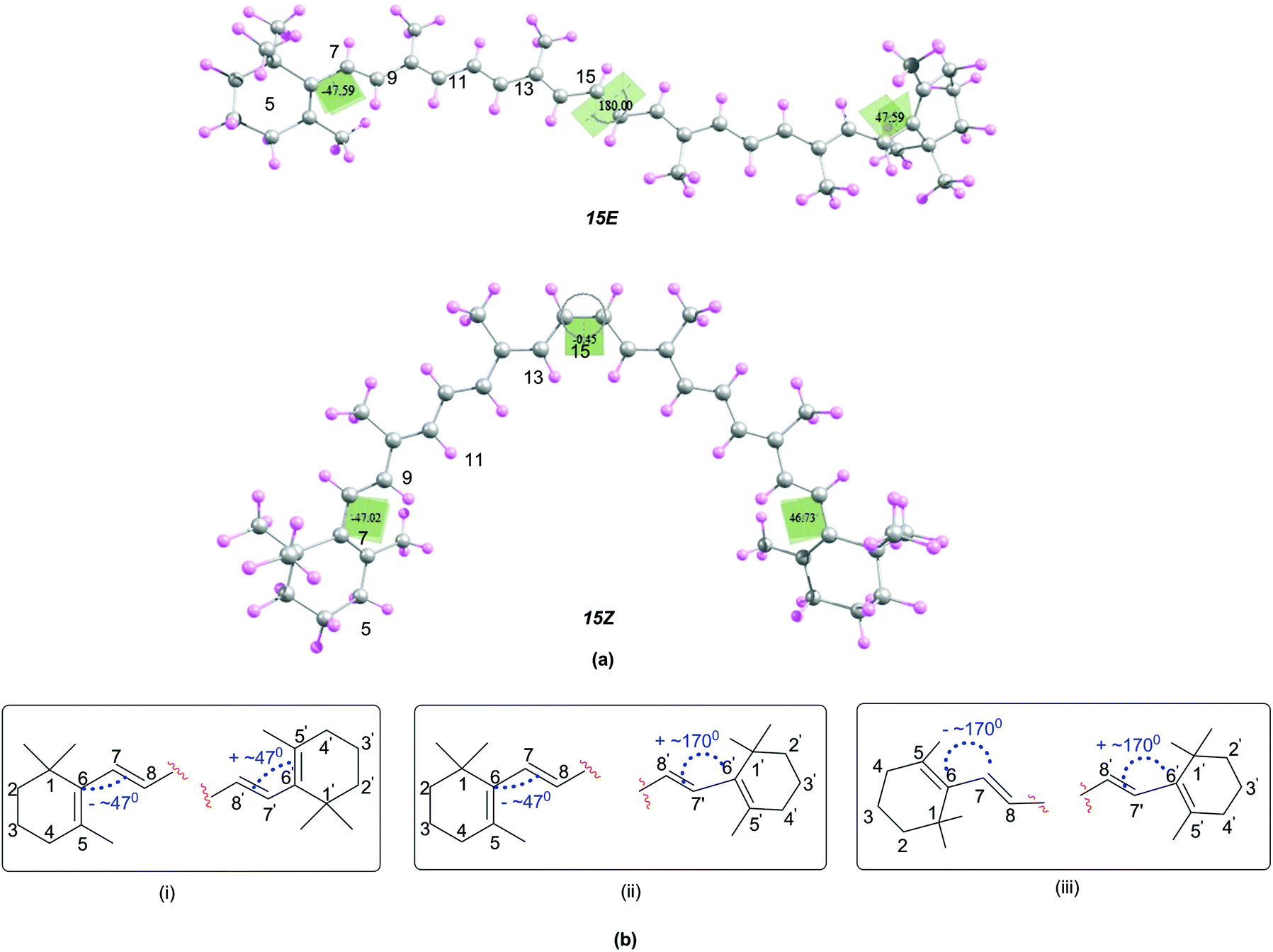 | ||
| Fig. 1 (a) Structures of the 15E and 15Z isomers of β-carotene optimized at B3LYP/6-31G(d) level (b) possible conformations due to the twisting of the end rings of β-carotene. | ||
The enthalpies of the rotamers with respect to the 15E and 15Z isomers in both gas and solution phases are illustrated in Fig. 2 and the corresponding thermochemical data are summarized in Table 2. The all-trans isomer (15E) is the most stable and it is interesting to note that the 15Z isomer obtained through rotation around the central C15–C15′ double bond is only 11.9 and 10.5 kJ mol−1, respectively less stable than the 15E isomer in the gas and the solution phases in terms of free energy. This compares to a free energy difference of +8.3 kJ mol−1 (+1.98 kcal mol−1) at the B3LYP/6-31+G(d,p)//B3LYP/6-31G(d) level reported in ref. 24. The 15E,9Z and 15E,13Z isomers obtained by CC double bond rotation are the next stable rotamers after the 15E isomer in both the gas and the solution phases. The next stable rotamers are obtained through rotation around the formal C8–C9 and C12–C13 single bonds of the polyene chain of β-carotene. These rotamers are of almost identical stability to the 15Z isomer. Rotation around the C10–C11 and C14–C15 bonds leads to 1,5-strain between methyl substituents and polyene methylene units and does, therefore, generate comparatively strained conformations which are the next stable conformers. Slightly higher we find the 15E,7Z and 15E,11Z conformers, which are less stable than the 15E isomer by 21.5 and 21.6 kJ mol−1, respectively in the gas phase and in solution. Surprisingly, the rotamer obtained by double rotations around the C9C10 and C9′C10′ (15E,9Z,9′Z) is more stable than the rotamers obtained from C–C single bond rotations in both the gas and solution phases (see Table 2). The rotamers obtained by C–C single and CC double bond rotations along the polyene chain of the 15Z isomer exhibit similar trends as compared to the rotamers of the 15E isomer in terms of stability. From Table 2 it is easily seen that the influence of polar solvation is not much pronounced in the relative enthalpy and free energy of the rotamers with respect to the 15E isomer.
