
Synthesis of alkynamides through reaction of alkyl- or aryl-substituted alkynylaluminums with isocyanates†
Soohong
Cho‡
a,
Yeonjoo
Lee‡
b,
Kyeongmin
Lee
a,
Hwiwoong
Lee
a,
Yunmi
Lee
 *a and
Byunghyuck
Jung
*b
*a and
Byunghyuck
Jung
*b
aDepartment of Chemistry, Kwangwoon University, Seoul 01897, Republic of Korea. E-mail: ymlee@kw.ac.kr
bDepartment of Emerging Materials Science, DGIST, Daegu 42988, Republic of Korea. E-mail: byunghyuck.jung@dgist.ac.kr
First published on 2nd December 2021
Abstract
An efficient and facile method for the preparation of alkynamides through Et3N-catalyzed alumination of alkyl- or aryl-substituted terminal alkynes with AlMe3 and sequential nucleophilic addition of in situ generated alkynylaluminums to isocyanates is described. This method has the merits of using readily available isocyanates and monosubstituted alkynes, easy access to organoaluminums, short reaction times, and high efficiency. A gram-scale synthesis of the desired alkynamide and its application to the formation of α-methylene-β-lactams demonstrates the synthetic utility of this method.
Introduction
Organoaluminums possess strong dual character, Lewis acidity owing to Al, and nucleophilicity because of the carbon atom bound to Al.1 This intrinsic reactivity affords various synthetic advantages to organoaluminums over other organometallic reagents (metal = Li, B, Mg, Zn, Sn, etc.), leading to their wide applications in organic synthesis. In particular, alkynylaluminums have been used to form Csp–Csp3, –Csp2, or –Csp bonds through cross-coupling reactions.2 They also undergo nucleophilic additions to carbonyls and sulfinyl aldimines.3 However, the use of alkynylaluminums for the construction of alkynamides has not been reported so far.Alkynamides are important building blocks for preparing potential drugs and functional materials as well as useful intermediates in synthetic organic chemistry.4 However, despite their wide range of synthetic applications, very few approaches have been developed for their synthesis compared to those for α,β-unsaturated amides or alkanamides.5 One conventional method for preparing alkynamides is a coupling reaction between alkynoic acids and primary or secondary amines in the presence of acid-activating reagents.6 However, it is challenging to synthesize some alkynoic acids and coupling reactions require a stoichiometric amount of activating reagent. Therefore, various alternative methods have been developed for the synthesis of alkynamides (Scheme 1). One such synthetic route involves Pd/Cu-catalyzed C–C bond formation between terminal alkynes and carbamoyl chlorides (Scheme 1a).7 Another method involves transition-metal-catalyzed oxidative aminocarbonylation of terminal alkynes or alkynoic acids with amines, including tertiary amines, in the presence of CO sources such as CO gas or Co2(CO)8 (Scheme 1b). Hoberg, Gabriele, Yamamoto, Bhanage, Xia, Kollár and their co-workers described the use of Ni- or Pd-catalysts for the aminocarbonylation of alkynes or haloalkynes using amines and carbon monoxide as the sources of amide moieties, respectively (Scheme 1b-i).8 Lee's group disclosed Pd-catalyzed decarboxylative aminocarbonylation using alkynoic acids, instead of terminal alkynes, and secondary or tertiary amines (Scheme 1b-ii).9 Dicobalt octacarbonyl (Co2(CO)8) was also used as an excellent source of carbonyl moiety. Wu et al. reported the synthesis of alkynamides from bromoalkynes, secondary amines, and Co2(CO)8 (Scheme 1b-iii).10 The other synthetic approach involves the use of isocyanides and water as the sources of amide moieties (Scheme 1c). Wang, Ji, and co-workers reported a cobalt-catalyzed three-component reaction of terminal alkynes with isocyanides and water to obtain alkynamides (Scheme 1c-i).11 The in situ generated Ag(I)-DPEphos complex from AgNO3 could activate arylacetylenes to promote the insertion of isocyanides into the Csp–H of terminal alkynes. Pd-catalyzed version of three-component reactions reported by Salehi et al. replaced terminal alkynes with gem-dibromoalkenes (Scheme 1c-ii).12 The reaction of gem-dibromoalkenes with water and isocyanides through palladium catalysis generated α-bromo acrylamides, followed by the elimination of HBr with cesium carbonate to provide the desired alkynamides.
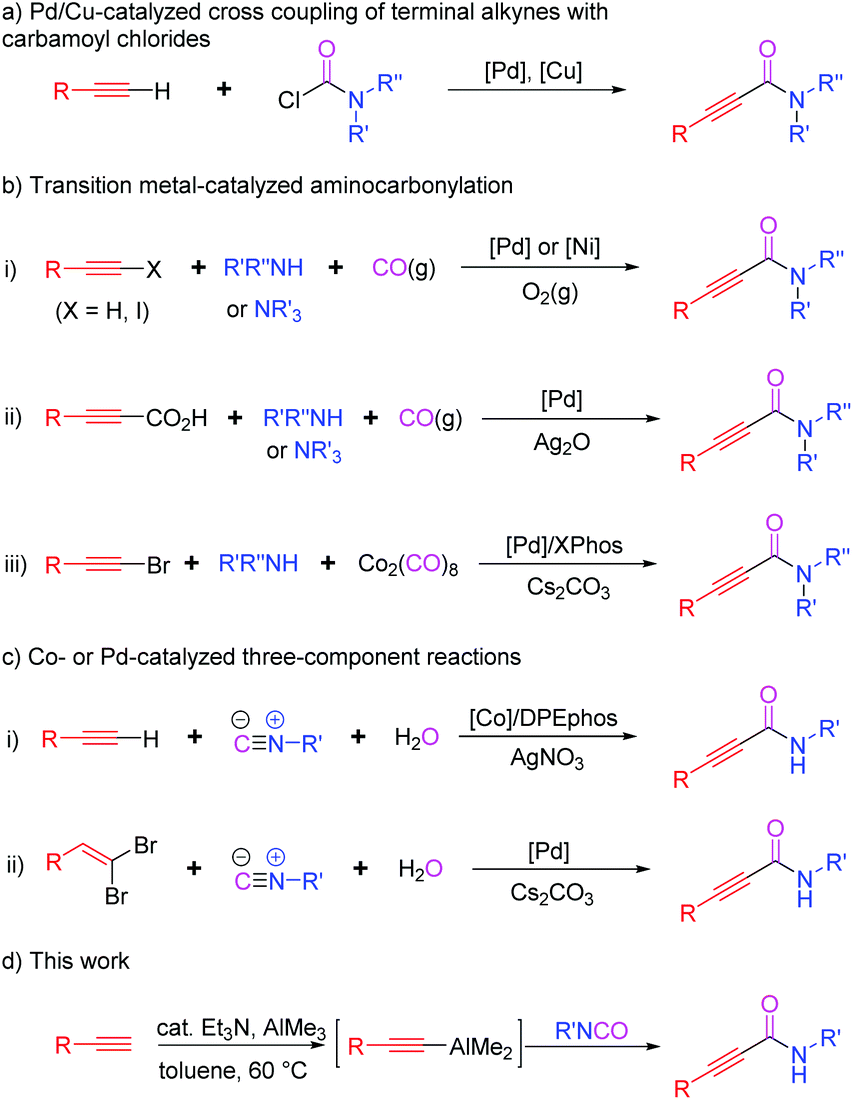 | ||
| Scheme 1 Synthesis of alkynamides. | ||
Although transition-metal catalysts show excellent performance, the development of a more efficient and practical method for the straightforward synthesis of alkynamides is still required. Moreover, as transition metals are costly and environment unfriendly, a transition metal-free method is highly desirable. Recently, we reported the one-pot synthesis of α-silyl-α,β-unsaturated amides through solvent-controlled stereoselective hydroalumination of Si-substituted arylacetylenes with diisobutylaluminum hydride (DIBAL-H) and the concomitant stereospecific reaction of in situ generated alkenylaluminums with isocyanates.13 Based on our previous work, we propose that the reaction of easily accessible isocyanates with organoaluminum reagents starting from terminal alkynes could be a transition-metal-free and efficient synthetic route for the construction of alkynamides. To the best of our knowledge, despite the broad applicability of isocyanates and organoaluminums, Al-mediated amidation of terminal alkynes with isocyanates have not been reported yet. Although protocols for the reaction of isocyanates with alkynyllithiums or -magnesiums for the synthesis of alkynamides are known, the substrate scope has not been investigated in detail, showing very limited examples.14 Herein, we describe the straightforward amidation of terminal alkynes through in situ generation of aluminum acetylides and their successive nucleophilic addition to isocyanates (Scheme 1d). Synthesis of a wide range of functionalized alkynamides, gram-scale synthesis, and β-lactam formation from alkynamides have also been discussed to demonstrate their synthetic utility.
