
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emerging investigator series: aqueous-phase processing of atmospheric aerosol influences dissolution kinetics of metal ions in an urban background site in the Po Valley†
Chiara
Giorio
 *ab,
Sara
D'Aronco
ab,
Valerio
Di Marco
b,
Denis
Badocco
b,
Francesco
Battaglia‡
ab,
Lidia
Soldà
b,
Paolo
Pastore
b and
Andrea
Tapparo
b
*ab,
Sara
D'Aronco
ab,
Valerio
Di Marco
b,
Denis
Badocco
b,
Francesco
Battaglia‡
ab,
Lidia
Soldà
b,
Paolo
Pastore
b and
Andrea
Tapparo
b
aYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB21EW, UK. E-mail: chiara.giorio@atm.ch.cam.ac.uk
bDipartimento di Scienze Chimiche, Università degli Studi di Padova, Via Marzolo 1, 35131 Padova, Italy
First published on 10th May 2022
Abstract
Metals are an important atmospheric aerosol component; their impacts on health and the environment depend also on their solubility, dissolution kinetics and chemical form in which they are present in the aerosol (e.g., oxidation state, inorganic salt or oxide/hydroxide, organic complex). In this study, we investigated the impact of fog processing on the solubility and dissolution of metals in PM2.5 samples collected in an urban background site in Padova (Italy). For each sample, we determined the solubility and dissolution kinetics of 17 elements in a solution simulating fog water in the winter season in the Po Valley (pH 4.7, T 5 °C, and water content ∼0.5 g m−3). We also determined water-soluble inorganic and organic compounds having ligand properties. We used the model E-AIM IV to calculate the aerosol liquid water (ALW) content and pH, and we used the model Visual MinteQ to determine the speciation picture of the most important elements under conditions of both deliquescent aerosol (ALW and pH calculated using E-AIM IV, ambient temperature) and simulated fog. We found that the dissolution of Al, Cu, and Fe metal ions, predicted to be largely coordinated with organic compounds under fog conditions, was either immediate or considerably faster in samples collected on days with observed fog events compared with those collected on days having drier conditions. For readily soluble elements, such as As, Cd, Cr, Sr, and Zn, such an effect was not observed. Our study highlights the importance of coordination chemistry in atmospheric aerosol and fog in determining the bioavailability of particle-bound metals.
Environmental significanceMetals in atmospheric aerosols are known to be a key component exerting adverse health effects. This study investigates fog processing of atmospheric aerosol, particularly the coordination chemistry between metals and short-chain organic acids, in an urban background environment, and its impact on the solubility and dissolution kinetics of particle-bound metals. We found that the solubility of metals increased upon fog processing except for Pb. We observed, for the first time, that the dissolution rate of metals predicted to coordinate with short-chain organic acids in fog water (e.g., Al, Cu, and Fe) is much faster in samples collected on days with recorded fog events. Our study highlights the importance of fog processing in influencing the bioavailability of particle-bound metals. |
1 Introduction
Air pollution is today a worldwide cause of health issues. Atmospheric aerosol, or particulate matter (PM), is a major determinant of air pollution known to be causing respiratory and cardiovascular diseases.1 In addition, a recent systematic review and meta-analysis found a significant association between PM2.5 exposure and neurodegenerative disorders (stroke, dementia, Alzheimer's disease and Parkinson's disease).2Insoluble PM, particularly nanoparticles, can exert adverse health effects.3 Particle toxicity depends also on their chemical composition, for example the presence of carcinogenic species such as polycyclic aromatic hydrocarbons.4,5 It has long been recognised that trace metals play a crucial role in aerosol toxicity.6,7 Trace metals in aerosol particles, their physico-chemical properties, chemical speciation, and reactivity have been studied extensively.8,9 In metals, the soluble fraction is the most bioavailable,10 not only the one able to undergo redox chemistry in lung fluids causing oxidative stress,11 but also the one that can cross the epithelial alveolar barrier and reach the bloodstream, potentially accessing cells and organs.12 Metal ions are also implicated in important atmospheric reactivity.8 Notable examples are the production of OH radicals through photolysis of iron(III) complexes13,14 and Fenton chemistry,15,16 the catalytic oxidation of sulphur(IV) to sulphur(VI),8,17,18 and the oxidation of water soluble organic compounds.19,20
For most metal ions, the solubility is dependent on their chemical form in the PM. As many water soluble organic and inorganic compounds in aerosols have coordinating properties towards metal ions, the solubility of the latter is affected by atmospheric aqueous-phase processing of aerosols, and by the stoichiometry and stability constants of the complexation reactions that can occur.21–24
Direct and indirect evidence have been provided for the formation of complexes between metal ions and ligands such as cyanides, oxalates, and macromolecules in deliquescent aerosols.21–24 Findings from Scheinhardt et al.24 pointed out that metal–ligand interactions may be an important phenomenon in deliquescent aerosols in the urban atmosphere in nine cities in Germany. Tapparo et al.21 found that Fe(III) can be almost entirely coordinated with oxalate in simulated rainwater in an urban background location in the Po Valley (Italy). Feng et al.25 found that many toxic metals were predominantly in a bioavailable form in Guangzhou (China), a significant fraction of which was found to be bound to organics.
The main factors governing the speciation and solubility of metals are (i) the amount of liquid water, (ii) the pH, (iii) the oxidation state of the metals, and (iv) the presence of ligands. Spokes et al.26 found pH-dependent solubilities for Al, Fe, and Mn metal ions. They also evidenced that around 50% of soluble Fe was in the oxidation state +2 and that Mn dissolution was dependent on the reduction of Mn(IV) to Mn(II).26 Other studies found that Fe(II) was either the dominant species in the soluble fraction,17,18,27,28 the main contributor (80%) to the readily soluble fraction of Fe,29 or equally present together with Fe(III).17,18,29 Li et al.30 found a logarithmic inverse relationship between trace element concentrations and cloud liquid water content during dust storms in southern China. They also observed a highly pH dependent concentration of trace elements, with minimum concentrations at a threshold of pH ∼5.0. Their speciation results showed that Fe(II), Zn(II), Pb(II), and Cu(II) were present predominantly as free ions (80.7–96.3%), while 71.7% of Fe(III) and 71.5% of Al(III) were complexed with oxalate and fluoride, respectively.30 Fang et al.31 and Baker et al.32 found enhanced metal solubilities linked with processing in acidic sulphate aerosols, most likely due to the low pH induced by sulphuric acid rather than the presence of sulphate itself.31,32 Shahpoury et al.11 investigated proton-driven vs. ligand-driven dissolution of Fe in various sites in Canada, but they could not separate the two effects due to a correlation between higher oxalate contents and lower pH in their samples.
While it has been demonstrated that atmospheric organic complexation impacts Fe solubility33,34 and that pH impacts the dissolution kinetics of dusts,35,36 to the authors' knowledge, no one has ever investigated the effect of atmospheric organic complexation on the kinetics of dissolution of metal ions.
The goal of this study was to establish if there is an impact of aqueous-phase processing of aerosols on the solubility and dissolution kinetics of particle-bound elements. PM2.5 samples were collected in an urban background site in Padova (Italy), in the Po Valley, under different conditions of humidity and temperature, and on days characterised by fog formation. Samples were analysed by inductively coupled plasma mass spectrometry (ICP-MS), to determine as many elements as possible, whereas organic/inorganic ligands were determined by ion chromatography (IC). Dissolution kinetics of the elements were studied in a simulated fog water (leaching solution of H2SO4 and HNO3 at pH = 4.7, T = 5 °C) that resembles the typical winter environmental conditions in the Po Valley. A multidimensional speciation study was performed to determine the stoichiometry and the concentrations of metal–ligand complexes and free metal ions in the deliquescent aerosol and in the simulated fog.
2 Methods
2.1 Chemicals
All reagents were of analytical grade and were used as purchased.Sulphuric acid (95–97%, Fluka) and nitric acid (68.5–69.5%, Aristar for trace analysis, VWR) were used for preparing solutions. Rhenium standard for ICP (1001 ± 5 mg L−1, 2% HNO3, Sigma Aldrich), germanium standard for ICP (1005 ± 2 mg L−1, 2% HNO3, Sigma Aldrich), and terbium standard for ICP (1002 ± 5 mg L−1, 2% HNO3, Sigma Aldrich) were used as internal standards for element quantification. External calibration was performed by diluting the CCS-5 for ICP (100.00 ± 0.70 μg mL−1 of B, Ge, Hf, Mo, Nb, P, Re, S, Sb, Si, Sn, Ta, Ti, W, and Zr, 1.2% (v/v) HF + 7.43% (v/v) HNO3, Inorganic Ventures) and the IV-ICPMS-71A (10 ± 0.08 μg mL−1 of Ag, Al, As, B, Ba, Be, Ca, Cd, Ce, Co, Cr, Cs, Cu, Dy, Er, Eu, Fe, Ga, Gd, Ho, K, La, Lu, Mg, Mn, Na, Nd, Ni, P, Pb, Pr, Rb, S, Se, Sm, Sr, Th, Tl, Tm, U, V, Yb, and Zn, Inorganic Ventures) calibrating standard solutions.
Primary standards for IC analysis, including methanesulphonic acid used as both a primary standard and eluent for cation analysis, were purchased from Sigma-Aldrich®.
Ultrapure water was produced with a Millipore Plus System (Milan, Italy, resistivity 18.2 Ω cm−1).
2.2 Sample collection
PM2.5 samples were collected (24 h sampling time, from 9 AM to 9 AM of the next day) from 1st February 2019 to 14th March 2019 (15 filters) at the sampling site located on the 6th floor of the Department of Chemical Sciences at the University of Padova (Italy),37 using an Aircube He pump fitted with a PM2.5 certified selector (CEN standard method UNI-EN 14907), and working at a constant flow rate of 2.3 m3 h−1. The samples were collected on Teflon membranes (PALL, fiber film, Ø 47 mm, 2.0 μm pore size) pre-cleaned by sonicating 3 times for 15 min in ultrapure water. Weighing of the filters was carried out before and after sampling, after conditioning at a temperature of 20 ± 1 °C and relative humidity of 50 ± 5% for at least 48 h, as in previous studies.21,37,38Each filter was cut into three parts (a portion of 3/4 and two of 1/8), where the first part was used for measurements of element dissolution kinetics, and the other two parts were used for determining the total content of the elements and the soluble ions. The filter samples were then stored in a refrigerator (−18 °C) until analysis.