 | ||
| Fig. 2 Relative enthalpies (ΔH298) for the isomers of β-carotene in the gas phase and in aqueous solution. | ||
| Isomers | B3LYP/MBa | B3LYP/MBb | ||
|---|---|---|---|---|
| Gas phase | Water | |||
| ΔH298 | ΔG298 | ΔH298 | ΔG298 | |
| a B3LYP/MB = B3LYP/6-311++G(d,p)//B3LYP/6-31G(d). b Geometries optimized at B3LYP/6-31G(d) level in the gas phase and energetics in water are obtained from these gas phase geometries at B3LYP/6-311++G(d,p) level using the PCM solvent model for water. Note that the energetics obtained at B3LYP/6-311++G(d,p)//PCM(H2O)B3LYP/6-31G(d) and PCM(H2O)/B3LYP/6-311++G(d,p)//PCM(H2O)B3LYP/6-31G(d) levels in the gas and solvent phases respectively, does not alter the energetics given in the above Table. The energetics obtained with these methods is given in Table S26.† | ||||
| All-trans (15E) | 0.0 | 0.0 | 0.0 | 0.0 |
| 15E,9Z | +4.1 | +4.5 | +7.1 | +7.4 |
| 15E,13Z | +4.2 | +5.2 | +4.8 | +5.8 |
| 15E, 9Z,9′Z | +7.0 | +6.5 | +12.1 | +11.5 |
| 15E,8Z | +13.2 | +12.3 | +13.9 | +13.0 |
| 15E,12Z | +14.9 | +12.2 | +14.4 | +12.4 |
| 15E,14Z | +20.4 | +20.8 | +20.5 | +20.8 |
| 15E,10Z | +20.9 | +22.7 | +23.0 | +24.8 |
| 15E,7Z | +21.5 | +24.1 | +19.5 | +21.4 |
| 15E,11Z | +21.6 | +20.8 | +21.4 | +24.9 |
| 15,15′-cis (15Z) | +10.2 | +11.9 | +9.0 | +10.5 |
| 15Z,9Z | +13.8 | +14.9 | +16.4 | +17.5 |
| 15Z,13Z | +14.7 | +14.1 | +13.8 | +13.2 |
| 15Z,8Z | +23.4 | +23.0 | +23.6 | +20.8 |
| 15Z,12Z | +24.5 | +23.9 | +26.0 | +22.7 |
| 15Z,10Z | +30.9 | +33.1 | +32.6 | +34.1 |
| 15Z,7Z | +31.3 | +33.3 | +31.1 | +30.0 |
| 15Z,11Z | +31.6 | +31.3 | +28.8 | +28.8 |
As is readily seen in Fig. 2, enthalpy differences between isomers of the 15E and 15Z families are largely similar in solution as in the gas phase. The relative order of stability of the studied isomers and conformers of β-carotene in the gas phase is 15E > 15E,9Z > 15E,13Z > 15E,9Z,9′Z > 15E,8Z > 15E,12Z > 15E,14Z > 15E,10Z, 15E,7Z and 15E,11Z in the trans form and 15Z > 15Z,9Z > 15Z,13Z > 15Z,8Z > 15Z,12Z > 15Z,10Z, 15Z,7Z and 15Z,11Z in the cis form.
C. H-Atom abstraction reactions from β-carotene by the OH radical
H-Atom abstraction reactions by the hydroxyl radical HO have been studied for the C4, C5–Me, C9–Me and C7 positions in β-carotene generating the radicals shown in Fig. 3. Depending on the conformational flexibility available to the product radicals and the β-carotene parent, two limiting scenarios may be envisaged for the calculation of reaction enthalpies: (a) in situations of high conformational flexibility and rapid conformational interchange, reaction enthalpies ΔH298 may be determined assuming all reactants and products to be in