
Interfacial tension driven adsorption of MnO2 nanoparticles at the liquid/liquid interface to tailor ultra-thin polypyrrole sheets†
Subin Kaladi
Chondath
a,
Abhishek Pradeep Kumar
Sreekala
a,
Chalikkara
Farzeena
b,
Subramanyan Namboodiri
Varanakkottu
 c and
Mini Mol
Menamparambath
*a
c and
Mini Mol
Menamparambath
*a
aDepartment of Chemistry, National Institute of Technology Calicut, Calicut-673601, Kerala, India. E-mail: minimol@nitc.ac.in
bSchool of Materials Science and Engineering, National Institute of Technology Calicut, Calicut-673601, Kerala, India
cDepartment of Physics, National Institute of Technology Calicut, Calicut-673601, Kerala, India
First published on 28th July 2022
Abstract
An emerging aspect of research is designing and developing fully tunable metamaterials for various applications with fluid interfaces. Liquid/liquid interface-assisted methods represent an efficient and facile route for synthesizing two-dimensional (2-D) thin films of potential materials. The underlying mechanism behind thin film formation at the liquid/liquid interface involves the preferential adsorption of nano-sized particles at the interface to minimize high interfacial tension. Here, a water/chloroform interface-assisted method is employed for the one-pot synthesis of highly crystalline polypyrrole/manganese dioxide (PPy/MnO2) sheets. The temporal evolution in the dynamic interfacial tension (from 32 mN m−1 to 17 mN m−1) observed in pendant drop tensiometry proved the preferential adsorption of MnO2 atttached PPy oligomers at the water/chloroform interface. An ultra-thin sheet-like morphology and uniform distribution of ∼6 nm highly crystalline MnO2 nanoparticles are evidenced by transmission and atomic force microscopy techniques. The predominance of interfacial polymerization in retaining the electrochemical activity of the PPy/MnO2 sheets is elucidated for the electrochemical detection of nicotine. This study opens a new avenue for the realization of ultra-thin sheets of polymer–nanomaterial hybrids, enabling applications ranging from new classes of sensors to optics.
1. Introduction
The competency of nanoparticle-laden fluid interfaces to configure functional architectures has received immense attention for their applications in electro-optics,1,2 catalysis,3,4 sensors,5–8 oil-spill recoveries,9,10 and more. The major challenge in organizing a single type of anisotropic nanoparticle into distinct super lattices11,12 over a large area was accomplished with liquid/liquid (L/L) interfaces. The adsorption of nanoparticles (NPs) at interfaces is a thermodynamically favorable and spontaneous process since the NPs tend to minimize the interfacial tension, interfacial area, and interfacial energy (Helmholtz free energy).13–16 The adsorption/desorption behavior of NPs across the L/L interface is governed by variation in interfacial free energies as validated using molecular dynamic simulations.17 Furthermore, the stability of the assembled NPs determines the stabilization of the interface of two immiscible liquids.13,18 The relationship between the particle size (R), the wettability of the particle surface (θ), the interfacial tension (IFT, γ), and the change in the Helmholtz free energy (ΔE) is postulated in the Pieranski equation (ΔE = −πR2γ (1 + cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ)2).19,20 The immobilization of NP assemblies at the L/L interface is achieved by the cross-linking agents or catalysts dispersed in either of the two phases.21 Another alternative method to immobilize the NP assemblies at the L/L interface is to entrap NPs in potential polymeric films formed in situ at the interface.22 The preferential adsorption of NP-attached oligomers at the interface and subsequent product confinement led to the formation of ultra-thin films with attached nano-sized particles.23–25 Thus, a unique one-pot synthesis of polymer thin films with encapsulated NPs is an effective strategy to confine NPs at the L/L interface or tune the polymer morphology to ultra-thin sheets.
θ)2).19,20 The immobilization of NP assemblies at the L/L interface is achieved by the cross-linking agents or catalysts dispersed in either of the two phases.21 Another alternative method to immobilize the NP assemblies at the L/L interface is to entrap NPs in potential polymeric films formed in situ at the interface.22 The preferential adsorption of NP-attached oligomers at the interface and subsequent product confinement led to the formation of ultra-thin films with attached nano-sized particles.23–25 Thus, a unique one-pot synthesis of polymer thin films with encapsulated NPs is an effective strategy to confine NPs at the L/L interface or tune the polymer morphology to ultra-thin sheets.
The paramount importance of L/L interface-assisted methods in processing and preparing potential materials is rooted in their ability to synthesize ultra-thin two-dimensional (2-D) films25,26 or reconfigure self-assembly on demand.1,27 The nanostructure tuning to ultra-thin 2-D films at the L/L interface is impelled by the self-organization of reactive molecular species at the interface of two immiscible liquids.24–26 The conventional methods are tedious to synthesize thin films due to the material characteristics (insolubility, dispersibility, transparency, and homogeneity) or stringent deposition conditions.22 The L/L interface-assisted method ensures the facile deposition of thin films of single or multi-component materials that are effortlessly transferred to any substrates (plastic or flexible substrates).22 The L/L interface-assisted synthesis route allows control over the thickness, homogeneity, and transparency of the thin film under ambient temperature and pressure conditions with a simple and economically viable procedure.22
To meet the challenges that lie ahead for targeted applications, thin films of various nanostructures are prepared by the self-assembly of these nanostructures, selectively dispersed in any of the two immiscible solvents.22,28,29 An interfacial film formation technique was employed to fabricate MXene thin films for ultra-compact passive photonic diodes.30 The in situ synthesis of multi-component (nanocomposite) materials is intricate to perform under ambient conditions, while the L/L interface-assisted method proved to be successful. Furthermore, the insolubility, infusibility, and dispersibility of reactive species limit the in situ distribution of nanostructures in a polymer matrix using conventional chemical or electrochemical routes. On the other hand, conductive polymers composited with multidimensional nanomaterials yielded free-standing thin films for several novel applications.31
Here, we investigated the self-assembly of in situ grown PPy/MnO2 at the water/chloroform interface, tuning the polymer composites to ultra-thin 2-D sheets without any hard or sacrificial templates. Experimental observations proved that the high interfacial tension at the water/chloroform interface acts as a driving force for the preferential adsorption of MnO2-bound oligomers. The dynamic interfacial tension of the water/chloroform interface as a function of time evidenced the stabilization of the interface with a persistent decrease in the IFT as the polymerization progressed at the interface. The water/chloroform interface acts as a soft template for the self-assembly of MnO2 attached PPy oligomers, leading to the confinement and tuning of PPy/MnO2 composites to ultra-thin sheets. The transmission electron microscopic images showed the growth and confinement of amorphous network-like structures to ultra-thin PPy/MnO2 sheets during the polymerization. The transition of diffused rings to bright Bragg spots in electron diffraction further confirmed the evolution of amorphous MnO2 agglomerates to highly crystalline MnO2 NPs. MnO2 NPs are widely accepted for electrochemical storage applications, and numerous works have been devoted to synthesizing diverse MnO2 nanostructures for energy applications.32,33 Herein, the uniform encapsulation of MnO2 NPs in PPy/MnO2 sheets was further explored for the electrochemical sensing of nicotine, a significant ingredient in cigarettes.34 The composites of PPy/MnO2 with multi-walled carbon nanotubes (MWCNTs) were used as electrochemical sensors for nicotine detection. The printable nature of the composite inks of PPy/MnO2 with MWCNTs was also tested on screen-printed electrodes and subsequently validated for electrochemical activity in nicotine oxidation.