2.3 Dissolution kinetics measurements
Dissolution kinetics of the elements were determined using the same procedure described by Di Marco et al. (2020).38 Briefly, the leaching solution had a computed pH of 4.7 (checked experimentally using a pH meter, Bench Meter pH 50+ with a 201T DHS Electrode, XS Instruments) and contained H2SO4 at a concentration of 5.3 × 10−6 M, HNO3 at a concentration of 9.4 × 10−6 M, and the internal standards for ICP-MS (72Ge, 159Tb, and 183Re) at a concentration of 25 μg L−1. For each experiment, the solution (40 mL) was loaded into a water-jacketed cell (Metrohm) having a volume capacity of 150 mL. A constant stirring speed (200 rpm) and temperature (5.0 ± 0.1 °C) were set and maintained using a magnetic stirrer and a HaakeF3 cryostat, respectively. Mixing time in the cell was evaluated in a previous study and found to be <2 min.38 A 3/4 piece of filter was loaded in a glass holder and inserted in the cell. Samples (0.75 mL aliquots) were taken from the solution at the following times: 0, 2, 4, 6, 8, 10, 12, 14, 16, 20, 24, 28, 32, 60, 120, 180, and 240 min. The withdrawn samples were then transferred to 15 mL Falcon tubes (prewashed with 2 mL of ∼0.001 M H2SO4 solution, rinsed with water and left to dry) and 27 μL of concentrated (∼69%) HNO3 were added to obtain a final concentration of HNO3 of 3.45% w/w before analysis by ICP-MS. Kinetics experiments were also performed on a blank filter. As the extraction volume decreased over time during the experiments, due to sample withdrawals, elements were leached in a solution that gradually became more and more concentrated. The concentration values obtained by ICP-MS were therefore corrected to the values which would have been obtained if no sample withdrawals were performed from the cell. The corrected concentration corresponding to the xth withdrawal (cx) was obtained using the formula reported by Di Marco et al.,38 where V0 is the starting volume and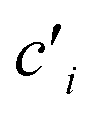 is the concentration obtained by ICP-MS analysis after the ith withdrawal:
is the concentration obtained by ICP-MS analysis after the ith withdrawal: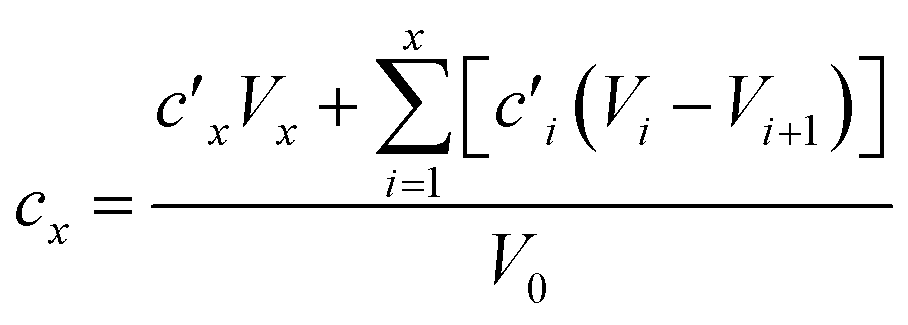
2.4 Sample preparation for the determination of the total amount of elements
Samples (1/8 portions) were extracted using a two-step procedure as used in a previous study.38 In the first step, each filter sample was placed in a 15 mL Falcon tube to which 3 mL of diluted HNO3 (3.45% w/w), also containing an internal standard for ICP-MS (159Tb 20 μg L−1), were added. The samples were then heated in a bain-marie (100 °C) for 2 h and then analysed by ICP-MS. Subsequently, the filters were removed, rinsed with ultrapure water, and placed in cleaned Falcon tubes. In the second step, the filter samples were extracted in 3 mL of concentrated HNO3 (∼69% w/w) in a bain-marie (100 °C) for 1 h. For this second step, sample extracts were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 with ultrapure water containing an internal standard for ICP-MS (72Ge 25 μg L−1) to obtain a final concentration of HNO3 of 3.45% w/w before analysis in ICP-MS.
:20 with ultrapure water containing an internal standard for ICP-MS (72Ge 25 μg L−1) to obtain a final concentration of HNO3 of 3.45% w/w before analysis in ICP-MS.
2.5 ICP-MS analysis
Analysis of elements was performed using an ICP-MS Agilent series 7700x (Agilent Technologies International Japan, Ltd., Tokyo, Japan) using instrumental operating conditions as reported elsewhere.38,39 Quantification was performed by external calibration using the isotopes 72Ge, 159Tb, and 183Re as internal standards.2.6 Determination of cations, anions, and short-chain organic acids
1/8 filter samples were extracted in 3 mL of ultrapure water for 24 h at 20 ± 1 °C, filtered (0.2 μm RC membrane, Phenomenex 4 mm syringe filters) and analysed (injection volume 10 μL) using a Dionex IC system equipped with a GP50 Gradient Pump, an EG40 eluent generation system fitted with a DionexEGC III KOHRFICTM eluent generator cartridge, an LC25 oven, and an ED40 Electrochemical Detector (in conductometric detection mode).For the analysis of cations (Na+, NH4+, K+, Mg2+, and Ca2+), a Dionex™ IonPac™ CS12A (2 × 250 mm) separation column fitted with a Dionex™ IonPac™ CG12A (2 × 50 mm) guard column was used. Isocratic elution was performed at room temperature (∼20 °C) with a 20 mM solution of methanesulphonic acid and a flow rate of 300 μL min−1. The signal was suppressed using a DionexCERS 500 (2 mm) self-regenerating suppressor (suppression current 50 mA).
The analysis of anions (Cl−, NO2−, NO3−, SO42−, and PO43−) and organic acids (formic, acetic, succinic, malonic, oxalic, and methanesulphonic acids) was carried out according to the procedure detailed by Tapparo et al. (2020),21 by using a Dionex™ IonPac™ AS11-HC (2 × 250 mm) separation column fitted with an AG11-HC (2 × 50 mm) guard column. The signal was suppressed using a DionexAERS 500 (2 mm) self-regenerating suppressor (suppression current 100 mA).
External calibration was performed daily with standard solutions in the range of 0.1–50 mg L−1 for each analyte in ultrapure water.
2.7 Statistical analysis
All data were checked for outliers using the Tukey's rule. Significance of the dissolution kinetics profiles (as varying element amounts leached in solution over time) was verified using the Spearman test. A 95% significance level was considered for all statistical tests.2.8 Determination of aerosol liquid water content and pH
Average aerosol liquid water (ALW) content and pH were calculated using the comprehensive version of the model E-AIM IV.40–42 Averaged relative humidity (RH) and temperature over the 24 h sampling period were used for the calculation as well as aerosol concentrations of NH4+, Na+, Cl−, NO3−, and SO42−. Ion balance was achieved by adding an appropriate amount of H+ or by converting an appropriate amount of NH4+ into NH3. Acetic acid, succinic acid, malonic acid, and oxalic acid were also included in the calculation and allowed to dissociate in solution.43 Partitioning equilibria with gas components and solid precipitation were also considered.2.9 Speciation calculations
Chemical speciation of metal ions was computed using the model Visual MinteQ 3.1 (available online at https://vminteq.lwr.kth.se/), which is a chemical equilibrium model for the calculation of metal speciation in solution and solubility equilibria. Visual MinteQ was used to determine the chemical form of metal ions in deliquescent aerosol particles and in simulated fog. For the speciation the following species were considered: Na+, NH4+, K+, Mg2+, Ca2+, Cl−, NO3−, SO42−, PO43−, succinate, malonate, oxalate, acetate, Al, Cu, Ni, Mn, Cr, Fe, Pb, and Zn. The oxidation state of metal ions was assumed to be the following: Al(III), Cu(II), Ni(II), Mn(II), Cr(III), Pb(II), and Zn(II). In the case of Fe, we assumed that Fe(II) and Fe(III) were present in the ratio 1:1. The speciation model was run according to the procedure described by Scheinhardt et al.24 When specific chemical species were predicted to be present in oversaturation conditions, the calculation was repeated allowing precipitation. Precipitation was ignored for rare or aged solids, such as hematite, goethite, lepidocrocite, magnetite, maghemite, cupric ferrite, anglesite, strengite, alunite, and Fe(OH)2.7Cl0.3(s), as they are unlikely to form in deliquescent aerosol particles and fogs.
For the speciation in deliquescent aerosol, liquid phase concentrations of the dissolved complex-forming compounds were calculated using the ALW obtained from E-AIM IV assuming initially a complete dissolution. The pH was fixed at the value calculated by E-AIM IV and the mean temperature over the 24 h sampling time was used. For metal ions, ICP-MS measurements at t = 2 min of the kinetics experiments were used (i.e., the readily soluble fraction).
For the speciation in simulated fog, the final conditions of the kinetics experiments (pH 4.7, T = 5 °C, a final water content of 0.5 g m−3 in sample air, representative of average fog conditions in the winter season) were used and the concentrations of metal ions were calculated by averaging the values obtained at t > 60 min of the kinetics experiments (as an estimate of the concentration at t = ∞).