Results and discussion
We initiated our study using phenylacetylene (1a) as a model substrate, trimethylaluminum as the alumination source, and phenyl isocyanate (3a) as an amide source (Table 1). When dimethylaluminum phenylacetylide 2a generated in situ from Et3N-catalyzed alumination of 1a with AlMe3 under the reaction conditions described previously by Micouin et al.15 was treated with isocyanate 3a in hexanes at 60 °C, the desired alkynamide 4aa was obtained in 72% yield along with double-alkyne addition product 5aa in 25% yield; no methyl addition product was observed (entry 1). The initial exploratory screening suggested that the use of 1.2 equiv. of 2a and reduced reaction temperature were sufficient to complete the reaction while suppressing the generation of by-product 5aa without compromising the efficiency (entries 2–5). The coordinating solvent, tetrahydrofuran, was not effective in the nucleophilic addition of 2a to 3a and toluene was selected as the optimal solvent (entries 6–9). Alkyne 1a was successfully transformed to the desired alkynamide 4aa in >98% yield when amidation was performed in toluene at −78 °C for 1 h (entries 10–13).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 2a (equiv.) | Solvent | Conc. (M) | Temp. (°C) | Time (h) |
4aa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5aa (%)b :5aa (%)b |
| a Reaction conditions: alkyne 1a (1.1 equiv. based on AlMe3), Et3N (10 mol% based on AlMe3), AlMe3 (0.36–0.60 mmol), alkynyl–Al (2, 0.36–0.60 mmol), PhNCO (3a, 0.30 mmol), and solvent (0.05–0.1 M) under N2. b Determined by 1H NMR spectrum analysis using 1,3,5-trimethoxybenzene as an internal standard. | ||||||
| 1 | 2.0 | Hexanes | 0.05 | 60 | 2 | 72:25 |
| 2 | 2.0 | Hexanes | 0.05 | 22 | 2 | 81:11 |
| 3 | 1.5 | Hexanes | 0.05 | 22 | 2 | 80:8 |
| 4 | 1.2 | Hexanes | 0.05 | 22 | 2 | 80:6 |
| 5 | 1.2 | Hexanes | 0.05 | 0 | 2 | 85:5 |
| 6 | 1.2 | Hexanes | 0.1 | 0 | 2 | 85:4 |
| 7 | 1.2 | CH2Cl2 | 0.1 | 0 | 2 | 80:4 |
| 8 | 1.2 | THF | 0.1 | 0 | 2 | <2 |
| 9 | 1.2 | Toluene | 0.1 | 0 | 2 | 90:3 |
| 10 | 1.2 | Toluene | 0.1 | −78 | 2 | >98:<2 |
| 11 | 1.2 | Toluene | 0.1 | −78 | 1 | >98:<2 |
| 12 | 1.2 | Toluene | 0.1 | −78 | 30 min | 97:<2 |
| 13 | 1.2 | Toluene | 0.1 | −78 | 10 min | 78:<2 |

With the optimized reaction conditions in hand, we explored the substrate scope of the isocyanates, as shown in Scheme 2. Amidation reactions of in situ generated phenylacetyl–Al reagent 2a with aryl isocyanates bearing an electron-donating methoxy group (3b) or strong electron-withdrawing groups such as –CN (3f) and –CF3 (3g) at the para-position of the benzene ring proceeded efficiently with similar performance to that of 3a. Additional observations of the smooth conversion of aryl isocyanates substituted with halogens (3c–e) at the para-position indicated that the efficiency was not affected by the electronic circumstances of the isocyanates. Various para-substituted aryl alkynamides, 4ab–4ag, were obtained in high yields. Aryl isocyanate 3l with a meta-methoxy group also worked well in this amidation, affording the desired alkynamide 4al in 91% yield. In contrast, the reaction of isocyanates 3i–k bearing ortho-substituted aryl moieties was sluggish under the standard reaction conditions. An elevated reaction temperature of −20 °C was required for efficient amidation with 3i, producing the desired alkynamide 4ai in 98% yield (vs. 54% yield at −78 °C). The ester group in the para- or ortho-position of the phenyl isocyanate was well tolerated by the nucleophilic addition of aluminium acetylide 2a, delivering the desired products 4ah and 4ak in 66% and 76% yields, respectively. Notably, amidation of 2a with 3h required higher reaction temperature to improve the yield of 4ah due to the poor solubility of 3h (66% yield at 0 °C vs. 28% yield at −78 °C). More sterically demanding isocyanates 3m (mesityl), 3n (2,6-(i-Pr)2C6H3), and 3o (1-naphthyl) were smoothly converted to the corresponding amides 4am-o in 72–98% yield under the modified reaction conditions. Reaction temperatures higher than −20 °C were not used because of the increase in double-alkyne addition by-products, which decreased the yield of the desired products. Aliphatic isocyanates were also suitable for amidation with alkynylaluminum reagents. The reaction of 1a with isocyanates bearing a sterically less hindered benzyl (3p) or furfuryl substituent (3q) provided the corresponding amides 4ap (89%) and 4aq (69%) under the established reaction conditions; however, sterically demanding isocyanate 3r bearing a cyclohexyl group required an elevated reaction temperature to afford the desired amide 4ar in 91% yield. In particular, reaction with tert-butyl isocyanate 3s at −20 °C was not selective and efficient, allowing the formation of the desired amide 4ar in 36% yield along with the double-alkyne addition product 5ar (28%) and the methyl-addition product (18%). The complete transformation of an isocyanate with a chloroethyl substituent into amide 4at (98%) showed tolerance to alkyl halides under the reaction conditions.
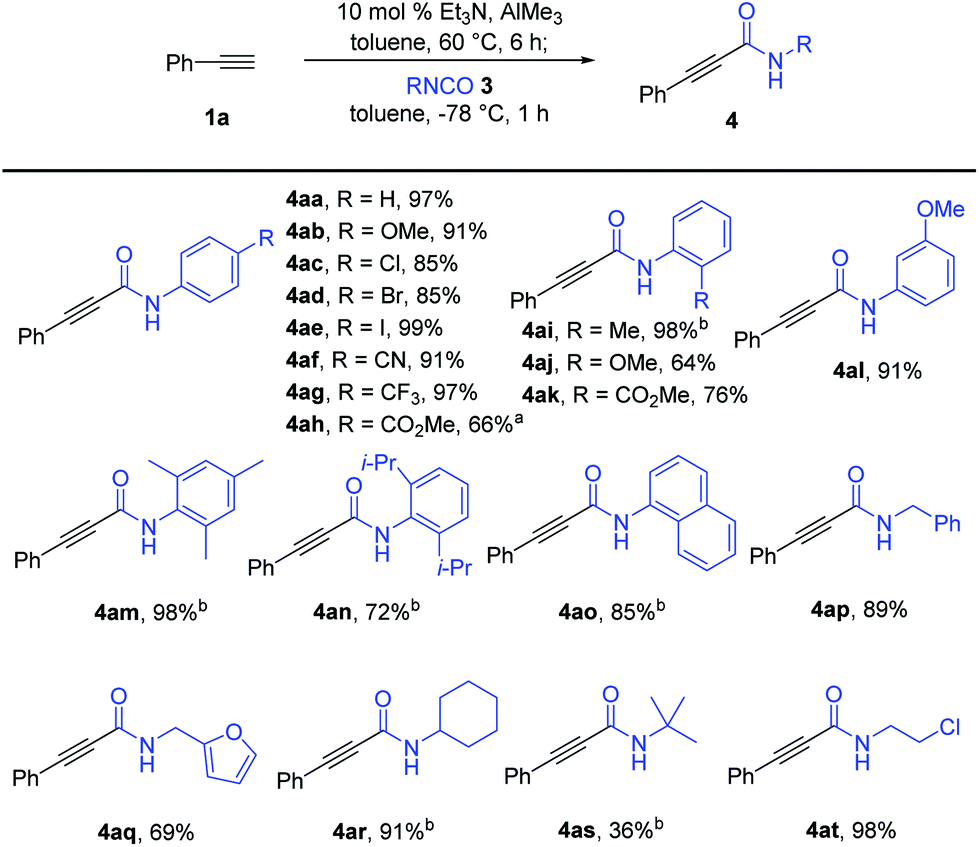 | ||
| Scheme 2 Synthesis of alkynamides with various isocyanates. Reaction conditions: phenylacetylene 1a (0.396 mmol), Et3N (10 mol% based on AlMe3), AlMe3 (0.360 mmol), RNCO (3, 0.300 mmol), and toluene (0.1 M) under N2. Yield of isolated products. aAmidation was carried out at 0 °C. bAmidation was carried out at −20 °C for 2 h. | ||
Next, we investigated the generality of aryl-substituted acetylenes for the construction of alkynamides, as depicted in Scheme 3. Et3N-catalyzed alumination of various aryl acetylenes with methoxy, methyl, tert-butyl, fluoro, chloro, and bromo groups at the para-, meta-, or ortho-positions of the benzene ring smoothly produced aluminum acetylides, which directly reacted with phenyl isocyanate 3a, affording the corresponding alkynamides 4ba–4ga and 4la–4qa with high efficiency. However, aryl acetylenes bearing para-cyano (1h), para-trifluoromethyl (1i) and para-ester(1j) substituents were not suitable for amide synthesis under the established reaction conditions. Generation of para-cyano-substituted phenylethynyl–Al reagent 2h was problematic because of the poor solubility of aryl acetylene 1h. Zhou et al. reported the synthesis of alkynylaluminum reagent 2i with a para-CF3-substituted phenyl group and its application to Ni-catalyzed cross-coupling reactions.16 However, the nucleophilic addition of the corresponding aluminum acetylide 2i to 3a did not proceed, presumably because of the low nucleophilicity of 2i. In addition, the reason why the amidation with aluminum acetylide 2j bearing a CO2Me group did not occur was presumed to be that the undesirable coordination of the carbonyl group to the aluminum acetylide 2j would cause the aluminum center to be coordinatively saturated, thereby interfering with the coordination of the alkynyl–Al to the isocyanate. Transformation of aryl acetylene with a meta-methoxy substituent was not efficient in producing the amide product 4ka and only 49% yield was obtained. It was noteworthy this methodology enabled efficient synthesis of bromo-substituted alkynamides such as 4ga and 4pa (79–96% yields), which were challenging to prepare from lithium- or magnesium acetylides due to labile bromo groups substituted on arylacetylenes during the generation of acetylides. Acetylenes bearing a sterically demanding naphthalene ring 1r and heteroaromatic rings 1s–t, such as thienyl and benzofuranyl moieties, were successfully converted to the corresponding amides 4ra–4ta in 78–98% yield. However, the reaction of acetylene with para-toluenesulfonyl-protected indole group afforded 4ua in 31% yield with reduced efficiency, probably owing to the undesirable coordination of alkynyl–Al to the sulfone group.
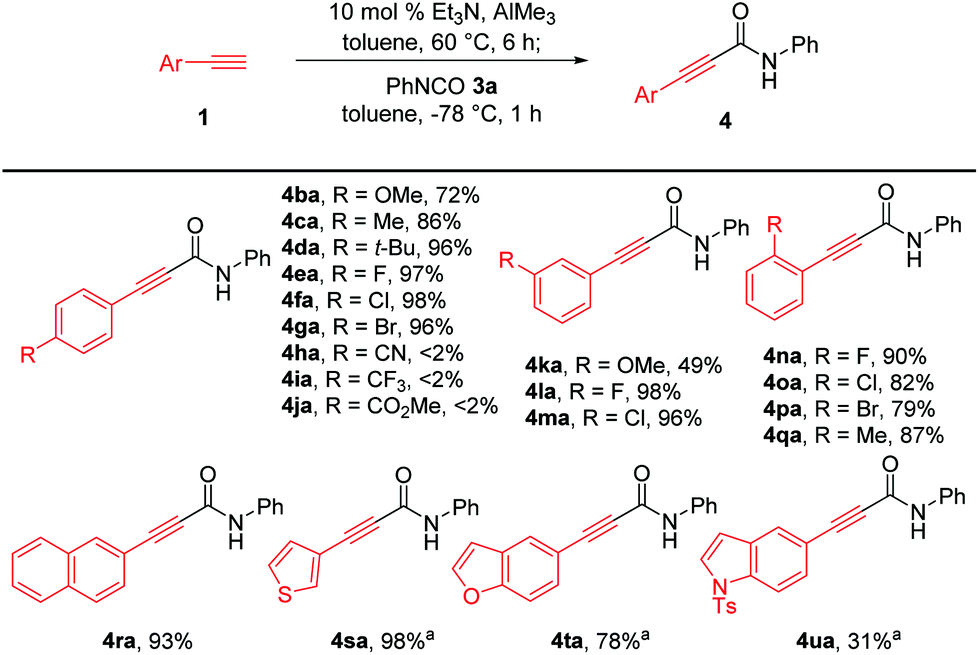 | ||
| Scheme 3 Synthesis of alkynamides with various aryl-substituted acetylenes. Reaction conditions: arylacetylene 1 (0.396 mmol), Et3N (10 mol% based on AlMe3), AlMe3 (0.360 mmol), PhNCO (3a, 0.300 mmol), and toluene (0.1 M) under N2. Yield of isolated products. aAmidation was carried out at −20 °C for 2 h. | ||
As illustrated in Scheme 4, terminal alkynes 6 bearing alkyl substituents were also effective for the synthesis of the corresponding alkynamides 7. Alkyl-substituted acetylenes with n-hexyl 6a, n-tridecyl 6b, i-pentyl 6c, phenethyl 6d, benzyl 6e, and cyclohexylmethyl 6f groups underwent alumination and subsequent amidation with phenyl isocyanate (3a), delivering the desired alkynamides 7a–f in excellent yields (88–97%). Sterically demanding alkyl-substituted acetylenes 6g–i also participated well in this transformation. The alumination/amidation reactions were compatible with functional groups such as a chloro and silyl ether moiety on the alkyne substrates, affording amides 7j–k in 66–89% yields. Notably, the amidation of 2-propynylbenzene (6e) afforded a mixture of alkynamide product 7e and the inseparable by-product of allenylamide 7ea in 63% and 32% yields, respectively. We observed that the alumination of 6e with AlMe3 under typical Micouin reaction conditions17 generated only alkynylaluminum reagents, not allenylaluminum intermediates. Therefore, we assumed that the isomerization occurred after the formation of the desired alkynamide 7e owing to the increased acidity of the benzyl C–H proton containing the electron-withdrawing –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CONHPh moiety.