their overall most stable conformation; (b) in the second limiting scenario conformational interchange is slow on the time scale of the hydrogen abstraction reaction, and different reaction enthalpies ΔH298 will therefore result for different conformers of the β-carotene parent. The analysis of both scenarios is based on gas phase enthalpies calculated at (U)B3LYP/6-311++G(d,p)//(U)B3LYP/6-31G(d) level. The impact of polar solvation is analyzed through addition of aqueous solvation free energies obtained from PCM/UAHF/(U)B3LYP/6-311++G(d,p) single point calculations. In scenario (a) the β-carotene parent is found in the most stable all-trans (15E) conformation. The most stable conformation of the product radicals vary as a function of radical position and reaction medium and are therefore given in Fig. 3 together with the actual reaction enthalpies.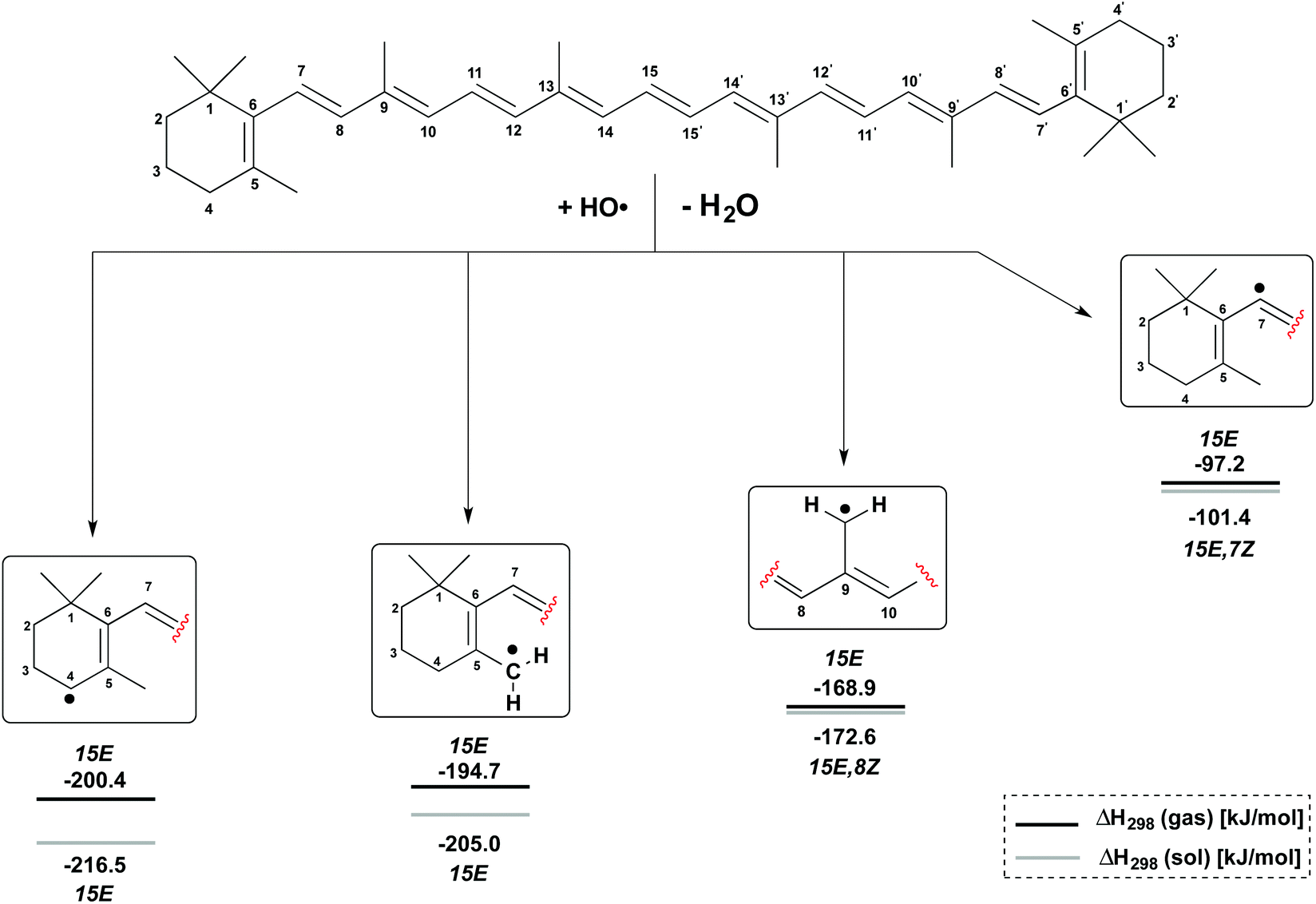 | ||
| Fig. 3 Gas and solution phase reaction enthalpies for hydrogen abstraction from β-carotene by hydroxyl radical HO. | ||
From Fig. 3 it is easily seen that H-atom abstraction is most exothermic from the C4 position in β-carotene (ΔH298 = −200.4 kJ mol−1), which is thus the most reactive site for oxidation reactions.17 This is closely followed by hydrogen abstraction from the C5–methyl group with ΔH298 = −194.7 kJ mol−1, while the C9–methyl group is much less reactive with ΔH298 = −168.9 kJ mol−1. In a qualitative sense these reaction enthalpies mirror the stabilities of the product radicals varying in the “resonance length” of the attached polyene chain and the number of substituents located at the (formal) radical center. All three product radicals are highly spin delocalized with the unpaired spin distributed over most of the polyene chain, and the drawing of localized radicals in Fig. 3 merely serves to indicate the location of H-atom abstraction. This is in contrast to the C7 radical, where hydrogen abstraction occurs from a C(sp2)–H bond, and the unpaired spin is almost completely located at the reacting C7 carbon atom. This reaction is much less favourable than the other three options with ΔH298 = −97.2 kJ mol−1 and has been included here as a less favourable reference to the other H-atom abstractions at the terminal positions in β-carotene. The inclusion of aqueous solvation effects leads to a small almost constant increase in reaction energies, mostly due to higher solvation energies of the water product relative to the hydroxyl radical. Solvation energies of β-carotene and the four radicals considered here are, on the other hand, so similar that no major changes in positional selectivity occur through polar solvation.