2. Experimental section
2.1. Chemicals and reagents
Pyrrole (Sigma Aldrich, 131709, 98%), nicotine (Sigma Aldrich, 8.20877, ≥99%), potassium permanganate (Merck Millipore, 105082, ACS, Reag. Ph Eur), and chloroform (Fischer Scientific, 33515, 99.5%) were used as received. All the reagents were of analytical purity, and deionized (DI) water (ELGA Purelab Quest UV, 18.2 MΩ) was used throughout the synthesis.2.2. Synthesis of PPy/MnO2 sheets at the water/chloroform interface
Pyrrole polymerization was performed at the water/chloroform interface at room temperature (RT) using potassium permanganate (KMnO4) as the oxidant. The molar ratio of pyrrole and KMnO4 was fixed as 1:1. The monomer pyrrole and oxidant KMnO4 of 0.01 M concentration were prepared respectively in chloroform (15 mL) and water (12 mL). A low-density aqueous layer was evenly placed on the top of a higher density organic layer of 78.5 cm2 to retain a flat liquid/liquid interface throughout the polymerization. The interfacial polymerization of pyrrole was carried out at room temperature under a non-stirring condition for about 15 hours. As shown in Fig. 1A and Fig. S1,† during the initial stages of the polymerization (up to 7.5 hours), the products, such as radical cations, oligomers, and short-chain polymers, exhibited more affinity towards the aqueous phase. Hence a uniform dispersion of products at the aqueous phase was observed for up to 7.5 hours. However, visible product confinement at the water/chloroform interface was observed after 7.5 hours. Hence the polymerization was continued for up to 15 hours. The as-synthesized product was washed several times with a mixture of DI water and ethanol and vacuum filtered, followed by vacuum drying for 24 hours at room temperature. It resulted in a powder form, as shown in Fig. S2.† The concentration of the monomer and oxidant was also varied to 0.02 M and 0.005 M, respectively, and control experiments were performed while retaining the monomer/oxidant ratio as 1:1. Pure PPy was also synthesized in a water/chloroform bisolvent medium using ammonium persulfate as the oxidant.25
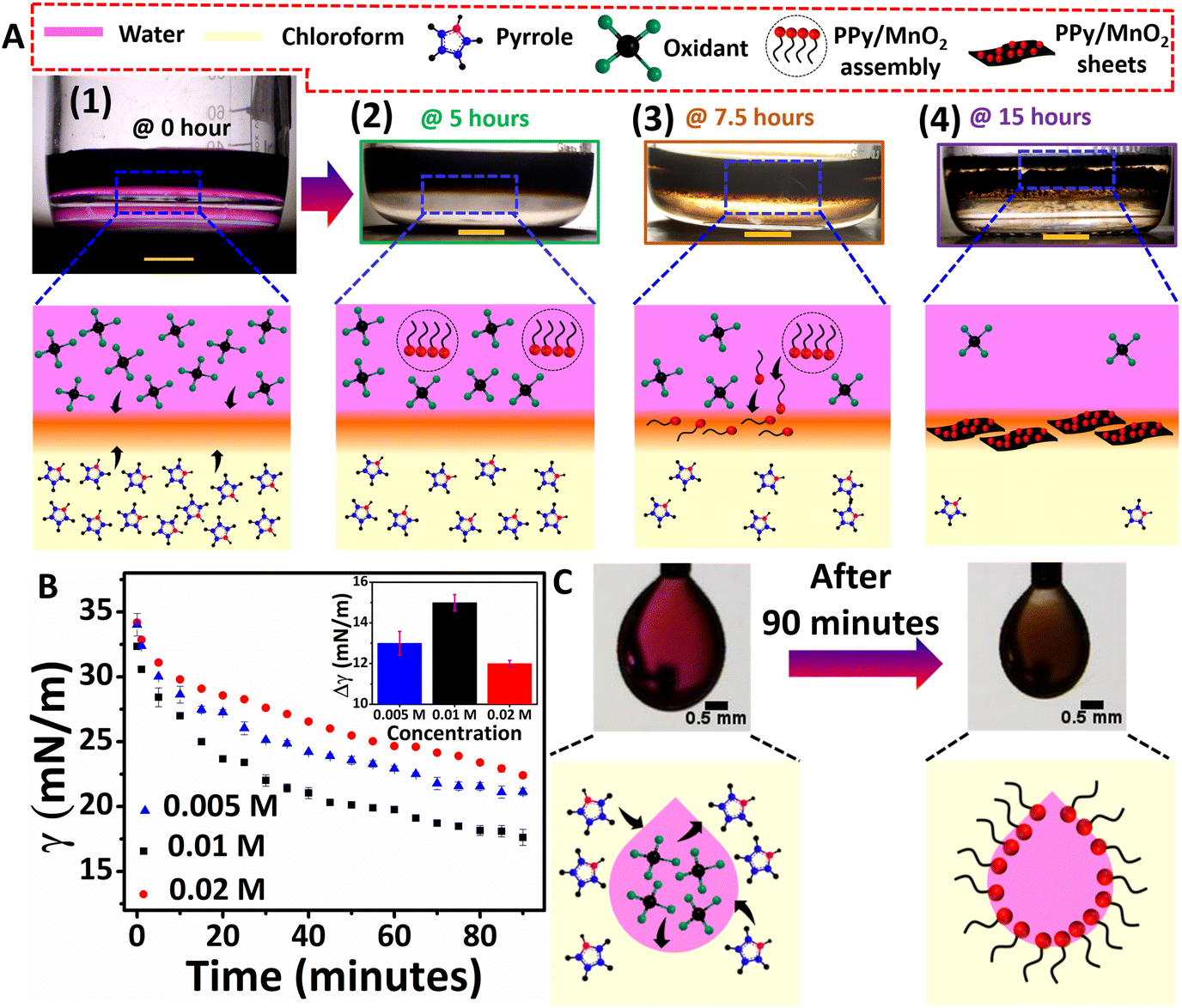 | ||
| Fig. 1 (A) Photographic images and schematic illustration of the interface-assisted polymerization of PPy/MnO2 at different time intervals. Scale bar: 1 cm. Steps 1–4 highlight the adsorption of NP-attached oligomers/short-chain polymers at the interface to form microscale PPy/MnO2 sheets. Step 2 exhibits a dark coloration in the aqueous phase (KMnO4 dissolved in water) due to the immediate transfer of MnO2 attached oligomers towards the polar aqueous phase. However, steps 3 and 4 indicate the adsorption of MnO2 attached oligomers/short-chain polymers towards the interface and the eventual formation of 2-D PPy/MnO2 sheets. (B) The IFT was measured as a function of time for pendant drops corresponding to three sets of monomer/oxidant concentrations (0.005 M, 0.01 M, and 0.02 M). The inset shows the variation of IFT as a function of monomer/oxidant concentration. (C) The photographic images show the pendant drop of 0.01 M oxidant solution suspended in 0.01 M monomer dispersion at zero and 90 minutes, respectively. The decrease in the IFT due to the adsorption of PPy/MnO2 nanostructures at the oxidant drop and monomer solution interface is illustrated schematically. | ||
2.3. Synthesis of PPy/MnO2 spheres in the aqueous phase
Pyrrole polymerization was performed in the aqueous phase at room temperature using KMnO4 as the oxidant. The molar ratio of pyrrole and KMnO4 was fixed as 1:1. A 0.01 M aqueous solution of pyrrole and KMnO4 was prepared in 15 mL and 12 mL of deionized water, respectively. The above two solutions were mixed to form the reaction medium and the reaction was carried out under a non-stirring condition for about 15 hours. The final products were dispersed in the aqueous medium at the end of the reaction. The products were directly taken out from the reaction medium for morphological studies. After 15 hours, the as-synthesized product was filtered and washed several times with deionized water and ethanol. After purification, the PPy/MnO2 spheres in their powder form were obtained and dried under vacuum at room temperature.
2.4. Instrumentation
Six identical reaction mixtures were formulated to collect samples at six different time intervals for ultraviolet-visible (UV-Vis) absorption (Shimadzu UV 2600), Raman (Horiba Labram HR Evolution confocal Raman spectrometer), and transmission electron microscopy (HRTEM, JEOL/JEM 2100) characterization studies. The sample collection methods for UV-Vis absorption and Raman analysis are as follows. The aqueous dispersion (300 μL) was collected from three identical reaction mixtures at 2.5, 5, and 7.5 hours intervals and further diluted by adding 3 mL of DI water. The products confined at the interface (for 10, 12.5, and 15 hours intervals) were carefully lifted and separated using a custom-made L-shape. It was then transferred to 3 mL of DI water. A thorough mixing was performed before the measurements to retain the dispersion of the products at the aqueous phase during the measurement. The morphological evolution of PPy/MnO2 sheets during the polymerization was probed using HRTEM by collecting products at different time intervals and redispersing in ice-cold water to quench the polymerization. Hereafter, the aqueous sample solutions were drop-cast on copper grids for HRTEM studies.Further morphological and physico-chemical properties of PPy/MnO2 were characterized using the as-synthesized product after 15 hours (filtered and washed several times with deionized water and ethanol). Field emission scanning electron microscopy (FESEM, FEI-Quanta FEG 200F), HRTEM with energy dispersive spectroscopy (EDS), and elemental mapping (Thermofischer Talos F200 S) were employed to obtain electron microscopy images and the elemental composition. The crystallinity of the PPy/MnO2 powder sample was analyzed using the powder XRD technique (Bruker D8 advance with Cu Kα radiation, scan rate: 0.013 steps per 26 to 32 s). The PPy/MnO2 films confined at the interface were transferred to mica sheets for atomic force microscopy (AFM, Bruker) by simply lifting the substrate in the film direction. The surface chemistry of the PPy/MnO2 samples was analyzed using XPS (PHI 5000 Versa Probe II, ULVAC-PHI Inc., USA) equipped with a micro-focused (200 μm, 15 kV) monochromatic Al-Kα X-ray source (hν = 1486.6 eV). Survey scans were recorded with an X-ray source power of 50 W and a pass energy of 187.85 eV. High-resolution spectra of the major elements were recorded at 46.95 eV pass energy. XPS data were processed using PHI's Multipak software. The binding energy was referenced to the C 1s peak at 284.8 eV.
2.5. Measurement of dynamic interfacial tension
The pendant drop method was used to measure the interfacial tension between water and chloroform. The aqueous KMnO4 droplet (8 μL) was suspended vertically using a syringe needle into a 1 × 1 cm2 glass cuvette containing pyrrole in chloroform. The images of the pendant drops were captured using a digital microscope at a rate of 20 frames per minute. The images were fitted with the ImageJ pendant drop plugin. The temporal evolution of the interfacial tension was measured for aqueous KMnO4 drops of varying concentrations (0.005 M, 0.01 M, and 0.02 M) suspended in pyrrole dispersions (concentration range: 0.005 M–0.02 M).2.6. Electrochemical detection of nicotine using cyclic voltammetry
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) scans were recorded using the Origalys OGF500 instrument. Electrochemical investigations were conducted on both modified glassy carbon electrodes (GCE) and modified screen-printed electrodes (SPE). 0.1 M phosphate buffer solution of pH 7.0 was used throughout the work. The electrochemical detection of nicotine was conducted in 1 mM nicotine solution.To enhance the electrical conductivity and hence the electrochemical activity of PPy/MnO2, MWCNTs of high aspect ratio (1250–6500) were added as a conductive filler. A stable dispersion of MWCNTs was prepared by sonicating MWCNTs in water with the help of a 5% hydroxypropyl methylcellulose (HPMC) solution. The composites of PPy/MnO2 sheets with different concentrations of MWCNTs (1–5 wt%) were prepared by sonicating the required amount of PPy/MnO2 with MWCNTs, and their electrochemical activity was recorded by CV experiments in the presence of 5 mM K4[Fe(CN)6] in 0.1 M KCl. The electrochemical activity of the GCE modified with a mixture of pure PPy and MWCNT (PPy/MWCNT) was also investigated by a similar method.
