
Ag@Pd bimetallic structures for enhanced electrocatalytic CO2 conversion to CO: an interplay between the strain effect and ligand effect†
 a
Yimin A.
Wu
*a
a
Yimin A.
Wu
*a
Abstract
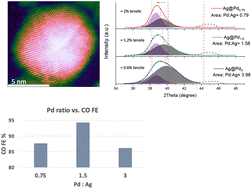
Electrochemical CO2 reduction reactions provide a promising path to effectively convert CO2 into valuable chemicals and fuels for industries. Among the many CO2 conversion catalysts, Pd stands out as a promising catalyst for effective CO2 to CO conversion. Here, using the misfit strain strategy, Ag@Pd bimetallic nanoparticles with different Pd overlayer contents were prepared as CO2 reduction catalysts. By varying the Pd overlayer content, all the Ag@Pd bimetallic nanoparticles exhibited superior CO2 conversion performance over their Pd and Ag nanoparticle counterparts. An optimal Pd-to-Ag ratio of 1.5 : 1 yielded the highest CO faradaic efficiency of 94.3% at −0.65 V vs. RHE with a high CO specific current density of 3.9 mA cm−2. It was found that the Pd content can substantially affect the interplay between the strain effect and ligand effect, resulting in optimized binding properties of the reaction intermediates on the catalyst surface, thereby enhancing the CO2 reduction performance.
- This article is part of the themed collection: CO2 capture and conversion
 Please wait while we load your content...
Please wait while we load your content...