Reaction enthalpies for C4–H-atom abstraction for selected β-carotene conformers (gas phase and aqueous solution) are given in Fig. 4. Even though the 15E isomer of β-carotene is the most stable, the H-atom abstraction from the 15E,8Z and 15E,12Z rotamers, which are 13.2 kJ mol−1 and 14.9 kJ mol−1 less stable than the 15E isomer in closed shell form, are more thermodynamically favourable with an exothermicity of −211 kJ mol−1 in the gas phase. Following these rotamers, the H-atom abstraction from C4 position of 15E,14Z rotamer is the next thermodynamically favourable reaction which is ∼6 kJ mol−1 less exothermic than the 15E,8Z and 15E,12Z rotamers. The 15E,7Z, 15E,10Z and 15E,11Z rotamers rank next with an exothermicity of −202 kJ mol−1 for C4–H-atom abstraction. These are actually the least stable single-flip rotamers of β-carotene, but nevertheless exhibit almost the same exothermicity for C4–H-atom abstraction as that of the 15E isomer at 200 kJ mol−1. Even less exothermic are C4–H-atom abstractions from the 15E, 13Z, 15E, 9Z and 15E, 9Z, 9′Z rotamers. Turning to C4–H-atom abstractions from the 15Z isomers of β-carotene, we find the 15Z,8Z rotamer to be most reactive with a reaction enthalpy of −209 kJ mol−1, which is closely matched by the 15Z,12Z rotamer at −208 kJ mol−1. This is followed by the 15Z,11Z, 15Z,10Z, 15Z, 15Z,13Z, 15Z,9Z and 15Z,7Z rotamers. The 15Z,11Z rotamer is 31.6 kJ mol−1 less stable than the 15Z isomer of β-carotene, but shows ∼2 kJ mol−1 more exothermicity towards C4–H-atom abstraction. The C4–H-atom abstraction from the 15Z isomer is 10 kJ mol−1 less exothermic than that from the 15Z,8Z rotamer. The 15Z,13Z, 15Z,9Z and 15Z,7Z rotamers are less exothermic by ∼14 kJ mol−1 when compared with that of the 15Z,8Z rotamer. All taken together we find the 8Z rotamers in the 15E and the 15Z conformational families to be most reactive towards HO-induced gas phase C4–H-atom abstraction.
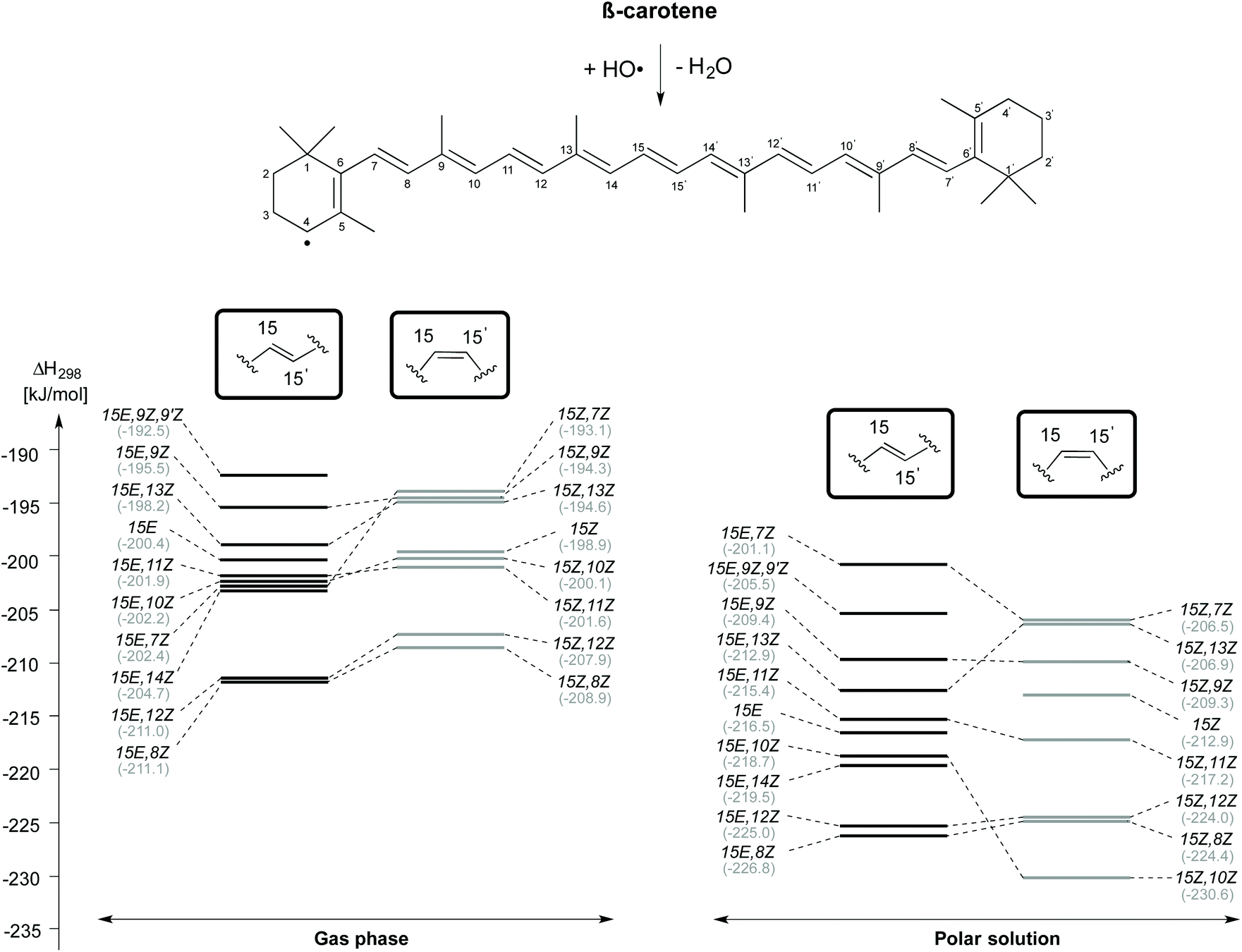 | ||
| Fig. 4 Reaction enthalpies (ΔH298) for hydrogen atom abstraction from the C4 position of cis/trans isomers of β-carotene in the gas phase and in aqueous solution. | ||
In aqueous solution, the C4–H atom abstractions from β-carotene are comparatively more exothermic and exoergic than those reactions in the gas phase. The trend in the conformational spacing in terms of enthalpy of the C4–H atom abstraction reactions is slightly altered through solvation effects. The H-atom abstraction from the C4 position of 15E,8Z and 15Z,10Z rotamers obtained from their respective counterparts are quite exothermic at −226.8 and −230.6 kJ mol−1, respectively. The C4–H atom abstraction from the 15E,7Z and 15Z,7Z rotamers are the least exothermic channels.
