
Domino Michael/Michael reaction catalyzed by switchable modularly designed organocatalysts†
Ramarao
Parella
,
Satish
Jakkampudi
,
Pranjal
Bora
,
Nagaraju
Sakkani
 and
John C.-G.
Zhao
*
and
John C.-G.
Zhao
*
Department of Chemistry, University of Texas at San Antonio, One UTSA Circle, San Antonio, Texas 78249-069, USA. E-mail: cong.zhao@utsa.edu
First published on 6th December 2021
Abstract
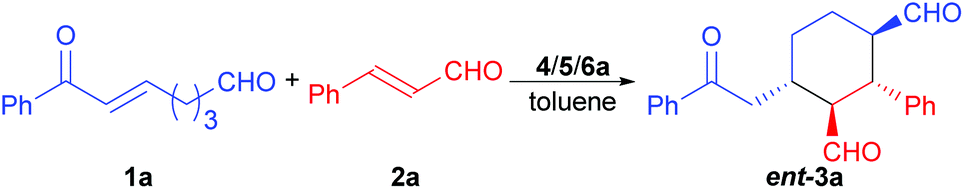
The domino Michael/Michael reaction between (E)-7-aryl-7-oxohept-5-enals and trans-cinnamaldehydes was investigated by using modularly designed organocatalysts (MDOs). It was found that both the enamine and iminium catalytic modes of the MDOs are switchable and can be individually switched on and off by using appropriate combinations of the precatalyst modules and the reaction conditions. When both the enamine and iminium catalysis modes of the MDOs are switched on, the desired domino reaction products can be obtained in good yields and stereoselectivities under optimized conditions.
Introduction
In a living cell, numerous reactions occur simultaneously. To ensure that all these reactions are not interfering with each other, these reactions must proceed with exact spatial and temporal control.1 To achieve that, the enzymes that catalyze these reactions are often regulated by feedback loops or triggers, which means their catalytic activities must be switchable and the switching must be reversible.1 The regulation of an enzyme by binding an effector molecule at a site other than the enzyme's active site is known as an allosteric regulation, which can be either an allosteric activation or an allosteric inhibition.2 Simple chemical systems that mimic enzyme reactivities have been actively pursued by chemists in the past decades with the goals of understanding the fundamental questions regarding enzyme activation and developing synthetically useful catalysts inspired by the enzymes. As a result of these efforts, many switchable catalytic systems based on the allosteric activation mode have been reported.3 Light, small molecules, pH (H+), temperature, and ions/metals have been used as the allosteric activation effectors. Scheme 1 below shows how allosteric regulation works in general. The off-state catalyst may be directly turned on by the allosteric activator, which then catalyzes the desired reaction (Scheme 1, top figure). Alternatively, the catalyst may be first turned off by an inhibitor, and then turned on through the removal of the inhibitor by the activator (Scheme 1, middle figure).4 Of course, switchable catalytic systems can also be designed based on the allosteric inhibition mode, in which the catalysis of the catalyst can be turned off by an allosteric inhibitor (Scheme 1, bottom figure).5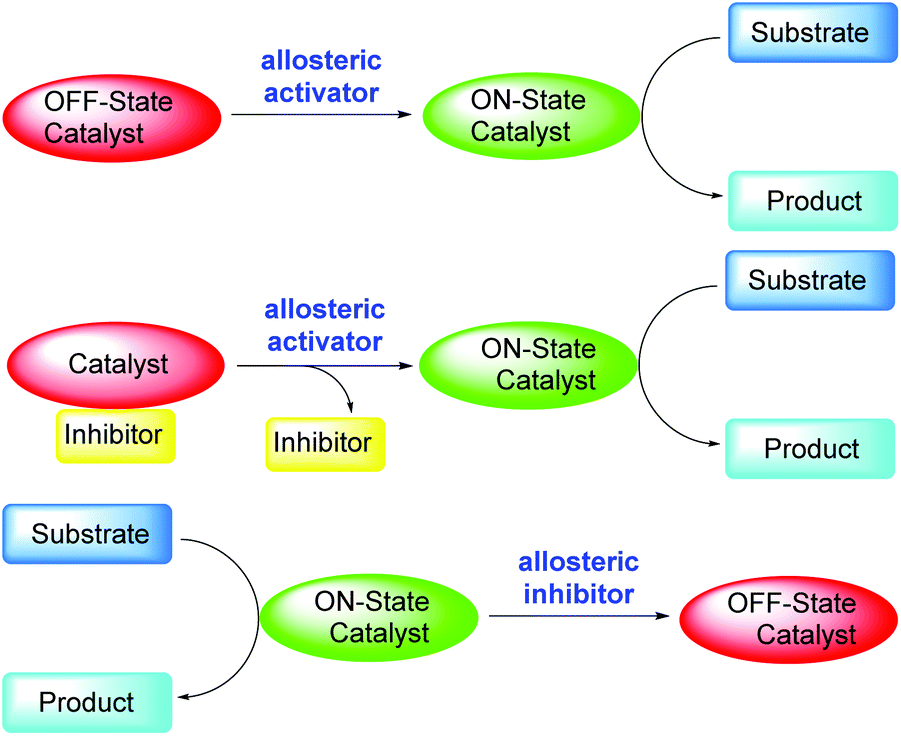 | ||
| Scheme 1 Reported allosteric regulations of switchable catalysts via an allosteric activator or an allosteric inhibitor. | ||
Since List and Barbas reported the first example of a proline-catalyzed intermolecular aldol reaction in 2000,6 many proline derivatives have been developed in the last two decades as organocatalysts to achieve a plethora of stereoselective transformations.7 Nonetheless, the allosteric regulations of the enamine or iminium catalysis of an amine catalyst is seldom studied in the past. To our knowledge, only Leigh and Leung have reported achiral rotaxane-based acyclic secondary amine catalyst, in which both the enamine and iminium catalysis can be subjected to allosteric inhibition simultaneously by protons8 due to the conformational change of the catalyst after protonation.8
Most recently, we reported that modularly designed organocatalysts (MDOs),9 which form in the reaction media via the self-assembly of cinchona alkaloid derivatives (such as QDT) and amino acids (such as L-Pro), are switchable catalysts.9f The iminium catalysis mode of the MDOs that is inhibited by the cinchona alkaloid module (i.e., QDT) can be switched on by adding an appropriate acid and, upon the switch-on, these MDOs can be used for catalyzing a diastereodivergent9g domino Mannich condensation/Michael/Michael reaction10 between ketones and trans-cinnamaldehydes (Scheme 2, top equation).9f