3 Results and discussion
3.1 PM2.5 composition and environmental conditions
The period of the sampling campaign was characterised by dry conditions (no precipitations recorded), RH ranging between 36% and 100%, average daily temperature between 3.3 °C and 11.4 °C, average daily dew point temperatures (calculated with the method from Lawrence44) between −4.0 °C and 6.9 °C, and calm to light wind (up to ∼2 m s−1). Fog events were observed mostly in the morning (February 1 and 20, March 4, 2019 (Fig. 1a)) but could not be distinguished during the night. On most days, environmental conditions favoured aqueous-phase processing of PM allowing coordination chemistry to occur in either deliquescent aerosol particles or fog droplets.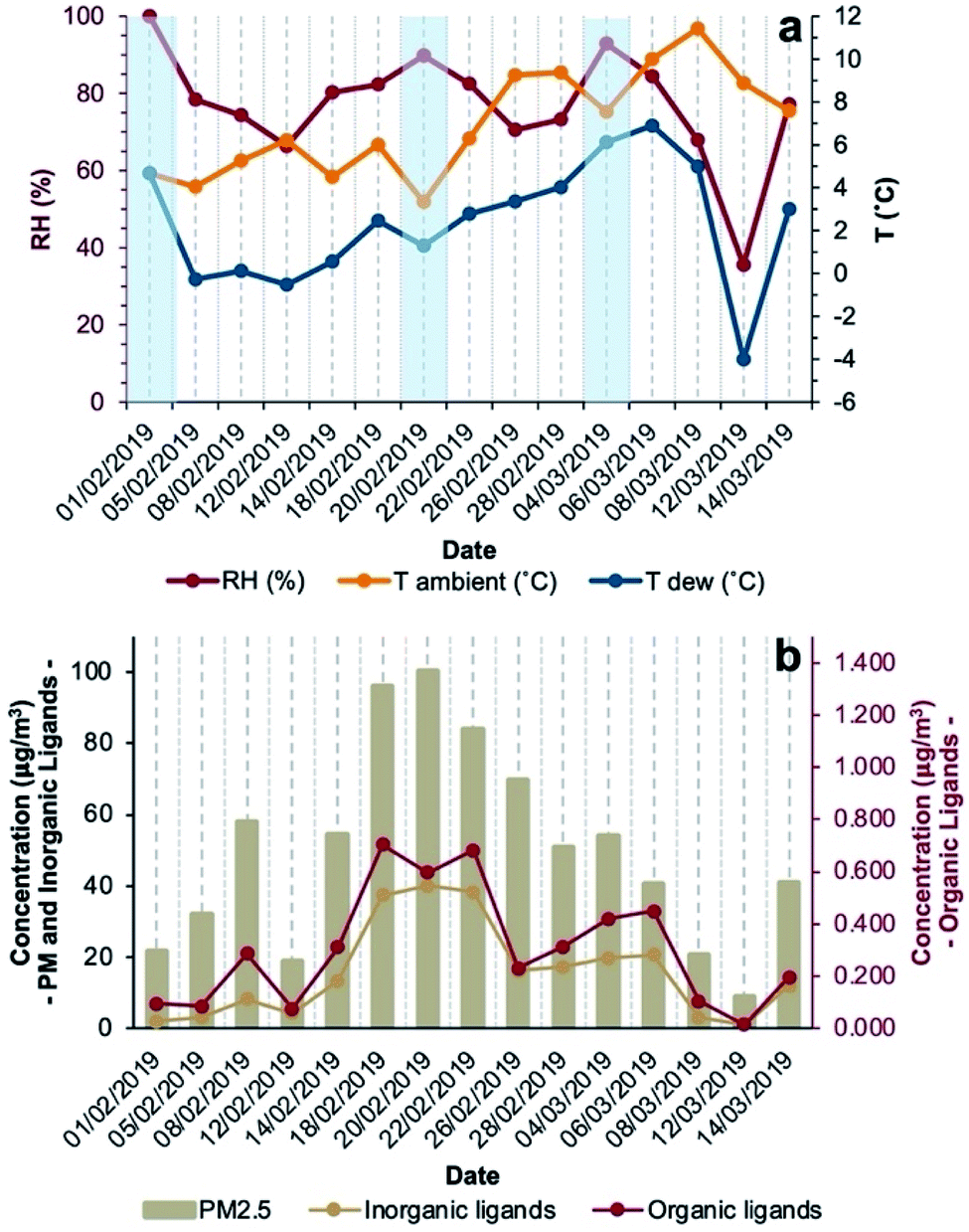 | ||
| Fig. 1 Time series of RH, ambient T and dew T (a) and the concentrations of PM2.5, inorganic and organic ligands (b) during the sampling campaign in Padova (Italy) in the winter of 2019. The light blue rectangles indicate the dates on which fog events were observed. The inorganic ligands represent the sum of Cl−, NO3−, SO42−, and PO43−; the organic ligands represent the sum of oxalate, malonate, succinate, and acetate. | ||
During the sampling campaign (February–March 2019), PM2.5 concentrations were considerably high, often exceeding EU target limits,45 with a median daily concentration of 51 μg m−3 (interquartile range (IQR) of 37 μg m−3) and a maximum daily concentration of 100 μg m−3 (Fig. 1b and Table S1, ESI†). Both meteorological and air quality conditions are typical of the Po Valley (Italy) in the winter season. Among the determined aerosol components, the most abundant was NO3− with a median contribution of 21% (IQR = 22%) of particle mass followed by NH4+ and SO42− contributing 7.0% (IQR = 8.3%) and 3.7% (IQR = 2.1%), respectively. Among metals (Table S2†), the most abundant was Fe (median 1.2%, IQR 3.8%), followed by Zn (median 0.27%, IQR 1.23%) and Al (median 0.04%, IQR 0.14%). Inorganic species (Cl−, NO3−, SO42−, and PO43−) with ligand capabilities accounted for 24%, while organic ligands (oxalate, succinate, malonate, and acetate) for 0.5% of the aerosol mass. In days characterised by fog events, concentrations of inorganic ligands ranged between 2.0 and 40 μg m−3 while organic ligand concentrations ranged between 0.095 and 0.60 μg m−3 (Fig. 1b).
3.2 Element dissolution kinetics and solubility
Dissolution kinetics were studied in a solution simulating fog water (see Section 2.3 for details on the composition of the leaching solution). The simulated fog had a pH of 4.7, a typical value for fog water in the Po Valley,46 which was adjusted using sulfuric and nitric acids given that sulphate and nitrate are the main anions observed in fog water. All other chemical components of the simulated fog were only those leached from the aerosol samples during the kinetics experiments. The composition of the simulated fog was therefore as close as possible to real fog samples from the same location except for volatile and semi-volatile components that would partition into the fog water from the gas phase. Time-dependent leaching from the PM collected on filters to the solution simulating fog water was investigated for each element in each sample by performing Spearman correlation tests.As reported in previous studies,36,38,47,48 the leaching rate is determined by the dissolution of the elements from the particle surface, according to the first order kinetics law. Therefore, we fitted the kinetics data using the following integral equation (with direct weighing based on the timestamp to account for unequally spaced data points):
| Cx = Ce − Ae−kt | (1) |
Most elements followed a first order kinetics (Fig. 2), as expected, with the kinetics constants on the order of 10−1 to 10−2 min−1 (Tables 1, S3 and S4†). In our study, average k values were generally 2–3 times higher than the results obtained by Di Marco et al.38 at the same location. Ba and Mn were the only two elements for which a first order kinetics was observed in all 15 samples (Fig. 2a and b). For most of the elements, a first order kinetics was observed in some but not in all samples. For the samples in which kinetics behaviour was not observed, elements were leached in solution immediately as their concentration was detected appreciably above detection limits (see Section 3.4). This result is surprising as Di Marco et al.38 observed that a given element either presented or did not present clear kinetics behaviour, and none of the elements showed inconsistent behaviour across different samples. Our results show that the solubility of the metals is different from sample to sample (Table 1), and most probably depends on their chemical form; thus, the kinetics behaviour is different in different PM samples. It is also worth noticing that for all elements, even when a time-dependent leaching was observed, a non-negligible or sometimes a major fraction of the element was immediately released in solution as the measured concentration at t = 0 min was not zero (Fig. 2b, c, Tables 1, S5 and S6†), consistent with the results from Di Marco et al.38
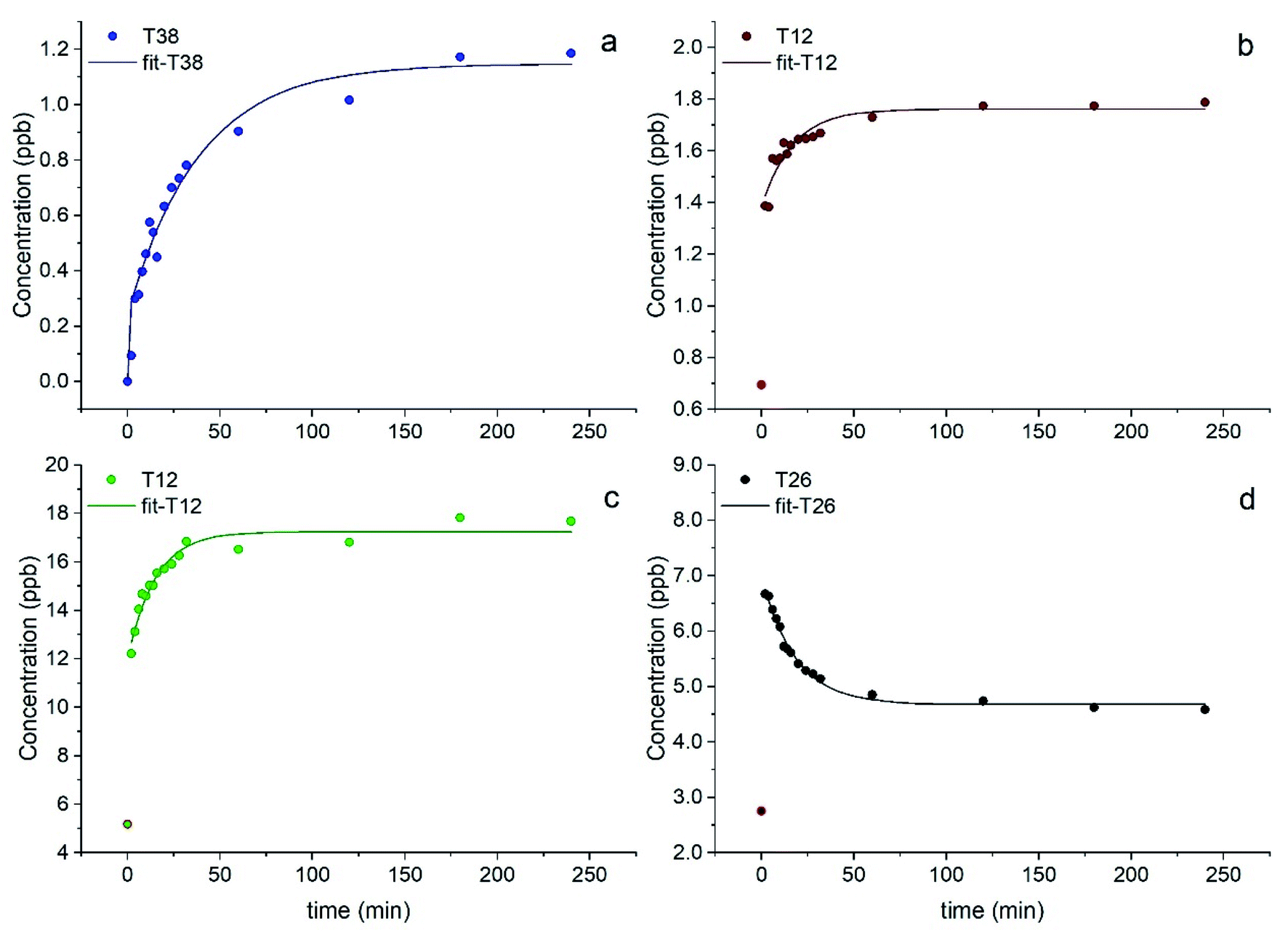 | ||
| Fig. 2 Examples of dissolution kinetics in simulated fog for the elements Ba (a), Mn (b), Zn (c), and Pb (d). The red-edge dots at t = 0 min were excluded from the fitting as the solution was not homogeneous. Samples T12, T26, and T38 are those collected on 01/02/2019, 22/02/2019, and 12/03/2019, respectively. | ||
| Element | k (min−1) (this study) | t 1/2 (min) (this study) | N | S 0 (%) (this study) | S ∞ (%) (this study) | k (min−1) (Di Marco et al.38) | t 1/2 (min) (Di Marco et al.38) | S ∞ (%) (Di Marco et al.38) |
|---|---|---|---|---|---|---|---|---|
| a Pb dissolution over time was characterised by a non-monotonic trend. | ||||||||
| Al | 0.033 (0.040) | 41 (27) | 5 | 44 (54) | 57 (60) | 0.0185 (0.0036) | 37.5 (7.3) | 12 (66) |
| As | 0.053 (0.069) | 27 (15) | 7 | 40 (39) | 46 (44) | — | — | 28 (28) |
| B | 0.10 (0.11) | 21 (24) | 4 | 56 (49) | 71 (54) | |||
| Ba | 0.015 (0.010) | 69 (47) | 15 | 18 (18) | 87 (59) | 0.0192 (0.0019) | 36.1 (3.6) | 37 (55) |