H-Atom abstraction from the CH3 group attached to the C5 position of β-carotene forming the C5–methyl radical and H2O has also been studied for different rotamers of β-carotene and the reaction enthalpies are presented in Fig. 5. As observed for H-atom abstraction from the C4 position, the C5-CH3 H-atom abstraction from the 15E,8Z rotamer is more exothermic than that from all other rotamers, both in the gas phase (at −204.9 kJ mol−1) as well as in solution (at −215.4 kJ mol−1). The enthalpic order of rotamer reactivity for C5-CH3 H-atom abstraction in the 15E isomer group in the gas phase is 15E,8Z > 15E,12Z > 15E,14Z > 15E,11Z > 15E,10Z > 15E > 15E,13Z > 15E,7Z > 15E,9Z and that for the 15Z isomer group is 15Z,11Z > 15Z > 15Z,10Z > 15Z,8Z > 15Z,12Z > 15Z,13Z > 15Z,7Z > 15Z,9Z. Polar solvation enhances the exothermicity of C5-CH3 H-atom abstractions when compared to the gas phase. Given that the enthalpy of the reaction of the rotamers towards H-atom abstraction is calculated with respect to their closed shell precursors, the exothermicity increases substantially. In the case of H-atom abstraction from the C5–Me group of the 15Z isomer, the enthalpy of the reaction calculated with respect to closed shell 15Z isomer is −191.4 and −202.9 kJ mol−1, respectively in the gas and aqueous phases, whereas the corresponding reaction enthalpies calculated for the most stable 15E isomer of β-carotene are −194.7 and −205.0 kJ mol−1, respectively in the gas and solution phases. Taken together the results for the C4–H and C5-CH3 H-atom abstraction reactions presented in Fig. 4 and 5 clearly indicate that conformer-specific reactions are more exothermic and thus potentially more relevant for determining the antioxidant properties of β-carotene.
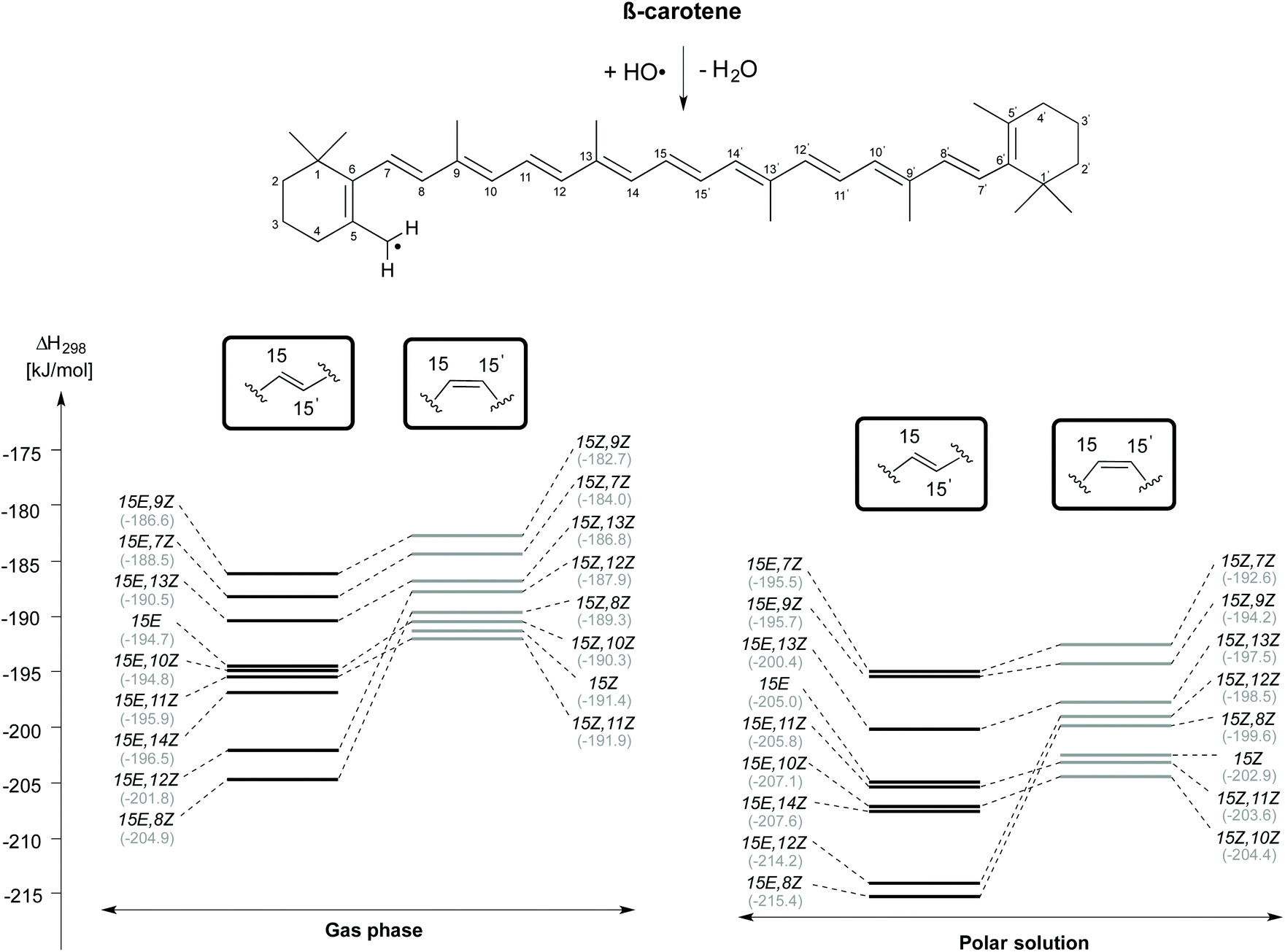 | ||
| Fig. 5 Reaction enthalpies (ΔH298) for hydrogen atom abstraction from the C5-CH3 position of cis/trans isomers of β-carotene in the gas phase and in aqueous solution. | ||
H-Atom abstraction reactions from the methyl substituent attached to the C9 position of β-carotene has also been studied in a confomer-specific way. As illustrated in Fig. 6, the C9-CH3 H-atom abstraction is thermodynamically most favourable in the rotamer obtained by C8–C9 rotation in both the 15E (15E,8Z) and 15Z (15Z,8Z) isomers in the gas and solvent phases. This is followed by C9-CH3 H-atom abstraction from the 15E,9Z and 15Z,9Z rotamers, which ranked lowest in the C4 and C5-CH3 H-atom abstraction reactions. The least exothermic gas phase C9-CH3 H-atom abstractions are due to 15E,11Z rotamer in the trans form and the 15Z isomer in the cis form. In solution the least exothermic channels are those resulting from 15E,11Z rotamer in trans form and 15Z,13Z rotamer in the cis form. Polar solvation only slightly alters the reactivity trends observed for C9-CH3 H-atom abstractions in gas phase.