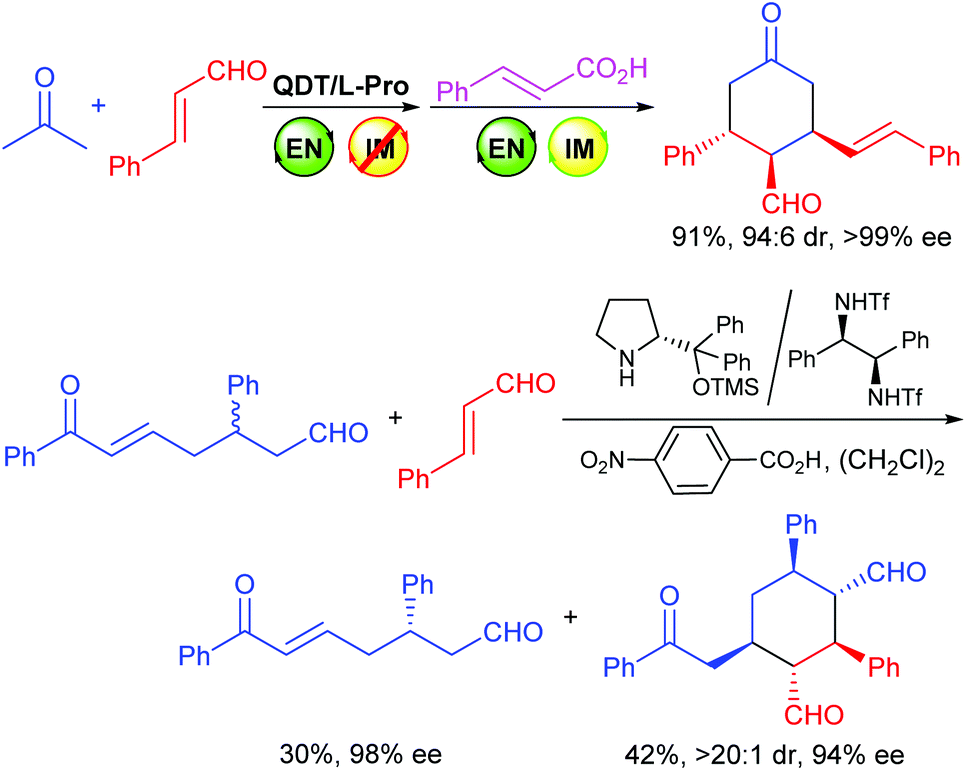 | ||
| Scheme 2 Some reported examples related to the current study. | ||
On the other hand, despite the advances in organocatalysis, the organocatalytic asymmetric Michael addition of ketones or aldehydes to α,β-unsaturated aldehydes via the enamine/iminium catalysis remains a challenging task,9f,11,12 and only a few examples are available.9f,11 In this respect, Xu and coworkers have reported a kinetic resolution of racemic enonals via an organocatalyzed asymmetric domino Michael/Michael reaction with cinnamaldehydes using a combination of the Jørgensen-Hayashi catalyst and a diamine-derived cocatalyst, involving both the enamine and iminium activations of the catalyst (Scheme 2, bottom equation).11b Hong and coworkers have also reported a domino Michael/Michael/aldol condensation reaction between enonals and enals.11c Inspired by these results, we studied the domino Michael/Michael reaction of achiral enonals 1, with which we have developed many useful synthetic methods,9 with trans-cinnamaldehydes using our switchable MDOs as the catalyst, and discovered that both the enamine and iminium catalytic modes can be individually switched on and off if appropriate combinations of precatalyst modules and/or reaction conditions are employed. Herein we wish to report the details of our findings.
Results and discussion
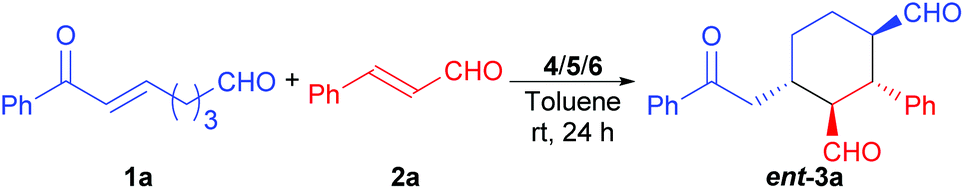
The achiral enonal compound 1a and cinnamaldehyde (2a) were adopted as the substrates for the stereoselective synthesis of cyclohexanedial ent-3a (Table 1), since, as in Xu's case,11b the domino Michael/Michael reaction between 1a and 2a requires both enamine and iminium catalysis. First, we conducted some experiments to determine whether an acid is indeed needed to switch on the iminium catalysis of the MDO. As the results in Table 1 show. When the MDO of 4a/5a (QDT/L-Pro, Scheme 4) was applied together with the acid 6a at room temperature, the desired 1,3-cyclohexanedial ent-3a was obtained in 53% yield, 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 dr, and 58% ee after 24 h, suggesting both the enamine and iminium catalysis was working under these conditions (entry 1). In contrast, the control reaction without adding any acid gave only trace amount of product under otherwise identical conditions (entry 2). These results confirm that the presence of an acid is crucial for the success of this domino Michael/Michael reaction. Further control reactions also revealed that the combination of 4a (QDT) and 6a (entry 3), and 5a (L-Pro) and 6a (entry 4) failed to catalyze the reaction. Similarly, the individual modules 4a (entry 5) and 5a (entry 6), and the acid 6a (entry 7) also failed to catalyze the reaction. Similar results were also obtained with the MDO of 4b/5a (entries 8–14), except that a better product yield (77%), dr (89:11), and ee value (78%) were obtained with this MDO in the presence of the acid 6a (entry 8). Since we have demonstrated that MDOs (such as 4a/5a) is able to catalyze enamine-mediated reaction without any acid,9 the failure of these MDOs to catalyze the domino Michael/Michael reaction without an acid (entries 2 and 9) is most likely because the iminium catalysis mode of these catalysts is not working without an acid cocatalyst.9f The above results again show that inhibited iminium catalysis can be switched on by adding the acid cocatalyst (Scheme 3, upper equation).9f
:20 dr, and 58% ee after 24 h, suggesting both the enamine and iminium catalysis was working under these conditions (entry 1). In contrast, the control reaction without adding any acid gave only trace amount of product under otherwise identical conditions (entry 2). These results confirm that the presence of an acid is crucial for the success of this domino Michael/Michael reaction. Further control reactions also revealed that the combination of 4a (QDT) and 6a (entry 3), and 5a (L-Pro) and 6a (entry 4) failed to catalyze the reaction. Similarly, the individual modules 4a (entry 5) and 5a (entry 6), and the acid 6a (entry 7) also failed to catalyze the reaction. Similar results were also obtained with the MDO of 4b/5a (entries 8–14), except that a better product yield (77%), dr (89:11), and ee value (78%) were obtained with this MDO in the presence of the acid 6a (entry 8). Since we have demonstrated that MDOs (such as 4a/5a) is able to catalyze enamine-mediated reaction without any acid,9 the failure of these MDOs to catalyze the domino Michael/Michael reaction without an acid (entries 2 and 9) is most likely because the iminium catalysis mode of these catalysts is not working without an acid cocatalyst.9f The above results again show that inhibited iminium catalysis can be switched on by adding the acid cocatalyst (Scheme 3, upper equation).9f