| Cd | 0.22 (0.33) | 8.3 (6.8) | 9 | 27 (29) | 35 (38) | 0.0271 (0.0029) | 25.6 (2.7) | 62 (26) |
| Co | 0.10 (0.10) | 13 (20) | 4 | 17 (18) | 19 (21) | |||
| Cr | 0.070 (0.047) | 17 (16) | 5 | 13 (19) | 13 (18) | 0.0079 (0.0016) | 88 (18) | 26 (65) |
| Cu | 0.030 (0.030) | 48 (39) | 13 | 26 (15) | 33 (19) | 0.0135 (0.0023) | 51.3 (8.7) | 22 (13) |
| Fe | 0.032 (0.022) | 52 (65) | 14 | 3.7 (3.4) | 5.1 (4.6) | 0.0143 (0.0025) | 48.5 (8.4) | 14 (15) |
| Ga | 0.038 (0.024) | 27 (20) | 11 | 24 (30) | 39 (39) | |||
| Mn | 0.087 (0.089) | 24 (33) | 15 | 31 (12) | 36 (14) | 0.0247 (0.0020) | 28.1 (2.2) | 44 (13) |
| Ni | 0.030 (0.012) | 29 (16) | 10 | 9 (10) | 12 (13) | |||
| Pba | 31 (14) | 36 (22) | 39 (30) | |||||
| Rb | 0.13 (0.13) | 10.6 (7.3) | 5 | 78 (23) | 81 (24) | 0.0463 (0.0050) | 15.0 (1.6) | 88 (19) |
| Sr | 0.079 (0.082) | 20 (18) | 7 | 44 (27) | 58 (36) | 0.0098 (0.0016) | 71 (12) | 41 (113) |
| V | 0.041 (0.052) | 37 (29) | 8 | 55 (31) | 66 (30) | 0.0218 (0.0019) | 31.7 (2.7) | 52 (27) |
| Zn | 0.128 (0.086) | 9.2 (8.3) | 14 | 36 (29) | 45 (36) | — | — | 102 (80) |
In the case of Pb, concentration trends were non-monotonic, showing an increase in the initial part of the experiment but then decreased over time (Fig. 2d), as also observed by Di Marco et al.38 and Mackey et al.49 It can be hypothesised that Pb may be present in the aerosol particles in a readily soluble form. Then, it may react in the simulated fog water with other species released from the PM itself (e.g., cyanide, sulphate), forming insoluble products and lowering its concentration in solution over time. For this reason, kinetic data were not fitted for Pb.
The solubility data obtained in this study (Table 1) were generally similar to those reported by Di Marco et al.38 but with some notable exceptions, namely Ba, for which the soluble fraction was 87% on average in this study compared to the 37% from Di Marco et al.,38 and Zn for which the soluble fraction was 45% compared to the 100% from Di Marco et al.38 The increased solubility of Ba may be attributable to the presence of BaSO4, which most probably dissolves under higher water content, e.g., under fog conditions (see Section 3.4). In contrast, the lower soluble fraction of Zn may be associated with the presence of ZnO which is a refractory form of Zn used in industrial applications (e.g., paint, galvanization, water repellents).50 A similarly lower soluble fraction of Zn was found previously by Gioda et al.51 in Rio de Janeiro (Brazil). The soluble fraction of Al was larger in this study (57%) than that in previous studies at the same location (12%)38 and elsewhere (2–8%).32 We hypothesize that the larger soluble fraction of Al observed in this study may be attributable to aqueous-phase processing of aerosol, leading to the formation of soluble Al–organic complexes (see Section 3.3). The water soluble fractions for Cu, Fe, and Mn were in line with previous studies.31,32,38,52
3.3 Metal speciation in deliquescent aerosol and simulated fog water
For the most abundant metals, we calculated their speciation in the deliquescent aerosol and in the simulated fog water in order to investigate the contributing species to their readily-soluble and soluble fractions.We hypothesised that the readily-soluble fraction of each element is made of a combination between readily-soluble primarily emitted species and species that underwent aqueous-phase processing in the atmosphere, either in deliquescent aerosol or in fog droplets (for days in which fog events were observed).
Concerning aqueous-phase processing in deliquescent aerosol, the ALW content estimated using the model E-AIM IV ranged from 1.9 μg m−3 on 8 March 2019 to 197 μg m−3 on 20 February 2019, excluding 12 March 2019 for which ambient conditions were dry (RH was below 40%) and for this reason the ALW could not be calculated (Fig. 3). Therefore, the ambient conditions under which the samples were collected spanned from dry (non-deliquescent aerosol) to humid (deliquescent aerosol), including fog formations. The computed pH values (from E-AIM IV) were highly acidic in all samples, ranging between −0.5 and 3 (Fig. 3). Generally, acidity was inversely proportional to ALW content but with some exceptions for which a low ALW (<25 μg m−3) but a relatively high pH (>2) were computed. These exceptions were towards the last days of the campaign, when temperatures were higher (9 °C on average in the period 8–14 March 2019, campaign average 7 °C) and the NH4+/NO3− w/w was higher (0.46 on average in the period 8–14 March 2019, campaign average 0.38).
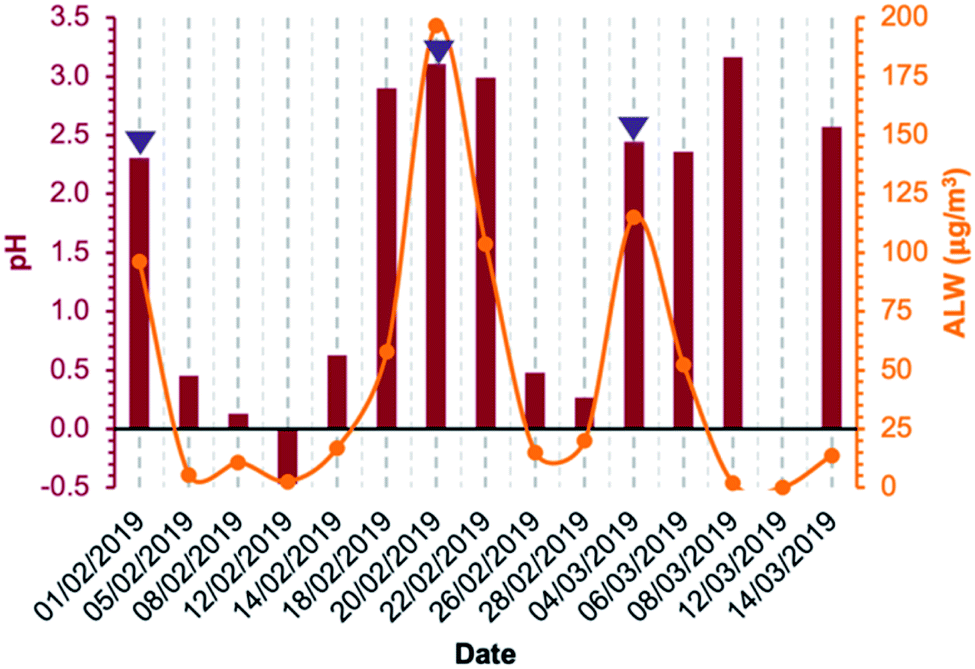 | ||
| Fig. 3 Aerosol liquid water (ALW) content and aerosol pH calculated using the model E-AIM IV based on the bulk chemical composition of PM2.5, and average RH and T over the 24 h sampling period. The blue triangles indicate the days with observed fog formation. | ||
The speciation picture of metal ions in deliquescent aerosol depended upon ALW content, pH, and the amount of inorganic and organic ligands present in the samples (Fig. 4 and Table S7†). These three factors, while always playing a role in determining metal speciation, contributed differently to the speciation of different metals. For example, on the drier day when ALW and pH could be computed (8 March 2019), precipitations of Fe(III) as ferrihydrite (Fe2O3·0.5H2O) and Al(III) as aluminium hydroxide sulphate (AlOHSO4(s)) were calculated to occur (Table S8†). A fraction of Mn(II) was also predicted to form a solid precipitate of MnHPO4(s) (Table S8†); however, its formation did not seem to be linked to ALW content. MnHPO4(s) was predicted to form even on days with high ALW and its formation seemed to be driven by higher concentrations of Mn and phosphate combined with relatively low acidic conditions (pH > 2). Calcium was predicted to precipitate as calcium oxalate (CaC2O4·H2O(s)) and calcium sulphate (CaSO4(s)), with the total precipitated fraction ranging between 28% and 90% (Table S8†). The fraction of calcium forming a solid precipitate showed a high day-to-day variability and seemed to be controlled by a combination of three factors (pH, ALW content, and concentrations of oxalate and sulphate).