 | ||
| Fig. 6 Reaction enthalpies (ΔH298) for hydrogen atom abstraction from the C9-CH3 position of cis/trans isomers of β-carotene in the gas phase and in aqueous solution. | ||
In terms of their overall conformational stability as well as their reactivity in hydrogen abstractions from the C4, C5-CH3 and C9-CH3 positions, the 15E,7Z and 15Z,7Z rotamers ranked among the less relevant conformational options. This is not so in H-atom abstractions from the C7-position, where these rotamers show the highest exothermicities (Fig. 7). The lowest reactivities for C7–H-atom abstractions are found for the 15E and 15Z,13Z rotamers in the gas phase, and for the 15E,14Z and 15Z rotamers in solution. The C7–H-atom abstraction for the 15E,7Z rotamer is exothermic by −118.2 and −121.1 kJ mol−1, respectively in gas and aqueous phases, whereas the exothermicities for the 15E isomer amount to −96.7 and −101.3 kJ mol−1, respectively. From the results obtained for C9-CH3 and C7–H-atom abstraction reactions, it is well understood that the flipping along the polyene chain plays an important role in determining its reactivity.
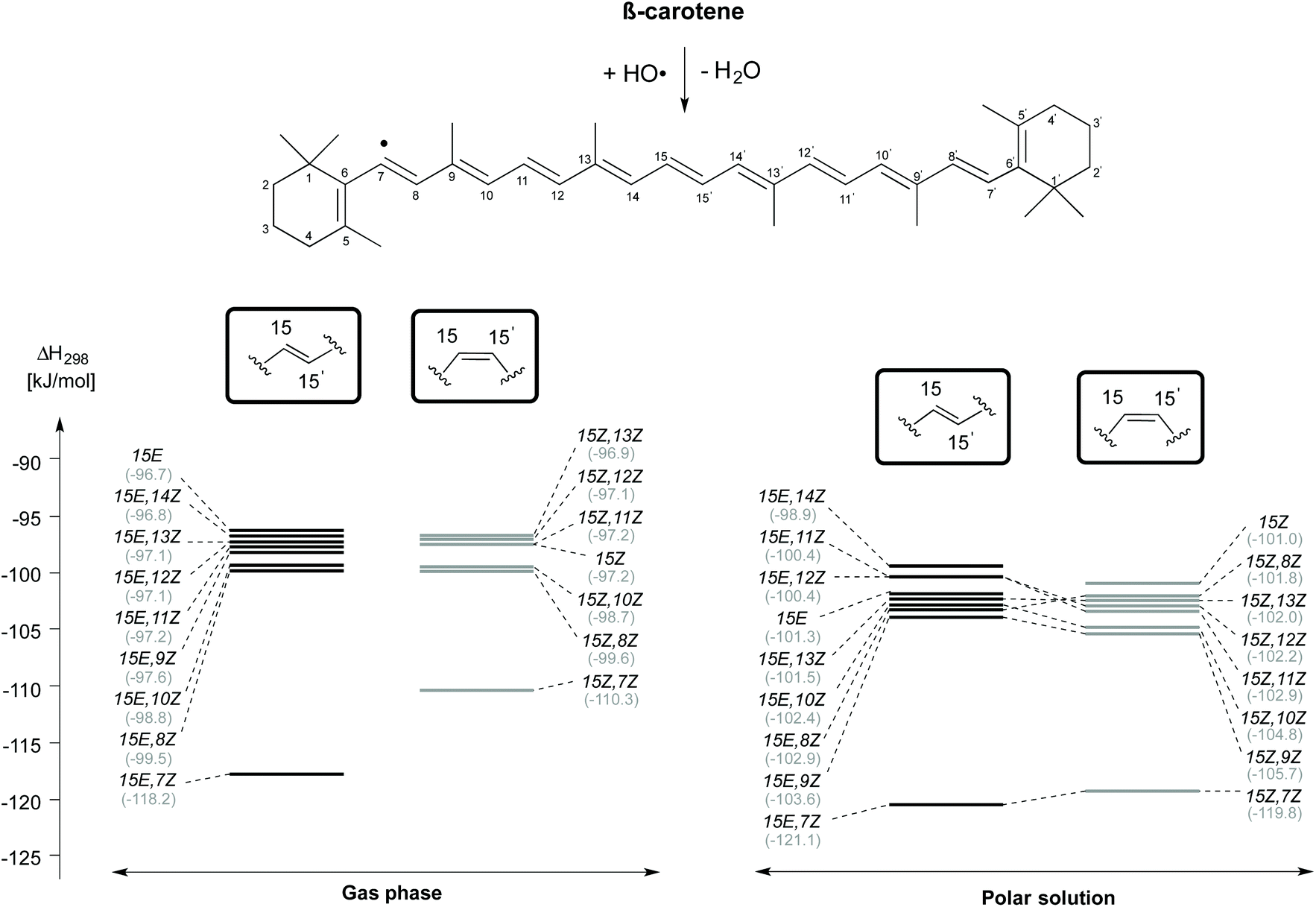 | ||
| Fig. 7 Reaction enthalpies (ΔH298) for hydrogen atom abstraction from the C7 position of cis/trans isomers of β-carotene in the gas phase and in aqueous solution. | ||
The study by Galano et al., which concerned the H-atom abstraction from the C4 position of β-carotene by HO2 radical, reported an exothermicity of −59.4 kJ mol−1 in water.17 That these reactions are much less exothermic as compared to those described in Fig. 4 for hydroxyl radicals is simply due to the much smaller gas phase O–H bond dissociation energy (BDE) in hydrogen peroxide (HOOH, +366.06 ± 0.29 kJ mol−1)44 as compared to the value in water (H2O, +497.1 ± 0.3 kJ mol−1).44 The trend in the enthalpy of these reactions is the same as that observed for free energies of reaction. The calculated thermochemistry of the H-atom abstraction reactions shows that these reactions occur more favourably in solution than in the gas phase, which is in accordance with the earlier studies on H-atom abstraction and electron transfer reactions in β-carotene.13