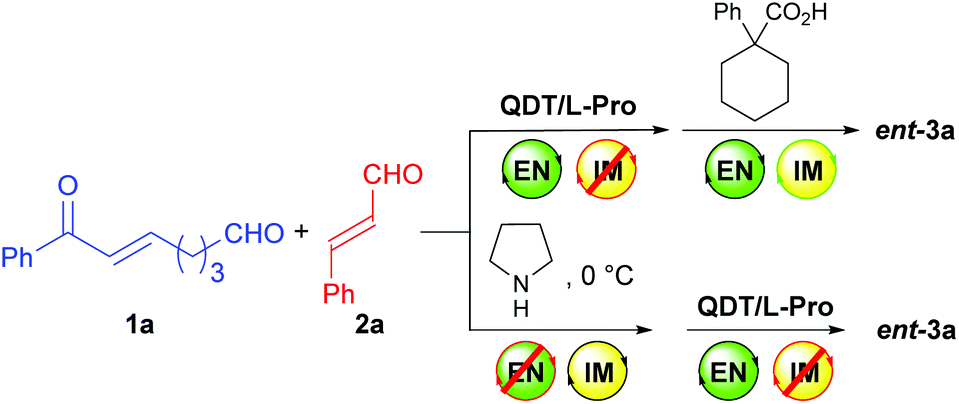 | ||
| Scheme 3 Switching on and off the iminium and enamine catalytic modes. | ||
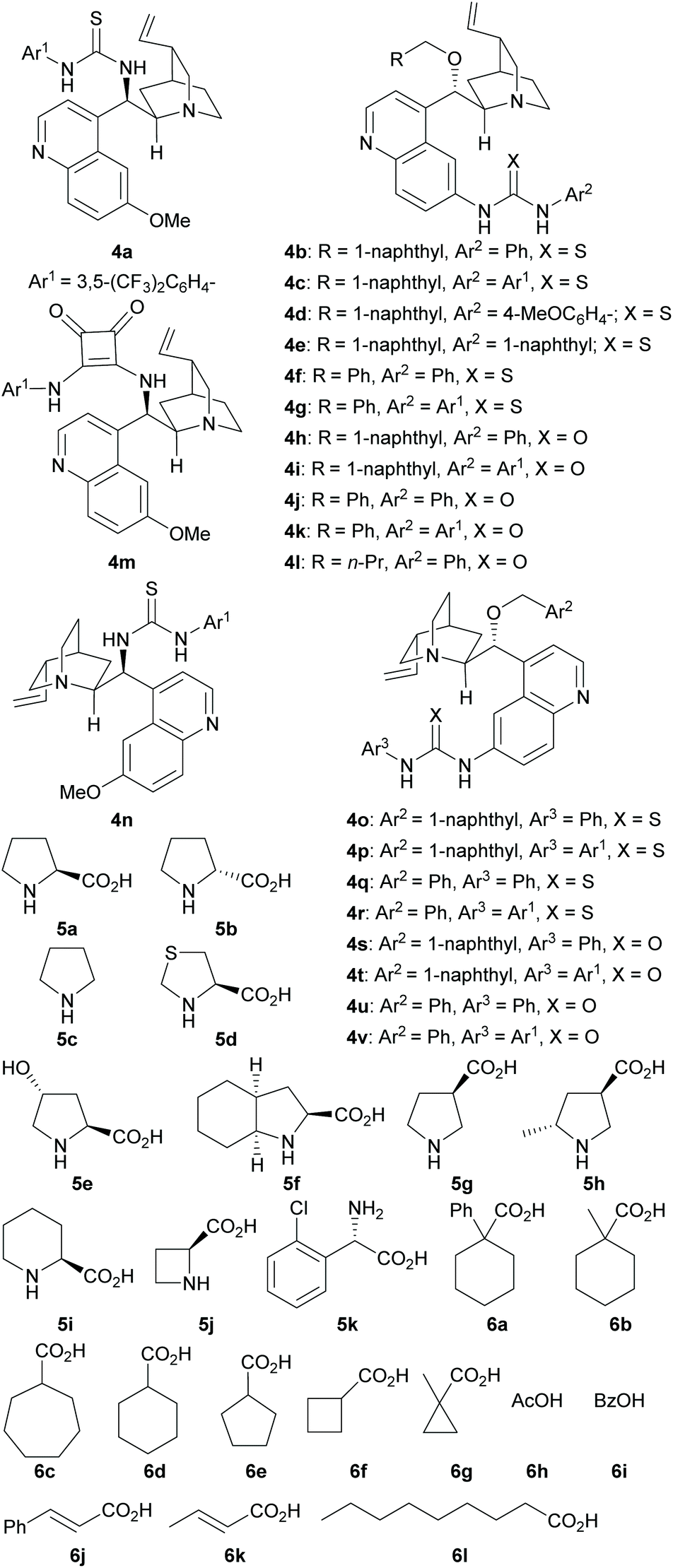 | ||
| Scheme 4 Structure of the catalyst modules and acids used in this study. | ||
a
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Precatalyst modules | Acid | Yieldb (%) | drc | eed (%) | |
| a Unless otherwise specified, all reactions were carried out with 1a (0.12 mmol), 2a (0.10 mmol), the precatalyst modules (4a, 5a, and/or 5c, 0.010 mmol each, 10 mol%), and the acid catalyst 6a (0.010 mmol, 10 mol%) in dry toluene (1.0 mL) at room temperature. b Yield of the isolated major diastereomeric product after column chromatography. c Determined by 1H NMR analysis of the crude product. d Determined by HPLC analysis on a ChiralPak IC column. e No reaction. f Carried out at 0 °C. | ||||||
| 1 | 4a | 5a | 6a | 53 | 80:20 |
58 |
| 2 | 4a | 5a | — | Trace | — | — |
| 3 | 4a | — | 6a | NRe | ||
| 4 | — | 5a | 6a | NRe | ||
| 5 | 4a | — | — | NRe | ||
| 6 | — | 5a | — | NRe | ||
| 7 | — | — | 6a | NRe | ||
| 8 | 4b | 5a | 6a | 77 | 89:11 |
78 |
| 9 | 4b | 5a | — | Trace | — | — |
| 10 | 4b | — | 6a | NRe | ||
| 11 | — | 5a | 6a | NRe | ||
| 12 | 4b | — | — | NRe | ||
| 13 | — | 5a | — | NRe | ||
| 14 | — | — | 6a | NRe | ||
| 15 | — | 5c | — | 69 | 95:5 |
0 |
| 16 | 4a | 5c | — | 57 | 93:7 |
2 |
| 17f | — | 5c | — | NRe | ||
| 18f | 4a | 5c | — | Trace | — | — |
| 19 | — | 5a + 5c | — | 68 | 92:8 |
3 |
| 20f | — | 5a + 5c | — | Trace | — | — |
| 21 | 4a | 5a + 5c | — | 85 | 99:1 |
14 |
| 22f | 4a | 5a + 5c | — | 65 | 83:17 |
17 |
Pyrrolidine (5c) is a good catalyst for both enamine and iminium catalysis. To further establish that the reaction indeed goes via the enamine and iminium catalysis,12 we tried the reaction with 5c as the catalyst, and the desired racemic product 3a was obtained in 69% yield and 95:5 dr (entry 15). The use of 5c together with 4a (QDT) did not show any improvement in terms of product yield and stereoselectivities (entry 16). These results exclude a possible involvement of the enolate mechanism in this reaction, because, if an enolate mechanism is responsible for the observed formation of 3a, adding QDT should have facilitated the domino reaction since QDT is a stronger base than 5c. It is surprising that when these reactions were repeated at 0 °C with 5c or 4a/5c as the catalyst, no desired domino reaction was observed (entries 17 and 18). These surprising failures may be due to the switch-off of the enamine, the iminium, or both the enamine and iminium catalysis modes by this lower temperature. To find out what is actually switched off by the lower temperature, we conducted some additional control reactions. It was found that, while the combination of 5a (L-Pro) and 5c catalyzes the reaction at rt (entry 19), it also failed at 0 °C (entry 20). Only when 4a (QDT), 5a (L-Pro), and 5c were used together, the desired domino reaction would proceed again at 0 °C (entry 22). We have shown above that MDO of 4a/5a can't catalyze the iminium catalysis, but it is known to catalyze the enamine-mediated reactions.9 Thus, it can be concluded that the iminium catalysis mode of 5c must still work at 0 °C; otherwise, we should not be able to obtain any product with 4a/5a/5c at this temperature (entry 22). Thus, what actually is switched off at 0 °C must be the enamine catalysis mode of 5c. In summary, we have showed that the enamine catalysis mode of pyrrolidine (5c) can be switched off by a lower temperature of 0 °C. With this interesting property of 5c, we can build a ternary system (such as 4a/5a/5c) at 0 °C, in which pyrrolidine (5c) is responsible for the iminium catalysis only and the MDO (such as 4a/5a) is responsible for the enamine catalysis only (Scheme 3, lower equation). Nevertheless, since this ternary system leads to a poor enantioselectivity for the desired domino product (entries 21 and 22), we focused on the first approach (the switch-on by an acid cocatalyst) in our further optimizations.