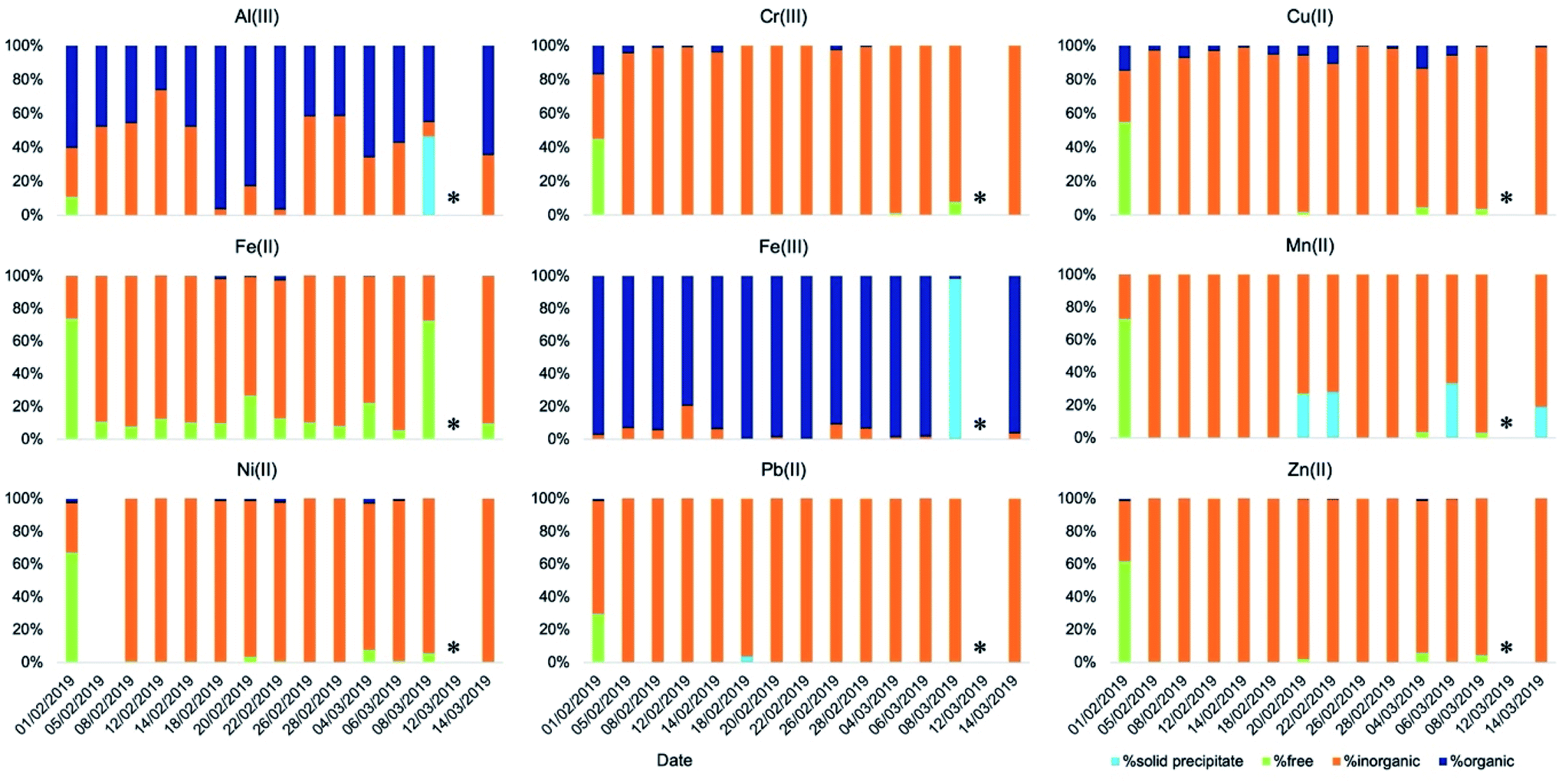 | ||
| Fig. 4 Speciation of readily soluble metals (t = 2 min) in ALW obtained using the model Visual MinteQ. | ||
A larger proportion of free metal ions was calculated for the sample collected on 1 February 2019. This day was characterised by a relatively high ALW content (96 μg m−3), a pH of 2.3, and relatively low ligand concentrations (2.0 μg m−3 and 0.095 μg m−3 for inorganics and organics, respectively). Fe(III) was a notable exception, as we predicted it to be bound mainly to organic compounds (Fig. 4).
On most of the days, either ALW was lower or the content of inorganic anions was higher. Under these conditions, metal ions were predominantly present in solution bound to inorganic ligands (Fig. 4). The most abundant species were NiNO3+, AlSO4+, CrSO4+, CuNO3+, Cu(NO3)2(aq), FeCl+, FeSO4(aq), FeH2PO4+, ZnSO4(aq), ZnNO3+, Zn(NO3)2(aq), and Pb(NO3)2(aq) (Table S7†).
Fe(III) and Al(III) were predicted to be predominantly bound to organics (Fig. 4 and Table S7†). The dominant species were complexes between Al and oxalate (AlC2O4+ and Al(C2O4)2−), Al and malonate (AlC3H4O4+ and Al(C3H4O4)2−), Fe(III) and oxalate (FeC2O4+ and Fe(C2O4)2−), and Fe(III) and malonate (FeC3H4O4+ and Fe(C3H4O4)2−). While Fe(III) was consistently predicted to be bound predominantly to organic compounds, the fraction of Al(III) bound to either inorganic or organic compounds was predicted to change from sample to sample (Fig. 4). The formation of complexes between Al(III) and organic compounds seemed to be favoured on days with larger concentrations of organic compounds and relatively high aerosol pH. On days with higher concentrations of organic compounds and higher ALW content, a small fraction of Fe(II) was also bound to oxalate (Fig. 3 and 4). A non-negligible fraction of Cu(II) was bound to organics (both oxalate and malonate), Ni(II) with oxalate, and a small fraction of Cr(III) formed complexes with acetate (Fig. 4 and Table S7†).
The simulated fog water used in our kinetics experiments, a good representation of fog in the Po Valley during the winter season,38,46 creates different conditions for the aqueous-phase processing compared to deliquescent aerosol. Average fog water content used was around 0.5 g m−3, 3–6 orders of magnitude more than that predicted in deliquescent aerosol. This means that dissolved species were 3–6 orders of magnitude more diluted. While the temperature (T = 5 °C) used was in the range of that of the aerosol samples, a pH of 4.7 is considerably less acidic than the values computed for deliquescent aerosol. As a consequence, the speciation of the metals is considerably different under the fog conditions (Fig. 5 and Table S9†) compared with that under the deliquescent aerosol conditions (Fig. 4 and Table S7†).
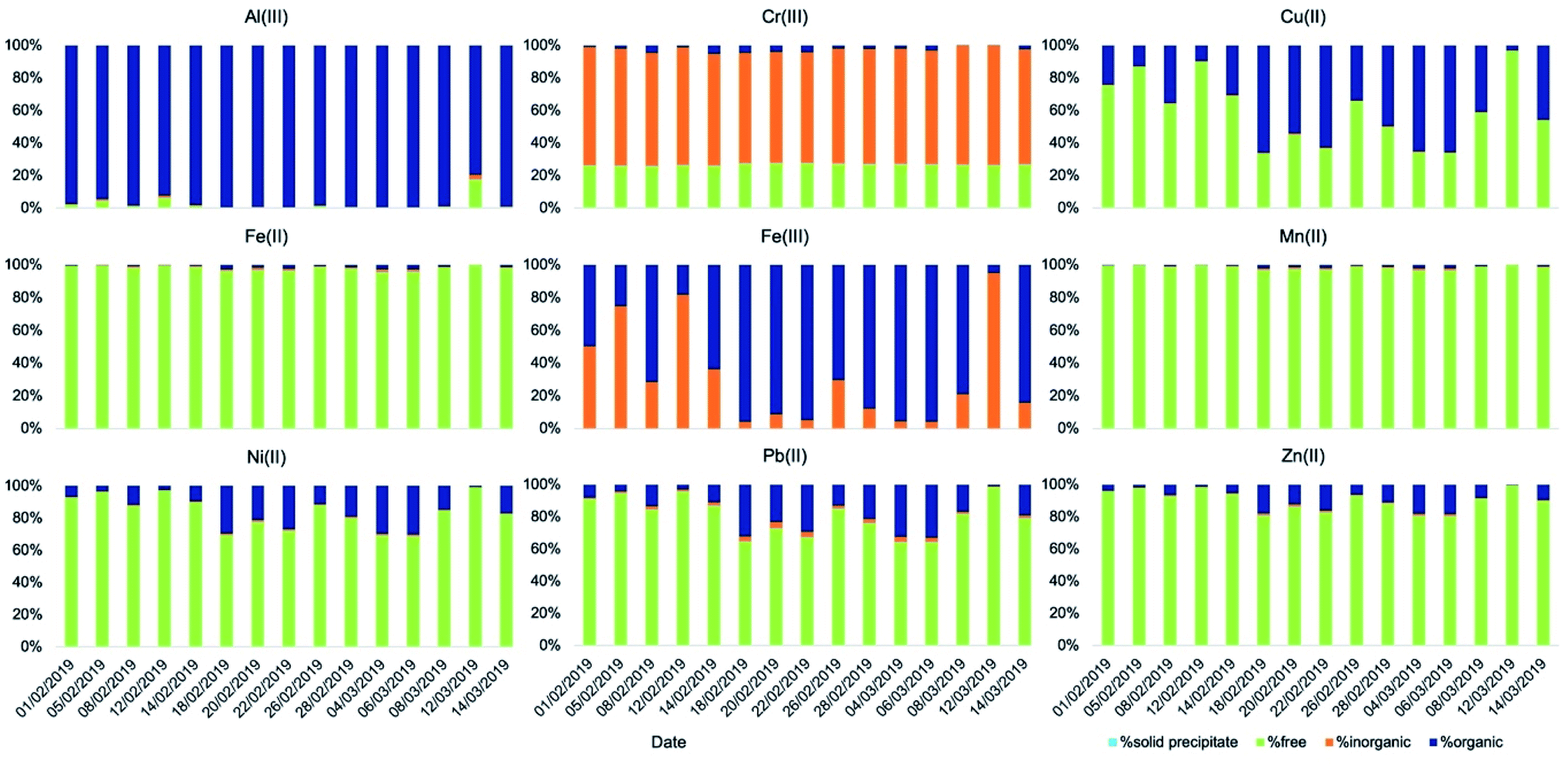 | ||
| Fig. 5 Speciation of soluble metals (t = ∞) in simulated fog (T = 5 °C, pH = 4.7) obtained using the model Visual MinteQ. | ||
One major difference is the presence of a higher fraction of free metal ions particularly for Fe(II), Mn(II), Ni(II), Pb(II), and Zn(II), likely because of dilution due to the larger water content. Under these conditions, speciation results also indicated the absence of precipitates for all metals, including calcium.
Another notable difference is that a smaller fraction of Fe(III) is bound to oxalate and malonate and it is present instead as hydroxide (Fe(OH)2+ and Fe(OH)2+). In contrast, a small but larger fraction of Fe(II) compared to what was found in the deliquescent aerosol is coordinated with oxalate under these conditions.