IV. Conclusions
In line with earlier studies we confirm that hydrogen abstractions from the allylic C4 and C5-CH3 positions in β-carotene are the most exothermic and thus most relevant for assessing the antioxidant activity of β-carotene through this reaction channel. The conformer-specific exothermicities from these positions are found to be larger than those obtained from conformationally averaged enthalpy values, and the 15E,8Z and 15Z,10Z rotamers appear to be especially relevant. The least favourable are H-atom abstractions from C9-CH3 and C7 positions. In these latter reactions the 15E,8Z and 15E,7Z rotamers offer the weakest C–H bonds. Aqueous solvation makes HO-radical mediated H-atom abstraction reactions from β-carotene uniformly more favourable than in the gas phase. At least for attack at the C4 position, it makes the 15E,8Z and 15Z,10Z isomers similarly reactive, which is in remarkable contrast to the conformationally averaged reactivity data presented in Fig. 3. Whether or not the conformer-specific properties determined here can help to rationalize the antioxidative effects observed through in vivo experiments depend on several other factors, the conformer-specific bioavailability being one of the more relevant.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by BASF is gratefully acknowledged. We furthermore thank Dr A. Schäfer and Dr P. Deglmann (BASF) for helpful discussions relating to this work.References
- N. I. Krinsky, Philos. Trans. R. Soc. London, Ser. B, 1978, 284, 581–590 CAS.
- N. I. Krinsky, Pure Appl. Chem., 1979, 51, 649–660 CAS.
- N. H. Jensen, A. B. Nielsen and R. Wilbrandt, J. Am. Chem. Soc., 1982, 104, 6117–6119 CrossRef CAS.
- R. J. Cogdell, Philos. Trans. R. Soc. London, Ser. B, 1978, 284, 569–579 CAS.
- G. W. Burton and K. U. Ingold, Science, 1984, 224, 569–573 CrossRef CAS.
- R. Edge, D. J. McGarvey and T. G. Truscott, J. Photochem. Photobiol., B, 1997, 41, 189–200 CrossRef CAS.
- N. E. Polyakov, T. V. Leshina, T. A. Konovalova and L. D. Kispert, Free Radical Biol. Med., 2001, 31, 398–404 CrossRef CAS.
- T. Polivka and V. Sundstrom, Chem. Rev., 2004, 104, 2021–2071 CrossRef CAS PubMed.
- C. A. Rice-Evans, J. Sampson, P. M. Bramley and D. E. Holloway, Free Radical Res., 1997, 26, 381–398 CrossRef CAS PubMed.
- S. A. Everett, M. F. Dennis, K. B. Patel, S. Maddix, S. C. Kundu and R. L. Willson, J. Biol. Chem., 1996, 271, 3988–3994 CrossRef CAS PubMed.
- D. Baker, E. Krol, N. Jacobsen and D. Liebler, Chem. Res. Toxicol., 1999, 12, 535–543 Search PubMed.