C–CONHPh moiety.
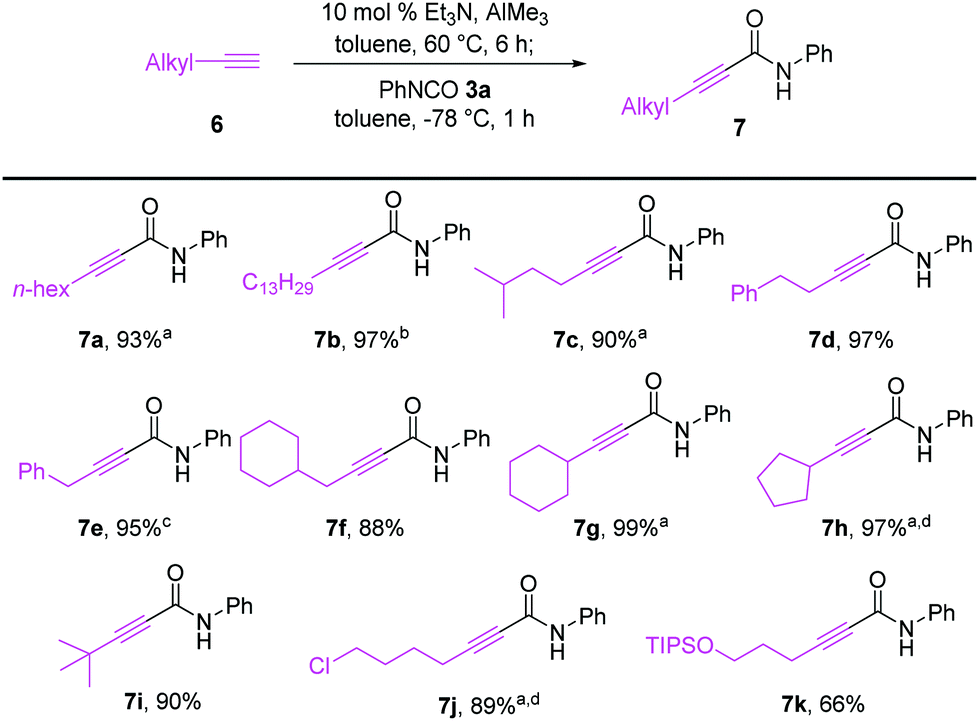 | ||
| Scheme 4 Synthesis of alkynamides with various alkyl-substituted acetylenes. Reaction conditions: alkylacetylene 6 (0.396 mmol), Et3N (10 mol% based on AlMe3), AlMe3 (0.360 mmol), PhNCO (3a, 0.30 mmol) solvent (0.1 M) under N2. Yield of isolated products. aAmidation was carried out at −20 °C for 2 h. bUse of 2 equiv. of alkynyl–Al (0.60 mmol). cThe inseparable mixture of alkynamide (7e, 63%) and allenylamide (7ea, 32%) was obtained in a total yield of 95%. dUse of 1.5 equiv. of alkynyl–Al (0.45 mmol). | ||
Synthesis of β-lactams 9a–c through base-promoted α-addition of activated C–C triple bonds on alkynamides 4 highlighted the usefulness of our method. β-Lactams such as penicillin, cephamycins, and carbapenems are ubiquitous in antibiotics.18 α-Methylene-β-lactams such as asparenomycin A have shown interesting biological activities.19 Alkynamides 4, synthesized by alumination/amidation of terminal alkynes with isocyanates, were smoothly transformed into α-methylene-β-lactams in 72–81% yield using 3 equiv. of potassium carbonate in the presence of 20 mol% of potassium iodide (Scheme 5).20
 | ||
| Scheme 5 Synthesis of β-lactams with alkynamides. | ||
The feasibility of gram-scale synthesis was also investigated (Scheme 6). Transformation of aryl acetylene 1l (8.32 mmol, 1.00 g) bearing a meta-fluoro group was smoothly performed using 7.57 mmol of AlMe3 and 6.31 mmol of phenyl isocyanate 3a to give the desired 4la in 89% yield (1.34 g).
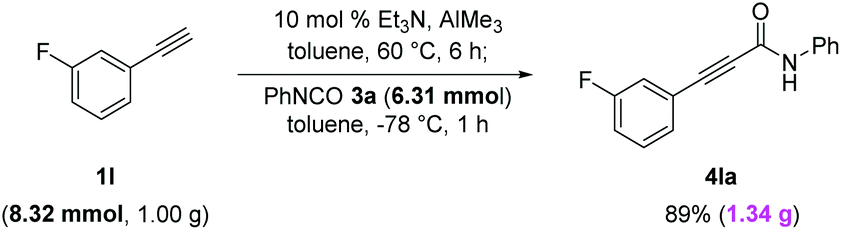 | ||
| Scheme 6 Gram-scale synthesis of alkynamide 4la. | ||
Conclusions
We successfully developed a mild and efficient method for one-pot synthesis of alkynamides from readily available terminal alkynes and isocyanates through Et3N-catalyzed alumination using AlMe3 and amidation of alkynylaluminums with isocyanates. A broad range of alkynamides were selectively obtained in moderate and excellent yields. Gram-scale synthesis was also achieved. Application of the resulting alkynamides to the synthesis of biologically interesting α-methylene-β-lactams highlighted the synthetic utility of our method. Additional investigation on this strategy using Al-based nucleophiles and isocyanates, as well as the synthesis of biologically-active molecules are ongoing.Experimental section
General information
Infrared (IR) spectra were recorded on an ABB MB3000 FT-IR spectrophotometer (νmax in cm−1). The bands were characterized as strong (s), medium (m), and weak (w). The 1H NMR spectra were recorded using a JEOL JNM-AL400 spectrometer (400 MHz). The chemical shifts (ppm) from tetramethylsilane were reported using solvent resonance as the internal standard (CDCl3: δ 7.27 ppm and DMSO-d6: δ 2.50 ppm). The data were reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet), coupling constants (Hz), and integration. The 13C NMR spectra were recorded on a JEOL JNM-AL400 (100 MHz) spectrometer with complete proton decoupling. The chemical shifts (ppm) from tetramethylsilane were reported with solvent resonance as the internal standard (CDCl3: δ 77.00 ppm and DMSO-d6: δ 39.51 ppm). High-resolution mass spectrometry (HRMS) was performed at the Korea Basic Science Institute for technical assistance using electron ionization (EI) or electrospray ionization (ESI) time-of-flight mass spectrometry.Unless otherwise noted, all reactions were carried out in distilled solvents under an atmosphere of dry N2 in oven-dried (130 °C) glassware. Triethylamine and hexanes were purified by distillation from CaH2 immediately prior to use. Toluene was purified by distillation from sodium benzophenone ketyl immediately prior to use, unless otherwise specified. Trimethylaluminum was purchased from Sigma-Aldrich Corporation and used as received. A variety of isocyanates and acetylenes were purchased from TCI, Alfa Aesar, and Sigma-Aldrich and used as received. All work-up and purification procedures were carried out using reagent-grade solvents in air. Various alkyne substrates were prepared according to previously reported experimental procedures.21
Representative experimental procedure for the synthesis of alkynamides
:hexanes = 1:5) to obtain the desired product 4aa (64.6 mg, 0.292 mmol, 97% yield) as a white solid. This compound has been previously reported and the spectra data match described.221H NMR (CDCl3, 400 MHz): δ 7.95 (br s, 1H), 7.61 (d, J = 7.8 Hz, 2H), 7.56–7.54 (m, 2H), 7.43 (t, J = 7.3 Hz, 1H), 7.37–7.32 (m, 4H), 7.15 (t, J = 7.3 Hz, 1H); 13C NMR (CDCl3, 100 MHz): δ 151.1, 137.3, 132.6, 130.3, 129.1, 128.5, 124.9, 112.0, 119.9, 85.8, 83.4.
:hexanes = 1:5). This compound has been previously reported and the spectra data match described.231H NMR (CDCl3, 400 MHz): δ 8.68 (br s, 1H), 8.19 (s, 1H), 8.10 (dd, J = 7.8, 1.8 Hz, 2H), 7.70 (m, 2H), 7.63–7.61 (m, 2H), 7.50–7.45 (m, 6H), 7.41 (t, J = 7.8 Hz, 2H), 7.19 (t, J = 7.8 Hz, 1 H); 13C NMR (CDCl3, 100 MHz): δ 161.6, 143.8, 137.7, 134.5, 131.4, 130.4, 130.3, 129.4, 129.0, 128.7, 128.5, 124.5, 121.9, 119.8, 114.4, 100.5, 84.9.
:hexanes = 1:3). This compound has been previously reported and the spectra data match described.241H NMR (CDCl3, 400 MHz): δ 8.34 (br s, 1H), 7.53 (d, J = 9.1 Hz, 2H), 7.49 (d, J = 8.2 Hz, 2H), 7.39 (t, J = 7.3 Hz, 1H), 7.30 (t, J = 7.3 Hz, 2H), 6.83 (d, J = 9.1 Hz, 2H), 3.76 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 156.6, 151.2, 132.5, 130.5, 130.0, 128.4, 121.8, 120.0, 114.1, 85.6, 83.5, 55.3.
:hexanes = 1:2). This compound has been previously reported and the spectra data match described.241H NMR (CDCl3, 400 MHz): δ 7.71 (br s, 1H), 7.57 (d, J = 6.9 Hz, 2H), 7.54 (d, J = 8.7 Hz, 2H), 7.46 (t, J = 7.3 Hz, 1H), 7.39 (t, J = 6.9 Hz, 2H), 7.32 (d, J = 8.7 Hz, 2H); 13C NMR (CDCl3, 100 MHz): δ 150.9, 135.8, 132.6, 130.5, 129.9, 129.2, 128.6, 121.1, 119.7, 86.2, 83.1.