All other metals have also a larger fraction bound to organics compared to that under deliquescent aerosol conditions. Al(III) is almost entirely bound to oxalate with a small fraction bound to malonate. Cu(II) is consistently more bound to oxalate with a small fraction bound to malonate. For the other metals the fraction bound to organics is smaller but significant: Cr(III) with acetate, and Ni(II), Pb(II), and Zn(II) with oxalate.
3.4 Possible impact of aqueous-phase processing on the dissolution kinetics and solubility of metals
In order to assess the impact of aqueous-phase processing on the dissolution kinetics of metals we compared the dissolution rates (in terms of t1/2) of metals obtained at different ALW contents, in particular, those obtained at high ALW (>50 μg m−3) and at lower ALW (<50 μg m−3), at different pH (pH > 2 vs. pH < 1), and at different concentrations of organic ligands (>150 ng m−3vs. <150 ng m−3) (Fig. S1†).In general, the average t1/2 of all metal ions was demonstrated to be independent of the ALW content, because even when a correlation was apparent (e.g., for Al, Ba, Fe, Ga, Sr, and Zn, t1/2 was lower on average for samples with a high ALW content, while for As, Cr and V the t1/2 was higher), it was not statistically significant (p-value > 0.05) as the sample-to-sample variability was very high. The half-life also appeared to be independent of pH and organic ligand content. Similarly, we compared solubilities at t0 and t∞ under the same set of conditions (Fig. S2 and S3†) but, also in this case, no significant differences could be observed. The reason for this lack of correlation with individual factors (ALW content, pH, and organic content) may be due to the fact that they all control metal speciation (see Section 3.3). In addition, the calculated speciation picture of the metals (Fig. 4) is qualitatively similar across the different samples except for one sample with an extremely low ALW (8 March 2019, ALW = 1.9 μg m−3) and for another sample collected under dry conditions (12 March 2019, RH < 40%) for which no speciation picture could be calculated. The drier day on which ALW and pH could be computed (8 March 2019) coincided with the sample for which the lowest dissolution rate was found (t1/2 = 81 min) for Al. For this sample, the speciation of Al(III) showed the precipitation of aluminium hydroxide sulphate (AlOHSO4(s)) in deliquescent aerosol thus explaining a lower dissolution rate. However, for the same sample, the speciation in deliquescent aerosol showed the precipitation of Fe(III) as ferrihydrite (Fe2O3·0.5H2O) but, in this case, the Fe dissolution rate was high (t1/2 = 10 min). This result suggests that Fe(II) may be the dominant Fe species in the soluble fraction at least for this sample. This result is not surprising considering that previous studies found that Fe(II) was either the dominant species in the soluble fraction18,27,28 or equally present together with Fe(III)18,29 but the main contributor (80%) to the readily soluble fraction of Fe.29
In contrast, the speciation pictures obtained under simulated fog conditions (Fig. 5) are considerably different from those obtained under deliquescent aerosol conditions (Fig. 4). To test whether the fog processing would have an impact on the dissolution kinetics of the metals we compared the kinetic profiles obtained on days with no recorded fog events and the kinetic profiles obtained on days with recorded fog events and high organic content (Fig. 6).
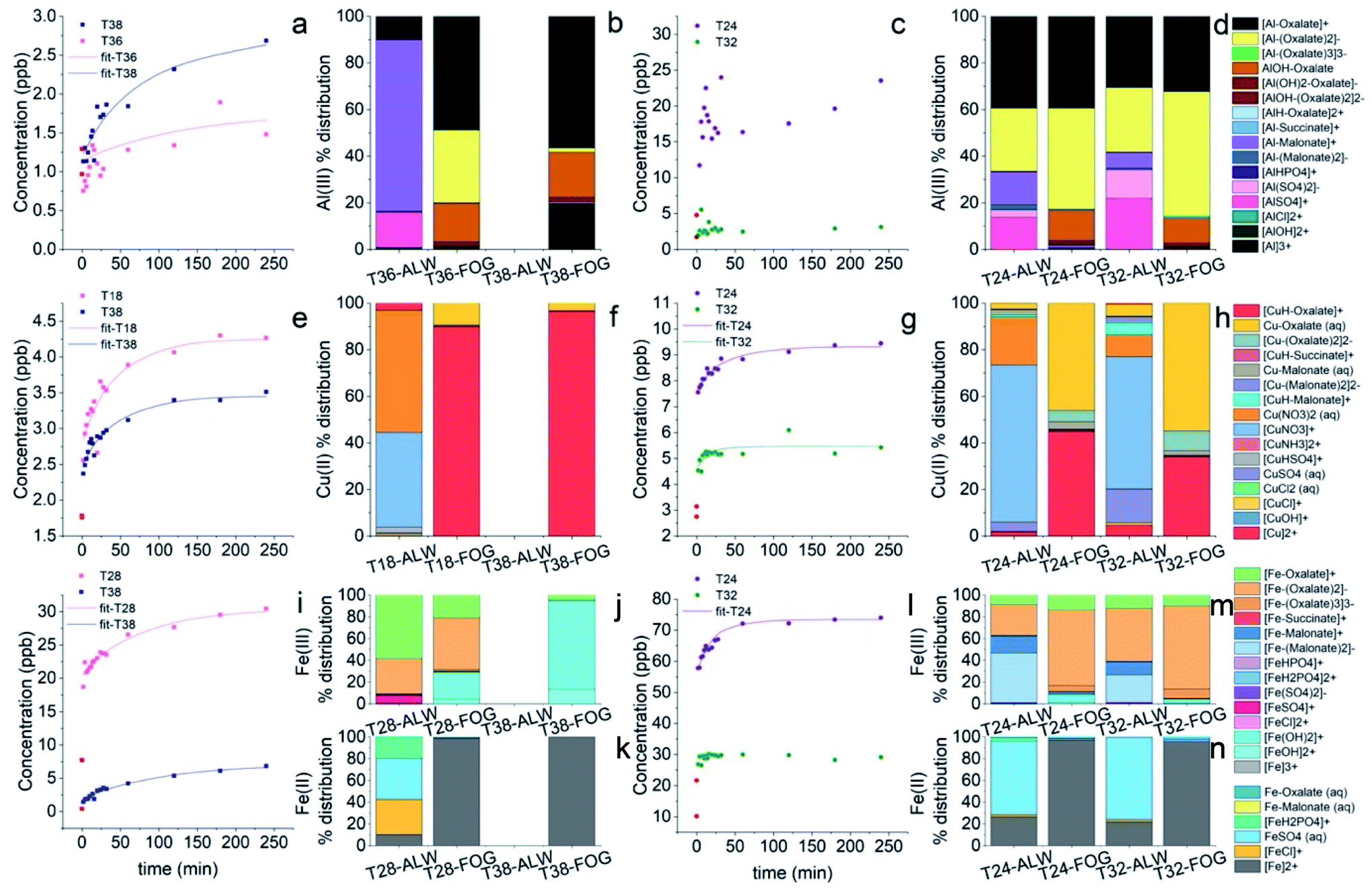 | ||
| Fig. 6 Examples of dissolution kinetics for two samples at lower RH for Al (a), Cu (e), and Fe (i). The corresponding detailed speciation picture in ALW and in simulated fog for Al(III) (b), Cu(II) (f), Fe(III) (j), and Fe(II) (k). Examples of dissolution kinetics for two samples at high RH for Al (c), Cu (g), and Fe (l). The corresponding detailed speciation picture in ALW and in simulated fog for Al(III) (d), Cu(II) (h), Fe(III) (m), and Fe(II) (n). The red dots at t = 0 min were excluded from the fitting as the solution was not homogeneous. Samples T18, T24, T28, T32, T36, and T38 are those collected on 12/02/2019, 20/02/2019 (fog event), 26/02/2019, 04/03/2019 (fog event), 08/03/2019, and 12/03/2019, respectively. | ||
We observed the largest effect on Al (Fig. 6a–d), for which on days with recorded fog events the soluble fraction of Al was immediately released in solution and no kinetics profile was observed, while on the days with drier conditions its dissolution rate was the slowest (t1/2 > 50 min). In the case of Cu (Fig. 6e–h), the fastest dissolution rates were obtained on days with recorded fog events and higher organic content (t1/2 of 22.8 ± 4.2 min and 10.6 ± 6.5 min on 20 February 2019 and 4 March 2019, respectively), whereas slower dissolution rates were measured on other days (average t1/2 ∼ 50 min) although with a high variability. In the case of Fe (Fig. 6i–n), on days with recorded fog events we observed either an immediate release in solution of Fe (4 March 2019) or a fast release but with a time-dependent profile (t1/2 = 14.5 ± 3.2 min). The faster dissolution rates for Fe and Cu on days with fog events and higher organic content could be partly explained by the formation of metal–ligand complexes with short-chain dicarboxylic acids (Fig. 5). However, the influence of other organic compounds on their dissolution rates cannot be excluded. For example, it has been observed that siderophores produced by microorganisms isolated in cloud water can strongly bind iron, even more strongly than oxalate, and can affect its redox behaviour.53–55
For more readily soluble elements, such as As, Zn, Cd, Cr, and Sr, we did not observe an impact of aqueous-phase processing on their dissolution rates, while a high sample-to-sample variability on days with fog events was evidenced for some metals such as V, Mn, and Ba. According to our calculations, V does not coordinate with organics, while only a minor fraction of Mn was found to coordinate with organics in the fog water (Fig. 5). Ba was predicted to simply solubilise in the available water (BaSO4(s) in deliquescent aerosol and Ba2+ in solution in fog water) rather than converting into a more readily soluble species (speciation results not shown). These results show that for metals for which a large fraction was predicted to coordinate with organic compounds in fog water (Fig. 5) the dissolution rates were faster on days with recorded fog events and high organic content. In contrast, for elements for which either only a minor fraction was predicted to coordinate with organic compounds in fog water or none, the dissolution rates were not affected by fog processing.