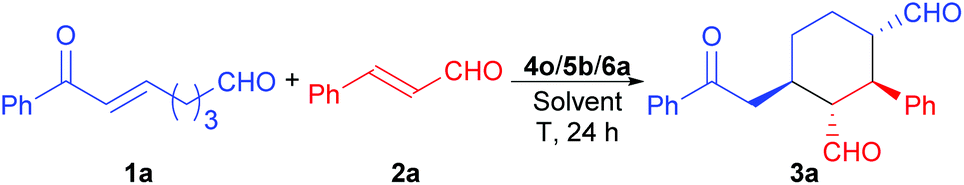
While the MDOs 4a/5a and 4b/5a both yield the desired domino Michael product ent-3a in the presence of acid 6a (Table 2, entries 1 and 2), the latter gives higher product yield and stereoselectivities (entry 2). Therefore, precatalyst module 4b was selected for the screening of the other amino acid precatalyst modules 5 (Scheme 4). As the results in Table 2 show, except for 5g (entry 7), which gave a much lower ee value of the product ent-3a than 5a did (entry 2), all the other amino acid precatalysts are not reactive at all (entries 3–6 and 8–11). Since 5a gave the best product ee values (entry 2), it was chosen for the screening the cinchona alkaloid-derived precatalyst modules 4 (Scheme 4). For quinidine-derived 6′-thioureas (4c–4g), they all led to the formation of the desired product in quite similar yields and stereoselectivities (entries 12 and 14–16), except for the module 4d (entry 13), which gave only trace amount of product. Similar results were also obtained by using the quinidine-derived 6′-ureas (4h–4l, entries 17–21). While some of these modules (such as 4c, 4h, 4j, and 4l) produces slightly better diastereoselectivities than that of 4b, the ee values obtained are usually lower. In contrast, quinidine-derived squaramide 4m (entry 22) gave a much lower ee value of the product than QDT (4a) and the 6′-thiourea 4b do. Next, quinine-derived thiourea (4n, QNT) and 6′-(thio)ureas were screened as the modules. It was found that when QNT (4n) was used together with 5a as the MDO, no desired product was obtained (entry 23). In contrast, when QNT (4n) was used together with D-Pro (5b) to form the MDO, the desired product was obtained in a decent yield and good stereoselectivities (entry 24). Comparing these results with those in entries 2 and 3, it is clear that there is a strong match-mismatch between the amino acid module and the cinchona alkaloid module9a in this reaction: L-Pro (5a) matches with quinidine derivatives, while D-pro (5b) matches with quinine derivatives.9a Because of this match-mismatch effect, the diastereodivergent catalysis9 we normally observed with the MDO-catalysis is not possible. Further screening revealed that the MDO of 4o/5b, which is the pseudo-enantiomer of 4b/5a, yielded the highest yield and stereoselectivities of the desired domino product (entry 25). The other quinine-derived 6′-thiourea modules (4p–4r) or 6′-urea modules (4s–4v) all yielded worse stereoselectivities (entries 26–32). It should be pointed out that the opposite enantiomer (i.e., 3a) was obtained as the major product when 5b (D-Pro) was used as the amino acid module and, therefore, the product absolute stereochemistry is completely determined the amino acid module. Next, the acid cocatalyst (Scheme 4) was screened, and it turned out that worse results than that of 6a in terms of both the product yields and stereoselectivities were obtained for all of the other cycloalkane carboxylic acids (6b–6g), acetic acid (6h), benzoic acid (6i), trans-cinnamic acid (6j), crotonic acid (6k), and nonanoic acid (6l) (entries 33–42 vs. entry 25).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Modules/acid | Yieldb (%) | drc | eed (%) | ||
| a All reactions were carried out with 1a (0.12 mmol), 2a (0.10 mmol), the precatalyst modules 4 and 5 (0.010 mmol each, 10 mol%), and the acid cocatalyst 6 (0.010 mmol, 10 mol%) in dry toluene (1.0 mL) at room temperature for 24 h. b Yield of the isolated major diastereomeric product after flash column chromatography. c Determined by 1H NMR analysis of the crude reaction product. d Determined by HPLC analysis on a ChiralPak IC column. e The opposite enantiomer was obtained as the major product. | ||||||
| 1 | 4a | 5a | 6a | 53 | 80:20 |
58 |