:hexanes = 1:2). This compound has been previously reported and the spectra data match described.251H NMR (CDCl3, 400 MHz): δ 7.92 (br s, 1H), 7.73 (d, J = 8.7 Hz, 2H), 7.64 (d, J = 8.7 Hz, 2H), 7.58 (d, J = 6.9 Hz, 2H), 7.48 (t, J = 7.6 Hz, 1H), 7.40 (t, J = 7.6 Hz, 2H); 13C NMR (CDCl3, 100 MHz): δ 151.0, 141.3, 133.4, 132.7, 130.8, 128.7, 119.7, 119.4, 118.6, 107.8, 87.1, 82.8.
:hexanes = 1:5). This compound has been previously reported and the spectra data match described.261H NMR (CDCl3, 400 MHz): δ 8.00 (br s, 1H), 7.73 (d, J = 8.7 Hz, 2H), 7.60 (d, J = 8.7 Hz, 2H), 7.56 (d, J = 7.3 Hz, 2H), 7.47 (tt, J = 7.4, 1.3 Hz, 1H), 7.40–7.36 (m, 2H); 13C NMR (CDCl3, 100 MHz): δ 151.7, 140.3, 132.6, 130.6, 128.6, 126.6 (q, JC–F = 32.5 Hz), 126.4 (q, JC–F = 3.90 Hz), 123.9 (q, JC–F = 271.6 Hz), 119.5, 119.5, 86.7, 83.0.
:hexanes = 1:5). mp 148–149 °C; IR (neat): 3279 (w), 2206 (m), 1705 (s), 1636 (m), 1597 (m), 1535 (s), 1404 (m), 1273 (s), 1111 (m), 756 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.58 (br s, 1H), 8.00 (d, J = 8.7 Hz, 2H), 7.71 (d, J = 8.7 Hz, 2H), 7.47 (d, J = 7.3 Hz, 2H), 7.40 (t, J = 7.8 Hz, 1H), 7.30 (t, J = 7.6 Hz, 2H), 3.90 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 166.6, 151.3, 141.7, 132.5, 130.8, 130.4, 128.5, 125.9, 119.5, 119.1, 86.6, 83.1, 52.1; HRMS (ESI) m/z: [M + H]+ calcd for C17H14NO3 280.0974, found 280.0974.
:hexanes = 1:5). This compound has been previously reported and the spectra data match described.241H NMR (CDCl3, 400 MHz): δ 7.86 (d, J = 7.8 Hz, 1H), 7.59 (d, J = 7.3 Hz, 2H), 7.55 (br s, 1H), 7.45 (t, J = 7.3 Hz, 1H), 7.38 (t, J = 7.8 Hz, 2H), 7.28–7.21 (m, 2H), 7.12 (t, J = 7.8 Hz, 1H), 2.34 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 151.3, 134.8, 132.6, 130.5, 130.2, 129.2, 128.5, 126.8, 125.8, 123.3, 119.9, 85.6, 83.4, 17.8.
:hexanes = 1:5). mp 93–94 °C; IR (neat): 3310 (w), 2206 (m), 1651 (m), 1528 (s), 1458 (s), 1335 (m), 1173 (m), 1026 (s), 741 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.39 (dd, J = 7.8, 1.4 Hz, 1H), 8.18 (br s, 1H), 7.61 (d, J = 6.9 Hz, 2H), 7.47–7.38 (m, 3H), 7.10 (td, J = 7.8, 1.4 Hz, 1H), 6.99 (t, J = 7.8 Hz, 1H), 6.92 (d, J = 8.2 Hz, 1H), 3.93 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 150.7, 147.6, 132.6, 130.2, 128.5, 127.2, 124.4, 121.1, 120.3, 120.1, 110.0, 85.2, 83.7, 55.7; HRMS (ESI) m/z: [M + H]+ calcd for C16H14NO2 252.1025, found 252.1025.
:hexanes = 1:10). IR (neat): 2924 (br), 2854 (m), 2314 (w), 2206 (w), 1693 (s), 1670 (m), 1616 (m), 1585 (m), 1558 (m), 1519 (s), 1450 (s), 1435 (m), 1296 (s), 1246 (s), 1188 (w), 1161 (m), 1087 (m), 1026 (w), 964 (w), 802 (w) cm−1; 1H NMR (CDCl3, 400 MHz): δ 11.48 (br s, 1H), 8.72 (d, J = 7.0 Hz, 1H), 8.06 (t, J = 7.0 Hz, 1H), 7.64–7.56 (m, 3H), 7.45–7.36 (m, 3H), 7.14 (t, J = 7.0 Hz, 1H), 3.96 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 168.5, 151.3, 140.8, 134.7, 132.8, 130.9, 130.2, 128.5, 123.2, 120.9, 120.0, 115.0, 85.5, 83.9, 52.4; HRMS (EI) m/z: [M]+ calcd for C17H13NO3 279.0895, found 279.0892.
:hexanes = 1:4). This compound has been previously reported and the spectra data match described.241H NMR (CDCl3, 400 MHz): δ 7.83 (br s, 1H), 7.56 (d, J = 8.0 Hz, 2H), 7.44 (t, J = 8.0 Hz, 1H), 7.39–7.32 (m, 3H), 7.24 (t, J = 8.0 Hz, 1H), 7.08 (d, J = 7.4 Hz, 1H), 6.71 (dd, J = 8.0, 2.0 Hz, 1H), 3.81 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 160.1, 151.0, 138.5, 132.6, 130.3, 129.8, 128.5, 120.0, 112.1, 110.7, 105.7, 85.7, 83.4, 55.3.
:hexanes = 1:5). Compound exists as a 9:1 mixture of rotamers. This compound has been previously reported and the spectra data match described.251H NMR (DMSO-d6, 400 MHz), a 9:1 mixture of rotamers: δ 10.10 (s, 0.9H), 9.57 (s, 0.1H), 7.64 (d, J = 8.0 Hz, 1.8H), 7.55–7.47 (m, 2.7H), 7.42 (d, J = 8.0 Hz, 0.1H), 7.36–7.33 (m, 0.2H), 7.11 (d, J = 8.0 Hz, 0.2H), 6.98 (s, 0.2H), 6.90 (s, 1.8H), 2.25 (s, 0.3H), 2.22 (s, 2.7H), 2.21 (s, 0.6H), 2.13 (s, 5.4H); 13C NMR (DMSO-d6, 100 MHz): δ 150.9, 136.2, 134.9, 132.3, 131.2, 130.5, 129.1, 128.5, 119.8, 84.1, 83.8, 20.6, 18.1.
:hexanes = 1:5). Compound exists as a 4:1 mixture of rotamers. This compound has been previously reported and the spectra data match described.271H NMR (DMSO-d6, 400 MHz), a 4:1 mixture of rotamers: δ 10.20 (s, 0.8H), 9.77 (s, 0.2H), 7.68 (d, J = 7.6 Hz, 0.4H), 7.66 (d, J = 7.6 Hz, 1.6H), 7.55–7.47 (m, 2.4H), 7.42–7.35 (m, 0.8H), 7.29 (t, J = 7.4 Hz, 0.8H), 7.24 (d, J = 7.9 Hz, 0.4H), 7.18 (d, J = 7.8 Hz, 1.6H), 3.16 (sept, J = 6.9 Hz, 0.4H), 3.07 (sept, J = 6.9 Hz, 1.6H), 1.19–1.12 (m, 12H); 13C NMR (DMSO-d6, 100 MHz), a 4:1 mixture of rotamers: δ 155.5, 152.1, 147.0, 145.9, 132.4, 132.2, 131.9, 131.0, 130.5, 129.1, 129.0, 128.7, 128.2, 123.3, 123.2, 119.8, 119.4, 88.6, 84.1, 84.0, 82.6, 28.3, 28.2, 23.9, 23.7, 23.3, 23.1.
:1 mixture of rotamers. The crude product was purified using silica gel column chromatography (EtOAc:hexanes = 1:7). This compound has been previously reported and the spectra data match described.241H NMR (DMSO-d6, 400 MHz): δ 10.92 (br s, 1H), 8.06 (d, J = 7.8 Hz, 1H), 7.97 (d, J = 7.3 Hz, 1H), 7.85 (d, J = 8.2 Hz, 1H), 7.71 (d, J = 6.4 Hz, 2H), 7.67 (d, J = 7.3 Hz, 1H), 7.61–7.50 (m, 6H); 13C NMR (DMSO-d6, 100 MHz): δ 151.6, 133.7, 132.3, 130.5, 129.1, 128.2, 127.9, 126.4, 126.3, 126.2, 125.6, 122.8, 122.7, 119.8, 84.8, 84.2.
:1 mixture of rotamers. The crude product was purified using silica gel column chromatography (EtOAc:hexanes = 1:5). This compound has been previously reported and the spectra data match described.281H NMR (CDCl3, 400 MHz), a 9:1 mixture of rotamers: δ 7.51 (d, J = 6.9 Hz, 2H), 7.42–7.37 (m, 1H), 7.35–7.27 (m, 7H), 6.59 (br s, 0.9H), 6.33 (br s, 0.1H), 4.67 (d, J = 6.4 Hz, 0.2H), 4.52 (d, J = 5.9 Hz, 1.8H); 13C NMR (CDCl3, 100 MHz): δ 153.3, 137.2, 132.4, 130.0, 128.7, 128.4, 127.8, 127.6, 120.0, 85.3, 83.1, 44.0.
:1 mixture of rotamers. The crude product was purified using silica gel column chromatography (EtOAc:hexanes = 1:2). This compound has been previously reported and the spectra data match described.291H NMR (CDCl3, 400 MHz), a 9:1 mixture of rotamers: δ 7.57 (d, J = 6.9 Hz, 0.2H), 7.52 (d, J = 6.9 Hz, 1.8H), 7.43–7.32 (m, 4H), 6.39 (br s, 1H), 6.34–6.33 (m, 1H), 6.30–6.29 (m, 1H), 4.67 (d, J = 6.4 Hz, 0.2H), 4.54 (d, J = 5.9 Hz, 1.8H); 13C NMR (CDCl3, 100 MHz): δ 153.2, 150.2, 142.2, 132.3, 129.9, 128.3, 119.9, 110.4, 107.8, 85.1, 82.6, 36.5.
:hexanes = 1:5). Compound exists as a 9:1 mixture of rotamers. This compound has been previously reported and the spectra data match described.301H NMR (CDCl3, 400 MHz), a 9:1 mixture of rotamers: δ 7.51 (d, J = 7.3 Hz, 2H), 7.36 (d, J = 6.4 Hz, 1H), 7.32 (t, J = 6.7 Hz, 2H), 6.07 (br s, 1H), 3.91–3.82 (m, 0.9H), 3.80–3.72 (m, 0.1H), 1.97 (d, J = 12.4 Hz, 2H), 1.72 (dt, J = 13.2, 3.5 Hz, 2H), 1.63–1.59 (m, 1H), 1.40–1.31 (m, 2H), 1.25–1.15 (m, 3H); 13C NMR (CDCl3, 100 MHz): δ 152.5, 132.4, 129.9, 128.4, 120.3, 84.1, 83.3, 48.8, 32.8, 25.4, 24.7.