Concerning element solubility (Fig. 7), the impact of fog processing varies depending on the element. For some of them, we observed a minimum impact, as their soluble fraction was readily soluble in all samples whether it was a major fraction, e.g., Rb (∼80%), or a minor fraction, e.g., Cr (∼13%) and Ni (∼10%). For most of the elements, the solubility and increase of solubility due to fog processing presented a high sample-to-sample variability. The metal ion for which the fog processing impacts the solubility the most is Ba. Ba solubility is associated with the relatively slow dissolution of BaSO4 due to the increase in water content. In the case of Pb, fog processing decreased Pb solubility most likely due to the precipitation of PbSO4 (non-monotonic trend of the time-dependent dissolution).
 | ||
| Fig. 7 Increase in % solubility from t = 2 min (S0) to t > 60 min (S∞) in simulated fog water for all the elements determined in the aerosol samples. | ||
4 Conclusions
We investigated the impact of aqueous-phase processing and metal speciation on the solubility and dissolution kinetics of metals in aerosols from an urban background location in a northern Italian valley. We observed a high sample-to-sample variability in terms of content of metals and ligands, environmental conditions from dry (RH < 40%) to humid conditions (RH up to 100% and fog formations), and aerosol pH (from −0.5 to 3). High sample-to-sample variability was also observed for metal solubilities (in terms of % of soluble fraction) and dissolution kinetics (in terms of kinetics constants and dissolution half-time) in simulated fog water (pH 4.7, T = 5 °C, water content corresponding to ∼0.5 g m−3).Concerning solubility, fog processing increased the soluble fraction of most metals, with the largest effect observed for Ba. We found that Ba solubility was associated with the dissolution of BaSO4 in the fog water due to increased water content. For Pb, we observed an opposite effect, with a decrease in its solubility due to fog processing which is most likely associated with precipitation of PbSO4 as also observed in previous studies.38,49
Dissolution kinetics were generally characterised by a readily soluble fraction immediately released in solution (t < 2 min) and a soluble fraction following a first-order dissolution kinetics. The controlling factors of the dissolution kinetics of the metals were ALW content, aerosol pH, and the concentration of organic ligands. However, their contribution could not be separated. In fact, the speciation picture of the metals in deliquescent aerosol across the different samples was qualitatively very similar, with only a couple of exceptions for which a statistically significant comparison could not be made. In contrast, the speciation picture under fog conditions was considerably different from that obtained under deliquescent aerosol conditions. We compared the dissolution kinetics on days with observed fog events with that on days with drier conditions. The metals affected the most by the aqueous-phase processing are those that are not readily soluble in their primarily emitted form and that are predicted to coordinate with organics in the fog water. The largest effects were observed for Al, Cu, and Fe. For more readily soluble elements, such as As, Zn, Cd, Cr, and Sr, we did not observe any impact of aqueous-phase processing while a high sample-to-sample variability on days with fog events was observed for other metals such as Ba, V, and Mn. These three, however, were not predicted to coordinate with organic compounds significantly.
Our study highlights the impact that coordination chemistry in fog water has, not only on the solubility of particle-bound metals, but also on their dissolution rate. In fact, we observed that fog processing can accelerate dissolution rates of metals that form complexes with short-chain dicarboxylic acids in fog water. Further studies are necessary to determine whether this increase in solubility and dissolution rate would make metals more prone to causing oxidative stress in lung fluids, where the aqueous environment is considerably more complex and could change the chemical form of the metals compared with that found in the atmosphere.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded by the Supporting TAlent in ReSearch@University of Padova STARS-StGMOCAA awarded by the University of Padova to CG and a BP Next Generation fellowship awarded by the Yusuf Hamied Department of Chemistry at the University of Cambridge to CG. The authors are grateful to Gianni Formenton (ARPA Veneto, the Environmental Regional Agency) for providing data on environmental conditions and to Leia Pryke for helpful revision of the manuscript.References
- M. Shiraiwa, K. Ueda, A. Pozzer, G. Lammel, C. J. Kampf, A. Fushimi, S. Enami, A. M. Arangio, J. Fröhlich-Nowoisky, Y. Fujitani, A. Furuyama, P. S. J. Lakey, J. Lelieveld, K. Lucas, Y. Morino, U. Pöschl, S. Takahama, A. Takami, H. Tong, B. Weber, A. Yoshino and K. Sato, Aerosol Health Effects from Molecular to Global Scales, Environ. Sci. Technol., 2017, 51(23), 13545–13567, DOI:10.1021/acs.est.7b04417.
- P. Fu, X. Guo, F. M. H. Cheung and K. K. L. Yung, The Association between PM2.5 Exposure and Neurological Disorders: A Systematic Review and Meta-Analysis, Sci. Total Environ., 2019, 655, 1240–1248, DOI:10.1016/j.scitotenv.2018.11.218.
- O. Schmid and T. Stoeger, Surface Area is the Biologically Most Effective Dose Metric for Acute Nanoparticle Toxicity in the Lung, J. Aerosol Sci., 2016, 99, 133–143, DOI:10.1016/j.jaerosci.2015.12.006.
- M. Tobiszewski and J. Namieśnik, PAH Diagnostic Ratios for the Identification of Pollution Emission Sources, Environ. Pollut., 2012, 162, 110–119, DOI:10.1016/j.envpol.2011.10.025.
- K. Srogi, Monitoring of Environmental Exposure to Polycyclic Aromatic Hydrocarbons: A Review, Environ. Chem. Lett., 2007, 5(4), 169–195, DOI:10.1007/s10311-007-0095-0.
- World Health Organization, Health Risks of Heavy Metals from Long-Range Transboundary Air Pollution,Copenhagen, Denmark, 2007 Search PubMed.
- X. Luo, Z. Zhao, J. Xie, J. Luo, Y. Chen, H. Li and L. Jin, Pulmonary Bioaccessibility of Trace Metals in PM 2.5 from Different Megacities Simulated by Lung Fluid Extraction and DGT Method, Chemosphere, 2019, 218, 915–921, DOI:10.1016/j.chemosphere.2018.11.079.
- L. Deguillaume, M. Leriche, K. Desboeufs, G. Mailhot, C. George and N. Chaumerliac, Transition Metals in Atmospheric Liquid Phases: Sources, Reactivity, and Sensitive Parameters, Chem. Rev., 2005, 105(9), 3388–3431, DOI:10.1021/cr040649c.
- I. Grgić, Metals in Aerosols, Environ. Chem. Aerosols, 2009, 117–139, DOI:10.1002/9781444305388.ch5.
- S. G. Klein, T. Serchi, L. Hoffmann, B. Blömeke and A. C. Gutleb, An Improved 3DTetraculture System Mimicking the Cellular Organisation at the Alveolar Barrier to Study the Potential Toxic Effects of Particles on the Lung, Part. FibreToxicol., 2013, 10(1), 31, DOI:10.1186/1743-8977-10-31.
- P. Shahpoury, Z. W. Zhang, A. Arangio, V. Celo, E. Dabek-Zlotorzynska, T. Harner and A. Nenes, The Influence of Chemical Composition, Aerosol Acidity, and Metal Dissolution on the Oxidative Potential of Fine Particulate Matter and Redox Potential of the Lung Lining Fluid, Environ. Int., 2021, 148, 106343, DOI:10.1016/j.envint.2020.106343.
- F. J. Kelly and J. C. Fussell, Size, Source and Chemical Composition as Determinants of Toxicity Attributable to Ambient Particulate Matter, Atmos. Environ., 2012, 60, 504–526, DOI:10.1016/j.atmosenv.2012.06.039.
- Y. Zuo and J. Holgne, Formation of Hydrogen Peroxide and Depletion of Oxalic Acid in Atmospheric Water by Photolysis of Iron(III)-Oxalato Complexes, Environ. Sci. Technol., 1992, 26(5), 1014–1022, DOI:10.1021/es00029a022.
- C. Weller, S. Horn and H. Herrmann, Effects of Fe(III)-Concentration, Speciation, Excitation-Wavelength and Light Intensity on the Quantum Yield of Iron(III)-Oxalato Complex Photolysis, J. Photochem. Photobiol., A, 2013, 255, 41–49, DOI:10.1016/j.jphotochem.2013.01.014.
- M. I. Litter and M. Slodowicz, An Overview on Heterogeneous Fenton and PhotoFenton Reactions Using Zerovalent Iron Materials, J. Adv. Oxid. Technol., 2017, 20(1), 2016016, DOI:10.1515/jaots-2016-0164.
- R. F. Hems, J. S. Hsieh, M. A. Slodki, S. Zhou and J. P. D. Abbatt, Suppression of OH Generation from the Photo-Fenton Reaction in the Presence of α-Pinene Secondary Organic Aerosol Material, Environ. Sci. Technol. Lett., 2017, 4(10), 439–443, DOI:10.1021/acs.estlett.7b00381.
- I. Grgić, A. Dovžan, G. Berčič and V. Hudnik, The Effect of Atmospheric Organic Compounds on the Fe-Catalyzed S(IV) Autoxidation in Aqueous Solution, J. Atmos. Chem., 1998, 29(3), 315–337, DOI:10.1023/A:1005918912994.
- I. Grgić, M. Poznič and M. Bizjak, S(IV) Autoxidation in Atmospheric Liquid Water: The Role of Fe(II) and the Effect of Oxalate, J. Atmos. Chem., 1999, 33(1), 89–102, DOI:10.1023/A:1006141913681.
- D. A. Thomas, M. M. Coggon, H. Lignell, K. A. Schilling, X. Zhang, R. H. Schwantes, R. C. Flagan, J. H. Seinfeld and J. L. Beauchamp, Real-Time Studies of Iron Oxalate-Mediated Oxidation of Glycolaldehyde as a Model for Photochemical Aging of Aqueous Tropospheric Aerosols, Environ. Sci. Technol., 2016, 50(22), 12241–12249, DOI:10.1021/acs.est.6b03588.
- A. S. Wozniak, R. U. Shelley, S. D. McElhenie, W. M. Landing and P. G. Hatcher, Aerosol Water Soluble Organic Matter Characteristics over the North Atlantic Ocean: Implications for Iron-Binding Ligands and Iron Solubility, Mar. Chem., 2015, 173, 162–172, DOI:10.1016/j.marchem.2014.11.002.
- A. Tapparo, V. Di Marco, D. Badocco, S. D'Aronco, L. Soldà, P. Pastore, B. M. Mahon, M. Kalberer and C. Giorio, Formation of Metal–Organic Ligand Complexes Affects Solubility of Metals in Airborne Particles at an Urban Site in the Po Valley, Chemosphere, 2020, 241, 125025, DOI:10.1016/j.chemosphere.2019.125025.
- C. Giorio, D. Marton, G. Formenton and A. Tapparo, Formation of Metal–Cyanide Complexes in Deliquescent Airborne Particles: A New Possible Sink for HCN in Urban Environments, Environ. Sci. Technol., 2017, 51(24), 14107–14113, DOI:10.1021/acs.est.7b03123.
- J. Wei, H. Yu, Y. Wang and V. Verma, Complexation of Iron and Copper in Ambient Particulate Matter and Its Effect on the Oxidative Potential Measured in a Surrogate Lung Fluid, Environ. Sci. Technol., 2019, 53(3), 1661–1671, DOI:10.1021/acs.est.8b05731.