- A. Arora, C. Willhite and D. Liebler, Carcinogenesis, 2001, 22, 1173–1178 CrossRef CAS.
- J. P. Ceron-Carrasco, A. Bastida, A. Requena, J. Zuniga and B. A. Miguel, J. Phys. Chem. B, 2010, 114, 4366–4372 CrossRef CAS.
- Y. Gao, A. L. Focsan, L. D. Kispert and D. A. Dixon, J. Phys. Chem. B, 2006, 110, 24750–24756 CrossRef CAS PubMed.
- A. Martínez and A. Barbosa, J. Phys. Chem. B, 2008, 112, 16945–16951 CrossRef.
- A. Martínez, M. A. Rodríguez-Girones and A. Barbosa, J. Phys. Chem. A, 2008, 112, 9037–9042 CrossRef.
- A. Galano and M. Francisco-Márquez, J. Phys. Chem. B, 2009, 113, 11338–11345 CrossRef CAS.
- A. Martínez, R. Vargas and A. Galano, Theor. Chem. Acc., 2010, 127, 595–603 Search PubMed.
- A. Galano, R. Vargas and A. Martínez, Phys. Chem. Chem. Phys., 2010, 12, 193–200 RSC.
- A. Martínez, R. Vargas and A. Galano, J. Phys. Chem. B, 2009, 113, 12113–12120 CrossRef.
- Y. Koyama, M. Kito, T. Takii, K. Saiki, K. Tsukida and J. Yamashita, Biochim. Biophys. Acta, Bioenerg., 1982, 680, 109–118 CrossRef CAS.
- W. von Doering, C. Sotiriou-Leventis and W. R. Roth, J. Am. Chem. Soc., 1995, 117, 2747–2757 CrossRef CAS.
- S. Schlucker, A. Szeghalmi, M. Schmitt, J. Popp and W. Kiefer, J. Raman Spectrosc., 2003, 34, 413–419 CrossRef.
- E. Hernandez-Marin, A. Galano and A. Martinez, J. Phys. Chem. B, 2013, 117, 4050–4061 CrossRef CAS.
- T. Polívka and V. Sundström, Chem. Rev., 2004, 104, 2021–2071 CrossRef PubMed.
- B. Durbeej and L. A. Eriksson, Phys. Chem. Chem. Phys., 2004, 6, 4190–4198 RSC.
- J. P. Cerón-Carrasco, A. Requena and C. M. Marian, Chem. Phys., 2010, 373, 98–103 CrossRef.
- J. Hioe and H. Zipse, Radical Stability - Thermochemical Aspects, in Encyclopedia of Radical in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, John Wiley & Sons Ltd, Chichester, UK, 2012, p. 449–476 Search PubMed.
- M. L. Coote, C. Y. Lin and H. Zipse, The Stability of Carbon-Centered Radicals, ed. M. D. E. Forbes, Carbon-Centered Free Radicals and Radicals Cations, John Wiley & Sons, 2010, pp. 83–104 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chem. Acc., 1973, 28, 213–222 Search PubMed.
- R. Krishnan, J. S. Binkley and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- D. C. Graham, A. S. Menon, L. Goerigk, S. Grimme and L. Radom, J. Phys. Chem. A, 2009, 113, 9861–9873 CrossRef CAS.
- A. G. Baboul, L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 1999, 110, 7650–7657 CrossRef CAS.
- D. J. Henry, C. J. Parkinson and L. Radom, J. Phys. Chem. A, 2002, 106, 7927–7936 CrossRef CAS.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822–2827 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
- R. Cammi and J. Tomasi, J. Comput. Chem., 1995, 16, 1449–1458 CrossRef CAS.
- (a) H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CAS; (b) H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, MOLPRO, version 2012.1, a package of ab initio programs Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- L. Sandhiya and H. Zipse, Chem. – Eur. J., 2015, 21, 14060–14067 CrossRef CAS.
- S. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, 2007 Search PubMed.
- M. O. Senge, H. Hope and K. M. Smith, Z. Naturforsch., C: J. Biosci., 1992, 47, 474–476 CrossRef CAS.
- H. Hashimoto, T. Yoda, T. Kobayashi and A. J. Young, J. Mol. Struct., 2002, 604, 125–146 CrossRef CAS.
- H.-R. Sliwka, T.-B. Melø, B. J. Foss, S. H. Abdel-Hafez, V. Partali, G. Nadolski, H. Jackson and S. F. Lockwood, Chem. – Eur. J., 2007, 13, 4458–4466 CrossRef CAS.
- F. Himo, J. Phys. Chem. A, 2001, 105, 7933–7937 CrossRef CAS.
- P. Faller, T. Maly, A. W. Rutherford and F. MacMillan, Biochemistry, 2001, 40, 320–326 CrossRef CAS.
- Active Thermochemical Tables (ATcT), version 1.122, available at http://atct.anl.gov/, using the following heat of formation values: ΔfH°(propene) = +19.99 ± 0.22 kJ mol−1; ΔfH°(allyl radical) = +167.84 ± 0.54 kJ mol−1; ΔfH°(water) = −241.834 ± 0.027 kJ mol−1; ΔfH°(HO˙ radical) = +37.490 ± 0.027 kJ mol−1.
Footnotes |
| † Electronic supplementary information (ESI) available: Structures and energetics of all the reactive species optimized at various levels of theory. See DOI: 10.1039/d1ob01723c |
| ‡ Present Address: CSIR-National Institute of Science Communication and Policy Research, New Delhi, India. |
| This journal is © The Royal Society of Chemistry 2022 |