:hexanes = 1:5). This compound has been previously reported and the spectra data match described.111H NMR (CDCl3, 400 MHz): δ 7.52 (dt, J = 6.9, 1.4 Hz, 2H), 7.43–7.38 (m, 1H), 7.43–7.33 (m, 2H), 7.77 (br s, 1H), 1.42 (s, 9H); 13C NMR (CDCl3, 100 MHz): δ 152.5, 132.4, 129.8, 128.5, 120.4, 84.0, 82.5, 52.4, 28.6.
:hexanes = 1:5). mp 87–88 °C; IR (neat): 3212 (br), 3039 (w), 2214 (m), 1682 (w), 1616 (s), 1550 (s), 1489 (s), 1423 (s), 1369 (s), 1315 (s), 1254 (s), 1227 (s), 1188 (s), 1157 (m), 1068 (m), 1045 (m), 1026 (m), 952 (m), 918 (m), 898 (w), 756 (s), 725 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.54 (d, J = 7.5 Hz, 2H), 7.44–7.41 (m, 1H), 7.38–7.34 (m, 2H), 6.48 (br s, 1H), 3.69 (pent, J = 5.1 Hz, 4H); 13C NMR (CDCl3, 100 MHz): δ 153.5, 132.5, 130.2, 128.5, 119.9, 85.5, 82.5, 43.4, 41.5; HRMS (EI) m/z: [M]+ calcd for C11H10ClNO 207.0451, found 207.0454.
:hexanes = 1:5). This compound has been previously reported and the spectra data match described.241H NMR (CDCl3, 400 MHz): δ 7.69 (br s, 1H), 7.58 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 8.3 Hz, 2H), 7.35 (t, J = 7.7 Hz, 2H), 7.15 (t, J = 7.7 Hz, 1H), 6.89 (d, J = 8.3 Hz, 2H), 3.84 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 161.2, 151.3, 137.4, 134.4, 129.1, 124.8, 119.9, 114.3, 111.7, 86.4, 82.8, 55.4.
:hexanes = 1:5). This compound has been previously reported and the spectra data match described.241H NMR (CDCl3, 400 MHz): δ 7.87 (br s, 1H), 7.59 (d, J = 8.0 Hz, 2H), 7.45 (d, J = 8.0 Hz, 2H), 7.34 (t, J = 8.0 Hz, 2H), 7.17–7.13 (m, 3H), 2.38 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 151.2, 140.8, 137.4, 132.5, 129.3, 129.1, 124.8, 119.9, 116.8, 86.2, 83.1, 21.6.
:hexanes = 1:5). mp 110–111 °C; IR (neat): 3271 (w), 2955 (m), 2214 (m), 1643 (s), 1597 (s), 1551 (s), 1497 (m), 1442 (s), 1327 (s), 1250 (m), 748 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.40 (br s, 1H), 7.64 (d, J = 8.2 Hz, 2H), 7.46 (d, J = 8.3 Hz, 2H), 7.34–7.29 (m, 4H), 7.12 (t, J = 7.3 Hz, 1H), 1.30 (s, 9H); 13C NMR (CDCl3, 100 MHz): δ 153.8, 137.5, 132.4, 129.0, 129.0, 125.5, 124.7, 120.0, 116.8, 86.3, 83.1, 34.9, 31.0; HRMS (ESI) m/z: [M + H]+ calcd for C19H20NO 278.1545, found 278.1548.
:hexanes = 1:5). mp 117–118 °C; IR (neat): 3286 (br), 3271 (w), 2920 (w), 2245 (w), 2214 (m), 1635 (s), 1597 (m), 1527 (s), 1496 (s), 1481 (m), 1442 (s), 1400 (w), 1319 (m), 1303 (w), 1219 (s), 1184 (m), 1153 (m), 1080 (w), 964 (m), 902 (w), 883 (w), 837 (s), 817 (w), 756 (m), 732 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.10 (br s, 1H), 7.61 (d, J = 8.0 Hz, 2H), 7.52–7.48 (m, 2H), 7.34 (t, J = 8.0 Hz, 2H), 7.15 (t, J = 8.0 Hz, 1H), 7.02 (t, J = 8.0 Hz, 2H); 13C NMR (CDCl3, 100 MHz): δ 163.7 (d, JC–F = 252 Hz), 151.1, 137.3, 134.8 (d, JC–F = 9.0 Hz), 129.1, 124.99, 120.0, 116.1 (d, JC–F = 3.0 Hz), 116.1 (d, JC–F = 23.0 Hz), 84.8, 83.3; 19F NMR (CDCl3, 376 MHz): δ −106.95; HRMS (EI) m/z: [M]+ calcd for C15H10FNO 239.0746, found 239.0745.
:hexanes = 1:5). This compound has been previously reported and the spectra data match described.14a1H NMR (CDCl3, 400 MHz): δ 8.07 (br s, 1H), 7.60 (d, J = 7.4 Hz, 2H), 7.50 (dd, J = 8.6, 5.5 Hz, 2H), 7.34 (t, J = 8.0 Hz, 2H), 7.15 (t, J = 7.4 Hz, 1H), 7.03 (t, J = 8.6 Hz, 2H); 13C NMR (CDCl3, 100 MHz): δ 150.9, 137.3, 136.6, 133.7, 129.1, 128.9, 125.0, 120.0, 118.3, 84.6, 84.2.
:hexanes = 1:5). mp 162–163 °C; IR (neat): 3302 (s), 2206 (s), 1636 (s), 1597 (m), 1520 (s), 1481 (m), 1435 (s), 1396 (m), 1311 (s), 1242 (m), 1173 (m), 1065 (m), 1003 (m), 964 (m), 825 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.01 (br s, 1H), 7.59 (d, J = 7.8 Hz, 2H), 7.48 (d, J = 8.7 Hz, 2H), 7.38–7.32 (m, 4H), 7.16 (t, J = 7.3 Hz, 1H); 13C NMR (CDCl3, 100 MHz): δ 150.9, 137.2, 133.9, 131.9, 129.1, 125.0, 125.0, 120.0, 118.8, 84.6, 84.3; HRMS (ESI) m/z: [M + H]+ calcd for C15H11BrNO 300.0024, found 300.0025.
:hexanes = 1:5). mp 119–120 °C; IR (neat): 3070 (w), 2206 (s), 1705 (w), 1597 (s), 1543 (s), 1489 (s), 1443 (s), 1327 (s), 1250 (s), 1165 (m), 1080 (w), 1034 (m), 987 (m), 903 (m), 849 (m) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.94 (br s, 1H), 7.60 (d, J = 7.8 Hz, 2H), 7.35 (t, J = 7.8 Hz, 2H), 7.26 (t, J = 8.2 Hz, 1H), 7.17–7.14 (m, 2H), 7.07 (s, 1H), 6.98 (dd, J = 8.2, 2.2 Hz, 1H), 3.79 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 159.3, 151.0, 137.3, 129.6, 129.0, 125.0, 124.9, 120.8, 119.9, 117.1, 117.1, 85.7, 83.1, 55.3; HRMS (ESI) m/z: [M + H]+ calcd for C16H14NO2 252.1025, found 252.1025.
:hexanes = 1:5). mp 95–96 °C; IR (neat): 3230 (br), 2924 (m), 2214 (m), 1654 (m), 1581 (s), 1546 (s), 1485 (s), 1442 (s), 1327 (s), 1292 (s), 1249 (s), 1211 (s), 1141 (w), 1099 (m), 1080 (m), 1030 (m), 983 (m), 898 (w), 879 (m), 786 (s), 752 (s), 736 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.91 (br s, 1H), 7.59 (d, J = 8.0 Hz, 2H), 7.37–7.33 (m, 4H), 7.23 (d, J = 9.1 Hz, 1H), 7.18–7.14 (m, 2H); 13C NMR (CDCl3, 100 MHz): δ 162.2 (d, JC–F = 246 Hz), 150.7, 137.2, 130.3 (d, JC–F = 8.0 Hz), 129.1, 128.5 (d, JC–F = 4.0 Hz), 125.1, 121.7 (d, JC–F = 10 Hz), 120.0, 119.2 (d, JC–F = 22 Hz), 117.7 (d, JC–F = 22 Hz), 84.1, 83.9; 19F NMR (CDCl3, 376 MHz): δ −111.74; HRMS (EI) m/z: [M]+ calcd for C15H10FNO 239.0746, found 239.0749.
:hexanes = 1:5). mp 95–96 °C; IR (neat): 3250 (br), 2218 (m), 1743 (w), 1631 (s), 1593 (s), 1550 (s), 1500 (s), 1442 (s), 1330 (s), 1257 (s), 1242 (s), 1180 (s), 1080 (m), 1033 (w), 968 (s), 948 (m), 906 (m), 867 (m), 790 (s), 756 (s), 717 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.99 (br s, 1H), 7.53 (d, J = 8.0 Hz, 2H), 7.43 (s, 1H), 7.38–7.31 (m, 2H), 7.28 (t, J = 8.0 Hz, 2H), 7.21 (t, J = 7.4 Hz, 1H), 7.09 (t, J = 7.4 Hz, 1H); 13C NMR (CDCl3, 100 MHz): δ 150.7, 137.2, 134.4, 132.2, 130.7, 130.5, 129.8, 129.1, 125.0, 121.6, 120.0, 84.2, 84.0; HRMS (EI) m/z: [M]+ calcd for C15H10ClNO 255.0451, found 255.0450.
:hexanes = 1:5). mp 129–130 °C; IR (neat): 3271 (w), 2955 (m), 2214 (m), 1643 (s), 1597 (s), 1551 (s), 1497 (m), 1442 (s), 1327 (s), 1250 (m), 748 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.98 (br s, 1H), 7.60 (d, J = 8.2 Hz, 2H), 7.54 (t, J = 7.3 Hz, 1H), 7.45–7.39 (m, 1H), 7.35 (t, J = 8.0 Hz, 2H), 7.17–7.11 (m, 3H); 13C NMR (CDCl3, 100 MHz): δ 163.3 (d, JC–F = 254.3 Hz), 150.7, 137.2, 134.4, 132.2 (d, JC–F = 8.7 Hz), 129.1, 125.0, 124.3 (d, JC–F = 3.9 Hz), 120.0, 115.7 (d, JC–F = 21.2 Hz), 108.8 (d, JC–F = 15.4 Hz), 87.9 (d, JC–F = 3.9 Hz), 79.1; HRMS (ESI) m/z: [M + H]+ calcd for C15H11FNO 240.0825, found 240.0824.
:hexanes = 1:5). This compound has been previously reported and the spectra data match described.311H NMR (CDCl3, 400 MHz): δ 8.19 (br s, 1H), 7.62 (d, J = 7.8 Hz, 2H), 7.52 (dd, J = 7.8, 1.8 Hz, 1H), 7.40 (d, J = 7.7 Hz, 1H), 7.35–7.31 (m, 3H), 7.21 (td, J = 7.8, 1.4 Hz, 1H), 7.14 (t, J = 7.8 Hz, 1H); 13C NMR (CDCl3, 100 MHz): δ 150.8, 137.3, 136.8, 134.4, 131.2, 129.4, 129.0, 126.6, 124.9, 120.1, 120.0, 87.7, 82.1.