- S. Scheinhardt, K. Müller, G. Spindler and H. Herrmann, Complexation of Trace Metals in Size-Segregated Aerosol Particles at Nine Sites in Germany, Atmos. Environ., 2013, 74, 102–109, DOI:10.1016/j.atmosenv.2013.03.023.
- X. D. Feng, Z. Dang, W. L. Huang and C. Yang, Chemical Speciation of Fine Particle Bound Trace Metals, Int. J. Environ. Sci. Technol., 2009, 6(3), 337–346, DOI:10.1007/bf03326071.
- L. J. Spokes, T. D. Jickells and B. Lim, Solubilisation of Aerosol Trace Metals by Cloud Processing: A Laboratory Study, Geochim.Cosmochim.Acta, 1994, 58(15), 3281–3287, DOI:10.1016/0016-7037(94)90056-6.
- J. Mao, S. Fan and L. W. Horowitz, Soluble Fe in Aerosols Sustained by Gaseous HO2 Uptake, Environ. Sci. Technol. Lett., 2017, 4(3), 98–104, DOI:10.1021/acs.estlett.7b00017.
- B. J. Majestic, J. J. Schauer and M. M. Shafer, Application of Synchrotron Radiation for Measurement of Iron Red-Ox Speciation in Atmospherically Processed Aerosols, Atmos. Chem. Phys., 2007, 7(10), 2475–2487, DOI:10.5194/acp-7-2475-2007.
- L. Deguillaume, K. V. Desboeufs, M. Leriche, Y. Long and N. Chaumerliac, Effect of Iron Dissolution on Cloud Chemistry: From Laboratory Measurements to Model Results, Atmos. Pollut. Res., 2010, 1(4), 220–228, DOI:10.5094/APR.2010.029.
- T. Li, Y. Wang, J. Zhou, T. Wang, A. Ding, W. Nie, L. Xue, X. Wang and W. Wang, Evolution of Trace Elements in the Planetary Boundary Layer in Southern China: Effects of Dust Storms and Aerosol-Cloud Interactions, J. Geophys. Res., 2017, 122(6), 3492–3506, DOI:10.1002/2016JD025541.
- T. Fang, H. Guo, L. Zeng, V. Verma, A. Nenes and R. J. Weber, Highly Acidic Ambient Particles, Soluble Metals, and Oxidative Potential: A Link between Sulfate and Aerosol Toxicity, Environ. Sci. Technol., 2017, 51(5), 2611–2620, DOI:10.1021/acs.est.6b06151.
- A. R. Baker, M. Li and R. Chance, Trace Metal Fractional Solubility in Size-Segregated Aerosols from the Tropical Eastern Atlantic Ocean, Global Biogeochem. Cycles, 2020, 34(6), 1–9, DOI:10.1029/2019GB006510.
- R. Paris and K. V. Desboeufs, Effect of Atmospheric Organic Complexation on Iron-Bearing Dust Solubility, Atmos. Chem. Phys., 2013, 13(9), 4895–4905, DOI:10.5194/acp-13-4895-2013.
- R. Paris, K. V. Desboeufs and E. Journet, Variability of Dust Iron Solubility in Atmospheric Waters: Investigation of the Role of Oxalate Organic Complexation, Atmos. Environ., 2011, 45(36), 6510–6517, DOI:10.1016/j.atmosenv.2011.08.068.
- K. V. Desboeufs, R. Losno and J. L. Colin, Relationship between Droplet pH and Aerosol Dissolution Kinetics: Effect of Incorporated Aerosol Particles on Droplet pH during Cloud Processing, J. Atmos. Chem., 2003, 46(2), 159–172, DOI:10.1023/A:1026011408748.
- K. V. Desboeufs, R. Losno, F. Vimeux and S. Cholbi, The pH-Dependent Dissolution of Wind-Transported Saharan Dust, J. Geophys. Res.: Atmos., 1999, 104(D17), 21287–21299, DOI:10.1029/1999JD900236.
- C. Giorio, A. Tapparo, M. L. Scapellato, M. Carrieri, P. Apostoli and G. B. Bartolucci, Field Comparison of a Personal Cascade Impactor Sampler, an Optical Particle Counter and CEN-EU Standard Methods for PM10, PM2.5 and PM1 Measurement in Urban Environment, J. Aerosol Sci., 2013, 65, 111–120, DOI:10.1016/j.jaerosci.2013.07.013.
- V. Di Marco, A. Tapparo, D. Badocco, S. D'Aronco, P. Pastore and C. Giorio, Metal Ion Release from Fine Particulate Matter Sampled in the Po Valley to an Aqueous Solution Mimicking Fog Water: Kinetics and Solubility, Aerosol Air Qual. Res., 2020, 20(4), 720–729, DOI:10.4209/aaqr.2019.10.0498.
- C. Giorio, S. Pizzini, E. Marchiori, R. Piazza, S. Grigolato, M. Zanetti, R. Cavalli, M. Simoncin, L. Soldà, D. Badocco and A. Tapparo, Sustainability of Using Vineyard Pruning Residues as an Energy Source: Combustion Performances and Environmental Impact, Fuel, 2019, 243, 371–380, DOI:10.1016/j.fuel.2019.01.128.
- A. S. Wexler, Atmospheric Aerosol Models for Systems Including the Ions H+, NH4+, Na+, SO42−, NO3−, Cl−, Br−, and H2O, J. Geophys. Res., 2002, 107(D14), 4207, DOI:10.1029/2001JD000451.
- S. L. Clegg, P. Brimblecombe and A. S. Wexler, Thermodynamic Model of the System H+–NH4+–SO42−–NO3−–H2O at Tropospheric Temperatures, J. Phys. Chem. A, 1998, 102(12), 2137–2154, DOI:10.1021/jp973042r.
- S. L. Clegg, K. S. Pitzer and P. Brimblecombe, Thermodynamics of Multicomponent, Miscible, Ionic Solutions.Mixtures Including Unsymmetrical Electrolytes, J. Phys. Chem., 1992, 96(23), 9470–9479, DOI:10.1021/j100202a074.
- S. L. Clegg, J. H. Seinfeld and E. O. Edney, Thermodynamic Modelling of Aqueous Aerosols Containing Electrolytes and Dissolved Organic Compounds. II. An Extended Zdanovskii–Stokes–Robinson Approach, J. Aerosol Sci., 2003, 34(6), 667–690, DOI:10.1016/S0021-8502(03)00019-3.
- M. G. Lawrence, The Relationship between Relative Humidity and the Dewpoint Temperature in Moist Air: A Simple Conversion and Applications, Bull. Am. Meteorol. Soc., 2005, 86(2), 225–233, DOI:10.1175/BAMS-86-2-225.
- The European Parliament and the Council of the European Union, Directive 2008/50/EC of the European Parliament and of the Council of 21 May 2008 on Ambient Air Quality and Cleaner Air for Europe, 2008,pp. 1–44 Search PubMed.
- L. Giulianelli, S. Gilardoni, L. Tarozzi, M. Rinaldi, S. Decesari, C. Carbone, M. C. Facchini and S. Fuzzi, Fog Occurrence and Chemical Composition in the Po Valley over the Last Twenty Years, Atmos. Environ., 2014, 98, 394–401, DOI:10.1016/j.atmosenv.2014.08.080.
- U. M. Joshi, K. Vijayaraghavan and R. Balasubramanian, Elemental Composition of Urban Street Dusts and Their Dissolution Characteristics in Various Aqueous Media, Chemosphere, 2009, 77(4), 526–533, DOI:10.1016/j.chemosphere.2009.07.043.
- X. Huang, R. Betha, L. Y. Tan and R. Balasubramanian, Risk Assessment of Bioaccessible Trace Elements in Smoke Haze Aerosols versus Urban Aerosols Using Simulated Lung Fluids, Atmos. Environ., 2016, 125, 505–511, DOI:10.1016/j.atmosenv.2015.06.034.
- K. R. M. Mackey, C. TeChien, A. F. Post, M. A. Saito and A. Paytan, Rapid and Gradual Modes of Aerosol Trace Metal Dissolution in Seawater, Front. Microbiol., 2015, 6, 1–11, DOI:10.3389/fmicb.2014.00794.
- National Center for Biotechnology Information,PubChem Compound Summary for CID 14806, ZINC oxide, 2022,https://pubchem.ncbi.nlm.nih.gov/compound/ZINC-oxide, accessed 21 January 2022 Search PubMed.
- A. Gioda, B. S. Amaral, I. L. G. Monteiro and T. D. Saint'Pierre, Chemical Composition, Sources, Solubility, and Transport of Aerosol Trace Elements in a Tropical Region, J. Environ. Monit., 2011, 13(8), 2134–2142, 10.1039/c1em10240k.
- Y. Zhu, W. Li, Q. Lin, Q. Yuan, L. Liu, J. Zhang, Y. Zhang, L. Shao, H. Niu, S. Yang and Z. Shi, Iron Solubility in Fine Particles Associated with Secondary Acidic Aerosols in East China, Environ. Pollut., 2020, 264, 114769, DOI:10.1016/j.envpol.2020.114769.
- V. Vinatier, N. Wirgot, M. Joly, M. Sancelme, M. Abrantes, L. Deguillaume and A. M. Delort, Siderophores in Cloud Waters and Potential Impact on Atmospheric Chemistry: Production by Microorganisms Isolated at the Puy de Dôme Station, Environ. Sci. Technol., 2016, 50(17), 9315–9323, DOI:10.1021/acs.est.6b02335.
- M. Passananti, V. Vinatier, A. M. Delort, G. Mailhot and M. Brigante, Siderophores in Cloud Waters and Potential Impact on Atmospheric Chemistry: Photoreactivity of Iron Complexes under Sun-Simulated Conditions, Environ. Sci. Technol., 2016, 50(17), 9324–9332, DOI:10.1021/acs.est.6b02338.
- A. G. González, A. Bianco, J. Boutorh, M. Cheize, G. Mailhot, A.-M. Delort, H. Planquette, N. Chaumerliac, L. Deguillaume and G. Sarthou, Influence of Strong Iron-Binding Ligands on Cloud Water Oxidant Capacity, Sci. Total Environ., 2022, 829, 154642, DOI:10.1016/j.scitotenv.2022.154642.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2em00023g |
| ‡ Present address: Université de Paris and Univ Paris Est Creteil, CNRS, LISA, F-75013 Paris, France. |
| This journal is © The Royal Society of Chemistry 2022 |