| 2 | 4b | 5a | 6a | 77 | 89:11 |
78 |
| 3 | 4b | 5b | 6a | Trace | — | — |
| 4 | 4b | 5d | 6a | Trace | — | — |
| 5 | 4b | 5e | 6a | Trace | — | — |
| 6 | 4b | 5f | 6a | Trace | — | — |
| 7 | 4b | 5g | 6a | 81 | 93:7 |
9 |
| 8 | 4b | 5h | 6a | Trace | — | — |
| 9 | 4b | 5i | 6a | 0 | — | — |
| 10 | 4b | 5j | 6a | 0 | — | — |
| 11 | 4b | 5k | 6a | 0 | — | — |
| 12 | 4c | 5a | 6a | 84 | 96:4 |
61 |
| 13 | 4d | 5a | 6a | Trace | — | — |
| 14 | 4e | 5a | 6a | 66 | 87:13 |
68 |
| 15 | 4f | 5a | 6a | 78 | 89:11 |
77 |
| 16 | 4g | 5a | 6a | 70 | 84:16 |
69 |
| 17 | 4h | 5a | 6a | 79 | 96:4 |
63 |
| 18 | 4i | 5a | 6a | 80 | 94:6 |
68 |
| 19 | 4j | 5a | 6a | 76 | 93:7 |
63 |
| 20 | 4k | 5a | 6a | 50 | 70:30 |
68 |
| 21 | 4l | 5a | 6a | 87 | 94:6 |
73 |
| 22 | 4m | 5a | 6a | 70 | 83:17 |
35 |
| 23 | 4n | 5a | 6a | 0 | — | — |
| 24 | 4n | 5b | 6a | 56 | 82:18 |
55e |
| 25 | 4o | 5b | 6a | 89 |
95:5 |
78 |
| 26 | 4p | 5b | 6a | 85 | 94:6 |
72e |
| 27 | 4q | 5b | 6a | 88 | 98:2 |
77e |
| 28 | 4r | 5b | 6a | 83 | 94:6 |
73e |
| 29 | 4s | 5b | 6a | 33 | 86:14 |
65e |
| 30 | 4t | 5b | 6a | 27 | 71:29 |
65e |
| 31 | 4u | 5b | 6a | 35 | 88:12 |
70e |
| 32 | 4v | 5b | 6a | 66 | 72:28 |
39e |
| 33 | 4o | 5b | 6b | 63 | 75:25 |
62e |
| 34 | 4o | 5b | 6c | 62 | 83:17 |
75e |
| 35 | 4o | 5b | 6d | 65 | 80:20 |
71e |
| 36 | 4o | 5b | 6e | 72 | 93:7 |
74e |
| 37 | 4o | 5b | 6f | 76 | 90:10 |
77e |
| 38 | 4o | 5b | 6g | 57 | 71:29 |
70e |
| 39 | 4o | 5b | 6h | Trace | — | — |
| 40 | 4o | 5b | 6i | 63 | 93:7 |
78e |
| 41 | 4o | 5b | 6j | 60 | 60:40 |
75e |
| 42 | 4o | 5b | 6k | 60 | 88:12 |
78e |
| 43 | 4o | 5b | 6l | 73 | 90:10 |
73e |
The solvent used in this reaction was then optimized using the best catalyst system 4o/5b/6a. As the results in Table 3 show, all the other organic solvents we screened are inferior to toluene in terms of the product yield and/or stereoselectivities (entries 2–13). Among these solvents, xylene (entry 3), trifloromethylbenzene (entry 5), and ethereal solvents, such as THF (entry 6) and cyclopentenyl methyl ether (entry 7), are especially poor, since no desired product could be isolated from the reactions conducted in them. Finally, the reaction was carried out in toluene at 0 °C (entry 14). Although a slightly higher ee value of the product was obtained (84% ee), the yield and diastereoselectivity of this reaction dropped significantly.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Yieldb (%) | drc | eed (%) |
| a Unless otherwise indicated, all reactions were carried out with 1a (0.12 mmol), 2a (0.10 mmol), the precatalyst modules 4o and 5b (0.010 mmol each, 10 mol%), and the acid cocatalyst 6a (0.010 mmol, 10 mol%) in the specified dry solvent (1.0 mL) at room temperature for 24 h. b Yield of the isolated major diastereomeric product after flash column chromatography. c Determined by 1H NMR analysis of the crude reaction product. d Determined by HPLC analysis on a ChiralPak IC column. e The reaction was performed at 0 °C. | ||||
| 1 | Toluene | 89 | 95:5 |
78 |
| 2 | Benzene | 73 | 86:14 |
79 |
| 3 | Xylene | Trace | — | — |
| 4 | C6H5Cl | 50 | 72:28 |
76 |
| 5 | C6H5CF3 | Trace | — | — |
| 6 | THF | <5 | — | — |
| 7 | c-C5H9OMe | Trace | — | — |
| 8 | CH2Cl2 | 65 | 81:19 |
60 |
| 9 | CHCl3 | 75 | 90:10 |
76 |
| 10 | ClCH2CH2Cl | 71 | 88:12 |
68 |
| 11 | MeOH | 63 | 96:4 |
30 |
| 12 | MeCN | 85 | 97:3 |
51 |
| 13 | DMSO | 67 | 83:27 |
56 |
| 14 | Toluene | 40 | 61:39 |
84e |
Once the reaction conditions were fully optimized, the scope of this reaction was probed. As the results in Table 4 show, besides trans-cinnamaldehyde (2a, entry 1), cinnamaldehydes with a substituent on the phenyl ring are also good substrates for this reaction (entries 2–7). While most of the substituted cinnamaldehydes gave very similar product ee values, 4-methoxy-substituted cinnamaldehyde gave a much lower ee value of the product (entry 2); however, no trend was observed for the electronic effects of the substituents on the yields and stereoselectivities of this reaction. In contrast, the use of trans-crotonaldehyde in this reaction led to the formation of a complex reaction mixture (entry 8). On the other hand, enonals with a substituent on the para position