:hexanes = 1:5). mp 135–136 °C; IR (neat): 3217 (w), 3032 (w), 2214 (s), 1597 (s), 1543 (s), 1443 (s), 1319 (s), 1257 (s), 1188 (m), 1026 (w), 964 (m) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.88 (br s, 1H), 7.63–7.56 (m, 4H), 7.35 (t, J = 7.7 Hz, 2H), 7.32–7.28 (m, 2H), 7.16 (t, J = 7.8 Hz, 1H); 13C NMR (CDCl3, 100 MHz): δ 150.7, 137.2, 134.5, 132.6, 131.4, 129.1, 127.2, 126.2, 125.0, 122.3, 120.0, 87.0, 83.7; HRMS (ESI) m/z: [M + H]+ calcd for C15H11BrNO 300.0024, found 300.0023.
:hexanes = 1:5). This compound has been previously reported and the spectra data match described.321H NMR (CDCl3, 400 MHz): δ 8.21 (br s, 1H), 7.63 (d, J = 8.2 Hz, 2H), 7.48 (d, J = 7.8 Hz, 1H), 7.35–7.29 (m, 3H), 7.21 (d, J = 7.8 Hz, 1H), 7.14 (td, J = 7.5, 3.7 Hz, 2H), 2.47 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 151.3, 141.6, 137.5, 132.9, 130.2, 129.6, 129.0, 125.7 124.7, 120.0, 119.7, 87.1, 84.9, 20.5.
:hexanes = 1:7). mp 138–139 °C; IR (neat): 3228 (m), 2214 (m), 1713 (w), 1636 (s), 1520 (s), 1443 (m), 1319 (m), 1242 (m), 1165 (w), 1072 (w), 748 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.42 (br s, 1H), 8.00 (s, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.72–7.66 (m, 4H), 7.53–7.44 (m, 3H), 7.32 (t, J = 8.0 Hz, 2H), 7.13 (t, J = 7.3 Hz, 1H); 13C NMR (CDCl3, 100 MHz): δ 151.3, 137.5, 133.6, 133.6, 133.5, 129.1, 128.3, 128.0, 128.0, 127.8, 127.7, 126.8, 124.8, 120.0, 117.0, 86.3, 83.6; HRMS (ESI) m/z: [M + H]+ calcd for C19H14NO 272.1075, found 272.1075.
:hexanes = 1:5). mp 102–103 °C; IR (neat): 3271 (m), 3101 (w), 2924 (m), 2206 (s), 1713 (w), 1605 (s), 1543 (s), 1443 (m), 1319 (m), 1242 (m), 1157 (w), 1072 (w), 980 (w), 779 (m) cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.96 (br s, 1H), 7.68 (d, J = 7.8 Hz, 2H), 7.51 (d, J = 3.2 Hz, 1H), 7.30 (t, J = 8.0 Hz, 2H), 7.19 (dd, J = 5.0, 2.7 Hz, 1H), 7.12 (t, J = 7.6 Hz, 1H), 7.07 (dd, J = 4.8, 1.1 Hz, 1H); 13C NMR (CDCl3, 100 MHz): δ 151.6, 137.5, 132.7, 129.8, 128.8, 125.8, 124.7, 120.1, 118.9, 83.3, 81.6; HRMS (ESI) m/z: [M + H]+ calcd for C13H10NOS 228.0483, found 228.0483.
:hexanes = 1:5). mp 117–118 °C; IR (neat): 3244 (br), 2218 (m), 1639 (s), 1597 (m), 1550 (s), 1465 (m), 1442 (s), 1327 (s), 1257 (s), 1230 (m), 1111 (m), 1029 (m), 979 (w), 910 (w), 875 (m), 810 (m), 760 (s), 733 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.92 (br s, 1H), 7.82 (s, 1H), 7.68 (d, J = 2.2 Hz, 1H), 7.61 (d, J = 8.0 Hz, 2H), 7.48 (s, 2H), 7.35 (t, J = 7.8 Hz, 2H), 7.15 (t, J = 8.0 Hz, 1H), 6.69 (d, J = 2.2 Hz, 1H); 13C NMR (CDCl3, 100 MHz): δ 155.4, 151.5, 146.2, 137.6, 129.0, 128.8, 127.6, 126.3, 124.7, 120.0, 114.3, 111.8, 106.4, 86.6, 82.5; HRMS (EI) m/z: [M]+ calcd for C17H11NO2 261.0790, found 261.0791.
:hexanes = 1:5). mp 133–135 °C; IR (neat): 3233 (br), 2214 (m), 1639 (s), 1593 (s), 1535 (s), 1496 (m), 1442 (s), 1366 (s), 1327 (m), 1249 (s), 1169 (s), 1123 (s), 1080 (m), 983 (m), 894 (m), 810 (m), 752 (s), 725 (s), 705 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.96 (d, J = 8.6 Hz, 1H), 7.81 (br s, 1H), 7.77 (d, J = 7.9 Hz, 2H), 7.72 (s, 1H), 7.62–58 (m, 3H), 7.46 (dd, J = 8.6, 1.4 Hz, 1H), 7.35 (t, J = 7.5 Hz, 2H), 7.25 (d, J = 6.7 Hz, 2H), 7.15 (t, J = 7.5 Hz, 1H), 6.62 (d, J = 2.1 Hz, 1H), 2.36 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 151.2, 145.4, 137.4, 135.3, 134.9, 130.6, 130.0, 129.2, 128.6, 127.6, 126.8, 126.4, 124.8, 119.9, 114.6, 113.7, 108.6, 86.2, 82.9, 21.6; HRMS (EI) m/z: [M]+ calcd for C24H18N2O3S 414.1038, found 414.1039.
:hexanes = 1:5). IR (neat): 3271 (w), 3063 (w), 2932 (m), 2862 (w), 2230 (m), 1643 (s), 1597 (s), 1543 (s), 1443 (s), 1319 (s), 1257 (s), 756 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.82 (br s, 1H), 7.54 (d, J = 8.2 Hz, 2H), 7.32 (t, J = 8.0 Hz, 2H), 7.11 (t, J = 7.8 Hz, 1H), 2.31 (t, J = 7.1 Hz, 2H), 1.56 (quint, J = 7.4 Hz, 2H), 1.43–1.36 (m, 2H), 1.34–1.25 (m, 4H), 0.89 (t, J = 6.9 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 151.3, 137.5, 128.9, 124.6, 119.8, 88.7, 75.9, 31.2, 28.5, 27.6, 22.4, 18.6, 14.0; HRMS (ESI) m/z: [M + H]+ calcd for C15H20NO 230.1545, found 230.1546.
:hexanes = 1:10). mp 50–51 °C; IR (neat): 3270 (w), 2932 (m), 2862 (w), 1643 (s), 1597 (s), 1543 (s), 1443 (s), 1257 (s), 756 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.85 (br s, 1H), 7.54 (d, J = 7.8 Hz, 2H), 7.31 (t, J = 8.0 Hz, 2H), 7.31 (t, J = 7.3 Hz, 1H), 2.30 (t, J = 7.1 Hz, 2H), 1.56 (quint, J = 7.3 Hz, 2H), 1.40–1.36 (m, 2H), 1.32–1.24 (m, 18H), 0.89 (t, J = 6.9 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 151.2, 137.5, 128.9, 124.5, 119.8, 88.7, 75.9, 31.8, 29.6, 29.6, 29.5, 29.4, 29.3, 29.0, 28.9, 27.7, 22.6, 18.6, 14.1; HRMS (ESI) m/z: [M + H]+ calcd for C22H34NO 328.2640, found 328.2641.
:hexanes = 1:5). IR (neat): 3256 (w), 3063 (w), 2970 (m), 2870 (w), 2230 (m), 1643 (s), 1597 (s), 1543 (s), 1497 (m), 1443 (s), 1327 (s), 1257 (s), 1126 (s), 756 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.51 (d, J = 7.3 Hz, 2H), 7.39 (br s, 1H), 7.34 (t, J = 8.0 Hz, 2H), 7.13 (t, J = 7.3 Hz, 1H), 2.36 (t, J = 7.3 Hz, 2H), 1.72 (sept, J = 6.8 Hz, 1H), 1.50 (q, J = 7.3 Hz, 2H), 0.93 (d, J = 6.9 Hz, 6H); 13C NMR (CDCl3, 100 MHz): δ 151.0, 137.3, 129.1, 124.7, 119.8, 88.7, 77.2, 36.5, 27.3, 22.1, 16.7; HRMS (ESI) m/z: [M + H]+ calcd for C14H18NO 216.1388, found 216.1389.
:hexanes = 1:5). mp 79–80 °C; IR (neat): 3263 (w), 3063 (w), 2230 (m), 1643 (s), 1597 (s), 1543 (s), 1497 (s), 1443 (s), 1389 (s), 1319 (s), 1257 (s), 748 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.78 (br s, 1H), 7.54 (d, J = 8.2 Hz, 2H),7.36–7.31 (m, 4H), 7.28–7.26 (m, 1H), 7.25–7.22 (m, 2H), 7.13 (t, J = 7.3 Hz, 1H), 2.89 (t, J = 7.5 Hz, 2H), 2.61 (t, J = 7.5 Hz, 2H); 13C NMR (CDCl3, 100 MHz): δ 151.3, 139.7, 137.4, 128.9, 128.5, 128.3, 126.6, 124.6, 119.8, 87.5, 76.6, 33.8, 20.7; HRMS (ESI) m/z: [M + H]+ calcd for C17H16NO 250.1232, found 259.1233.  :7ea = 2:1) as a light yellow solid. The crude product was purified using silica gel column chromatography (EtOAc:hexanes = 1:5). A mixture of 7e and 7ea could not be separated. mp 111–112 °C; IR (neat): 3294 (s), 2230 (m), 1944 (w), 1628 (s), 1520 (s), 1443 (s), 1319 (s), 1250 (s), 1180 (m), 1072 (w), 1026 (w), 980 (w), 887 (m), 964 (m), 825 (s) cm−1; 1H NMR of 7e (CDCl3, 400 MHz): δ 7.69 (br s, 1H), 7.53 (d, J = 8.2 Hz, 2H), 7.39–7.29 (m, 7H), 7.14 (t, J = 7.3 Hz, 1H), 3.75 (s, 2H); 13C NMR of 7e (CDCl3, 100 MHz): δ 150.8, 137.3, 134.4, 129.1, 128.8, 128.0, 127.2, 124.8, 119.8, 85.6, 7.5, 25.0; HRMS (ESI) m/z: [M + H]+ calcd for C16H14NO 236.1075, found 236.1074.