3. Results and discussion
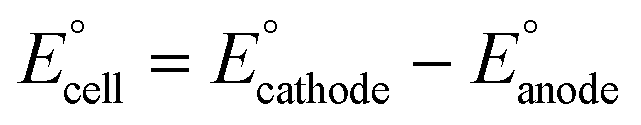
The present work uses a water/chloroform interface-assisted polymerization to synthesize PPy/MnO2 sheets (Fig. 1A). The fundamental idea of cationic type polymerization is based on the redox reaction between the monomer and oxidant to form a monomer radical cation.35 The spontaneity of the redox reaction between the monomer and oxidant depends on the decrease in the thermodynamic Gibbs free energy (ΔG°).24 Here, the monomer pyrrole and the oxidant KMnO4 form an electrode couple in which the pyrrole (Py) is oxidized to a pyrrole radical cation (Py˙+) (pyrrole works as the anode) and KMnO4 gets reduced to MnO2 (KMnO4 works as the cathode) as shown in eqn (1) and (2). From the electrochemical series, the standard reduction potentials of the pyrrole and KMnO4 are −0.9 V and 0.595 V, respectively.24,36 Hence the spontaneity of the initiation of polymerization (pyrrole to pyrrole radical cation conversion) is thermodynamically evaluated from the electrochemical cell potential (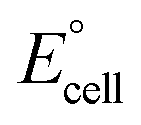 ) as follows:
) as follows:
Dissociation of KMnO4 in water is represented as KMnO4 → K+ + MnO4−
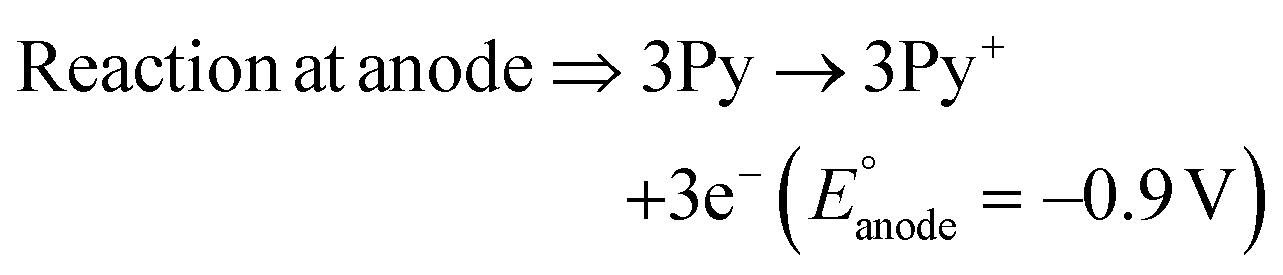 | (1) |
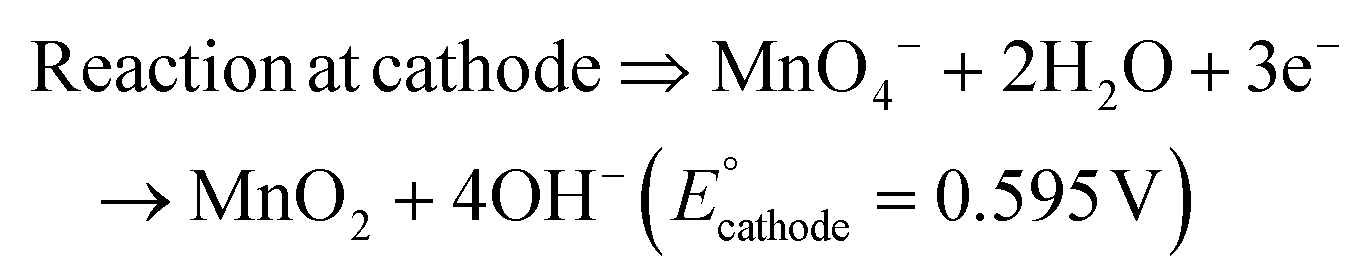 | (2) |
 | (3) |
 | (4) |
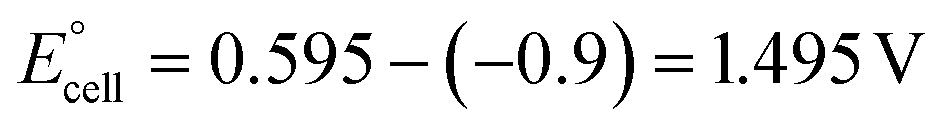 | (5) |
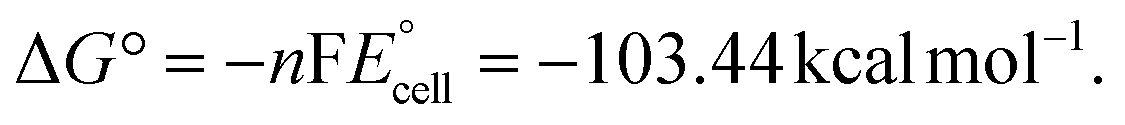 | (6) |
Thus, a significantly negative value of ΔG° (−103.44 kcal mol−1) predicts the feasibility of polymerization initiation, i.e., the spontaneous generation of pyrrole radical cations at the interface. The mechanism of the redox-initiated polymerization of pyrrole in the presence of KMnO4 to yield PPy/MnO2 is shown in Scheme 1.37
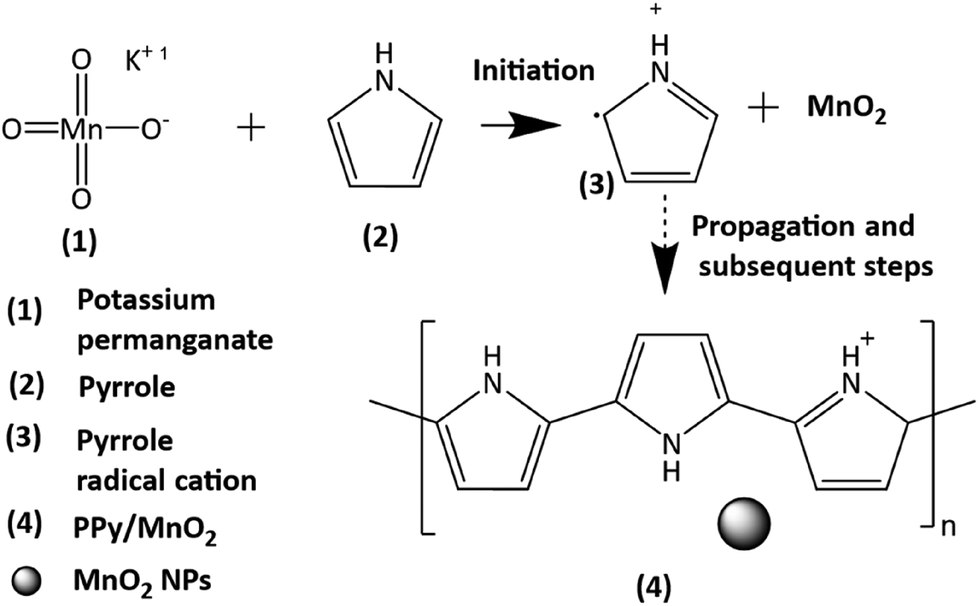 | ||
| Scheme 1 The mechanism of the redox initiated polymerization of a pyrrole monomer in the presence of KMnO4 as an oxidant to give PPy/MnO2. The mechanism involves the formation of a pyrrole radical cation in the initiation step and subsequently leads to the formation of PPy/MnO2 with attached MnO2 NPs. | ||
As shown in the photographic images, the initial stages of polymerization and the simultaneous adsorption of PPy/MnO2 at the 2-D interface of water and chloroform led to the confinement of 2-D PPy/MnO2 sheets within 15 hours. The 2-D L/L interface formed between water and chloroform serves as a reactive template for the organization of reactive molecules.38 Initially, the monomer pyrrole and oxidant KMnO4 meet at the interface, where they undergo a redox reaction to form pyrrole radical cations and MnO2 (eqn (1)–(3), Fig. 1A 1). The subsequent steps lead to the formation of charged PPy oligomers with attached MnO2 that are difficult to confine at the interface due to their continuous adsorption/desorption at the L/L interface.15,19,21,22,39,40 As per the Pieranski equation, the stabilization of the L/L interface by the minimization of Helmholtz free energy is influenced mainly by the size of the adsorbed particles.18 Hence, the abrupt nature of the L/L interface leads to continuous adsorption/desorption of smaller particles at the L/L interface;18 therefore, small-sized charged oligomers freely migrate towards the polar aqueous phase.13,15,41,42 The formation of intense dark coloration in the aqueous phase within 5 hours manifests PPy/MnO2 assembly formation within 5 hours, as shown in Fig. 1A 2. Within 7.5 hours of polymerization, a substantial increase in the chain length of oligomers or short-chain polymers and an increase in the size of attached MnO2 occur. The interface tends to adsorb MnO2 NP-attached PPy chains towards the interface to minimize the high interfacial tension and high interfacial free energy (Fig. 1A 3).18,24,43 The interfacial adsorption of MnO2 attached oligomers, or short-chain polymers, acts as the driving force for the confined 2-D growth of PPy sheets. The 2-D ordering of PPy chains and the layer-by-layer growth of π–π stacked 2-D aligned PPy chains at the water/chloroform interface resulted in PPy/MnO2 sheets at the end of 15 hours (Fig. 1A 4).44 The preferential adsorption of MnO2 attached PPy chains followed by jamming at the L/L interface could offer PPy sheets with uniformly distributed MnO2 NPs. The observation of transparency in the aqueous phase (upper layer) in the photographic image of Fig. 1A 4 implies the confinement of PPy/MnO2 sheets at the interface. The photographic images of the polymerization captured at 2.5, 10, and 12.5 hours during the reaction are shown in Fig. S1.† The control experiments of interfacial polymerization using lower (0.005 M) and higher (0.02 M) concentrations of the monomer and oxidant were performed (Fig. S3†). As observed in the photographic images (Fig. 1A 4 and Fig. S3A†), PPy/MnO2 illustrated final product confinement at the interface for lower concentrations (0.005 M and 0.01 M) of reactants. The final products were dispersed in the aqueous phase (Fig. S3B†) for higher concentrations of reactants (0.02 M). The top view photographic images in Fig. S4† demonstrated increased interfacial product confinement for a reaction conducted using 0.01 M concentrations of the monomer and oxidant. Since the middle concentration yielded a significant amount of product confinement at the interface, detailed investigations of nanoparticle-aided morphology tuning of polypyrrole were conducted using 0.01 M concentrations of reactants.
The adsorption of NP-attached oligomers at the interface results in the confinement of short-chain polymers, which may subsequently reduce the interfacial tension at the L/L interface. The pendant drop tensiometry studies evaluated the variation of interfacial tension due to the adsorption of products during the polymerization. The γ values were measured by suspending drops of different concentrations of oxidants (0.005 M, 0.01 M, and 0.02 M) in monomer dispersions (0.005 M, 0.01 M, and 0.02 M), and the results are summarized in Fig. 1B and C and Fig. S5.† The interfacial tension of pure water and chloroform was 34 mN m−1 under the same experimental conditions; the drops remained at the tip for less than 2 minutes. In the case of reaction mixtures of different concentrations, the pendant drops exhibited significant stability for up to 90 minutes, accompanied by a reduction of γ for all three concentrations (Fig. 1B and Fig. S5†). The pendant drops of 0.01 M concentration showed γ reduction from 32 mN m−1 to 17 mN m−1 (Fig. 1B). Similarly, 0.005 M and 0.02 M showed a γ reduction of 34 mN m−1 to 21 mN m−1 and 34 mN m−1 to 22 mN m−1 (Fig. 1B), respectively. Thus, as shown in the inset of Fig. 1B, the extent of decrease in IFT (Δγ) for 0.005 M, 0.01 M, and 0.02 M is 13 mN m−1, 15 mN m−1, and 12 mN m−1, respectively. The greater extent of IFT reduction could be attributed to the adsorption of nanoparticle-attached oligomers or short-chain polymers at the interface. A similar trend was reported for lignin nanoparticle adsorption for the 1-pentanol/water interface stabilization by IFT reduction during oil-spill recoveries.9 However, at a higher amount of product formation (for 0.02 M concentration), the IFT reduction is relatively minor. The preponderant solvent–polymer interaction as compared to the adsorption energy may promote the product confinement at the polar phase, as shown in Fig. S3B.† Therefore, it is confirmed that the monomer and oxidant molecules present in the respective phases initially meet at the interface and initiates polymerization, as shown in Fig. 1A 1 and C. Nano-sized PPy/MnO2 adsorbs at the interface as the reaction proceeds, and the product morphology gets tuned to 2-D sheets (Fig. 1C and 1A2–4).
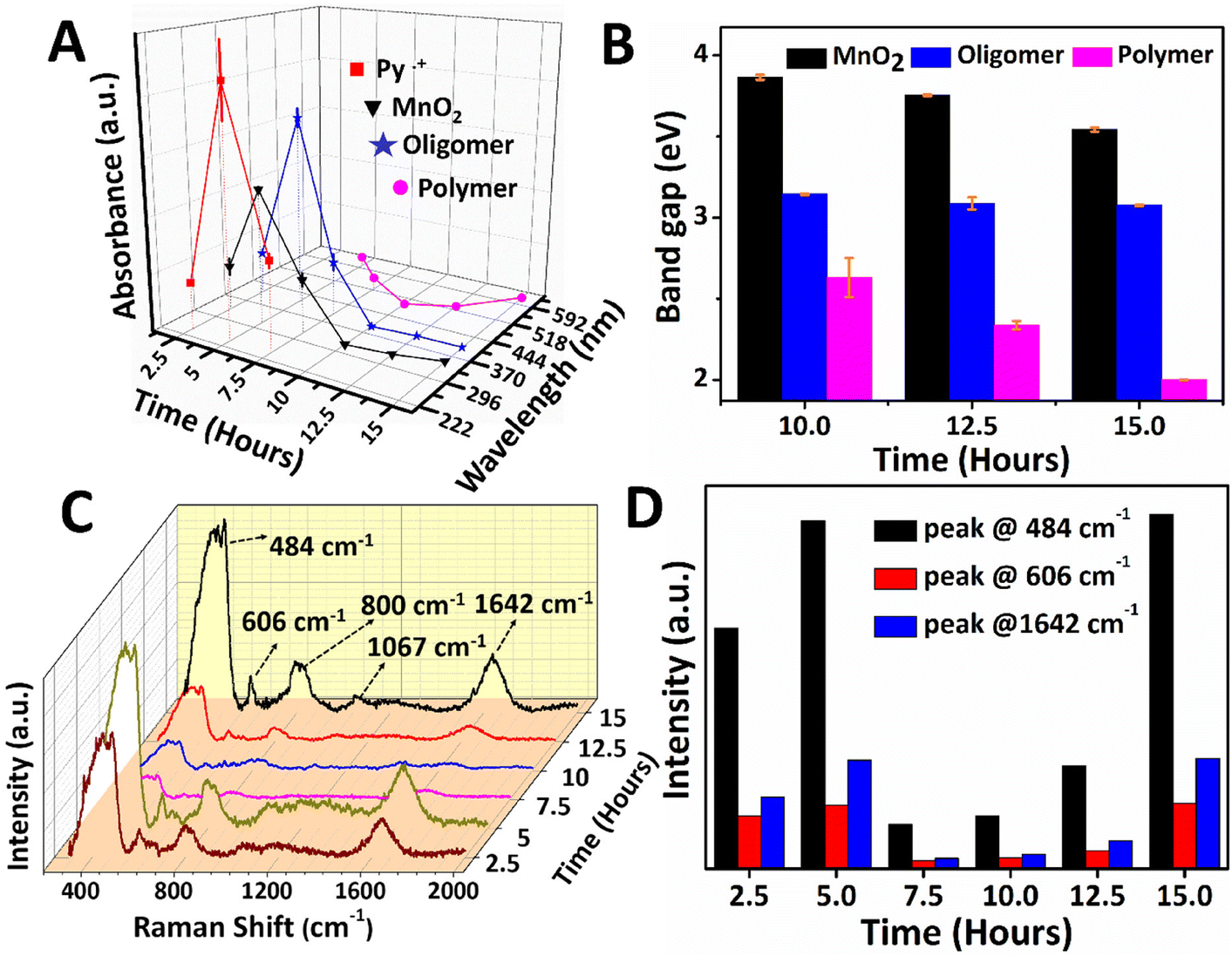 | ||
Fig. 2 (A) The absorbance and wavelength of UV-Vis absorption were plotted against the reaction time. (B) The band gap corresponding to the absorption peaks of the oligomer, the polymer, and MnO2 plotted against the reaction time. (C) Raman spectra of PPy/MnO2 were recorded at varying time intervals of 2.5 to 15 hours. Bar plots showing (D) the intensity corresponding to the peaks of α-MnO2 and the backbone stretching mode of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in PPy as a function of reaction time. C bonds in PPy as a function of reaction time. | ||
 | ||
| Fig. 3 High-resolution transmission electron microscopic images (series 1) and selected area electron diffraction images of PPy/MnO2 (series 2) recorded in a water/chloroform bisolvent medium at varying time intervals. The images A1 and A2, B1 and B2, C1 and C2, D1 and D2, E1 and E2, and F1 and F2 represent the reaction times of 2.5, 5, 7.5, 10, 12.5, and 15 hours, respectively. | ||
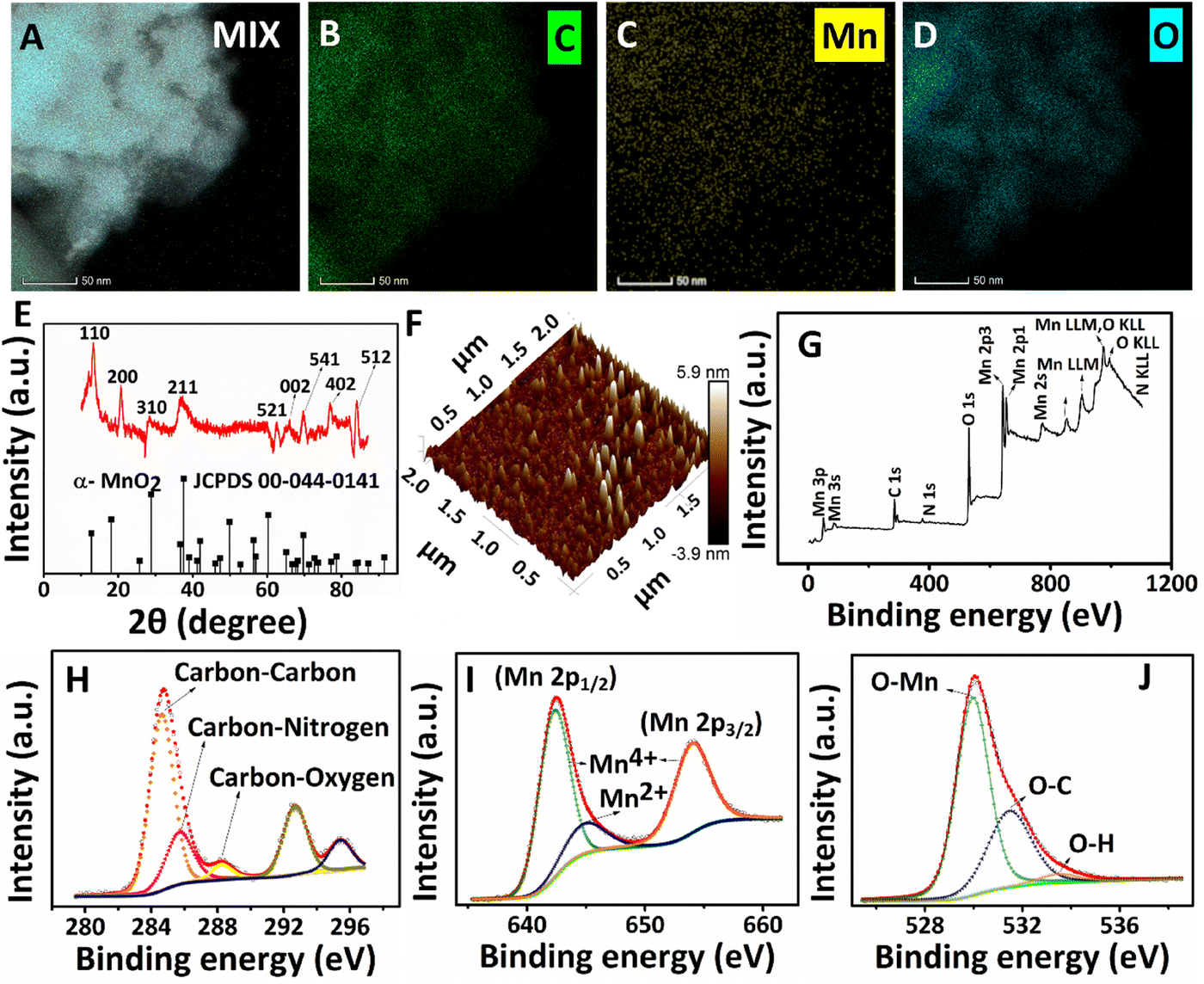 | ||
| Fig. 4 (A–D) HRTEM mapping images of PPy/MnO2 sheets synthesized in 15 hours. (A) Elemental mapping image of the mix with elements such as carbon (green), manganese (yellow), and oxygen (cyan). Elemental mapping of the individual elements, (B) carbon, (C) manganese, and (D) oxygen. (E) The XRD spectrum of PPy/MnO2 sheets synthesized in 15 hours. The peaks are indexed with the respective diffraction planes of α-MnO2 (JCPDS card no. 00-044-0141). (F) The 3-D atomic force microscopy image of PPy/MnO2 was recorded at 15 hours. The image clearly shows the sheet-like nature of PPy/MnO2 with submerged MnO2 NPs. (G) XPS survey scan spectrum of PPy/MnO2 sheets synthesized in 15 hours. (H–J) The high-resolution XPS spectra of C 1s, Mn 2p, and O 1s. | ||
The reaction mixture from the aqueous phase (2.5 to 7.5 hours) and the interface (10 to 15 hours) was collected and investigated using UV-Vis absorption spectroscopy, as shown in Fig. 2A and B. The absorbance variation and wavelength shift in the UV-Vis spectra of PPy/MnO2 was analyzed as a function of reaction time, and the results are shown in Fig. 2A. The relative trend in the absorption intensity for the samples synthesized in 2.5 to 7.5 hours follows a concentration dependence as per the Beer–Lambert law.45 The monomer absorption peak corresponding to the π to π* transition was observed at 230 nm (Fig. S6†). Hence, the absorption peak at 240–242 nm is assigned to Py˙+ which is detected in the reaction mixtures collected between 2.5 and 7.5 hours.46,47 The self-assembly of radical cations during the initial stages of polymerization is reflected in the absorbance profile. Maximum accumulation of Py˙+ adjacent to the interface was observed at 5 hours, and the subsequent decrease in the absorbance is due to the growth of polymeric chains. Furthermore, the absence of an absorption peak corresponding to Py˙+ after 7.5 hours is due to the total consumption of Py˙+ during the polymerization. The absorption peaks centered at 394–415 nm corresponding to the low molecular weight oligomers were detected in all the PPy/MnO2 samples (2.5 to 15 hours).46,47 Similarly, a maximum accumulation of oligomers was observed at 5 hours, and the subsequent decrease in the absorbance is due to the growth of polymeric chains. The bands attributed to the polar transitions of the PPy backbone are observed at 495–620 nm, which were absent at 2.5 hours. A distinguishable trend in the absorbance was not observed in the samples collected at 5–15 hours.46–48 However, a significant blue shift of 107 nm was observed for the PPy backbone absorptions for 5–10 hours, reflecting the random distribution of polymeric chains nearby the interface. Nonetheless, a red shift of 125 nm was observed from 10–15 hours due to the decrease of bandgap due to polymeric chain elongation.
The absorption peak corresponding to α-MnO2 at 315–350 nm (ref. 49 and 50) showed the maximum absorbance at 5 hours; however, the absorbance decreased as the reaction progressed. Furthermore, a red shift of 30 nm in the absorption wavelength could be due to the growth of MnO2 NPs. Since visible confinement of PPy/MnO2 was observed after 10 hours, the growth of MnO2 attached PPy chains was evaluated from the bandgap (E = hc/λ, λ = absorption wavelength) calculations, as plotted in Fig. 2B. The bandgap of oligomers remained constant after 10 hours, indicating the unequivocal conversion of oligomers to polymers. The apparent increase in the π conjugation length of oligomers and short-chain polymers in PPy/MnO2 after 10 hours decreased the bandgap, as observed in Fig. 2B.45,51,52 Furthermore, a consistent reduction in the bandgap of MnO2 observed within 10–15 hours predicts an increase in the size of MnO2 after 10 hours of polymerization.53
Additionally, the reaction mixtures collected at various time intervals were investigated using Raman spectroscopy, as shown in Fig. 2C. The Raman spectrum of PPy/MnO2 after 15 hours of reaction showed well-defined peaks corresponding to the PPy backbone and MnO2 NPs. The peaks at 484 cm−1 and 606 cm−1 correspond to α-MnO2,54,55 and the peaks at 800 cm−1, 1642 cm−1 and 1067 cm−1 can be attributed to the low-frequency C–C vibration,56 CC stretching mode48,57 and C–H in-plane bending vibrations of PPy,48,57 respectively. The intensity of Raman peaks is directly proportional to the concentration of active species.58 The peaks at 484 cm−1 and 606 cm−1 of α-MnO2, and 1642 cm−1 of PPy have remarkable intensity and consistency in all the samples. Hence these peaks were focused on investigating the mechanism of polymerization as a function of reaction time (Fig. 2C and D). The polymerized products are distributed in the aqueous phase for 2.5 to 7.5 hours, and the confinement becomes prominent from 10 hours. The MnO2 attached oligomers are initially formed at low concentrations at the interface within 2.5 hours. As a result, the Raman peaks corresponding to α-MnO2 and PPy are less intense, as shown in Fig. 2C and D. Due to the small size and high polarity, the MnO2 attached oligomers freely diffuse from the interface, leading to the formation of assemblies of MnO2 attached oligomers/short-chain polymers in the polar aqueous phase (Fig. 1A 2). Thus, after a reaction time of 5 hours, PPy/MnO2 is formed at the polar aqueous phase at a high concentration, and the intensity of the Raman peaks increases gradually (Fig. 2C and D). After 7.5 hours, the pervasive need for interfacial stabilization starts playing, and the interface begins to preferentially adsorb nano-sized MnO2 attached oligomers/short-chain polymers towards it (Fig. 1A 3). This led to a sudden decrease in the intensity of the Raman peaks, as can be seen in Fig. 2C and D. After 10 hours, the interface starts to confine the adsorbed nano-sized particles, and the 2-D growth of nano-sized MnO2 attached oligomers leads to the confinement of PPy/MnO2 sheets at the interface. The intensity of the Raman peaks shows a linear trend after 10 hours due to the possible increase in the concentration of PPy/MnO2 sheets formed at the interface (Fig. 2C and D). Furthermore, the interface promotes the layer-by-layer growth of sheets by π–π stacking interactions.44 The π–π interactions during the assembly of multilayer graphene sheets have been reported to furnish more intense Raman peaks.59
The practical 2-D nanostructural tuning of PPy/MnO2 nanocomposites was achieved by the combined effect of interfacial stabilization and the formation of NP-attached oligomers. The transmission electron microscopy images (Fig. 3) reveal the morphological evolution of PPy/MnO2 synthesized in the water/chloroform bisolvent medium over 15 hours. As indicated in TEM images (Fig. 3A1, B1, and C1), the agglomerated network-like structures strengthen the concept of self-assembly of MnO2 attached oligomers in the initial stages of polymerization as discussed in Fig. 1A 1–1A 3. However, as the reaction progressed from 10–15 hours, the evolution of 2-D sheet-like structures (Fig. 3D1, E1, and F1) further confirmed the confinement of oligomers or short-polymeric chains at the interface that tunes the polymers to the 2-D sheet-like structures. The TEM images show the 2-D sheet-like morphology of PPy/MnO2 formed at the interface for 10–15 hours. High magnification TEM images of all samples are shown in Fig. S7;† a relatively uniform distribution of MnO2 particles in the polymeric matrix is evident in Fig. 3F1 and Fig. S7F.† Furthermore, the corresponding selected area electron diffraction patterns support the formation of the tetragonal structure of α-MnO2.32,54,60,61 The planes (002), (200), (211), (220), (310), (400), (402), (411), (431), (440), (521), (541), (600), (602), (710), (730), and (741) are reported for α-MnO2 (JCPDS no. 00-044-0141).32,54,60,61 The planes corresponding to the polypyrrole backbone were not present in the samples. As the reaction progressed from 2.5 hours to 15 hours, the nature of diffraction patterns ranged from diffused rings to bright rings and bright Bragg spots analogous to a set of planes of α-MnO2. Furthermore, the TEM images and the diffraction patterns confirm the interface confinement-induced morphology and crystallinity tuning of PPy/MnO2 composites. This further confirms the synergistic effect of the high interfacial tension-induced confinement of MnO2 NPs, and confinement-induced morphology tuning. As reported previously, efficient interfacial stabilization is achieved for larger particles, which could be a possible reason for the visibly recognizable confinement of products only after 10 hours during the polymerization.18 Due to the varying size and shape of PPy/MnO2 agglomerates and evident transition from agglomerated network-like structures to a distinct 2-D sheet-like morphology, the SAED patterns of PPy/MnO2 synthesized at varying reaction times may lack a few identical diffraction planes. As observed in Fig. 3, a consistently observed highly intense diffraction peak of the (310) plane was chosen to compare crystallinity using SAED patterns. An S-shaped growth curve in the intensity of the (310) plane is provided in Fig. S8.† An inflection point was observed at 8 hours, and the intensity becomes constant after 10 hours of polymerization. Hence, the SAED investigations confirm the interfacial tension driven adsorption of MnO2 NPs and the π–π stacking of PPy/MnO2 sheets at the interface. The morphology of PPy/MnO2 composites synthesized in a single solvent was also analyzed and compared with that of PPy/MnO2 sheets prepared at the interface (Fig. S9†). The FESEM image in Fig. S9A† clearly shows the 2-D sheet-like morphology of PPy/MnO2 sheets synthesized at the water/chloroform interface. However, the PPy/MnO2 composites prepared in a single solvent medium exhibited an agglomerated spherical morphology (Fig. S9B†). This could be due to the three-dimensional diffusion of monomers and the resultant secondary growth of oligomers in the bulk aqueous phase.25 Thus, the superior nature of the L/L interface in fine-tuning the 2-D morphology is further validated.
The presence of characteristic elements (carbon, nitrogen, manganese, and oxygen) and the distribution of MnO2 NPs in the PPy/MnO2 sheets (after 15 hours) were further investigated with HRTEM images, elemental mapping images, and the EDS spectrum (Fig. 4A–D and Fig. S10 and S11†). The carbon and nitrogen represent the PPy backbone in 2-D sheets, and the corresponding elemental mapping images are shown in Fig. 4B and Fig. S10B.† The elemental mapping images (Fig. 4C and D) and the corresponding HRTEM images represent a uniform encapsulation of MnO2 NPs in PPy sheets (Fig. S10A, C and D†). The few-layer nature of PPy/MnO2 sheets is also visible in Fig. S10A, C and D,† where the MnO2 NPs exhibit less particle agglomeration. The uniform distribution of manganese and oxygen in PPy/MnO2 sheets is evident in Fig. 4C and D. The bright spots in the SAED pattern of Fig. 3F2 demonstrated the highly crystalline nature of PPy/MnO2 sheets synthesized in 15 hours. XRD studies further examined the long-range order crystallinity of the PPy/MnO2 sheets, and the spectrum is shown in Fig. 4E. The peak positions of the XRD spectrum match well with α-MnO2 (JCPDS card no. 00-044-0141). The peak corresponding to the Bragg reflections caused by the interplanar spacing of PPy chains is absent in the XRD spectrum.24 This could be due to the highly crystalline nature of MnO2 that subsides the diffractions caused by amorphous PPy.24 The peaks at 2θ = 13.461°, 20.874°, 28.829°, 37.507°, 62.818°, 69.688°, 77.101°, and 84.152° can be attributed to the Bragg diffractions caused by the (110), (200), (310), (211), (521), (541), (402) and (512) planes, respectively.32,54,60 The XRD spectrum (Fig. 4E) and SAED pattern (Fig. 3F2) of PPy/MnO2 sheets show dissimilar existence of diffraction planes. This could be because XRD records long-range order crystallinity over the bulk crystal, and SAED measures short-range order crystallinity in selected portions.24 The small shifts observed in the diffraction peaks could be due to the possible lattice deformations caused by the entrapment of MnO2 NPs inside the PPy sheet matrix.62 In addition, the shifts in the peak positions corresponding to the (110), (200), and (521) planes towards higher angles are observed as in Fig. 4E. This could be due to the compressive strain imposed by the amorphous PPy matrix on the crystalline MnO2 lattice that leads to a decrease in lattice spacing and an increase in Bragg angle.62,63 The XRD spectrum of PPy/MnO2 synthesized in a single solvent system demonstrated the characteristic crystalline peaks of MnO2 along with the peaks corresponding to Mn3O4 as impurity (Fig. S12†). The peaks at 2θ = 37.878°, 45.111°, 51.6°, 58.55°, 65.725°, 72.98°, 79.68°, and 87.078° are attributed to crystalline MnO2.64–67 The characteristic peaks of Mn3O4 are centered at 17.09°, 24.08°, and 30.83°.68,69 Thus, the L/L interface tunes the polymerization in a controlled manner, eliminating the formation of undesirable oxides of manganese and resulting in pure MnO2 NPs. The potential capability of the water/chloroform interface in thinning the PPy/MnO2 sheets to a few nanometres is also investigated using atomic microscopy images. The 3-D AFM image (Fig. 4F) and 2-D AFM image (Fig. S13A†) of PPy/MnO2 sheets synthesized in 15 hours exhibited densely populated MnO2 NPs submerged in PPy sheets over an area of 4 μm2. The surface profile plots attributed to the 2-D AFM image in Fig. S13A† predict an average thickness of ∼6 nm for ultra-thin PPy/MnO2 sheets (Fig. S13B†). The XPS spectrum shows the valence states and bonding nature of the characteristic elements of PPy/MnO2 sheets synthesized in 15 hours. The XPS survey scan spectrum reveals the existence of C, N, Mn, and O in PPy/MnO2 sheets (Fig. 4G). The high-resolution C 1s spectrum in Fig. 4H shows four prominent peaks at 284.7 eV, 285.7 eV, 288.25 eV, and 292.68 eV.70–72 The peaks at 284.7 eV, 285.7 eV, and 288.25 eV can be assigned to carbon–carbon, carbon–nitrogen, and carbon–oxygen, respectively.70–72 The peak at 292.68 eV corresponds to the carboxyl group that may be generated during the over-oxidation of the PPy backbone due to the oxidant KMnO4.71 The N 1s spectrum in Fig. S13C† shows the deconvoluted peak corresponding to pyrrolic nitrogen (–N–H).73 The core-level spectrum of Mn 2p shows a spin doublet of Mn 2p1/2 and Mn 2p3/2 located at 642.5 eV and 654.18 eV (Fig. 4I).70,74–76 The spin energy difference between these two peaks gives a binding energy of 11.68 eV, which confirms the presence of Mn4+ in PPy/MnO2.74 The spin doublets of Mn 2p1/2 and Mn 2p3/2 are further deconvoluted to few other peaks to confirm the possible valence states of Mn. The peaks at 642.45 eV and 654.18 eV are assigned to Mn4+.76,77 The deconvoluted Mn 2p spectrum also possesses a less intense peak at 644.9 eV corresponds to Mn2+.76,77 This indicates a small amount of Mn2+ formed during the reduction of KMnO4 to MnO2. The peak for O 1s at 532 eV can be deconvoluted into three prominent peaks at 530 eV, 531.45 eV, and 533.5 eV (Fig. 4J). The peak at 530 eV is assigned to the O–Mn bond, which further confirms the formation of MnO2.33,76,77 The low intense peak at 531.45 eV and 533.5 eV can be attributed to O–C and O–H bonds.33,76,77
The structure-related properties of PPy/MnO2 sheets can be utilized for various potential applications. The electrochemical detection of nicotine by interfacially synthesized PPy/MnO2 sheets was investigated, and the results are presented in Fig. 5. The CV scans of bare GCE and PPy/MnO2 modified GCE in 1 mM nicotine are shown in Fig. 5A. The inset in Fig. 5A shows the oxidation peaks for the bare GCE, and PPy/MnO2 modified GCE at 1.07 V (73 μA) and at 1.03 V (39 μA), respectively. To enhance the electrical conductivity and electrochemical activity of PPy/MnO2 sheets, a high aspect ratio MWCNT was incorporated into PPy/MnO2 sheets. MWCNTs as a conductive filler is highly promising because they promote enhanced electron transfer due to their high electrical conductivity and surface area.78 A required amount of PPy/MnO2 was mechanically mixed with varying wt% (1–5) of MWCNTs. The electrochemical activity of the as-prepared composites was examined using CV scans (Fig. 5B) in 5 mM K4[Fe(CN)6] in 0.1 M KCl, and the oxidation currents were compared (Fig. 5C) to evaluate the effect of MWCNTs in the composites. An optimum current (428 ± 9 μA) was exhibited on the addition of 3 wt% MWCNTs, and further addition decreased the oxidation current to 367 ± 1.5 μA. This could be due to particle agglomeration from dense MWCNT networks at an elevated amount of MWCNTs (Fig. 5C).79 Therefore, 3 wt% was identified as the optimum concentration required for the percolation threshold, and the composite with 3% MWCNTs was selected for the electrochemical detection of 1 mM nicotine (Fig. 5D). The PPy/MnO2 – 3% MWCNT modified GCE (Fig. 5D.i) showed an enhanced electrochemical activity with an onset oxidation potential of 0.68 V and a maximum current of 332 μA (inset of Fig. 5D).
 | ||
| Fig. 5 (A) Cyclic voltammograms of (i) bare GCE and (ii) PPy/MnO2 modified GCE in the presence of 1 mM nicotine solution recorded at a scan rate of 50 mV s−1. The inset shows the zoom-in view of the oxidation peaks. (B) Cyclic voltammograms of PPy/MnO2 – MWCNT modified GCE having different wt% of CNTs (i, ii, iii, iv, and v corresponding to 1, 2, 3, 4, and 5 wt% of MWCNTs) in the presence of 5 mM K4[Fe(CN)6] in 0.1 M KCl recorded at a scan rate of 50 mV s−1. (C) The corresponding anodic current is plotted against MWCNT content in PPy/MnO2 – MWCNTs. The mechanism of percolation in the PPy/MnO2 – MWCNT composite is illustrated schematically. (1) Before percolation, randomly oriented MWCNTs cannot form a conductive pathway. (2) At a critical concentration of 3 wt% of MWCNTs, maximum percolation is achieved by forming MWCNT networks. (3) After percolation, elevated amounts of MWCNTs led to dense or agglomerated CNT networks. (D) Cyclic voltammograms of (i) PPy/MnO2 – 3% MWCNTs, (ii) pure PPy, (iii) pure MWCNTs, and (iv) PPy – 3% MWCNT modified GCE in the presence of 1 mM nicotine at a scan rate of 50 mV s−1. The inset shows the zoom-in view of the oxidation peaks. (E) Schematic illustration of the electrochemical oxidation of nicotine using PPy/MnO2 – MWCNT sheets on a commercial SPE. (F) Cyclic voltammograms of bare SPE and PPy/MnO2 – 3% MWCNT modified SPE in the presence of 1 mM nicotine at a scan rate of 50 mV s−1. | ||
The electrochemical oxidation of nicotine occurs at a very high oxidation potential.80 The main challenge in developing electrochemical nicotine sensors is to decrease the high overpotential of nicotine oxidation and enhance the overall performance. Here, MnO2 NPs uniformly attached in PPy sheets as in Fig. 3F1 and Fig. S7F† act as catalysts for the oxidation of nicotine. The incorporation of a minimal amount of MWCNTs as a filler in PPy/MnO2 improved the performance of the electrochemical nicotine sensor with a 355% increase in the oxidation current at a low onset potential of 0.68 V. Furthermore, the electrochemical detection of nicotine using various other modified electrodes, such as pure PPy (Fig. 5D.ii), pure MWCNTs (Fig. 5D.iii), and PPy – 3% MWCNTs (Fig. 5D.iv) was also analyzed. These modified electrodes did not give apparent peaks corresponding to nicotine oxidation, as shown in the inset of Fig. 5D. The maximum anodic currents from the respective CV scans of the three potential electrodes (bare GCE, PPy/MnO2 modified GCE, and PPy/MnO2 – 3% MWCNT modified GCE) were compared and are presented in Fig. S14.† The differential pulse voltammograms of all modified electrodes in 1 mM nicotine were also recorded (Fig. S15†). This further confirms the exclusive catalytic activity of MnO2 NPs in the oxidation of nicotine.
The printable nature of PPy/MnO2–MWCNT composites on the SPE opens a new route for successfully preparing miniaturized electrode set-ups for nicotine detection. The electrochemical sensing of nicotine in commercial screen printed electrodes (Zensor TE100) was also tested. The schematic representation of nicotine oxidation by PPy/MnO2–MWCNTs on the SPE is shown in Fig. 5E. The electrochemical oxidation of nicotine is a two-electron mediated N-methyl hydroxylation reaction (Fig. 5E).80,81 The PPy/MnO2–3% MWCNT modified SPE showed a high oxidation current of 175 μA towards the electrochemical detection of 1 mM nicotine (Fig. 5F). The difference in the anodic current of commercial SPE compared to that of GCE may be attributed to the difference in the electrode combinations, design, and materials used. The DPV scans of bare and modified SPE are shown in Fig. S16.† Thus, the conductive composites of PPy/MnO2 sheets are ideal for successfully developing high-performance electrochemical nicotine sensors.
4. Conclusion
In summary, the high interfacial tension of the water/chloroform interface drives the interfacial adsorption of MnO2 attached PPy oligomers which led to the interface-assisted 2-D microstructure tuning of PPy/MnO2. Pendant drop tensiometry confirmed the gradual decrease of IFT (from 32 mN m−1 to 17 mN m−1) due to the adsorption of MnO2 attached PPy oligomers at the water/chloroform interface. The time-dependent self-assembly of MnO2 attached oligomers and polymerization to form PPy/MnO2 sheets were verified using UV-Vis and Raman spectroscopy. Transmission electron and atomic force microscopy techniques demonstrated the 2-D nature of ultra-thin PPy/MnO2 sheets with the uniform incorporation of submerged MnO2 NPs. The evolution of bright Bragg spots from diffusive rings confirmed the perpetual transition of amorphous to highly crystalline MnO2 NPs at the interface. The composites exhibiting a uniform distribution of ∼6 nm MnO2 NPs were composited with MWCNTs and identified as a potential electrochemical sensor for nicotine. The prepared electrodes exhibited a very high oxidation current of 332 μA at a low onset potential of 0.68 V. The printable nature of the composite inks of PPy/MnO2 with MWCNTs on a commercially available SPE was also assessed. It showed a high oxidation current of 175 μA towards the electrochemical detection of 1 mM nicotine. The proposed synthesis strategy may open new pathways to develop multifunctional polymer nanocomposite films with attractive morphologies or structure-related properties.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
MMM greatly acknowledges the funding from the Science and Engineering Research Board (EEQ/2019/000606) and DST-INSPIRE (DST/INSPIRE/04/2015/002050) by the Department of Science and Technology (DST), India. The authors thank SMSE-NIT Calicut for Raman facility. The authors express their gratitude toward STIC-CUSAT, Cochin for HRTEM and XRD analyses. The authors acknowledge the AFM facility at MRC, MNIT Jaipur and XPS facility at NIIST Trivandrum. The authors also acknowledge the HRTEM-EDS-mapping facility at C.R.F., CeNS Bengaluru.References
- Y. Montelongo, D. Sikdar, Y. Ma, A. J. S. McIntosh, L. Velleman, A. R. Kucernak, J. B. Edel and A. A. Kornyshev, Nat. Mater., 2017, 16, 1127–1135 CrossRef CAS PubMed.
- P. P. Fang, S. Chen, H. Deng, M. D. Scanlon, F. Gumy, H. J. Lee, D. Momotenko, V. Amstutz, F. Cortés-Salazar, C. M. Pereira, Z. Yang and H. H. Girault, ACS Nano, 2013, 7, 9241–9248 CrossRef CAS PubMed.
- Z. Chen, L. Zhou, W. Bing, Z. Zhang, Z. Li, J. Ren and X. Qu, J. Am. Chem. Soc., 2014, 136, 7498–7504 CrossRef CAS PubMed.
- Y. Zhang, R. Ettelaie, B. P. Binks and H. Yang, ACS Catal., 2021, 11, 1485–1494 CrossRef CAS.
- S. Yu, M. Lv, G. Lu, C. Cai, J. Jiang and Z. Cui, Langmuir, 2021, 37, 10683–10691 CrossRef CAS PubMed.
- S. Björkegren, M. C. A. Freixiela Dias, K. Lundahl, L. Nordstierna and A. Palmqvist, Langmuir, 2020, 36, 2357–2367 CrossRef PubMed.
- G. Ren, X. Zheng, H. Gu, W. Di, Z. Wang, Y. Guo, Z. Xu and D. Sun, Langmuir, 2019, 35, 13663–13670 CrossRef CAS PubMed.
- L. Li and W. S. Chin, ACS Appl. Mater. Interfaces, 2020, 12, 37538–37548 CrossRef CAS PubMed.
- J. G. Lee, L. L. Larive, K. T. Valsaraj and B. Bharti, ACS Appl. Mater. Interfaces, 2018, 10, 43282–43289 CrossRef CAS PubMed.
- O. Owoseni, E. Nyankson, Y. Zhang, S. J. Adams, J. He, G. L. McPherson, A. Bose, R. B. Gupta and V. T. John, Langmuir, 2014, 30, 13533–13541 CrossRef CAS PubMed.
- Y. H. Lee, W. Shi, H. K. Lee, R. Jiang, I. Y. Phang, Y. Cui, L. Isa, Y. Yang, J. Wang, S. Li and X. Y. Ling, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- Q. Wang, Z. Wang, Z. Li, J. Xiao, H. Shan, Z. Fang and L. Qi, Sci. Adv., 2017, 3, e170118 Search PubMed.
- Y. Lin, H. Skaff, T. Emrick, A. D. Dinsmore and T. P. Russell, Science, 2003, 299, 226–229 CrossRef CAS PubMed.
- R. M. E. Silva, R. Poon, J. Milne, A. Syed and I. Zhitomirsky, Adv. Colloid Interface Sci., 2018, 261, 15–27 CrossRef CAS PubMed.
- X. Hua, J. Frechette and M. A. Bevan, Soft Matter, 2018, 14, 3818–3828 RSC.
- E. Guzmán, I. Abelenda-Núñez, A. Maestro, F. Ortega, A. Santamaria and R. G. Rubio, J. Phys.: Condens. Matter, 2021, 33, 333001 CrossRef PubMed.
- S. Razavi, J. Koplik and I. Kretzschmar, Soft Matter, 2013, 9, 4585–4589 RSC.
- Y. Chai, J. Hasnain, K. Bahl, M. Wong, D. Li, P. Geissler, P. Y. Kim, Y. Jiang, P. Gu, S. Li, D. Lei, B. A. Helms, T. P. Russell and P. D. Ashby, Sci. Adv., 2020, 6, eabb8675 CrossRef CAS PubMed.
- J. Smits, F. Vieira, B. Bisswurn, K. Rezwan and M. Maas, Langmuir, 2019, 35, 11089–11098 CrossRef CAS PubMed.
- B. P. Binks and S. O. Lumsdon, Langmuir, 2000, 16, 8622–8631 CrossRef CAS.
- S. Shi and T. P. Russell, Adv. Mater., 2018, 30, 1800714 CrossRef PubMed.
- A. J. G. Zarbin, Mater. Horiz., 2021, 8, 1409–1432 RSC.
- S. K. Chondath and M. M. Menamparambath, Nanoscale Adv., 2021, 3, 918–941 RSC.
- S. K. Chondath, J. S. Gopinath, R. R. Poolakkandy, P. Parameswaran and M. M. Menamparambath, Macromol. Mater. Eng., 2022, 307, 2100705 CrossRef CAS.
- S. K. Chondath, R. R. Poolakkandy, R. Kottayintavida, A. Thekkangil, N. K. Gopalan, S. T. Vasu, S. Athiyanathil and M. M. Menamparambath, ACS Appl. Mater. Interfaces, 2019, 11, 1723–1731 CrossRef CAS PubMed.
- R. Rahman Poolakkandy, S. Kaladi Chondath, N. Puthiyottil, D. Davis and M. M. Menamparambath, Langmuir, 2020, 36, 872–879 CrossRef CAS PubMed.
- Y. K. Park and S. Park, Chem. Mater., 2008, 20, 2388–2393 CrossRef CAS.
- S. Biswas and L. T. Drzal, Nano Lett., 2009, 9, 167–172 CrossRef CAS PubMed.
- V. H. R. Souza, S. Husmann, E. G. C. Neiva, F. S. Lisboa, L. C. Lopes, R. V. Salvatierra and A. J. G. Zarbin, Electrochim. Acta, 2016, 197, 200–209 CrossRef CAS.
- Y. Dong, S. Chertopalov, K. Maleski, B. Anasori, L. Hu, S. Bhattacharya, A. M. Rao, Y. Gogotsi, V. N. Mochalin and R. Podila, Adv. Mater., 2018, 30, 1705714 CrossRef PubMed.
- R. V. Salvatierra, M. M. Oliveira and A. J. G. Zarbin, Chem. Mater., 2010, 22, 5222–5234 CrossRef CAS.
- M. Jayashree, M. Parthibavarman, R. BoopathiRaja, S. Prabhu and R. Ramesh, J. Mater. Sci.: Mater. Electron., 2020, 31, 6910–6918 CrossRef CAS.
- R. A. Davoglio, G. Cabello, J. F. Marco and S. R. Biaggio, Electrochim. Acta, 2018, 261, 428–435 CrossRef CAS.
- L. C. Tai, C. H. Ahn, H. Y. Y. Nyein, W. Ji, M. Bariya, Y. Lin, L. Li and A. Javey, ACS Sens., 2020, 5, 1831–1837 CrossRef CAS PubMed.
- O. N. Efimov and T. Vernitskaya, Russ. Chem. Rev., 1997, 66, 443–457 CrossRef.
- P. Vanysek, Electrochemical Series, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, 2002 Search PubMed.
- Y. Song, M. Shang, J. Li and Y. Su, Chem. Eng. J., 2021, 405, 127059 CrossRef CAS.
- P. Dallas and V. Georgakilas, Adv. Colloid Interface Sci., 2015, 224, 46–61 CrossRef CAS PubMed.
- S. K. Ghosh and A. Böker, Macromol. Chem. Phys., 2019, 220, 1900196 CrossRef.
- V. Garbin, J. C. Crocker and K. J. Stebe, J. Colloid Interface Sci., 2012, 387, 1–11 CrossRef CAS PubMed.
- X. Hua, M. A. Bevan and J. Frechette, Langmuir, 2016, 32, 11341–11352 CrossRef CAS PubMed.
- X. Hua, M. A. Bevan and J. Frechette, Langmuir, 2018, 34, 4830–4842 CrossRef CAS PubMed.
- G. Sun, Z. Yi and T. Ngai, Acta Phys.-Chim. Sin., 2020, 36, 1–13 CrossRef PubMed.
- D. Barpuzary, K. Kim and M. J. Park, ACS Nano, 2019, 13, 3953–3963 CrossRef CAS PubMed.
- G. A. Rance, D. H. Marsh, R. J. Nicholas and A. N. Khlobystov, Chem. Phys. Lett., 2010, 493, 19–23 CrossRef CAS.
- J. R. Guimarães, J. G. Amazonas, C. Alberto Brito Silva Junior, C. P. de Melo, B. Laks and J. del Nero, Mater. Sci. Eng. C, 2008, 28, 1076–1081 CrossRef.
- C. I. Simionescu, I. Cianga, M. Ivanoiu, A. Airinei, M. Grigoras and I. Radu, Eur. Polym. J., 1999, 35, 1895–1905 CrossRef CAS.
- A. Singh, Z. Salmi, N. Joshi, P. Jha and P. Decorse, RSC Adv., 2013, 3, 24567–24575 RSC.
- H. S. Roy, M. Y. A. Mollah, M. M. Islam and M. Susan, Polym. Bull., 2018, 75, 5629–5643 CrossRef CAS.
- A. V. Soldatova, G. Balakrishnan, O. F. Oyerinde, C. A. Romano, B. M. Tebo and T. G. Spiro, Environ. Sci. Technol., 2019, 53, 4185–4197 CrossRef CAS PubMed.
- R. Gutzler, Phys. Chem. Chem. Phys., 2016, 18, 29029–29100 RSC.
- M. C. Scharber and N. S. Sariciftci, Adv. Mater. Technol., 2021, 6, 2000857 CrossRef CAS.
- N. E. Makori, D. A. Oeba and C. O. Mosiori, Chem. Res. J., 2017, 5, 15–21 Search PubMed.
- G. Zhu, W. Zhu, Y. Lou, J. Ma, W. Yao, R. Zong and Y. Zhu, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed.
- J. Zhang, Y. Li, L. Wang, C. Zhang and H. He, Catal. Sci. Technol., 2015, 5, 2305–2313 RSC.
- H. Salah Abdullah, Int. J. Phys. Sci., 2012, 7, 5468–5476 Search PubMed.
- M. Trchová and J. Stejskal, J. Phys. Chem. A, 2018, 122, 9298–9306 CrossRef PubMed.
- B. P. Kafle, Chemical Analysis and Material Characterization by Spectrophotometry, Elsevier, New York, 2020 Search PubMed.
- Z. Ni, Y. Wang, T. Yu and Z. Shen, Nano Res., 2008, 1, 273–291 CrossRef CAS.
- X. Huang, D. Lv, H. Yue, A. Attia and Y. Yang, Nanotechnology, 2008, 19, 225606 CrossRef PubMed.
- M. Musil, B. Choi and A. Tsutsumi, J. Electrochem. Soc., 2015, 162, A2058–A2065 CrossRef CAS.
- L. R. Nivedita, A. Haubert, A. K. Battu and C. V. Ramana, Nanomaterials, 2020, 10, 1–24 CrossRef PubMed.
- T. Maimaitiyili, R. Woracek, M. Neikter, M. Boin, R. C. Wimpory, R. Pederson, M. Strobl, M. Drakopoulos, N. Schäfer and C. Bjerkén, Materials, 2019, 12, 667 CrossRef CAS PubMed.
- B. Mendoza-Sánchez, J. Coelho, A. Pokle and V. Nicolosi, Electrochim. Acta, 2015, 174, 696–705 CrossRef.
- C. Zhang, W. Chu, F. Chen, L. Li, R. Jiang and J. Yan, J. Rare Earths, 2020, 38, 70–75 CrossRef CAS.
- T. Shoji and T. Yamamoto, J. Electroanal. Chem., 1993, 362, 153–157 CrossRef CAS.
- J. Haines, J. M. Léger and S. Hoyau, J. Phys. Chem. Solids, 1995, 56, 965–973 CrossRef CAS.
- L. ben Said, A. Inoubli, B. Bouricha and M. Amlouk, Spectrochim. Acta, Part A, 2017, 171, 487–498 CrossRef CAS PubMed.
- J. B. Jasinski, D. Ziolkowska, M. Michalska, L. Lipinska, K. P. Korona and M. Kaminska, RSC Adv., 2013, 3, 22857–22862 RSC.
- S. Kumar and R. B. Choudhary, Mater. Sci. Semicond. Process., 2022, 139, 106322 CrossRef CAS.
- C. Wang, Z. Liu, Q. Wang, J. Guo, Q. Zhao and Y. Lu, J. Electroanal. Chem., 2021, 901, 115780 CrossRef CAS.
- B. Li, X. Wang, S. Hong, Q. Wang, L. Li, O. Eltayeb, C. Dong and S. Shuang, Food Funct., 2021, 12, 6334–6347 RSC.
- W. Yao, H. Zhou and Y. Lu, J. Power Sources, 2013, 241, 359–366 CrossRef CAS.
- D. E. Pacheco-catal and J. Uribe-calderon, Synth. Met., 2020, 269, 116541 CrossRef.
- C. Guo, S. Tian, B. Chen, H. Liu and J. Li, Mater. Lett., 2020, 262, 127180 CrossRef CAS.
- S. Chen, X. Shu, H. Wang and J. Zhang, J. Mater. Chem. A, 2019, 7, 19719–19727 RSC.
- Z. Mo, H. Xu, Z. Chen, X. She, Y. Song, J. Lian, X. Zhu, P. Yan, Y. Lei, S. Yuan and H. Li, Appl. Catal., B, 2019, 241, 452–460 CrossRef CAS.
- R. R. Poolakkandy, A. R. Neelakandan, M. F. Puthiyaparambath, R. G. Krishnamurthy, R. Chatanathodi and M. M. Menamparambath, J. Mater. Chem. C, 2022, 10, 3048–3060 RSC.
- T. Tarlton, E. Sullivan, J. Brown and P. A. Derosa, J. Appl. Phys., 2017, 121, 085103 CrossRef.
- X. Li, H. Zhao, L. Shi, X. Zhu, M. Lan, Q. Zhang and Z. Hugh Fan, J. Electroanal. Chem., 2017, 784, 77–84 CrossRef CAS.
- A. Karthika, P. Karuppasamy, S. Selvarajan, A. Suganthi and M. Rajarajan, Ultrason. Sonochem., 2019, 55, 196–206 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr02130g |
| This journal is © The Royal Society of Chemistry 2022 |