of the phenyl ring are all good substrates for this reaction, and the electronic effects of these substituents have minimal influence on the product yields and stereoselectivities (entries 9–13). When the same substituent (i.e., bromo) was placed instead in the meta position of the phenyl ring, there was almost no change in the product yields and ee values (entry 14 vs. entry 13), although the diastereoselectivity observed was slightly better for the meta-substituted substrate. Nonetheless, when this substituent was place in the ortho-position, only trace amount of the desired product was formed (entry 15). This negative result most likely was due to the steric effects of the ortho-bromo substituent.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | 3/Yieldb (%) | drc | eed (%) |
| a All reactions were carried out with 1 (0.12 mmol), 2 (0.10 mmol), the precatalyst modules 4o and 5b (0.010 mmol each, 10 mol%), and the acid cocatalyst 6a (0.010 mmol, 10 mol%) in dry toluene solvent (1.0 mL) at room temperature for 24 h. b Yield of the isolated major diastereomeric product after flash column chromatography. c Determined by 1H NMR analysis of the crude reaction product. d Determined by HPLC analysis. e A complex mixture was obtained. | ||||||
| 1 | Ph | Ph | 24 | 3a/89 | 95:5 |
78 |
| 2 | Ph | 4-MeOC6H4 | 72 | 3b/50 | 84:16 |
45 |
| 3 | Ph | 4-MeC6H4 | 48 | 3c/76 | 90:10 |
61 |
| 4 | Ph | 4-FC6H4 | 48 | 3d/70 | 90:10 |
79 |
| 5 | Ph | 4-ClC6H4 | 48 | 3e/65 | 84:16 |
67 |
| 6 | Ph | 4-BrC6H4 | 48 | 3f/72 | 88:12 |
62 |
| 7 | Ph | 4-CNC6H4 | 72 | 3g/56 | 80:20 |
76 |
| 8 | Ph | Me | 48 | 0e | — | — |
| 9 | 4-MeOC6H4 | Ph | 48 | 3h/68 | 92:8 |
80 |
| 10 | 4-MeC6H4 | Ph | 48 | 3i/72 | 92:8 |
66 |
| 11 | 4-FC6H4 | Ph | 48 | 3j/68 | 88:12 |
72 |
| 12 | 4-ClC6H4 | Ph | 48 | 3k/70 | 91:9 |
76 |
| 13 | 4-BrC6H4 | Ph | 48 | 3l/61 | 80:20 |
77 |
| 14 | 3-BrC6H4 | Ph | 48 | 3m/68 | 92:8 |
72 |
| 15 | 2-BrC6H4 | Ph | 48 | Trace | — | — |
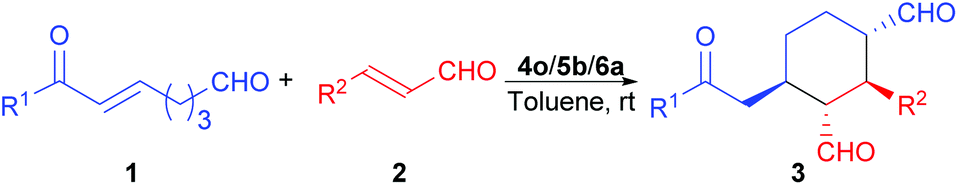
The relative stereochemistry of the reaction product was determined by COSY experiments and the coupling constants using compound 3l (for details, please see the ESI†). Since we could not grow a single crystal from the reaction products or their derivatives, the absolute stereochemistry of the reaction product was determined by comparing the major enantiomer obtained in our reaction and the major enantiomer obtained by using the Xu's catalytic system11b (for details, please see the ESI†). Based on the reported computational study of the MDO catalysis13 and our previous reported results,9 the following transition states were proposed to explain the stereochemical outcome of this reaction (Scheme 5). As shown in transition state 7, the enamine intermediate formed from 1a and D-Pro (5b) and the iminium intermediate formed from 2a and D-Pro (5b) self-assembled with the 6′-thiourea 4o through hydrogen bonds between the carboxylic acid group of the D-Pro moieties and the ammonium as well as the thiourea moieties of 4o. The attack of the (E)-enamine onto the Re face of α,β-unsaturated iminium intermediate from the back is favored, which leads to the formation the intermediate with an (S,S)-stereochemistry for the two newly formed stereogenic centers (please see structure 8). After the hydrolytic removal of one of the D-Pro moieties, the intermediate adopts a chair conformation and reassembles with 4o, with a new hydrogen bond being formed between the keto group of the enone and the ammonium of 4o (transition state 9). An intramolecular Michael addition of the enamine onto the enone moiety yields the expected product 3a after a hydrolytic removal of the protonated MDO (4o/5b/H+).
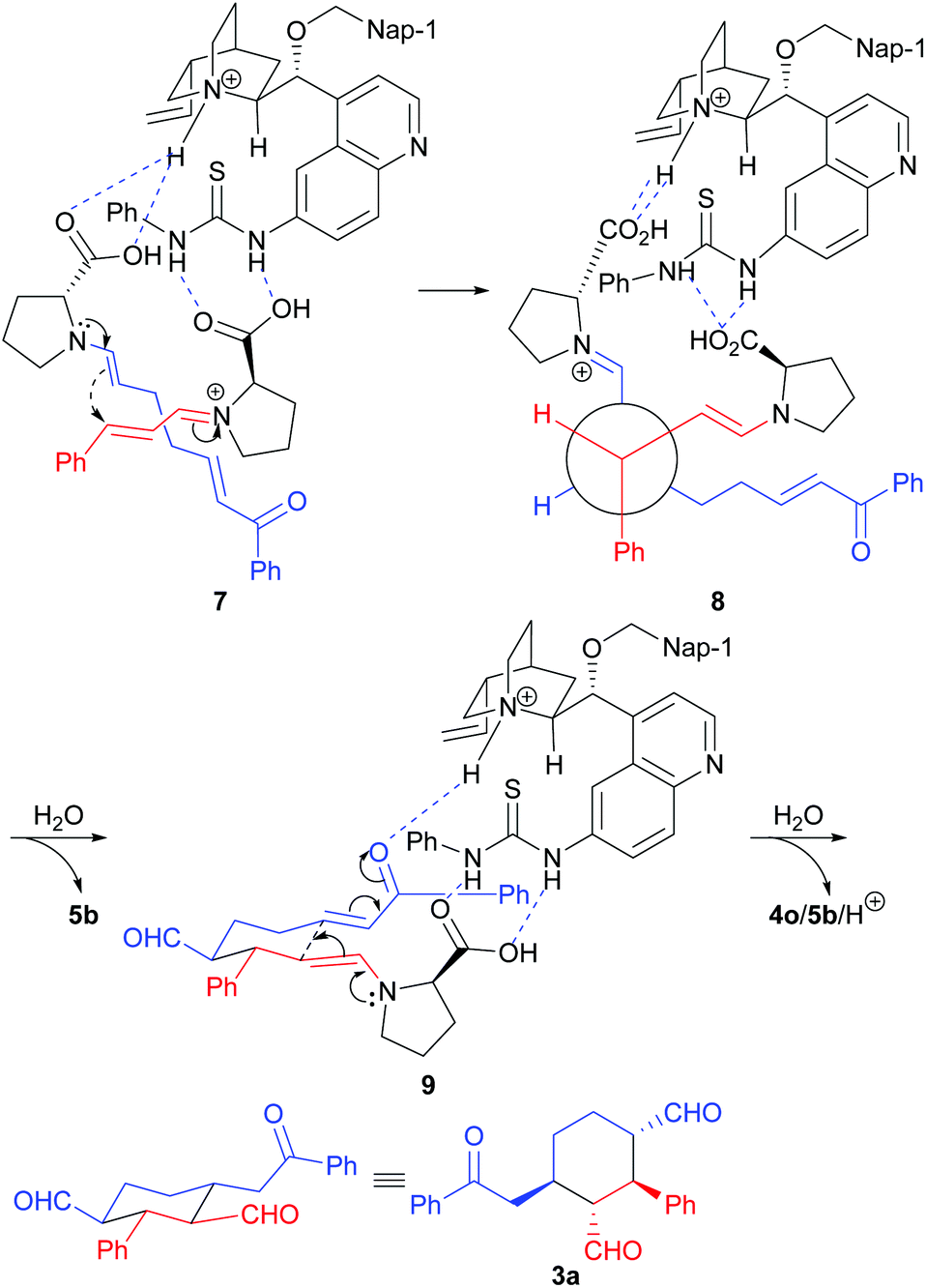 | ||
| Scheme 5 Proposed favored transition states for the formations of product 3a. | ||
Finally, some synthetic maneuvers were conducted to demonstrate the utility of the products. As shown in Scheme 6, selectively reactions of the two aldehyde groups in compound 3a with methyl (triphenylphosphoranylidene)acetate can be achieved: by using appropriate the loadings of the latter reagent, compounds 10 or 11 can be obtained with complete retention of the stereochemistry.
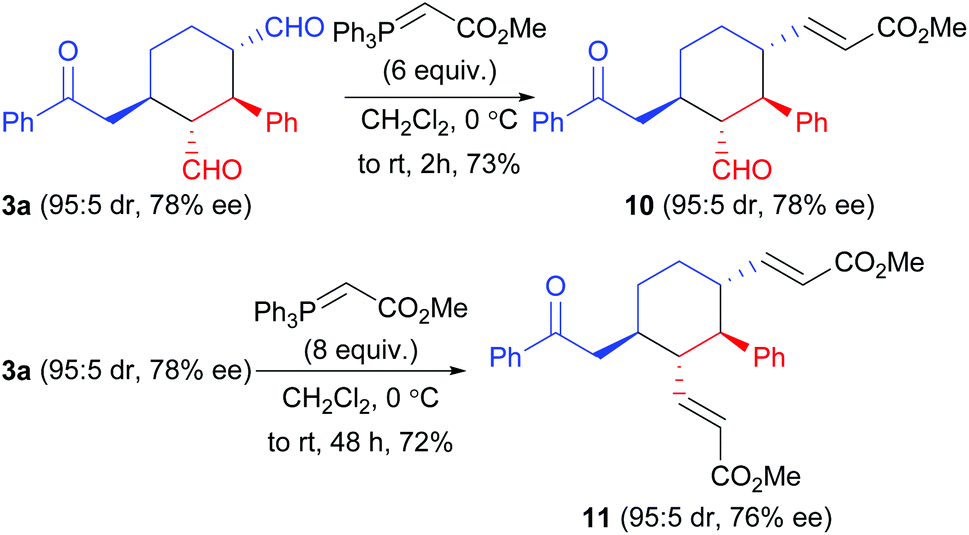 | ||
| Scheme 6 Derivatization of the reaction product 3a. | ||
Conclusions
In summary, we have demonstrated that the enamine catalysis mode of the pyrrolidine can be switched on or off by temperature, while the iminium catalysis mode of the modularly designed organocatalysts (MDOs), which is switched off by the cinchona module, can be switched on by an acid cocatalyst. Based on these properties, we have developed two catalytic systems for the simultaneous enamine and iminium activations: a ternary system containing the MDO and pyrrolidine and a binary MDO system plus an acid cocatalyst. With the more selective binary system, we have achieved good yields and stereoselectivities in the domino Michael/Michael reaction between (E)-7-aryl-7-oxohept-5-enals and trans-cinnamaldehydes under the optimized conditions.Experimental
General information
All reactions were conducted in 17 × 60 mm glass vials and monitored by TLC on silica gel plates (200 μm) and visualized by UV. Column chromatography was performed on silica gel (32–63 μ). 1H NMR spectra was recorded on a 500 MHz or a 300 MHz spectrometer (126 MHz or 75 MHz for 13C NMR). Infrared spectra were measured on a Bruker Vector 22 instrument. Enantiomeric excesses (ee) were determined by chiral HPLC analysis using a Shimadzu instrument. ChiralPak IC, ID, columns (4.6 mm × 250 mm) were purchased from Daicel Chemical Industries. HRMS were conducted by the RCMI Core Facilities, Department of Chemistry, UTSA. All starting materials used in this study are known compounds and were either commercially available or synthesized according to the literature procedures.14trans-Cinnamaldehyde (2a) was washed with saturated NaHCO3 solution and water, dried, and redistilled prior to use. All of the precatalyst modules, except for 4d, 4e, 4f, 4h, 4j, 4l, 4o, 4q, 4s, 4t, and 4u, are known compounds, and were either commercially available or synthesized according to the reported procedures.15 Those new modules were similarly synthesized from the corresponding cinchona-derived amines by reacting with the desired isocyanates or isothiocyanates.14 All the reagents were purchased from commercial sources and used as received. Solvents were dried according to standard protocols prior to use.Experimental procedures
:30 hexane/EtOAc to give product 3a–m.
:25 hexane/EtOAc to give product 10 (28.5 mg, 73% yield).
:25 hexane/EtOAc to give product 11 (32.2 mg, 72% yield).
:30 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 18.5 min, minor enantiomer: tR = 21.8 min.
:30 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 34.4 min, minor enantiomer: tR = 37.5 min.
:30 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 18.0 min, minor enantiomer: tR = 22.3 min.
:30 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 18.6 min, minor enantiomer: tR = 22.2 min.
:30 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 14.7 min, minor enantiomer: tR = 18.3 min.
:30 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 16.2 min, minor enantiomer: tR = 20.2 min.
:40 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 34.6 min, minor enantiomer: tR = 48.9 min.
:40 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 25.2 min, minor enantiomer: tR = 28.9 min.
:40 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 22.3 min, minor enantiomer: tR = 32.2 min.
:30 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 21.2 min, minor enantiomer: tR = 31.0 min.
:30 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 25.3 min, minor enantiomer: tR = 38.0 min.
:20 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 36.5 min, minor enantiomer: tR = 49.2 min.
:30 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 25.4 min, minor enantiomer: tR = 44.3 min.
:30 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 19.7 min, minor enantiomer: tR = 31.3 min.
:30 hexanes/i-PrOH at 1.0 mL min−1, λ = 254 nm), major enantiomer: tR = 25.4 min, minor enantiomer: tR = 43.4 min.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Welch Foundation (Grant No. AX-1593) and the National Science Foundation (Grant No. CHE-1664278) for the financial support of this project. We thank Dr Daniel Wherritt for help with the 2-D COSY experiments.Notes and references
- Molecular Switches, ed. B. L. Feringa and W. R. Browne, Wiley-VCH, Weinheim, 2011, vol. 1–2 Search PubMed.
- (a) Enzyme Dynamics and Regulation, ed. P. B. Chock, C. Y. Huang, C. L. Tsou and J. H. Wang, Springer, New York, 1988 Search PubMed; (b) T. Traut, Allosteric Regulatory Enzymes, Springer, New York, 2008 CrossRef.
- For reviews, see: (a) U. Lüning, Angew. Chem., Int. Ed., 2012, 51, 8163–8165 CrossRef; (b) V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC; (c) J. Choudhury, Tetrahedron Lett., 2018, 59, 487–495 CrossRef CAS.
- A. Ueno, K. Takahashi and T. Osa, J. Chem. Soc., Chem. Commun., 1980, 837–838 RSC.
- (a) L. Osorio-Planes, C. Rodríguez-Escrich and M. A. Pericàs, Org. Lett., 2014, 16, 1704–1707 CrossRef CAS; (b) Z. Dai, Y. Cui, C. Chen and J. Wu, Chem. Commun., 2016, 52, 8826–8829 RSC.
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- (a) A. Berkessel and H. Groeger, Asymmetric Organocatalysis, Wiley-VCH, Weinheim, 2005 CrossRef; (b) Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions and Applications, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2013 Search PubMed.
- (a) V. Blanco, A. Carlone, K. D. Hänni, D. A. Leigh and B. Lewandowski, Angew. Chem., Int. Ed., 2012, 51, 5166–5169 CrossRef CAS; (b) V. Blanco, D. A. Leigh, U. Lewandowska, B. Lewandowski and V. Marcos, J. Am. Chem. Soc., 2014, 136, 15775–15780 CrossRef CAS; (c) C.-S. Kwan, A. S. C. Chan and K. C.-F. Leung, Org. Lett., 2016, 18, 976–979 CrossRef CAS PubMed.
- For selected examples, see: (a) T. Mandal and C.-G. Zhao, Angew. Chem., Int. Ed., 2008, 47, 7714–7717 CrossRef CAS; (b) N. K. Rana, H. Huang and J. C.-G. Zhao, Angew. Chem., Int. Ed., 2014, 53, 7619–7623 CrossRef CAS; (c) H. Huang, S. Konda and J. C.-G. Zhao, Angew. Chem., Int. Ed., 2016, 55, 2213–2216 CrossRef CAS; (d) S. Jakkampudi, R. Parella, H. D. Arman and J. C.-G. Zhao, Chem. – Eur. J., 2019, 25, 7515–7520 CrossRef CAS; (e) S. Jakkampudi, S. Konda, H. Arman and J. C.-G. Zhao, Adv. Synth. Catal., 2020, 362, 2419–2426 CrossRef CAS; (f) P. Bora, S. Jakkampudi, R. Parella, N. Sakkani, Q. Dai, M. Bihani, H. D. Arman and J. C. G. Zhao, Chem. Commun., 2021, 57, 5334–5337 RSC. For a review on diastereodivergent catalysis, see: (g) M. Bihani and J. C.-G. Zhao, Adv. Synth. Catal., 2017, 359, 534–575 CrossRef CAS.
- For a review, see: T. Chanda and J. C.-G. Zhao, Adv. Synth. Catal., 2018, 360, 2–79 CrossRef CAS.
- (a) F.-Z. Bao, X.-B. Wang and L.-Y. Kong, Tetrahedron Lett., 2013, 54, 1405–1408 CrossRef CAS; (b) Z.-L. Jia, Y. Wang, G.-Q. Xu and P.-F. Xu, Chem. Commun., 2017, 53, 4938–4941 RSC; (c) B.-C. Hong, R. Y. Nimje and J.-H. Liao, Org. Biomol. Chem., 2009, 7, 3095–3101 RSC.
- Hayashi reported a binary catalyst system that was believed to catalyze the addition of ketones to α,β-enones via the enamine-iminium activation originally, only to find out that the reaction went through an enolate-iminium activation, see: N. Umekubo, T. Terunuma, E. Kwon and Y. Hayashi, Chem. Sci., 2020, 11, 11293–11297 RSC . For a reaction catalyzed by a similar binary system, see: V. Maurya, M. S. Kutwal and C. Appayee, Org. Lett., 2021, 23, 1566–1571 CrossRef CAS.
- B. Bhaskararao and R. B. Sunoj, Chem. Sci., 2018, 9, 8738–8747 RSC.
- For the synthesis of enonals 1, see: E. L. Richards, P. J. Murphy, F. Dinon, S. Fratucello, P. M. Brown, T. Gelbrich and M. B. Hursthouse, Tetrahedron, 2001, 57, 7771–7784 CrossRef CAS . For synthesis of enals 2, see: (a) L. Legoabe, J. Kruger, A. Petzer, J. J. Bergh and J. P. Petzer, Eur. J. Med. Chem., 2011, 46, 5162–5174 CrossRef CAS PubMed; (b) M. L. N. Rao, P. Dasgupta and V. N. Murty, RSC Adv., 2015, 5, 24834–24845 RSC; (c) M. K. Gupta, Z. Li and T. S. Snowden, J. Org. Chem., 2012, 77, 4854–4860 CrossRef CAS.
- (a) B. Vakulya, S. Varga, A. Csámpai and T. Soós, Org. Lett., 2005, 7, 1967–1969 CrossRef CAS; (b) Y. Liu, B. Sun, B. Wang, M. Wakem and L. Deng, J. Am. Chem. Soc., 2009, 131, 418–419 CrossRef CAS; (c) W. Yang and D.-M. Du, Org. Lett., 2010, 12, 5450–5453 CrossRef CAS; (d) S. Mitsumori, H. Zhang, P. H.-Y. Cheong, K. N. Houk, F. Tanaka and C. F. Barbas, J. Am. Chem. Soc., 2006, 128, 1040–1041 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob01991k |
| This journal is © The Royal Society of Chemistry 2022 |