:hexanes = 1:7). mp 70–71 °C; IR (neat): 3263 (w), 3063 (w), 2924 (m), 2854 (m), 2230 (m), 1636 (s), 1597 (s), 1543 (s), 1497 (s), 1443 (s), 1381 (s), 1319 (s), 1257 (s), 756 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.89 (br s, 1H), 7.54 (d, J = 8.2 Hz, 2H), 7.31 (t, J = 7.8 Hz, 2H), 7.11 (t, J = 7.3 Hz, 1H), 2.20 (d, J = 6.9 Hz, 2H), 1.81 (d, J = 11.9 Hz, 2H), 1.73–1.63 (m, 3H), 1.28–1.22 (m, 3H), 1.19–1.12 (m, 1H), 1.04–0.96 (m, 2H); 13C NMR (CDCl3, 100 MHz): δ 151.3, 137.5, 128.9, 124.6, 119.8, 87.7, 76.8, 36.7, 32.7, 26.3, 26.0, 25.9; HRMS (ESI) m/z: [M + H]+ calcd for C16H20NO 242.1545, found 242.1546.
:hexanes = 1:5). mp 108–109 °C; IR (neat): 3232 (w), 3063 (w), 2932 (m), 2854 (w), 2230 (m), 1636 (s), 1597 (s), 1543 (s), 1497 (m), 1443 (s), 1319 (s), 1257 (s), 756 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.90 (br s, 1H), 7.55 (d, J = 8.2 Hz, 2H), 7.31 (t, J = 7.8 Hz, 2H), 7.11 (t, J = 7.3 Hz, 1H), 2.51–2.45 (m, 1H), 1.86–1.80 (m, 2H), 1.75–1.67 (m, 2H), 1.56–1.44 (m, 2H), 1.35–1.25 (m, 4H); 13C NMR (CDCl3, 100 MHz): δ 151.4, 137.5, 128.9, 124.5, 119.9, 92.1, 75.8, 31.5, 28.8, 25.5, 24.6; HRMS (ESI) m/z: [M + H]+ calcd for C15H18NO 228.1388, found 228.1389.
:hexanes = 1:5). mp 143–144 °C; IR (neat): 3225 (w), 2962 (w), 2230 (m), 1638 (m), 1543 (s), 1443 (s), 1319 (s), 1257 (s), 910 (m) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.81 (br s, 1H), 7.52 (d, J = 7.8 Hz, 2H), 7.29 (t, J = 7.8 Hz, 2H), 7.09 (t, J = 7.4 Hz, 1H), 2.74–2.67 (m, 1H), 1.97–1.90 (m, 2H), 1.74–1.64 (m, 4H), 1.59–1.53 (m, 2H); 13C NMR (CDCl3, 100 MHz): δ 151.4, 137.5, 128.9, 124.5, 119.8, 92.6, 75.5, 33.1, 29.7, 25.1; HRMS (ESI) m/z: [M + H]+ calcd for C14H16NO 214.1232, found 214.1233.
:hexanes = 1:5). This compound has been previously reported and the spectra data match described.341H NMR (DMSO-d6, 400 MHz): δ 10.46 (br s, 1H), 7.58 (d, J = 7.8 Hz, 2H), 7.30 (t, J = 7.8 Hz, 2H), 7.06 (t, J = 7.5 Hz, 1H), 1.28 (s, 9H); 13C NMR (DMSO-d6, 100 MHz): δ 151.3, 137.3, 129.1, 129.1, 124.7, 119.9, 95.7, 30.1, 27.5.
:hexanes = 1:5). IR (neat): 3263 (m), 3063 (w), 2978 (w), 2230 (m), 1636 (s), 1597 (s), 1543 (s), 1497 (m), 1443 (s), 1319 (s), 1257 (s), 756 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.31 (br s, 1H), 7.55 (d, J = 8.2 Hz, 2H), 7.30 (t, J = 8.0 Hz, 2H), 7.11 (t, J = 7.8 Hz, 1H), 3.49 (td, J = 6.4, 2.3 Hz, 2H), 2.31 (td, J = 7.1, 2.3 Hz, 2H), 1.83 (quint, J = 6.9 Hz, 2H), 1.67 (quint, J = 7.3 Hz, 2H); 13C NMR (CDCl3, 100 MHz): δ 151.1, 137.3, 129.0, 124.7, 119.9, 87.6, 76.4, 44.2, 31.3, 24.8, 17.9; HRMS (ESI) m/z: [M + H]+ calcd for C13H15ClNO 236.0842, found 236.0843.
:hexanes = 1:5). mp 80–81 °C; IR (neat): 3256 (w), 2939 (m), 2862 (m), 2230 (m), 1636 (s), 1543 (s), 1443 (s), 1319 (s), 1257 (s), 1095 (s), 995 (w), 887 (m) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.51 (d, J = 8.2 Hz, 2H), 7.45 (br s, 1H),7.33 (t, J = 7.8 Hz, 2H), 7.13 (t, J = 7.4 Hz, 1H), 3.79 (t, J = 5.7 Hz, 2H), 2.49 (t, J = 7.3 Hz, 2H), 1.82 (quint, J = 6.5 Hz, 2H), 1.14–1.04 (m, 21H); 13C NMR (CDCl3, 100 MHz): δ 151.0, 137.4, 129.0, 124.7, 119.8, 88.4, 76.0, 61.5, 31.0, 18.0, 15.2, 11.9; HRMS (ESI) m/z: [M + H]+ calcd for C21H34NO2Si 360.2359, found 360.2360.
:7ea = 2:1) as a light yellow solid. The crude product was purified using silica gel column chromatography (EtOAc:hexanes = 1:5). A mixture of 7e and 7ea could not be separated. mp 111–112 °C; IR (neat): 3294 (s), 2230 (m), 1944 (w), 1628 (s), 1520 (s), 1443 (s), 1319 (s), 1250 (s), 1180 (m), 1072 (w), 1026 (w), 980 (w), 887 (m), 964 (m), 825 (s) cm−1; 1H NMR of 7e (CDCl3, 400 MHz): δ 7.69 (br s, 1H), 7.53 (d, J = 8.2 Hz, 2H), 7.39–7.29 (m, 7H), 7.14 (t, J = 7.3 Hz, 1H), 3.75 (s, 2H); 13C NMR of 7e (CDCl3, 100 MHz): δ 150.8, 137.3, 134.4, 129.1, 128.8, 128.0, 127.2, 124.8, 119.8, 85.6, 7.5, 25.0; HRMS (ESI) m/z: [M + H]+ calcd for C16H14NO 236.1075, found 236.1074.
:hexanes = 1:7). mp 70–71 °C; IR (neat): 3263 (w), 3063 (w), 2924 (m), 2854 (m), 2230 (m), 1636 (s), 1597 (s), 1543 (s), 1497 (s), 1443 (s), 1381 (s), 1319 (s), 1257 (s), 756 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.89 (br s, 1H), 7.54 (d, J = 8.2 Hz, 2H), 7.31 (t, J = 7.8 Hz, 2H), 7.11 (t, J = 7.3 Hz, 1H), 2.20 (d, J = 6.9 Hz, 2H), 1.81 (d, J = 11.9 Hz, 2H), 1.73–1.63 (m, 3H), 1.28–1.22 (m, 3H), 1.19–1.12 (m, 1H), 1.04–0.96 (m, 2H); 13C NMR (CDCl3, 100 MHz): δ 151.3, 137.5, 128.9, 124.6, 119.8, 87.7, 76.8, 36.7, 32.7, 26.3, 26.0, 25.9; HRMS (ESI) m/z: [M + H]+ calcd for C16H20NO 242.1545, found 242.1546.
:hexanes = 1:5). mp 108–109 °C; IR (neat): 3232 (w), 3063 (w), 2932 (m), 2854 (w), 2230 (m), 1636 (s), 1597 (s), 1543 (s), 1497 (m), 1443 (s), 1319 (s), 1257 (s), 756 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.90 (br s, 1H), 7.55 (d, J = 8.2 Hz, 2H), 7.31 (t, J = 7.8 Hz, 2H), 7.11 (t, J = 7.3 Hz, 1H), 2.51–2.45 (m, 1H), 1.86–1.80 (m, 2H), 1.75–1.67 (m, 2H), 1.56–1.44 (m, 2H), 1.35–1.25 (m, 4H); 13C NMR (CDCl3, 100 MHz): δ 151.4, 137.5, 128.9, 124.5, 119.9, 92.1, 75.8, 31.5, 28.8, 25.5, 24.6; HRMS (ESI) m/z: [M + H]+ calcd for C15H18NO 228.1388, found 228.1389.
:hexanes = 1:5). mp 143–144 °C; IR (neat): 3225 (w), 2962 (w), 2230 (m), 1638 (m), 1543 (s), 1443 (s), 1319 (s), 1257 (s), 910 (m) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.81 (br s, 1H), 7.52 (d, J = 7.8 Hz, 2H), 7.29 (t, J = 7.8 Hz, 2H), 7.09 (t, J = 7.4 Hz, 1H), 2.74–2.67 (m, 1H), 1.97–1.90 (m, 2H), 1.74–1.64 (m, 4H), 1.59–1.53 (m, 2H); 13C NMR (CDCl3, 100 MHz): δ 151.4, 137.5, 128.9, 124.5, 119.8, 92.6, 75.5, 33.1, 29.7, 25.1; HRMS (ESI) m/z: [M + H]+ calcd for C14H16NO 214.1232, found 214.1233.
:hexanes = 1:5). This compound has been previously reported and the spectra data match described.341H NMR (DMSO-d6, 400 MHz): δ 10.46 (br s, 1H), 7.58 (d, J = 7.8 Hz, 2H), 7.30 (t, J = 7.8 Hz, 2H), 7.06 (t, J = 7.5 Hz, 1H), 1.28 (s, 9H); 13C NMR (DMSO-d6, 100 MHz): δ 151.3, 137.3, 129.1, 129.1, 124.7, 119.9, 95.7, 30.1, 27.5.
:hexanes = 1:5). IR (neat): 3263 (m), 3063 (w), 2978 (w), 2230 (m), 1636 (s), 1597 (s), 1543 (s), 1497 (m), 1443 (s), 1319 (s), 1257 (s), 756 (s) cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.31 (br s, 1H), 7.55 (d, J = 8.2 Hz, 2H), 7.30 (t, J = 8.0 Hz, 2H), 7.11 (t, J = 7.8 Hz, 1H), 3.49 (td, J = 6.4, 2.3 Hz, 2H), 2.31 (td, J = 7.1, 2.3 Hz, 2H), 1.83 (quint, J = 6.9 Hz, 2H), 1.67 (quint, J = 7.3 Hz, 2H); 13C NMR (CDCl3, 100 MHz): δ 151.1, 137.3, 129.0, 124.7, 119.9, 87.6, 76.4, 44.2, 31.3, 24.8, 17.9; HRMS (ESI) m/z: [M + H]+ calcd for C13H15ClNO 236.0842, found 236.0843.
:hexanes = 1:5). mp 80–81 °C; IR (neat): 3256 (w), 2939 (m), 2862 (m), 2230 (m), 1636 (s), 1543 (s), 1443 (s), 1319 (s), 1257 (s), 1095 (s), 995 (w), 887 (m) cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.51 (d, J = 8.2 Hz, 2H), 7.45 (br s, 1H),7.33 (t, J = 7.8 Hz, 2H), 7.13 (t, J = 7.4 Hz, 1H), 3.79 (t, J = 5.7 Hz, 2H), 2.49 (t, J = 7.3 Hz, 2H), 1.82 (quint, J = 6.5 Hz, 2H), 1.14–1.04 (m, 21H); 13C NMR (CDCl3, 100 MHz): δ 151.0, 137.4, 129.0, 124.7, 119.8, 88.4, 76.0, 61.5, 31.0, 18.0, 15.2, 11.9; HRMS (ESI) m/z: [M + H]+ calcd for C21H34NO2Si 360.2359, found 360.2360.
Representative experimental procedure for the synthesis of β-lactams
:hexanes = 1:5) to obtain 4la (1.34 g, 5.62 mmol, 89% yield) as a yellow solid.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) (NRF-2020R1A2C1005817) and the DGIST R&D Program of the Ministry of Science and ICT (21-BRP-05). This work was also partially supported by a research grant from Kwangwoon University in 2020.Notes and references
- (a) L. Micouin, I. Marek and Z. Rappoport, The Chemistry of Organoaluminum Compounds, Wiley, 2017 Search PubMed; (b) S. Nagendran and H. W. Roesky, Organometallics, 2008, 27, 457–492 CrossRef CAS.
- (a) H. Ogawa, Z.-K. Yang, H. Minami, K. Kojima, T. Saito, C. Wang and M. Uchiyama, ACS Catal., 2017, 7, 3988–3994 CrossRef CAS; (b) L. Qinghan, J. Xin, W. Kun, L. Ruiqiang, Z. Zhihao, L. Meng and H. Zheyao, Mini-Rev. Org. Chem., 2021, 18, 213–220 CrossRef CAS.
- (a) S. Turcaud, F. Berhal and J. Royer, J. Org. Chem., 2007, 72, 7893–7897 CrossRef CAS PubMed; (b) P. von Zezschwitz, Synthesis, 2008, 1809–1831 CrossRef CAS; (c) C. Verrier, S. Carret and J.-F. Poisson, Org. Biomol. Chem., 2014, 12, 1875–1878 RSC.
- (a) P. A. Donets and E. V. Van der Eycken, Org. Lett., 2007, 9, 3017–3020 CrossRef CAS PubMed; (b) S. Eichner, T. Eichner, H. G. Floss, J. Fohrer, E. Hofer, F. Sasse, C. Zeilinger and A. Kirschning, J. Am. Chem. Soc., 2012, 134, 1673–1679 CrossRef CAS; (c) H. Nada, G. Eros and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 5611–5614 CrossRef; (d) M. Nogami, K. Hirano, K. Morimoto, M. Tanioka, K. Miyamoto, A. Muranaka and M. Uchiyama, Org. Lett., 2019, 21, 3392–3395 CrossRef CAS.
- (a) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3414 RSC; (b) R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS; (c) E. Massolo, M. Pirola and M. Benaglia, Eur. J. Org. Chem., 2020, 4641–4651 CrossRef CAS.
- (a) E. Valeur and M. Bardley, Chem. Soc. Rev., 2009, 38, 606–631 RSC; (b) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS.
- (a) T. Suda, K. Noguchi, M. Hirano and K. Tanaka, Chem. – Eur. J., 2008, 14, 6593–6596 CrossRef CAS; (b) Y. Lee, Y. Motoyama, K. Tsuji, S.-H. Yoon, I. Mochida and H. Nagashima, ChemCatChem, 2012, 4, 778–781 CrossRef CAS; (c) Y.-X. Xie, R.-J. Song, X.-H. Yang, J.-N. Xiang and J.-H. Li, Eur. J. Org. Chem., 2013, 5737–5742 CrossRef CAS; (d) J.-J. Wu, Y. Li, H.-Y. Zhou, A.-H. Wen, C.-C. Lun, S.-Y. Yao, Z. Ke and B.-H. Ye, ACS Catal., 2016, 6, 1263–1267 CrossRef CAS.
- (a) F. J. Fananas and H. Hoberg, J. Organomet. Chem., 1984, 277, 135–142 CrossRef CAS; (b) B. Gabriele, G. Salerno, L. Veltri and M. Costa, J. Organomet. Chem., 2001, 622, 84–88 CrossRef CAS; (c) Y. Izawa, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 2004, 77, 2033–2045 CrossRef CAS; (d) S. T. Gadge, M. V. Khedkar, S. R. Lanke and B. M. Bhanage, Adv. Synth. Catal., 2012, 354, 2049–2056 CrossRef CAS; (e) C. Zhang, J. Liu and C. Xia, Catal. Sci. Technol., 2015, 5, 4750–4754 RSC; (f) R. S. Mane and B. M. Bhanage, J. Org. Chem., 2016, 81, 4974–4980 CrossRef CAS; (g) P. Szuroczki, B. Boros and L. Kollár, Tetrahedron, 2018, 74, 6129–6136 CrossRef CAS.
- (a) P. A. Donets, K. Van Hecke, L. Van Meervelt and E. V. Van der Eycken, Org. Lett., 2009, 11, 3618–3621 CrossRef CAS PubMed; (b) J. Hwang, J. Choi, K. Park, W. Kim, K. H. Song and S. Lee, Eur. J. Org. Chem., 2015, 2235–2243 CrossRef CAS; (c) M. A. Idris, M. Kim, J. G. Kim and S. Lee, Tetrahedron, 2019, 75, 4130–4137 CrossRef CAS.
- Y. Dong, S. Sun, F. Yang, Y. Zhu, W. Zhu, H. Qiao and Y. Wu, Org. Chem. Front., 2016, 3, 720–724 RSC.
- R. Zhang, Z.-Y. Gu, S.-Y. Wang and S.-J. Ji, Org. Lett., 2018, 20, 5510–5514 CrossRef CAS PubMed.
- P. Salehi and M. Shiri, Adv. Synth. Catal., 2019, 361, 118–125 CrossRef CAS.
- H. Lee, S. Cho, Y. Lee and B. Jung, J. Org. Chem., 2020, 85, 12024–12035 CrossRef CAS.
- (a) D. Qian and J. Zhang, Chem. Commun., 2012, 48, 7082–7084 RSC; (b) L.-F. Yu, Y.-Y. Li, M.-B. Su, M. Zhang, W. Zhang, L.-N. Zhang, T. Pang, R.-T. Zhang, B. Liu, J.-Y. Li, J. Li and F.-J. Nan, ACS Med. Chem. Lett., 2013, 4, 475–480 CrossRef CAS; (c) C. Jiang, P. J. Young, S. Brown-Xu, J. C. Gallucci and M. H. Chisholm, Inorg. Chem., 2017, 56, 1433–1445 CrossRef CAS.
- For a plausible alumination pathway of terminal alkynes, see: (a) B. Wang, M. Bonin and L. Micouin, Org. Lett., 2004, 6, 3481–3484 CrossRef CAS; (b) Y. Zhou, T. Lecourt and L. Micouin, Adv. Synth. Catal., 2009, 351, 2595–2598 CrossRef CAS.
- H. Fang, Z. Yang, L. Zhang, W. Wang, Y. Li, X. Xu and S. Zhou, Org. Lett., 2016, 18, 6022–6025 CrossRef CAS.
- (a) P. B. Biradar and H.-M. Gau, Chem. Commun., 2011, 47, 10467–10469 RSC; (b) R. Kumar, S. Turcaud and L. Micouin, Org. Lett., 2014, 16, 6192–6195 CrossRef CAS; (c) C. Wen, C. Wu, R. Luo, Q. Li and F. Chen, Synthesis, 2021, 53, 3847–3861 CrossRef CAS.
- (a) R. P. Elander, Appl. Microbiol. Biotechnol., 2003, 61, 385–392 CrossRef CAS PubMed; (b) K.-F. Kong, L. Schneper and K. Mathee, APMIS, 2010, 118, 1–36 CrossRef CAS PubMed.
- (a) S. Mobashery, S. O. Meroueh and J. F. Fisher, Chem. Rev., 2005, 105, 395–424 CrossRef; (b) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Rev., 2007, 107, 4437–4492 CrossRef CAS; (c) G. S. Singh and S. Sudleesh, in Natural Lactones and Lactams: Synthesis, Occurrence and Biological Activity, ed. T. Janecki, Wiley-VCH, Weinheim, Germany, 2014, ch. 3, pp. 101–145 Search PubMed.
- L. Zhang, L. Ma, H. Zhou, J. Yao, X. Li and G. Qiu, Org. Lett., 2018, 20, 2407–2411 CrossRef CAS PubMed.
- (a) F. Punner and G. Hilt, Chem. Commun., 2012, 48, 3617–3619 RSC; (b) E. D. Slack, C. M. Gabriel and B. H. Lipshutz, Angew. Chem., Int. Ed., 2014, 53, 14051–14054 CrossRef CAS; (c) J. Nguyen, N. Duncan and G. Lalic, Eur. J. Org. Chem., 2016, 5803–5806 CrossRef CAS.
- Y. Xie, R. Song, X. Yang, J. Xiang and J. Li, Eur. J. Org. Chem., 2013, 5737–5742 CrossRef CAS.
- J. Liu, C. Schneider, J. Yang, Z. Wei, H. Jiao, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 371–379 CrossRef CAS PubMed.
- D. S. Ryabukhin, L. Y. Gurskaya, G. K. Fukin and A. V. Vasilyev, Tetrahedron, 2014, 70, 6428–6443 CrossRef CAS.
- M. Pramanik, K. Choudhuri, S. Chakraborty, A. Ghosh and P. Mal, Chem. Commun., 2020, 56, 2991–2994 RSC.
- D. A. Petrone, I. Franzoni, J. Ye, J. F. Rodriguez, A. I. Bahamonde and M. Lautens, J. Am. Chem. Soc., 2017, 139, 3546–3557 CrossRef CAS.
- C.-S. Wang, T. Roisnel, P. H. Dixneuf and J.-F. Soulé, Adv. Synth. Catal., 2019, 361, 445–450 CrossRef CAS.
- Y. Kawagoe, K. Moriyama and H. Togo, Tetrahedron, 2013, 69, 3971–3977 CrossRef CAS.
- Y. Yang, C. Fei, K. Wang, B. Liu, D. Jiang and B. Yin, Org. Lett., 2018, 20, 2273–2277 CrossRef CAS.
- X. Luo and P. Wang, Org. Lett., 2021, 23, 4960–4965 CrossRef CAS PubMed.
- S.-Q. Wang, X.-W. Yan, B.-L. Hu and X.-G. Zhang, Tetrahedron, 2020, 76, 131021, DOI:10.1016/j.tet.2020.131021.
- S.-Q. Wang, B.-L. Hu and X.-G. Zhang, Adv. Synth. Catal., 2019, 361, 1459–1462 CrossRef CAS.
- J. Li, D. Ding, L. Liu and J. Sun, RSC Adv., 2013, 3, 21260–21266 RSC.
- H. Adams, M. Baker, H. Hodson and M. J. Morris, Tetrahedron Lett., 2017, 58, 1685–1698 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C and 19F NMR for all compounds. See DOI: 10.1039/d1ob01990b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |