
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vitro study of polydopamine nanoparticles as protective antioxidant agents in fibroblasts derived from ARSACS patients†
Matteo
Battaglini
 *a,
Alessio
Carmignani
ab,
Chiara
Martinelli
a,
Jamila
Colica
a,
Attilio
Marino
a,
Stefano
Doccini
c,
Valentina
Mollo
d,
Francesca
Santoro
d,
Martina
Bartolucci
e,
Andrea
Petretto
e,
Filippo Maria
Santorelli
c and
Gianni
Ciofani
*a
*a,
Alessio
Carmignani
ab,
Chiara
Martinelli
a,
Jamila
Colica
a,
Attilio
Marino
a,
Stefano
Doccini
c,
Valentina
Mollo
d,
Francesca
Santoro
d,
Martina
Bartolucci
e,
Andrea
Petretto
e,
Filippo Maria
Santorelli
c and
Gianni
Ciofani
*a
aIstituto Italiano di Tecnologia, Smart Bio-Interfaces, Viale Rinaldo Piaggio 34, 56025 Pontedera, Italy. E-mail: matteo.battaglini@iit.it; gianni.ciofani@iit.it
bSant'Anna School of Advanced Studies, The Biorobotics Institute, Viale Rinaldo Piaggio 34, 56025 Pontedera, Italy
cIRCCS Fondazione Stella Maris, Molecular Medicine for Neurodegenerative and Neuromuscular Diseases Unit, Via dei Giacinti 3, 56128 Pisa, Italy
dIstituto Italiano di Tecnologia, Center for Advanced Biomaterials for Health Care, Largo Barsanti e Matteucci 53, 80125 Napoli, Italy
eIRCCS Istituto Giannina Gaslini, Core Facilities-Clinical Proteomics and Metabolomics, Via Gerolamo Gaslini 5, 16147 Genova, Italy
First published on 19th May 2022
Abstract
Reactive oxygen species (ROS) are active molecules involved in several biological functions. When the production of ROS is not counterbalanced by the action of protective antioxidant mechanisms present in living organisms, a condition of oxidative stress can arise with consequent damage to biological structures. The brain is one of the main ROS-generating organs in the human body, with the consequence that most of the neurological disorders are associated with an overproduction of ROS. Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) is a neurodegenerative disease associated with mutations in the sacsin gene (SACS). At cellular level, ARSACS is characterized by mitochondrial impairments, a reduction in bioenergetic processes, and by both an over-production of and an over-sensitivity to ROS. Several antioxidant molecules have been proposed as a potential treatment for ARSACS, such as idebenone and resveratrol. Polydopamine nanoparticles (PDNPs) gained significant attention in recent years owing to their peculiar physical/chemical properties, and especially because of their antioxidant activity. PDNPs have shown a great ROS scavenging capacity that, combined with their completely organic nature that grants them the ability to be degraded and excreted by living organisms, make them a promising candidate in the treatment of oxidative stress-related disorders. In this work, we assessed the effect of PDNPs on human fibroblasts derived from ARSACS patients in terms of antioxidant properties and protein expression. PDNP interaction with fibroblasts was analyzed in terms of biocompatibility, internalization and uptake pathway, reduction of ROS levels, prevention of ROS-induced apoptosis/necrosis, and protective action upon ROS-induced mitochondrial dysfunctions. Moreover, a complete proteomic analysis was performed. Altogether, our data showed that PDNPs can partially counteract ROS-induced damages in ARSACS patient-derived fibroblasts, making them a potential therapeutic candidate to treat – or at least to ameliorate – the condition of oxidative stress associated with ARSACS.
Introduction
Reactive oxygen species (ROS) are a class of highly reactive molecules involved in several pivotal physiological functions, such as cellular respiration.1 Under physiological conditions, ROS levels are kept under control by antioxidant enzymes like glutathione peroxidase, catalase, and superoxide dismutase, and by antioxidant molecules such as glutathione and vitamins.2 However, when the antioxidant defense mechanisms are overwhelmed by ROS, a condition of oxidative stress can occur.2 High and uncontrolled ROS levels can cause damages to biological molecules including proteins, membrane lipids, and oligonucleotides, leading to cellular misfunction and even to cell death.3,4 The brain, being responsible for the consumption of the 20% of the oxygen present in the human body, represents one of the main targets of ROS damage due both to its high metabolic activity and to the relatively low levels of antioxidant defense mechanisms present in neuronal cells.5 The intracellular organelles mainly responsible for ROS production are mitochondria, which represent also the main target of ROS-induced damage. It is therefore no surprising that both ROS-induced damage and mitochondrial dysfunctions are common hallmarks of neurological diseases.6Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) is a neurodegenerative disease characterized by pyramidal spasticity, cerebellar ataxia, loss of Purkinje cells, atrophy of peripheral nerves, and cognitive impairments.7–11 ARSACS disease has been related to mutations to the SACS gene encoding sacsin, a protein expressed on the mitochondrial surface and involved in mitochondrial dynamics.12,13 At cellular level, the alteration of sacsin expression has been linked to an increment of ROS levels, reduction of mitochondrial activity, lower ATP production, alteration of mitochondrial dynamics, and an increment of oxidative DNA damages.13 Currently, there are no therapies for ARSACS; however, the use of antioxidants able to counteract the high level of oxidative stress typical of the disease has been hypothesized as a feasible therapeutic strategy.13 Antioxidants have been in fact greatly investigated for several biomedical applications, including wound healing14,15 and as treatment for CNS disorders,16–19 and also in ARSACS they hold great promises: for example, Kageyama et al. showed as the treatment with antioxidants of Purkinje cells derived from DRP1 (a protein involved in mitochondrial division and linked to ARSACS12) KO mice prevented their ROS-induced cellular death.20
Polydopamine nanoparticles (PDNPs) have gained significant attention in recent years owing to their near-infrared (NIR) light photo-conversion abilities and to their strong antioxidant properties.21,22 PDNPs are rich in functional groups such as imine and catechols, which grant them the ability to scavenge a large variety of ROS. Moreover, being entirely composed of organic and naturally occurring molecules, they are highly biocompatible and biodegradable. PDNPs have known place in a large number of applications including the potential treatment of periodontal disease,22 the treatment of inflammation,23 the potential treatment of Parkinson's disease,24 and as an anti-cancer platform against several forms of neoplasia.25–28
In this work, we tested the effect of PDNPs on healthy and ARSACS patient-derived fibroblasts, in terms of protective antioxidant effect and of differential protein expression. We provided the first analysis of PDNPs on an ARSACS model, and, to the best of our knowledge, this is also the first work where PDNPs have been tested on human-derived primary cells through a combination of imaging studies and proteomic analysis. The hypothesis suggesting the exploitation of PDNPs as a potential treatment of ARSACS was supported by previous evidences demonstrating a positive effect of antioxidant treatments on ARSACS models,12 including works from our group,29,30 and on our previous experience in the application of PDNPs as neuroprotective agents.21 ARSACS patient-derived fibroblasts have been chosen as a model of disease based on previous reports where these cells demonstrated an altered phenotype in terms of mitochondrial organization and functionality.13 In particular, ARSACS patient-derived fibroblasts have been already used as a model of the disease because of similar alterations involving ROS production, ATP levels, mitochondrial dynamics, ROS-induced DNA damages, and overall energy metabolism.13 Biocompatibility, cellular uptake, and intracellular localization of PDNPs upon interaction with fibroblasts have been evaluated. Moreover, the ability of PDNPs to prevent damage induced by pro-oxidative stimulus has been analyzed in terms of overall ROS production, induction of cellular death, mitochondrial morphology disruption, loss of mitochondrial membrane potential, and overall protein expression.
Materials and methods
Synthesis of PDNPs
Synthesis of PDNPs was adapted from Bao et al.22 Briefly, 90 ml of Milli-Q water, 40 ml of ethanol, and 2 ml of ammonium hydroxide were mixed for 30 min under magnetic stirring (Sigma-Aldrich). Thereafter, a solution of 0.5 g of dopamine hydrochloride (Sigma-Aldrich) in 10 ml of Milli-Q water was added to the mixture and left under stirring overnight. The obtained mixture was then mixed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with pre-cooled ethanol at 4 °C and centrifuged at 8960g for 30 min. After centrifugation, the obtained supernatant was discarded, and the pellet was re-suspended in Milli-Q water and washed three times with centrifugation steps at 8960g for 30 min at 4 °C. Obtained PDNPs have been quantified following freeze-drying.
:1 with pre-cooled ethanol at 4 °C and centrifuged at 8960g for 30 min. After centrifugation, the obtained supernatant was discarded, and the pellet was re-suspended in Milli-Q water and washed three times with centrifugation steps at 8960g for 30 min at 4 °C. Obtained PDNPs have been quantified following freeze-drying.
Electron microscopy
PDNP size and morphology have been assessed by scanning electron microscopy (SEM). Briefly, 5 µl of PDNPs at 100 µg ml−1 were drop-cast on a piece of a silicon wafer, let dry, and then sputtered with gold using a Quorum Tech Q150RES Gold Sputter Coater with 30 mA for 60 s. SEM imaging was carried out with a Helios NanoLab 600i FIB/SEM (FEI) system. The average diameter of the PDNPs was measured through the software Gwyddion.Transmission electron microscopy (TEM) was carried out as a further characterization technique. Briefly, 10 µl of PDNPs at 100 µg ml−1 were drop-cast onto an ultrathin carbon-coated 150 mesh copper grid. Images were acquired in bright-field mode (JEOL JEM-1400Plus TEM), with a thermionic source (LaB6) operating at 120 kV.
Dynamic light scattering
The average hydrodynamic diameter, the average surface ζ-potential, and the polydispersity index (PDI) of PDNPs were analyzed through dynamic light scattering (DLS), by using a Malvern-Zetasizer Nano ZS90. Measurements were carried out at a concentration of 100 µg ml−1 of nanoparticles in water. For hydrodynamic diameter and PDI evaluation, disposable polystyrene cuvettes (Malvern Zetasizer Nano series) were used, while disposable folded capillary cells (Malvern Zetasizer Nano series) were exploited for surface ζ-potential measurements.Cell cultures
Human fibroblasts were collected with informed consent according to standard procedures for diagnostic (skin biopsies) and treated according to the standards of good clinical practice. Primary skin fibroblasts derived from four healthy controls and primary skin fibroblasts derived from four ARSACS patients were used. All cells were cultured in the same conditions using high-glucose Dulbecco's modified Eagle's medium (DMEM, Sigma-Aldrich) supplemented with 10% of heat-inactivated FBS (Gibco), 1 mM sodium pyruvate (Gibco), 100 IU ml−1 of penicillin (Gibco), and 100 µg ml−1 of streptomycin (Gibco).Viability assays
PDNPs effects were firstly assessed by using both Quant-iT™ PicoGreen dsDNA Assay Kit (Invitrogen) and Live/Dead assay (Thermo Fisher).For PicoGreen analysis, healthy primary fibroblasts were seeded in a 48-well plate (Corning) at 10000 cells per cm2 and let grow for 24 h. Thereafter, cells were incubated with PDNPs at different concentrations (0, 31.25, 62.5, 125, and 250 μg ml−1) for either 24 or 72 h. After the incubation with PDNPs, cells were washed with Dulbecco's phosphate-buffered saline (DPBS, Sigma), incubated in 100 μl of Milli-Q water, and subjected to three cycles of freeze/thaw (from −80 °C to 37 °C). Quant-iT™ PicoGreen dsDNA assay was carried out in Corning Costar 96-well black polystyrene plates following the manufacturer's instructions, being the fluorescence assessed with a Victor X3 Multilabel Plate Reader.
For live/dead assay, healthy primary fibroblasts were seeded in a 24-well plate (Corning) at 10000 cells per cm2 and let grow for 24 h. Thereafter, cells were incubated for 72 h with PDNPs at different concentrations (0, 12.5, 25, 50, 100 and 250 μg ml−1). After the incubation with PDNPs, cells were washed with DPBS and incubated for 20 min with phenol red-free medium supplemented with 5 μg ml−1 of Hoechst 33342 (Invitrogen), 4 μM of ethidium homodimer-1, and 2 μM of calcein-AM (all reagents from Thermo Fisher). After the staining, cells were washed again with DPBS, and the analysis was carried out by using a fluorescence microscope (Eclipse Ti, Nikon) equipped with a 10× objective, by counting the relative numbers of live cells (calcein-positive cells) and dead cells (ethidium homodimer-1-positive cells) in each condition.
Internalization analysis
Internalization of PDNPs in healthy primary fibroblasts was assessed through flow cytometry, confocal microscopy, and focus ion beam-scanning electron microscopy (FIB-SEM) milling and imaging.For fluorescence-based analysis, PDNPs have been stained using DiO dye (Vybrant™ Multicolor Cell-Labeling Kit, Thermo Fisher): 1 ml of milliQ water containing 10 mg ml−1 of PDNPs was mixed with 20 µM of DiO dye. The mixture was left under stirring for 2 h at 37 °C and obtained DiO-stained PDNPs were thereafter washed three times through centrifugation at 16602g.
For flow cytometry analysis, cells were seeded at 10000 cells per cm2 in 24-well plates (Corning). After 24 h, cultures were incubated with phenol red-free medium containing 100 μg ml−1 of DiO-stained PDNPs. Cells were detached through trypsinization at different time points (24, 48, and 72 h), and their fluorescence was assessed through flow cytometry (CytoFLEX platform, Beckman Coulter, FITC-A channel: λex 488 nm, λem 525 nm; ECD-A channel: λex 488 nm, λem 610 nm) and compared to the fluorescence level of control cells. Data were analyzed with the software CytExpert (Beckman Coulter).
For confocal analysis, cells have been seeded at 10000 cells per cm2 in 24-well μ-Plate Black (Ibidi), let grow for 24 h, and thereafter incubated with phenol red-free medium supplemented with 100 µg ml−1 of DiO-stained PDNPs. Cells were then fixed at different time points (24, 48, and 72 h) in paraformaldehyde (PFA): cultures have been washed in DPBS, incubated in a solution of PFA 4% in DPBS for 20 min at 4 °C, and then rinsed twice with DPBS. Fixed cells were then blocked with goat serum (GS, 10% in DPBS, Sigma) for 40 min and then incubated with GS supplemented with 2.5 μg ml−1 of TRITC-phalloidin (Sigma) and 5 μg ml−1 of Hoechst 33342 (Invitrogen) for 1 h. After the incubation, cultures were rinsed twice with DPBS (Sigma) and then imaged through confocal microscopy (C2s system, Nikon) with a 60× oil immersion objective. To assess intracellular localization of PDNPs, cells were seeded at 10000 cells per cm2 in Willco Petri dishes (GWST-3512), let grow for 24 h, and then incubated with phenol red-free medium containing 100 µg ml−1 of DiO stained PDNPs for 72 h. After the incubation with PDNPs, cells were washed twice in DPBS and then stained with either a solution containing 5 μM LysoTracker-Red (Thermo Fisher Scientific) and 5 μg ml−1 Hoechst 33342 (Invitrogen) for lysosomes imaging or with a solution containing 1 μM tetramethylrhodamine methyl ester (TMRM, Life Technologies) and 5 μg ml−1 Hoechst 33342 (Invitrogen) for mitochondrial imaging. After the incubation, all the cultures were rinsed twice with DPBS (Sigma) and then imaged at confocal microscope (C2s system, Nikon) with a 60× oil immersion objective.
Internalization of PDNPS was also assessed through FIB-SEM milling and imaging. Healthy fibroblasts were seeded at 10000 cells per cm2 on circular coverslip glass, let grown for 72 h, and then treated for further 72 h with PDNPs at 100 µg ml−1. Thereafter, cells were fixed in PFA 4% as already mentioned; samples were then fixed and stained following the ultra-thin plasticization protocol (UTP) as previously described.31,32 Briefly, after fixation in 2.5% glutaraldehyde (Electron Microscopy Sciences) prepared in 0.1 M sodium cacodylate (2 h at room temperature, Electron Microscopy Sciences) and washing in the same buffer (3 times, each for 10 min), specimens were incubated with 20 mM glycine in 0.1 M sodium cacodylate for 20 min on ice. Afterward, the staining was carried on by following a ROTO protocol.33 In detail, samples were post-fixed in 2% osmium tetroxide/1% potassium ferrocyanide (Electron Microscopy Sciences) in 0.1 M sodium cacodylate for 1 h, at 4 °C in the dark, (RO-step), then washed 3 times in chilled sodium cacodylate before incubation with 1% thiocarbohydrazide (TCH, Electron Microscopy Sciences) aqueous solution at room temperature. Specimens were washed 3 times in water; then, 1% osmium tetroxide water solution was added for 1 h, at 4 °C, in the dark. They were again rinsed in water, and thereafter the en-bloc staining with 1% uranyl acetate was performed. The incubation with uranyl acetate was performed overnight at 4 °C, and after several washing steps in chilled distilled water, 0.15% tannic acid aqueous solution was added for 3 min on ice before dehydration. The dehydration step was performed at 4 °C with an ethanol gradient (30%, 50%, 70%-2×, 95%-3×; absolute ethanol, Merck) and each step was carried out for 10 min. One more step in absolute ethanol was performed at room temperature before resin embedding; then, a mixture of low viscosity Spurr's resin (Electron Microscopy Science)/absolute ethanol was added with the following ratio: 1:3 (2 h); 1:2 (2 h); 1:1 (overnight); 2:1 (2 h); 3:1 (2 h). The mixture was replaced with absolute Spurr's resin for overnight incubation. Specimens were embedded in fresh resin two times (2 h for each step), and thereafter put in a vertical position for 2–3 h to remove resin in excess. Before polymerization, each sample was washed for 3 s with absolute ethanol and then moved in an oven at 70 °C, for 24 h. After polymerization, samples were mounted on a 12 mm aluminum stub (Agar Scientific) by using silver paste (RS Company). Before FIB-SEM milling and imaging (Helios CX5, Thermo Scientific), each sample was coated with 20 nm of gold by using a HR 208 Sputter coater (Cressington). The sample surface was scanned with an electron beam in a range of 2–5 kV by using a detector for the secondary electrons to identify the region of interest (ROI). A layer of 100 nm platinum was deposited via electron-beam by using a voltage of 2 kV and a current of 5 nA. Afterward, the stage was tilted at 52 °C and a second layer of platinum was deposited by using the ion beam assisted deposition at 30 kV/0.23 pA to reach a final thickness of 1.5 µm (ROI of 30 µm × 30 µm). FIB milling was then performed by fixing with a nominal depth of 5 µm for silicon, length of 30 µm and the width varied in a range of 10–15 µm. The ion milling was performed at 30 kV with a current in the range of 7.7 pA–0.79 nA, and the cross-section imaging was performed at 2 kV with a range of current of 0.17 nA–0.34 nA, in backscattered mode.
Protection against ROS
PDNP antioxidant properties have been assessed on four batches of healthy fibroblasts and four batches of fibroblasts derived from ARSACS patients. The same procedure was performed for all the 8 cultures. Cells were seeded at 10000 cells per cm2 in 24-well plates (Corning) and let grown for 24 h. Thereafter, a treatment with 100 µg ml−1 of PDNPs was performed; control cultures, without nanoparticles, have been considered as well. After 72 h, cells were stained in phenol red-free medium using 2.5 µM CellROX™ Green reagent (Invitrogen) for 30 min and detached. After resuspension in phenol red-free medium, cells from each experimental condition were divided into separate flow cytometry tubes and half of the obtained samples were treated with 2.5 mM tert-butyl hydroperoxide solution (TBH, Sigma Aldrich). The relative fluorescence intensity of all of the experimental conditions (Control, TBH, PDNPs, and PDNPs + TBH) was measured by flow cytometry at different time points after the addition of TBH (30 and 60 min; λex 488 nm, λem 525 ± 40 nm); data were analyzed with the CytExpert software (Beckman Coulter). A further experiment was carried out to compare the antioxidant effect of PDNPs with other compounds. In particular, healthy cells were seeded as previously described and pre-incubated for 72 h with 100 µg ml−1 of PDNPs, 100 µg ml−1 of control non-antioxidant lipid nanostructures (nanostructured lipid carriers, NLCs, prepared as previously described29), 5 µM of idebenone (Sigma),30 5 µM of tannic acid (Sigma),34 or 5 µM of ascorbic acid (Sigma).34 After the pre-incubation treatments, cells were stained with cellROX™ green reagent (Invitrogen) as previously described, resuspended in phenol-red free medium, and thereafter half samples treated with TBH 2.5 mM. As previously reported, the relative fluorescence intensity of all the experimental conditions (Control, PDNPs, NLCs, idebenone, tannic acid, ascorbic acid, TBH, PDNPs + TBH, NLCs + TBH, idebenone + TBH, tannic acid + TBH, L-ascorbic acid + TBH) was measured and analyzed through flow cytometry.
To assess the protective effect of PDNPs against ROS-induced cellular death, fibroblast cultures (4 healthy and 4 derived from ARSACS patients) have been seeded at 10000 cells per cm2 in 6-well plates and let grow for 24 h. Thereafter, cells were incubated with the cell culture medium doped with 100 µg ml−1 of PDNPs for 72 h (or without particles, as control). After the incubation with PDNPs, cells were incubated in the cell culture medium with or without 100 µM of TBH for 24 h; thereafter, cells were detached through trypsinization and stained with annexin V-FITC/propidium iodide (PI) using the FITC Annexin/Dead Cell Apoptosis Kit from Thermo Fisher; briefly, cells were re-suspended in annexin V-binding buffer containing 1 μg ml−1 of PI and annexin V-FITC 7 mM for 15 min (100 μl total volume). Afterward, 400 μl of annexin-binding buffer was added to each sample. The fluorescence levels of cells were measured through flow cytometry (for annexin V-FITC: λex 488 nm, λem 525 ± 40 nm; for PI: λex 488 nm, λem 610 ± 20 nm). Data were analyzed with the CytExpert software (Beckman Coulter).
Analysis of mitochondrial functionality
PDNP ability to prevent ROS-induced damage on mitochondrial morphology and membrane potential was assessed. Four batches of healthy fibroblasts, and fibroblasts derived from four ARSACS patients were seeded at 10000 cells per cm2 in Willco dishes (GWST-3512) and let grow for 24 h. Thereafter, cells were incubated with fresh medium either containing or not 100 µg ml−1 of PDNPs. After 72 h, cells were treated for 40 min in phenol red-free medium with or without 2.5 mM of TBH. Cells were then stained with 1 μM TMRM (Life Technologies) for 1 h and rinsed twice with DPBS (Sigma). Finally, cultures were incubated in phenol red-free high-glucose DMEM (supplemented with HEPES 25 mM, Thermo Fisher), and imaged through confocal microscopy (C2s system, Nikon). For each culture type, the elongation parameter of mitochondria was measured in function of the axis ratio of each individual mitochondrion.
For mitochondrial membrane potential analysis, after the staining with TMRM, a time-lapse acquisition was performed, taking images every 30 s for 900 s. At 300 s since the beginning of the acquisition, oligomycin 6 μM (Sigma) was added. For each culture type, the alteration of the mitochondrial membrane potential (ΔΨm) level induced by oligomycin was evaluated in each different experimental condition and expressed as % of the initial value. This procedure was carried out for each culture type (4 batches of healthy fibroblasts and 4 batches of fibroblasts derived from ARSACS patients). The relative fluorescence was analyzed frame by frame through NIS elements software by selecting several regions of interest (ROI), and by calculating the relative reduction in fluorescence levels as F/F0.
In vitro blood–brain barrier model
To preliminary assess the ability of PDNPs to cross the blood–brain barrier (BBB) and to reach the brain environment, we developed an in vitro model of the BBB based on the co-culture of brain endothelial cells and astrocytes. bEnd.3 (ATCC CRL-2299) mouse brain endothelial and C8-D1A (ATCC CRL-2541) mouse astrocytes were cultured in high-glucose DMEM (Sigma-Aldrich) supplemented with 10% of heat-inactivated FBS (Gibco), 1 mM sodium pyruvate (Gibco), 100 IU ml−1 of penicillin (Gibco), and 100 µg ml−1 of streptomycin (Gibco). The in vitro model of the BBB was set up by using a series of trans-wells with 3 μm diameter pores (Falcon®). Trans-wells were placed (inverted) into a Petri dish and C8D1A cells were seeded at 10000 cells per cm2 on the basolateral side, and let adhere for 24 h. Thereafter, trans-wells were inverted again, placed in a 24-well plate pre-filled with cell culture medium, and bEnd.3 cells were then seeded at 50000 cells per cm2 on the apical side; the formation of the barrier was allowed for 5 days. To assess the maturation of the BBB model, medium containing 250 μg ml−1 of FITC-dextran (70 kDa, Sigma) was administered to the apical side of both trans-wells seeded as previously described and “plain” trans-wells (without cells, used as controls). At each time point, 100 µl of medium was taken from the basolateral side of the insert and its fluorescence was measured through a Victor X3 Multilabel Plate Reader (PerkinElmer; λex 485 nm, λem 535 nm; samples were measured after 24 and 72 h from the administration of FITC-dextran). To assess the PDNP passage, cell culture medium doped with 50 μg ml−1 of PDNPs was added to the apical side of the trans-wells, either seeded or not with bEnd.3 and C8-D1A cells. After 24 and 72 h, 100 μl of cell culture medium were taken from the basolateral side of the insert and absorbance measured with a Victor X3 Multilabel Plate Reader (PerkinElmer), by exploiting a calibration curve obtained at 490 nm to quantify the amount of PDNPs that crossed the BBB model.35 To confirm the ability of PDNPs to be internalized by the cells forming the barrier, trans-well inserts supporting the cultures were treated with DiO-stained PDNPs for 72 h. Thereafter, cells were fixed, stained with TRITC-phalloidin (Sigma) and Hoechst 33342 (Invitrogen), and imaged through confocal microscopy (C2s system, Nikon) with a 60× oil immersion objective, as previously described for fibroblasts.
Proteomic analysis
For proteomic analyses, 4 batches of healthy fibroblasts and 4 batches of fibroblasts derived from ARSACS patients were seeded in Petri dishes at 10000 cells per cm2. Upon reaching approximately full confluency, cells were incubated for 72 h with cell culture medium doped (or not, as control) with 100 µg ml−1 of PDNPs. After incubation, cells were treated in part for 24 h with 100 µM of TBH and then detached through trypsinization, rinsed once in DPBS, and the obtained pellet was frozen at −80 °C. After thawing, samples were lysed, reduced, and alkylated in 50 μl LYSE buffer (Preomics) at 95 °C for 10 min and sonicated with an Ultrasonic Processor UP200St (Hielscher), 3 cycles of 30 s. Lysates samples were digested with 0.7 μg trypsin and 0.3 μg LysC overnight at 37 °C. To remove PDNPs, a 20 min centrifuge step at 20000 I has been performed, and the supernatants were processed with iST protocol.36
The resulting peptides were analyzed by a nano-UHPLC-MS/MS system using an Ultimate 3000 RSLC coupled to an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific Instrument). Elution was performed using a 200 cm uPAC C18 column (PharmaFluidics) mounted in the thermostated column compartment maintained at 50 °C. Firstly, a concentration gradient from 5% to 10% of buffer B was applied (80% acetonitrile and 20% H2O, 5% DMSO, 0.1% formic acid), coupled with a flow gradient from 750 nl min−1 to 350 nl min−1 for 15 min. Then, peptides were eluted with a 60 min non-linear gradient from 10% to 60% of buffer B at a constant flow rate of 350 nl min−1. Orbitrap detection was used for MS1 measurements at a resolving power of 120 K in a range between 375 and 1500 m/z and with a standard AGC target. Advanced Peak Detection was enabled for MS1 measurements. MS/MS spectra were acquired in the linear ion trap (rapid scan mode) after higher-energy C-trap dissociation (HCD) at a collision energy of 30% and with a Custom AGC target. For precursor selection, the least abundant signals in the three ranges 375–575 m/z, 574–775 m/z, and 774–1500 m/z were prioritized. Dynamic exclusion was set at 25 s.
MaxQuant software,37 version 1.6.17.0, was used to process the raw data. The false discovery rate (FDR) for the identification of proteins, peptides, and PSM (peptide-spectrum match) was set to 0.01. A minimum length of 6 amino acids was required for peptide identification. Andromeda engine, incorporated into MaxQuant software, was used to search MS/MS spectra against the Uniprot human database (release UP000005640_9606 October 2020). In the processing, the variable modifications were acetyl (Protein N-term), oxidation (M), and deamidation (NQ). carbamidomethyl (C) was selected as fixed modification. Algorithm MaxLFQ was chosen for the protein quantification with the activated option “match between runs” to reduce the number of the missing proteins. The intensity values were extracted and statistically evaluated using the ProteinGroup Table and Perseus software38 version 1.6.14.0. GO enrichment specific for skin fibroblast was obtained with the webserver HumanBase.39 The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE40 partner repository with the dataset identifier PXD032916.
Statistical analysis
The normality of data distributions was assessed with the Shapiro–Wilk normality test. Normally distributed data were analyzed via ANOVA followed by the LSD post-hoc test with Bonferroni correction. Non-normally distributed data were analyzed through Kruskal–Wallis test followed by the Wilcoxon signed-rank test. Normally distributed data were expressed as mean ± standard error, non-normally distributed data as median ± confidence interval, and represented in box plot. Significance was set for p < 0.05.Results and discussion
PDNP size and morphology
As shown in Fig. 1a and b, both SEM and TEM analyses confirmed the presence of homogeneous spherical nanostructures of an average size of 197 ± 25 nm. Fig. 1c and d reports DLS result, showing an average hydrodynamic diameter of 296.8 ± 2.7 nm with a polydispersity index of 0.025 ± 0.007 and an average surface Z-potential of −46.7 ± 0.3 mV. These results are in line with previous reports of PDNPs synthesis using an analogous fabrication procedure.21 In particular, PDNPs appear to be monodisperse and homogeneous.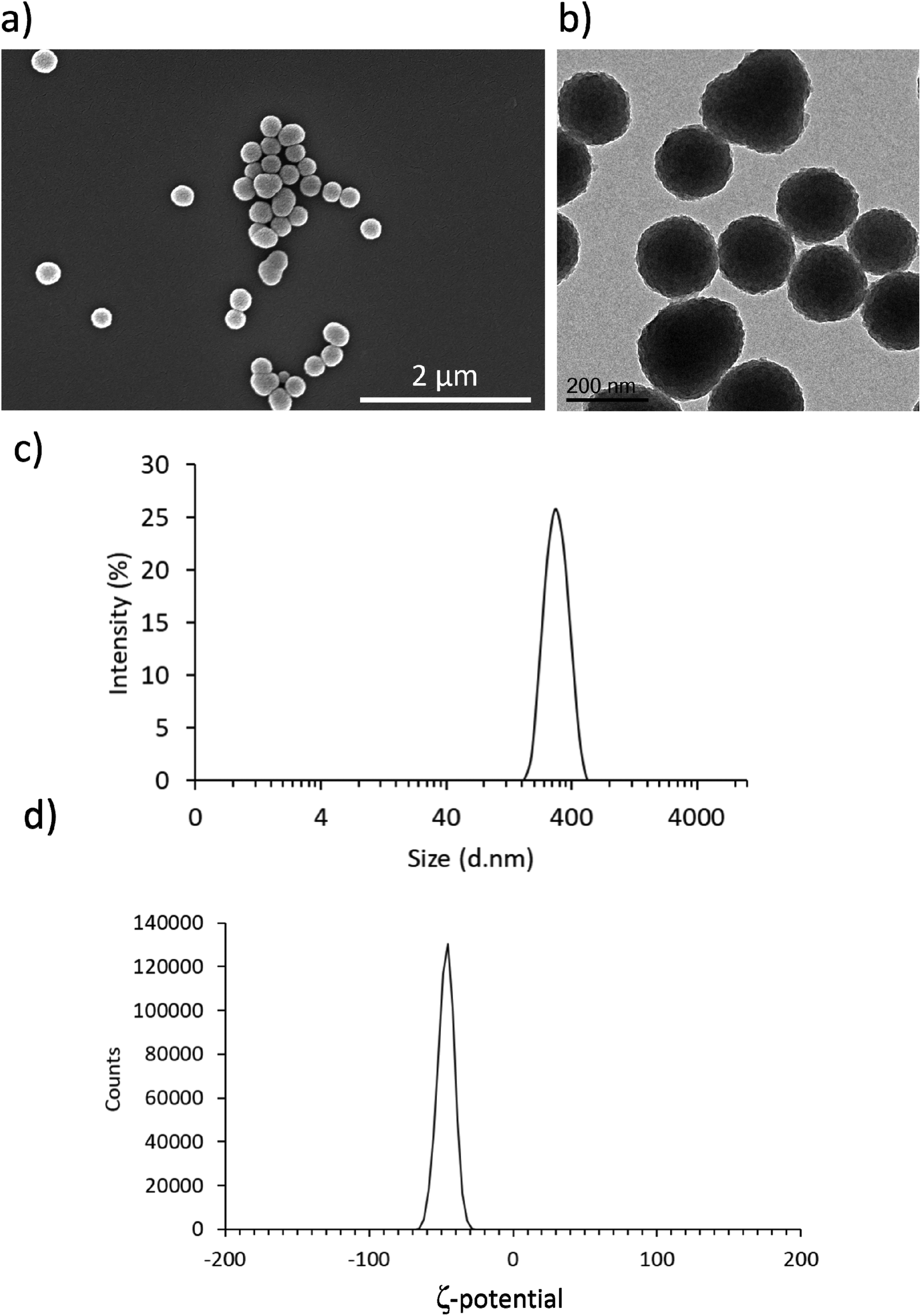 | ||
| Fig. 1 Characterization of PDNPs. Representative (a) SEM and (b) TEM images of PDNPs showing spherical morphology and uniform size. (c) Hydrodynamic diameter and (d) ζ-potential distributions of PDNP dispersions in water. | ||
Biocompatibility analysis
As shown in Fig. 2a, PicoGreen analysis demonstrated that PDNPs at various concentrations (0, 6.25, 12.5, 25, 50, 100, 250 µg ml−1) did not cause any statistically significant effect ondsDNA content at neither 24 h nor 72 h of treatment, indicating no harmful effects at these PDNP concentrations on cellular proliferation. The same treatment did not cause any statistically significant increment in the number of dead cells (propidium iodide-positive cells; quantitative analysis reported in Fig. 2b, representative fluorescence images in Fig. S1†).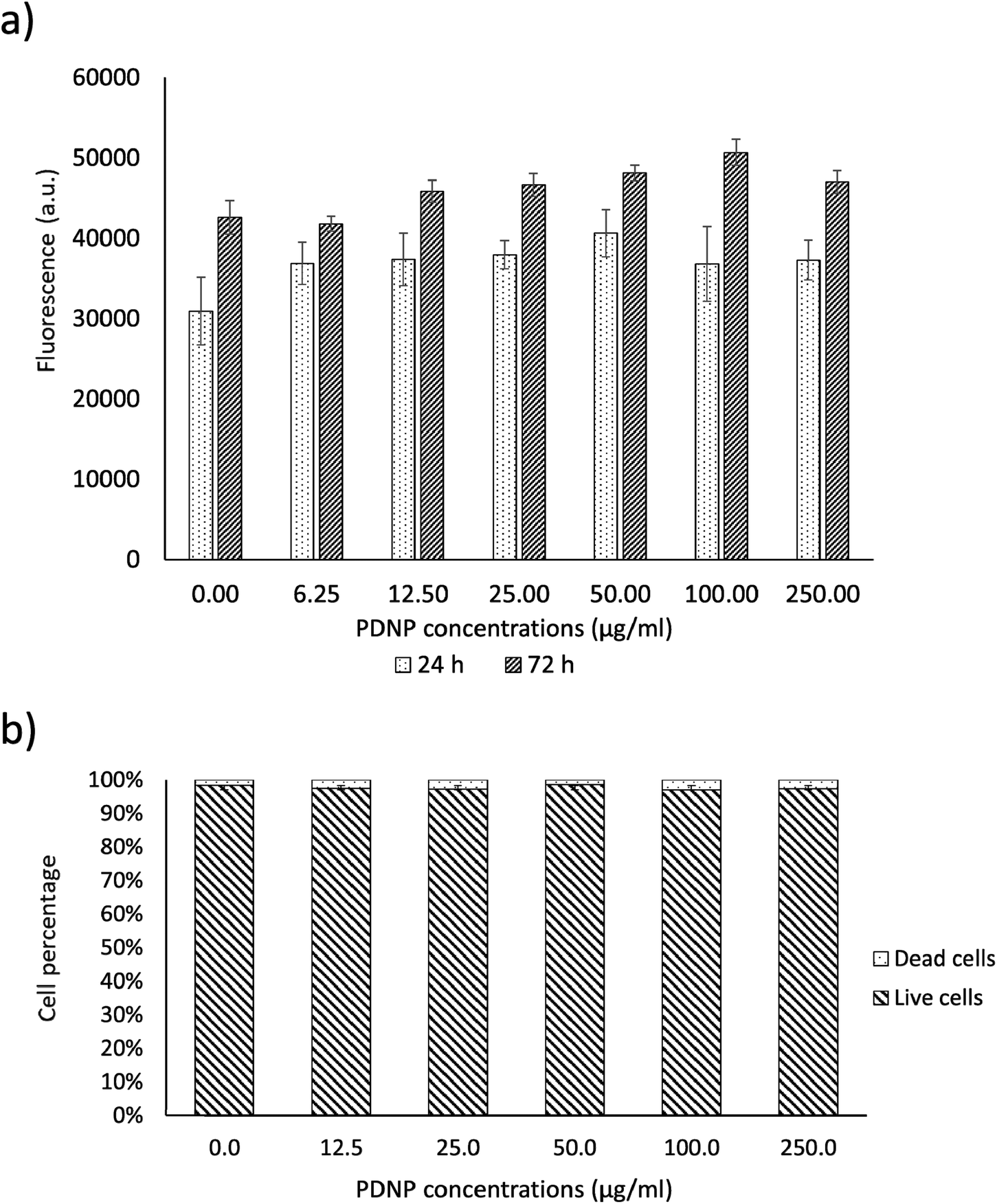 | ||
| Fig. 2 Analysis of PDNP biocompatibility on healthy fibroblasts. (a) PicoGreen™ analysis and (b) quantitative evaluation of live/dead assay. | ||
Polydopamine materials, and PDNPs in particular, are well known for their biocompatibility as demonstrated by several literature works.21,22 The concentration of 100 µg ml−1 was selected as a working value for the experiments described in the next sections, based on previous characterizations of the antioxidant properties of similar polydopamine nanostructures.21
Internalization analysis
Internalization analysis showed a relatively high level of internalization by fibroblasts as depicted in Fig. 3a. These results were confirmed through flow cytometry, the data of which are reported in Fig. 3b (blue trace referring to control cells, red trace to cells treated for 24 h, green trace to cells treated for 48 h, and pink trace to cells treated for 72 h). Fig. 3c shows the quantitative evaluation, reporting % of cells positive or negative for DiO-stained PDNPs (indicated as FITC+ and FITC-, respectively) in the control group and at the different incubation time points. In particular we observed 1.1 ± 1.0% FITC+ cells in control conditions, 67.3 ± 1.0% FITC+ cells after 24 h of incubation with DiO-PDNPs, 84.6 ± 1.6% FITC+ cells after 48 h of incubation with DiO-PDNPs, and 96.3 ± 1.0% FITC+ cells after 72 h of incubation with DiO-PDNPs. | ||
| Fig. 3 Internalization analysis of PDNPs in healthy fibroblasts. (a) Representative confocal fluorescence images showing internalization of DiO-stained PDNPs at different time points (24, 48 and 72 h; DiO-stained PDNPs in green, F-actin in red, nuclei in blue). In (b) and (c) flow cytometry analysis (representative fluorescence profile and quantitative analysis, respectively) of healthy fibroblasts incubated with DiO-stained PDNPs at different time points (24, 48 and 72 h), with respect to control cultures. | ||
Intracellular localization analysis is shown in Fig. 4; in particular, Fig. 4a shows representative confocal images relative to the co-localization of DiO-stained PDNPs with mitochondria (top panel) and lysosomes (bottom panel), while in Fig. 4b the quantitative analysis is reported, expressed as Pearson's correlation coefficient. We observed the absence of co-localization with mitochondria (Pearson's coefficient 0.01 ± 0.01), while a considerable quantity of PDNPs was found in lysosomes after 72 h of incubation (Pearson's coefficient 0.31 ± 0.07). The internalization of PDNPs inside fibroblast cells was also confirmed by SEM-FIB analysis, as shown in Fig. 4c. In particular, control cells analyzed trough SEM-FIB imaging (Fig. 4c, left panel) did not show the presence of PDNPs that were instead evident in the cytoplasm of cells pre-incubated with the particles (Fig. 4c, right panel), where PDNPs appear as electron dense spot distributed in the cytoplasm of cells. Data of intracellular localization are similar to what was previously reported by our group on other cell types, such as differentiated SH-SY5Y.21 It is worth mentioning that, despite the absence of PDNPs in mitochondria, we reported several beneficial effects of PDNPs over mitochondria and mitochondrial functions, as reported in the following paragraph. The observed protective effects are most probably due to an indirect antioxidant effect elicited by the particles, as discussed more in detail in the following sections. Furthermore, PDNP internalization in lysosomes could suggest an inactivation of their antioxidant activity, and the observed effects could appear contradictory. However, it is worth mentioning that not all the PDNPs are internalized inside of lysosomes, and a large portion appear to have a cytoplasmic distribution; moreover, lysosomes, and in particular their membranes, have been shown to be sensitive to ROS-induced damages, and several ROS-responsive genes such as the antioxidant lipocalin apolipoprotein D (ApoD) are involved in the protection of lysosomes.41
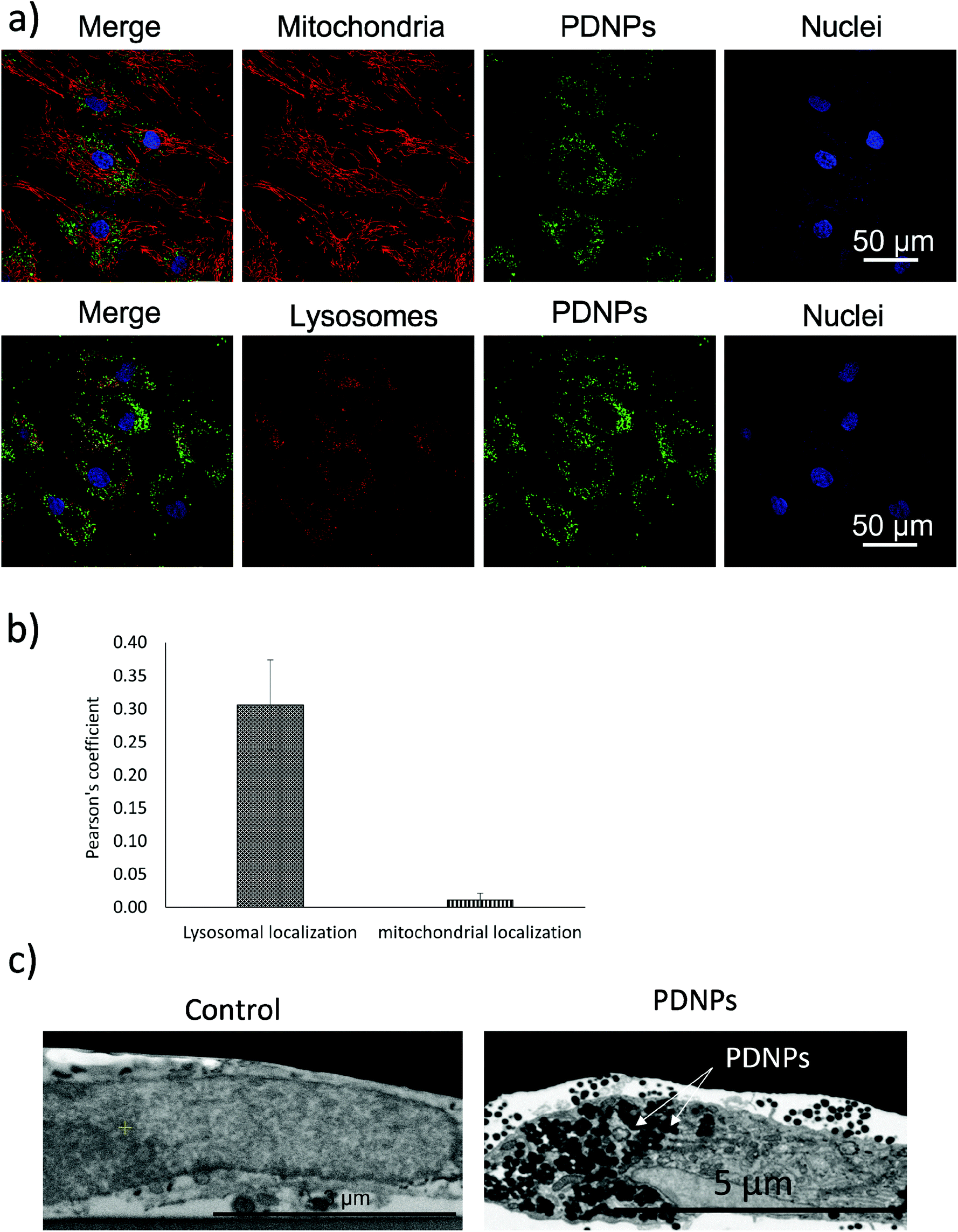 | ||
| Fig. 4 Analysis of the intracellular localization of PDNPs. (a) Representative confocal fluorescence images showing the intracellular localization of DiO-PDNPs with respect to mitochondria (upper panels: DiO-PDNPs in green, mitochondria in red, nuclei in blue) and lysosomes (lower panels: DiO-PDNPs in green, lysosomes in red, nuclei in blue). (b) Pearson's correlation coefficients between DiO-PDNPs and mitochondria (green column) or lysosomes (orange column) fluorescence signals. (c) Representative SEM-FIB images showing the internalization of PDNPs in healthy fibroblasts. On the left control cells, on the right cells incubated with PDNPs (white arrows indicate PDNPs correspondents to electron dense spot in the image). | ||
Antioxidant effects of PDNPs
The protective effects of PDNPs in terms of reduction of ROS level and prevention of apoptosis/necrosis induced by pro-oxidative stimulus were measured through flow cytometry on both healthy and ARSACS patient-derived cells. The results of both analyses are shown in Fig. 5 and 6, while representative histograms and dot plots of flow cytometry experiments are presented in Fig. S2–S5.† | ||
| Fig. 5 Analysis of the antioxidant properties of PDNPs in healthy fibroblasts and in ARSACS patient-derived fibroblasts (data were obtained from the analysis of cells derived from 4 healthy subjects and 4 ARSACS patients). The graphs show the % of ROS positive (ROS+) and ROS negative (ROS−) cells assessed by flow cytometry through CellRox™ staining at different time points (30 and 60 min) and following different treatments in (a) healthy fibroblasts and (b) ARSACS patient-derived fibroblasts (*p < 0.05, **p < 0.01, ***p < 0.001). | ||
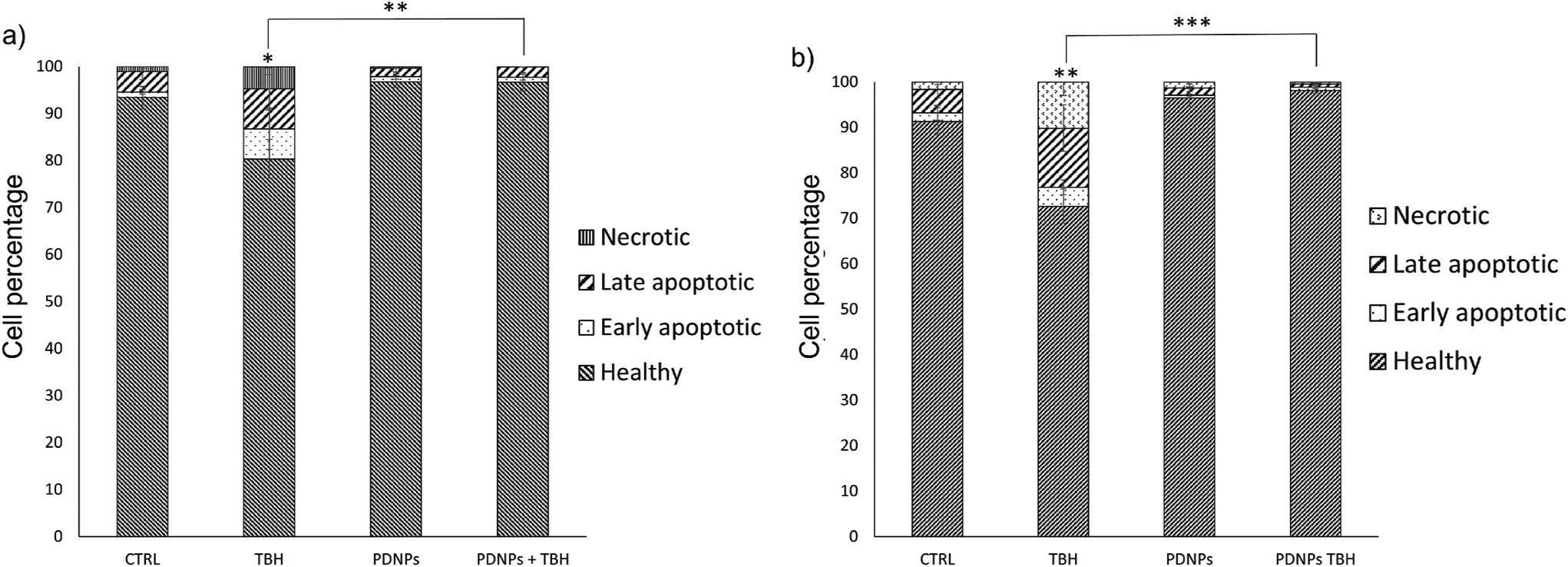 | ||
| Fig. 6 Evaluation of apoptosis/necrosis levels through flow cytometry upon annexin-V-FITC/PI staining. (a) Healthy fibroblasts and (b) ARSACS patient-derived fibroblasts having undergone different experimental treatments (* p < 0.05, ** p < 0.01, *** p < 0.001). | ||
The acute exposure to TBH caused a statistically significant increment (p < 0.05) of ROS levels after 60 min of treatment in healthy cells, and after 30 and 60 min in patients-derived cells (in particular the number of ROS+ cells in the case of healthy fibroblasts increased from 6.0 ± 0.3% in the control cultures to 22.2 ± 7.9% after 60 min of treatment with TBH, while in the case of ARSAC patient-derived fibroblasts the number of ROS+ cells increased from 5.6 ± 1.4% in the control to 14.7 ± 0.8% after 30 min of treatment with TBH and to 28.5 ± 3.8% after 60 min of treatment with TBH). This difference of sensitivity to pro-oxidant stimulus between healthy and patient-derived cells is probably due to the altered phenotype of ARSACs cells, as previously reported in the literature.13 The treatment with PDNPs (100 µg ml−1) caused a statistically significant reduction (p < 0.05) in the basal ROS level of both healthy and patients-derived cells at both time points: in particular, the number of ROS+ cells drop to approximately 0.1 ± 0.1% in each of these conditions. Moreover, the treatment with PDNPs also was able to prevent the increment in ROS caused by the treatment with TBH in both cell types with ROS+ cells in healthy and ARSACS patient-derived fibroblasts being equal to approximately 0.1 ± 0.1% at both 30 and 60 min. These analyses are in line with what we previously reported using PDNPs on differentiated SH-SY5Y, where the treatment with PDNPs was able to reduce intracellular basal ROS levels and to prevent the increment in ROS levels caused by the treatment with TBH.21 Results obtained with PDNPs are also comparable with our previous report regarding the use of idebenone-loaded nanostructured lipid carriers (IDE-NLCs) in ARSACS patient-derived fibroblasts.30 However, PDNPs appear to outperform IDE-NLCs in terms of antioxidant effects, most probably owing to the high content of polyphenol groups present on the surface of PDNPs, that act as ROS scavengers, thus reducing ROS basal levels and counterbalancing the increment of ROS caused by TBH treatment.
The treatment with low TBH concentration for longer periods (100 µM for 24 h) caused a statistically significant reduction of viable cells in both healthy and ARSACS patient-derived fibroblasts, with a statistically significant increment of either apoptotic or necrotic cells. This phenomenon was counterbalanced by the pre-treatment with 100 µg ml−1 of PDNPs. In particular, the healthy controls showed 93.4 ± 2.3% of viable cells, 1.2 ± 0.5% of early apoptotic cells, 4.4 ± 1.8% of late apoptotic cells, and 1.1 ± 0.4% of necrotic cells. The treatment with PDNPs provided a similar outcome to the controls with 96.8 ± 1.3% viable cells. The chronic treatment with TBH caused a statistically significant increment in the number of apoptotic and necrotic cells, and a consequent decrement of healthy cells (80.4 ± 4.1 of viable cells, 6.4 ± 4.4% of early apoptotic cells, 8.5 ± 4.5% of late apoptotic cells, and 4.8 ± 1.8% of necrotic cells). This increment in apoptotic and necrotic cells was prevented by the pre-treatment with 100 µg ml−1 of PDNPs (96.7 ± 1.9% of viable cells, 1.0 ± 0.6% of early apoptotic cells, 2.2 ± 1.3% of late apoptotic cells, and 0.1 ± 0.1% of necrotic cells). ARSACS patient-derived fibroblasts showed 91.3 ± 6.0% of viable cells, 1.9 ± 1.6% of early apoptotic cells, 5.2 ± 1.9% of late apoptotic cells, and 1.6 ± 0.6% of necrotic cells. Once again, the treatment with PDNPs showed a similar trend with 96.5 ± 2.0% of viable cells. The treatment with TBH caused a statistically significant increment in the number of apoptotic and necrotic cells, and a statistically significant reduction of viable cells (72.7 ± 3.8% of viable cells, 4.1 ± 1.7% of early apoptotic cells, 13.0 ± 5.1% of late apoptotic cells, and 10.2 ± 3.0% of necrotic cells), while the pre-treatment with PDNPs prevented this phenomenon, providing 98.2 ± 0.6% of viable cells, 0.7 ± 0.3% of early apoptotic cells, 0.7 ± 0.3% of late apoptotic cells, and 0.5 ± 0.2% of necrotic cells.
PDNPs antioxidant capacity was also compared to other antioxidant compounds (tannic acid, L-ascorbic acid, and idebenone) and with non-antioxidant lipid-based nanostructures (NLCs, as control) on healthy cells treated either treated or not with 2.5 mM TBH for 60 min (Fig. S6.† Only the treatment with PDNPs caused a statistically significant reduction of ROS+ cells in cultures in basal conditions (2.9 ± 0.4% in controls, 0.7 ± 0.2% in PDNPs-treated cultures, 3.1 ± 0.5% NLCs-treated cultures, 3.0 ± 0.3% in tannic acid-treated cultures, 3.7 ± 0.5% in L-ascorbic acid-treated cultures, and 2.8 ± 0.1% idebenone-treated cultures Moreover, again just PDNPs were able to significantly reduce the levels of oxidative stress in TBH-treated cells; in particular, we observed a percentage of ROS+ cells equal to 10.2 ± 1.1% in the case of cells treated with TBH, 1.0 ± 0.1% in the case of cells treated with both PDNPs and TBH, 8.0 ± 1.4% in the case of cells treated with both NLCs and TBH, 11.0 ± 2.2% in the case of cells treated with both tannic acid and TBH, 9.7 ± 1.1% in the case of cells treated with both L-ascorbic acid and TBH and 7.6 ± 0.4% in the case of cells treated with both idebenone and TBH. These control experiments confirm as PDNPs are able to outperform all the other tested antioxidant compounds, and, by comparing the effect of PDNPs with previously reported antioxidant nanostructures such as resveratrol- or idebenone-loaded nanoparticles,1,29 PDNPs are clearly able to scavenge intracellular ROS levels at significant higher extent.
ROS overproduction is known for causing damages to cellular components such as macromolecules (lipids, proteins, and nucleotides), membranes, and mitochondria. If ROS are left uncontrolled and the cellular damages accumulate without being repaired, the result of this chain of molecular events is commonly cell death either by apoptosis or necrosis. As shown from our data, PDNPs, being able to counteract the accumulation of ROS thanks to their antioxidant ability, can prevent ROS-induced cellular death. As it will be discussed in the following sections, this effect is probably due to the combination of an antioxidant direct effect of PDNPs, that reduce ROS levels and partially prevent cellular damage, and of an indirect effect upon protein expression. In particular, as discussed in the following proteomic analysis section, PDNPs appear to modulate a large variety of biological pathways, including antioxidant defense mechanisms and DNA repair systems that could contribute to the protective effect showed by PDNPs. It is clear that ROS overproduction is a common hallmark of ARSACS; however, the role of ROS in ARSACS is still unclear. In particular, in the case of fibroblasts derived from ARSACS patients, it has been reported how mutation at the level of SACS gene could cause a decreased mitochondrial respiration and a reduced activity of the respiratory chain with a consequent increment of ROS levels and DNA damage.13 In the case of ARSACS, ROS overproduction seems to be a consequence rather than the cause of the biological impairments caused by the altered synthesis of sacsin, in particular the already discussed mitochondrial defects putatively at the base of the Purkinje cell loss typical of the disease.11 However, it has also been suggested that ROS could be part of a self-sustaining cycle where sacsin under-expression causes mitochondrial impairments that in turn increase ROS levels leading to further mitochondrial impairments and to other forms of damages, eventually causing cellular death.13 Based on our analysis, PDNPs are probably not able to completely recover the altered phenotype of ARSACS patient-derived cells; however, they could be used as a protective agent able to prevent or at least halt the evolution of the disease.
Protective effect of PDNPs on mitochondria
The protective effect of PDNPs on mitochondria was measured both in terms of protection against morphological impairments caused by the treatment with a pro-oxidative stimulus (TBH 2.5 mM) and in terms of ability to prevent the loss of mitochondrial membrane potential in cells exposed to both pro-oxidative stimulus (TBH 2.5 mM) and an inhibitor of the respiratory chain (oligomycin 6 µM).In terms of morphology, we observed that the treatment with TBH caused in all conditions a statistically significant loss of elongation (lower elongation values) of the mitochondria, while the administration of PDNPs partially, yet significantly, recovered the elongation loss. This phenomenon was observed both in fibroblasts derived from healthy subjects (data shown in Fig. 7; the average median elongation values were equal to 2.3 ± 0.4 in control conditions, 2.3 ± 0.2 after treatment with PDNPs, 1.78 ± 0.1 after treatment with TBH, and 2.2 ± 0.2 in the case of cells pre-incubated with PNDPs and then treated with TBH), and in fibroblasts derived from ARSACS patients (data shown in Fig. 8; the average median elongation values were equal to 2.14 ± 0.3 in control conditions, 2.2 ± 0.3 after treatment with PDNPs, 1.68 ± 0.1 after treatment with TBH and 1.9 ± 0.1 in the case of cells pre-incubated with PNDPs and then treated with TBH).
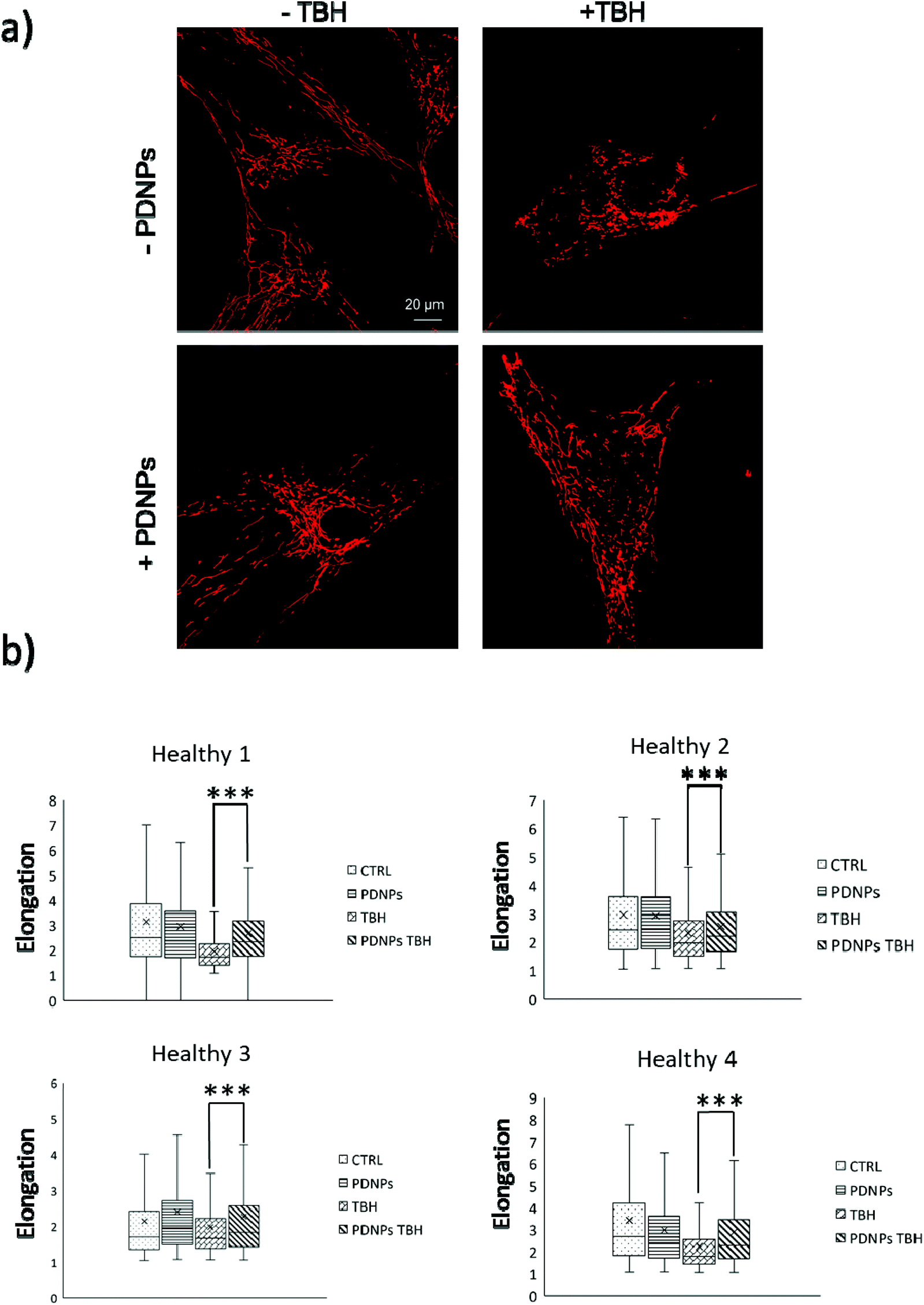 | ||
| Fig. 7 Analysis of mitochondrial morphology of healthy fibroblasts in different experimental conditions: (a) representative confocal images; (b) quantitative analysis of the elongation parameter (*p < 0.05, ***p < 0.001). | ||
 | ||
| Fig. 8 Analysis of mitochondrial morphology of ARSACS fibroblasts in different experimental conditions: (a) representative confocal images; (b) quantitative analysis of the elongation parameter (*p < 0.05, ***p < 0.001). | ||
Generally, healthy fibroblasts are characterized by a relatively large population of elongated tubular mitochondria and several reports in the literature describe how the overproduction of ROS can induce mitochondrial morphological aberration.21,42 In particular, the treatment with pro-oxidative stimuli has been shown to induce a change in the shape of mitochondria with significant enrichment of fragmented mitochondria characterized by a circular shape.21,42 PDNPs seem to be able to prevent the fragmentation caused by TBH on both healthy and ARSACS patient-derived cells. It is worth mentioning that PDNPs did not show any significant internalization in mitochondria as shown by the previously discussed confocal imaging analysis. Based on our data, the mitochondrial protective effect shown by PDNPs is probably due to an indirect antioxidant effect, where PDNPs reduce the amount of ROS generated by the pro-oxidant stimulus, thus protecting mitochondria from oxidative damage. Similar results have been already reported by our group on skin fibroblasts using cerium oxide nanoparticles.42
In the case of mitochondrial membrane potential analysis, we observed a further protective effect of PDNPs against the mitochondrial damage induced by TBH. In particular, as shown in Fig. 9, cells (healthy and ARSACS-derived) in the control condition and treated with PDNPs were able to maintain their mitochondrial potential levels (measured through variation of the fluorescence intensity of rhodamine and indicated in Fig. 9 by the blue and the orange trace, respectively), even after the addition of oligomycin. However, healthy cells showed a loss of fluorescence intensity upon TBH treatment after the addition of oligomycin, correlated with a loss in mitochondrial membrane potential (in particular fluorescence levels drop from 100% to 79 ± 5%, gray trace in Fig. 9a). This loss was almost completely absent in the case of cells treated with TBH but in the presence of both PDNPs (fluorescence intensity variation decreasing from 100% to 93 ± 2%, yellow trace in Fig. 9a). In the case of ARSACS patient-derived fibroblasts the treatment with TBH caused a continuous loss of fluorescence intensity correlated with a loss of mitochondrial membrane potential, which was exacerbated with the addition of oligomycin (a drop in fluorescence intensity from 100% to 71 ± 4% at the end, gray trace in Fig. 9b). Also in this case, the combined treatment with PDNPs partially prevented this loss: we observed a drop in fluorescence intensity from 100% to 93 ± 2% (yellow trace in Fig. 9b). Healthy cells are generally able to maintain mitochondrial potential levels even after exposure to oligomycin.42 The accumulation of ROS can cause severe damages on mitochondrial membranes with the consequence of making them unable to maintain their potential when exposed to oligomycin. We observed a peculiar behavior in the case of ARSACS patient-derived cells treated with TBH (Fig. 9b, gray trace) where a loss of membrane potential was observed from the very beginning of the analysis, with the addition of oligomycin causing an exacerbation of the phenomenon. We hypothesize that ARSACS cells, being more sensitive to oxidative stress-induced damage,13 accumulate severe mitochondrial damage after TBH treatment at an extent that makes them unable to maintain their mitochondrial membrane potential even before oligomycin addition. Once again, PDNPs showed protective effects, being able to prevent the membrane potential loss caused by the addition of oligomycin in TBH-treated cells. This effect is probably related to the previously discussed antioxidant capacities of PDNPs, that are able to decrease ROS generated upon TBH treatment, thus reducing mitochondrial damages and, as a consequence, providing an improved capacity to stabilize the membrane potential.
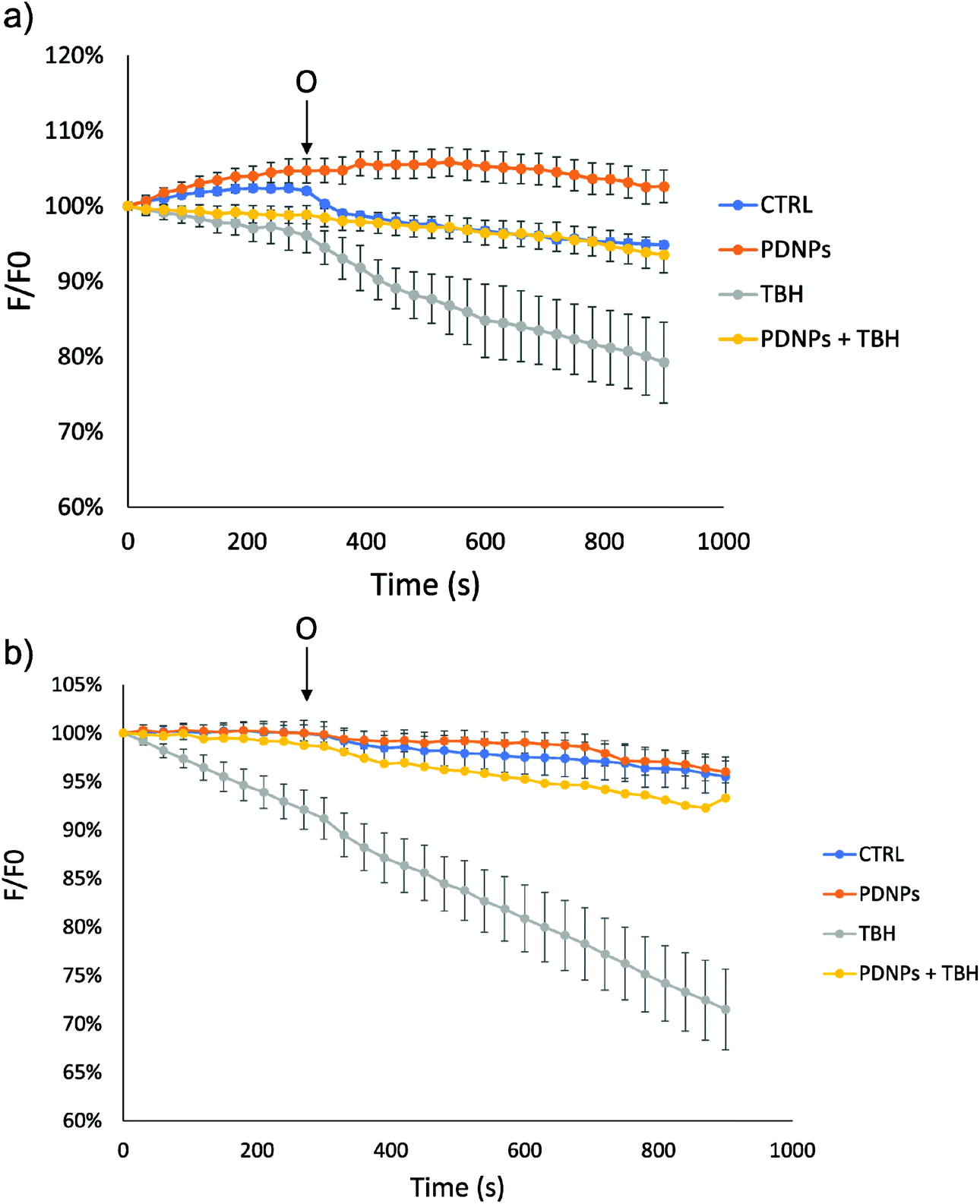 | ||
| Fig. 9 Analysis of mitochondrial membrane potential (ΔΨm, based on fluorescence analysis) in both (a) healthy and (b) ARSACS patient-derived fibroblasts in different experimental conditions. O: addition of 6 μM oligomycin. | ||
BBB in vitro model crossing
A trans-well model of BBB, based on endothelial cells and astrocytes co-cultures,30,43 has been set up to preliminary assess the BBB crossing ability of PDNPs. Upon verification of a satisfactory formation of a BBB model (about 80% of hindering of FITC-dextran 70 kDa crossing after 24 h, about 70% after 72 h), PDNPs have been administered on the apical side of the inserts to assess the ability of the particles to be internalized by brain endothelial cells and thereafter transported to the basolateral side of the in vitro BBB model. The results depicted in Fig. S7a† indicate how PDNPs are able to cross the barrier; in particular, we observed the passage of approximately 8 µg and 13 µg of PDNPs after 24 h and 72 h of administration, respectively. Moreover, confocal imaging reported in Fig. S7b,† showing the presence of a double layer of cells (bEnd.3 on the apical side and C8D1A on the basolateral side of the membrane, indicated as “top view” and “bottom view”) demonstrates the ability of PDNPs (in green) to be internalized by bEnd.3 cells (bottom panels of the figures).While collected findings are just preliminary data obtained through a simplified in vitro model of the BBB, lacking the complexity of an in vivo model, the observed BBB crossing ability of PDNPs is promising, and its future in vivo verification will represent a pivotal milestone for the clinical application of polydopamine-based nanomaterials in the treatment of CNS disorders.
Proteomic analysis
Proteomic analysis was carried out to unveil the cellular pathways influenced both by TBH and PDNPs treatment. The chronic treatment with TBH (100 µM for 24 h) caused a statistically significant change in the protein expression profile of both fibroblasts derived from healthy subjects and fibroblasts derived from ARSACS patients, while pre-treatment with PDNPs (100 µg ml−1 for 72 h) followed by the same pro-oxidative stimulus partially prevented this effect, maintaining stable the expression level of several proteins otherwise affected by TBH. Protein expression levels are shown in Fig. 10 for fibroblasts derived from healthy subjects and in Fig. 11 for fibroblasts derived from ARSACS patients, with over-expressed proteins represented in red and under-expressed proteins represented in blue. The complete list of proteins for each cluster of interest are presented in Table S1† (healthy cells) and Table S2† (ARSACS derived cells). Several GO biological terms grouped by biological function resulted enriched in the comparison between healthy fibroblasts in control conditions compared to healthy fibroblasts treated with PDNPs (Fig. 12a and Table 1), between cells derived from ARSACS patients with analogous cells treated with PDNPs (Fig. 12b and Table 2), between cells derived from healthy subjects treated with TBH with analogous cells pre-incubated with PDNPs and then treated with TBH (Fig. 12c and Table 3), and between cells derived from ARSACS patients treated with TBH with analogous cells pre-incubated with PDNPs and treated with TBH (Fig. 12d and Table 4). These four comparisons were chosen in order to understand the effects of PDNPs on protein expression in basal conditions (both on healthy and on ARSACS patient-derived cells), and in a situation of increased oxidative stress (again both on healthy and on ARSACS patient-derived cells).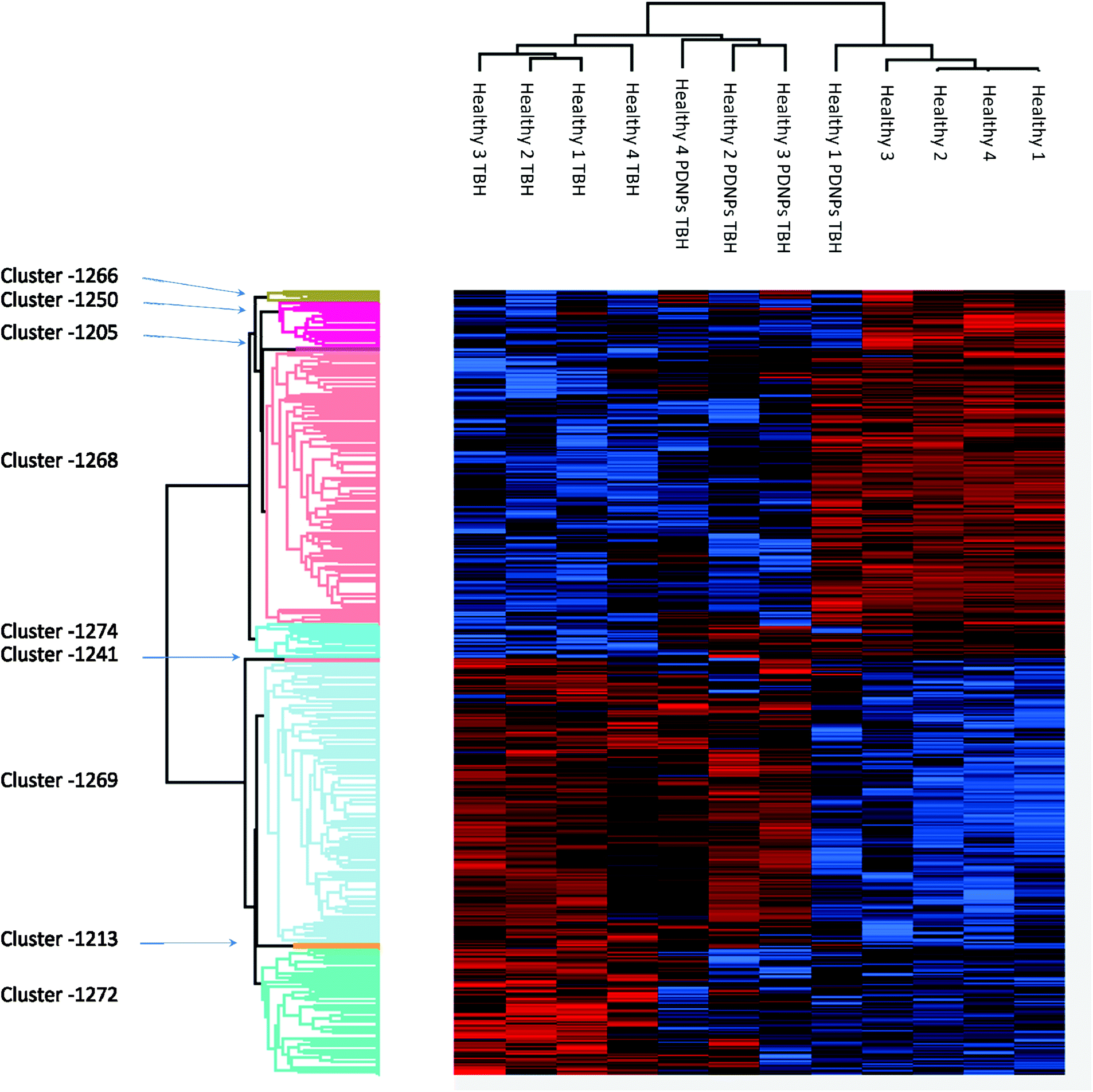 | ||
| Fig. 10 Heatmap showing the differential expression of various proteins in fibroblasts derived from four healthy subjects in different experimental conditions (control indicated as “healthy”, cells treated with 100 µM TBH for 24 h indicated as “healthy TBH”, and cells pre-incubated with 100 µg ml−1 of PDNPs for 72 h and consequentially treated with 100 µM of TBH for 24 h indicated as “healthy PDNPs TBH”). In red over-expressed protein, in blue under-expressed proteins; the single proteins present in each cluster are listed in Table S1.† | ||
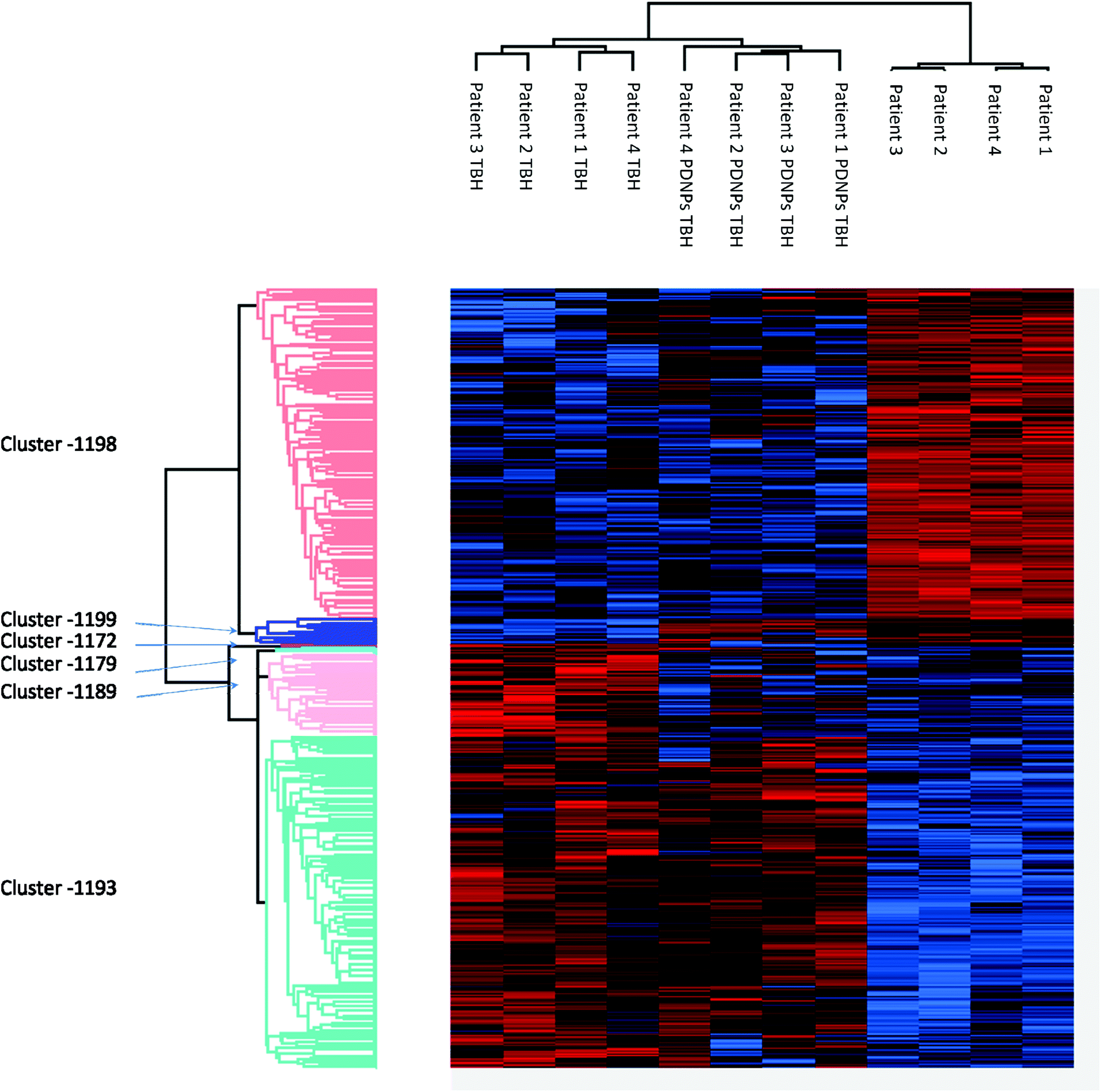 | ||
| Fig. 11 Heatmap showing the differential expression of various proteins in fibroblasts derived from four ARSACS patients in different experimental conditions (Control indicated as “patients”, cells treated with 100 µM TBH for 24 h indicated as “Patients TBH”, and cells pre-incubated with 100 µg ml−1 of PDNPs for 72 h and consequentially treated with 100 µM of TBH for 24 h indicated as “Patients PDNPs TBH”). In red over-expressed protein, in blue under-expressed proteins; the single proteins present in each cluster are listed in Table S1.† | ||
 | ||
| Fig. 12 GO enrichment for biological function in both healthy and ARSACS patient-derived fibroblasts. (a) Node map relative to healthy fibroblasts in control conditions vs. healthy fibroblasts incubated with 100 µg ml−1 of PDNPs; (b) node map relative to ARSACS patient-derived fibroblasts in control conditions vs. ARSACS patient-derived fibroblasts incubated with 100 µg ml−1 of PDNPs; (c) node map relative to healthy fibroblasts treated with TBH vs. healthy fibroblasts pre-incubated with 100 µg ml−1 of PDNPs and then treated with TBH; (d) node map relative to ARSACS patient-derived fibroblasts treated with TBH vs. ARSACS patient-derived fibroblasts pre-incubated with 100 µg ml−1 PDNPs and then treated with TBH. | ||
| Module | Top terms (max 10) | Q VAL | Genes | Terms |
|---|---|---|---|---|
| M1 | Cytoplasmic translation | <10−8 | 43 | 17 |
| Translation | <10−8 | |||
| Peptide biosynthetic process | <10−8 | |||
| Amide biosynthetic process | <10−8 | |||
| Peptide metabolic process | <10−8 | |||
| Ribosomal small subunit biogenesis | 0.00000003 | |||
| rRNA processing | 0.00000029 | |||
| rRNA metabolic process | 0.00000207 | |||
| Ribosome biogenesis | 0.00000250 | |||
| ncRNA processing | 0.00005382 | |||
| M2 | mRNA processing | 0.00138956 | ||
| mRNA splicing, via spliceosome | 0.00188861 | |||
| RNA splicing, via transesterifcation reactions with bulged adenosine as nucleophile | 0.00188861 | |||
| RNA splicing, via transesterifcation reactions | 0.00188861 | |||
| mRNA metabolic process | 0.00296335 | |||
| RNA splicing | 0.00674337 | |||
| Cholesterol biosynthetic process | 0.03670478 | |||
| Sterol biosynthetic process | 0.03670478 | |||
| Secondary alcohol biosynthetic process | 0.03670478 | |||
| Regulation of DNA replication | 0.03670478 | |||
| M3 | Activation of GTPase activity | 0.00954957 | 26 | 8 |
| Positive regulation of angiogenesis | 0.07869581 | |||
| Positive regulation of vasculature development | 0.07869581 | |||
| Positive regulation of GTPase activity | 0.09018516 | |||
| Positive regulation of cellular protein localization | 0.09261664 | |||
| Regulation of GTPase activity | 0.09310140 | |||
| Regulation of angiogenesis | 0.09310140 | |||
| Regulation of vasculature development | 0.09692422 | |||
| M4 | Symbiont process | 0.07869581 | 8 | 2 |
| Response to organonitrogen compound | 0.07876174 | |||
| M5 | Positive regulation of apoptotic process | 0.09310140 | 17 | 2 |
| Positive regulation of programmed cell death | 0.09310140 | |||
| Module | Top terms (max 10) | Q VAL | Genes | Terms |
|---|---|---|---|---|
| M1 | Cytoplasmic translation | 0.00000001 | 50 | 48 |
| Translation | 0.00000403 | |||
| Peptide biosynthetic process | 0.00000403 | |||
| Amide biosynthetic process | 0.00000909 | |||
| Peptide metabolic process | 0.00001803 | |||
| Ribosomal small subunit biogenesis | 0.00122840 | |||
| rRNA processing | 0.00148218 | |||
| rRNA metabolic process | 0.00284025 | |||
| Ribosome biogenesis | 0.00299515 | |||
| Ribonucleoprotein complex biogenesis | 0.00302824 | |||
| M2 | Cellular response to amino acid starvation | 0.00080248 | 2 | 14 |
| Negative regulation of autophagy | 0.00110716 | |||
| Cellular response to starvation | 0.00225552 | |||
| Response to starvation | 0.00228298 | |||
| Cellular response to extracellular stimulus | 0.00260054 | |||
| Cellular response to Nutrient levels | 0.00260054 | |||
| Response to nutrient levels | 0.00299515 | |||
| Response to extracellular stimulus | 0.00299515 | |||
| Regulation of autophagy | 0.00302824 | |||
| Negative regulation of cellular catabolic process | 0.00302824 | |||
| M3 | Negative regulation of binding | 0.00260054 | 58 | 344 |
| Receptor internalization | 0.00260054 | |||
| Negative regulation of interleukin-13 production | 0.00260054 | |||
| Negative regulation of interleukin-13 production | 0.00260054 | |||
| Positive regulation of cell migration | 0.00299515 | |||
| Smooth muscle cell proliferation | 0.00299515 | |||
| Regulation of smooth muscle cell proliferation | 0.00299515 | |||
| Positive regulation of cell motility | 0.00302824 | |||
| Regulation of cell fate specification | 0.00302824 | |||
| Positive regulation of cellular component movement | 0.00313336 | |||
| M4 | DNA replication | 0.00329726 | 73 | 123 |
| Positive regulation of nuclease activity | 0.00566404 | |||
| Regulation of DNA metabolic process | 0.00977086 | |||
| Protein folding | 0.01032849 | |||
| RNA export from nucleus | 0.01032849 | |||
| DNA recombination | 0.01138742 | |||
| mRNA processing | 0.01228949 | |||
| Regulation of DNA replication | 0.01373059 | |||
| Positive regulation of DNA replication | 0.01379163 | |||
| RNA transport | 0.01468916 | |||
| M5 | Regulation of focal adhesion assembly | 0.00409593 | 62 | 73 |
| Regulation of cell–substrate junction assembly | 0.00409593 | |||
| Regulation of adherens junction organization | 0.00451915 | |||
| Cell–substrate adherens junction assembly | 0.00526505 | |||
| Focal adhesion assembly | 0.00526505 | |||
| Cell–substrate junction assembly | 0.00582875 | |||
| Adherens junction assembly | 0.00582875 | |||
| regulation of cell junction assembly | 0.00720708 | |||
| Adherens junction organization | 0.00778497 | |||
| Regulation of cell–matrix adhesion | 0.01006351 | |||
| M6 | Regulation of DNA-templated transcription, elongation | 0.01009686 | 41 | 16 |
| DNA-templated transcription, elongation | 0.01095057 | |||
| Regulation of interleukin-6 production | 0.02465116 | |||
| Interleukin-6 production | 0.02515402 | |||
| Positive regulation of response to DNA damage stimulus | 0.02783404 | |||
| Histone lysine methylation | 0.02813321 | |||
| Peptidyl-lysine methylation | 0.03372173 | |||
| Histone methylation | 0.03509956 | |||
| Protein methylation | 0.04762668 | |||
| Protein alkylation | 0.04762668 |
| Module | Top terms (max 10) | Q VAL | Genes | Terms |
|---|---|---|---|---|
| M1 | DNA duplex unwinding | 0.00044961 | 5 | 11 |
| DNA geometric change | 0.00044961 | |||
| DNA conformation change | 0.00143292 | |||
| Positive regulation of chromosome organization | 0.00143292 | |||
| Double-strand break repair | 0.00144415 | |||
| Nucleic acid phosphodiester bond hydrolysis | 0.00144415 | |||
| Positive regulation of DNA metabolic process | 0.00144415 | |||
| Regulation of chromosome organization | 0.00264256 | |||
| Posttranscriptional regulation of gene expression | 0.00271200 | |||
| DNA repair | 0.00293709 | |||
| M2 | TOR signaling | 0.00126701 | 5 | 1 |
| Module | Top terms (max 10) | Q VAL | Genes | Terms |
|---|---|---|---|---|
| M1 | Translation | 0.00236699 | 8 | 5 |
| Amide biosynthetic process | 0.00236699 | |||
| Peptide biosynthetic process | 0.00236699 | |||
| Cytoplasmic translation | 0.00236699 | |||
| Peptide metabolic process | 0.00259519 | |||
| M2 | Neuron projection extension | 0.00822538 | 18 | 35 |
| Developmental cell growth | 0.00927676 | |||
| Developmental growth involved in morphogenesis | 0.00927676 | |||
| Developmental growth | 0.01500115 | |||
| Neuron projection morphogenesis | 0.01645356 | |||
| Plasma membrane bounded cell projection Morphogenesis | 0.01797199 | |||
| cell projection morphogenesis | 0.01797199 | |||
| Cell part morphogenesis | 0.01977233 | |||
| Regulation of angiogenesis | 0.02903029 | |||
| Cytokine-mediated signaling pathway | 0.02903029 | |||
| M3 | Divalent metal ion transport | 0.00822538 | 14 | 20 |
| Divalent inorganic cation transport | 0.00822538 | |||
| Protein glycosylation | 0.01319595 | |||
| Macromolecule glycosylation | 0.01319595 | |||
| Glycosylation | 0.01500115 | |||
| Glycoprotein biosynthetic process | 0.01797199 | |||
| Glycoprotein metabolic process | 0.02051793 | |||
| Calcium ion transmembrane transport | 0.02051793 | |||
| Regulation of actin flament-based process | 0.02599316 | |||
| Calcium ion transport | 0.02634459 | |||
| M4 | Carbohydrate derivative biosynthetic process | 0.01004057 | 4 | 1 |
| M5 | Cell morphogenesis involved in differentiation | 0.01253541 | 8 | 11 |
| Positive regulation of GTPase activity | 0.01319595 | |||
| Regulation of GTPase activity | 0.01500115 | |||
| Generation of neurons | 0.02051793 | |||
| Neurogenesis | 0.02051793 | |||
| Cell morphogenesis | 0.02051793 | |||
| Cellular component morphogenesis | 0.02075162 | |||
| Regulation of multi-organism process | 0.02075162 | |||
| Regulation of cellular protein localization | 0.02075162 | |||
| Intracellular protein transport | 0.02306617 | |||
| M6 | Response to oxidative stress | 0.01797199 | 15 | 2 |
| Response to drug | 0.02903029 |
The comparison between healthy cells in control conditions and healthy cells treated with PDNPs resulted in the enrichments of terms such as mRNA and protein synthesis and processing (Fig. 12a and Table 1, group M1 and M2), GTP related cellular activities (Fig. 12a and Table 1, group 3), and apoptosis related genes (Fig. 12a and Table 1, group M5). Concerning apoptosis related proteins, it is worth mentioning that the only two affected proteins were the high-mobility group box 1 protein (HMGB1) and Sin3A Associated Protein 18 (SAP18). HMGB1 is a protein involved in several biological functions including chromatin remodeling and inflammation.44 In particular, HMBG1 has been shown to be able to stimulate both cell survival and cell death depending on its redox status;45 SAP18 is instead a component of the histone deacetylase complex involved in the regulation of transcription.46 The evidence of the protective effect in terms of ROS production and apoptosis/necrosis levels of PDNPs, combined with the previously described biocompatibility tests and the fact that only two proteins with several different biological functions are present in the group M5 of Table 1 (GO terms related to apoptosis), suggest that PDNPs do not have pro-apoptotic effects upon healthy cells.
The comparison between cells derived from ARSACS patients treated and untreated with PDNPS resulted in the enrichment of terms such as protein synthesis and processing (Fig. 12b and Table 2, group M1), autophagy-related proteins (Fig. 12b and Table 2, group M2), cell motility, and interaction with external substrates (Fig. 12b and Table 2, groups M3 and M5), and processing/repair of nucleotides (Fig. 12b and Table 2, groups M4 and M6). The comparison between cells derived from healthy subjects and treated with TBH, and the same cells pre-incubated with PDNPs and then treated with TBH show a statistically significant GO enrichment in terms involved in DNA processing and DNA repair (Fig. 12c and Table 3). Lastly, the comparison between cells derived from ARSACS patients treated with TBH vs. the same cells pre-incubated with PDNPs and then treated with TBH showed a statistically significant enrichment in GO terms involved in protein synthesis and processing (Fig. 12d and Table 4, group M1), neuronal differentiation pathway (Fig. 12d and Table 4, group M2), cation transport and glycosylation (Fig. 12d and Table 4, groups M3 and M4), protein transport and GTP related cellular activities (Fig. 12d and Table 4, group M5), and response to external stimuli and oxidative stress (Fig. 12d and Table 4, group M6).
Several of the enriched GO groups have relevance in the context of ARCACS disease. Defective mitochondrial mRNA processing and maturation have been linked to cases of autosomal-recessive spastic ataxia,47,48 while DNA repair, replication, and processing together with protein synthesis, folding, and post-translational modifications are all been linked to various forms of autosomal recessive cerebellar ataxias.49 As previously discussed, all these molecular pathways are influenced by the treatment of ARSACS patient-derived cells with PDNPs (Fig. 12b and Table 2), hinting that PDNPs could potentially have a role in ameliorating some of the molecular impairments typical of the disease.
The GO enrichment depicted in Fig. 12 shows how the pre-incubation with PDNPs causes stimulation of molecular pathways involved in DNA repair and organization. ROS are known to be able to induce oxidative damage to DNAs even under physiological conditions:50 PDNPs could therefore partially prevent oxidative DNA damage by both directly acting as a ROS scavenger and by indirectly stimulating DNA repair molecular pathways. Cytoskeletal aberrations and autophagy impairments have also been identified in ARSACS cells and in cells with reduced production of the protein sacsin.51 As previously mentioned, the pre-incubation with PDNPs affected the protein expression levels of molecules involved in autophagy and catabolic pathways (Fig. 12b and Table 2, group M2). Lastly, PDNPs appear to stimulate relevant pathways in ARSACS cells exposed to the pro-oxidative stimulus, in particular proteins involved in ion metabolism, neuronal differentiation, glycosylation, protein synthesis and processes, GTP-related activities, and antioxidant defenses. The stimulation of antioxidant defenses hints once again at the possibility of PDNPs having a double effect as a direct antioxidant agent and as an indirect enhancer of physiological scavenger defense mechanisms. Altered metabolism of metal cations (such as iron) and calcium ions have been linked to several ataxia forms such as Friedreich's ataxia;52,53 glycosylation impairments are linked to a large variety of congenital neurological disorders including various forms of ataxia.54 Overall, the treatment with PDNPs in ARSACS patient-derived cells affects several biological pathways with considerable relevance in potential antioxidant-based therapies for many forms of neurological disorders.
Conclusions
PDNPs demonstrated interesting antioxidant properties, being able to reduce oxidative stress levels in both healthy and ARSACS patient-derived cells. Moreover, PDNPs demonstrated the ability to partially prevent the damage induced by ROS in terms of induction of apoptosis/necrosis, disruption of mitochondrial morphology, and loss of mitochondrial potential. Finally, PDNPs demonstrated the ability to partially counteract the alteration of protein expression induced by a pro-oxidative stimulus, while activating various metabolic pathways involved in biosynthetic processes, DNA repair, and antioxidant defense mechanisms. Altogether, these data lay a promising foundation for future analysis involving the use of PDNPs as a potential treatment for ARSACS. Further analysis to characterize the molecular pathways influenced by PDNPs, also involving the use of in vivo models, will be however a mandatory step for the exploitation of PDNPs in clinical applications.Author contributions
Dr Matteo Battaglini prepared and characterized the nanostructures, performed in vitro test and microscopy analysis, and wrote the manuscript; Mr Alessio Carmignani performed and analyzed biocompatibility assays; Dr Chiara Martinelli performed oxidative stress measurement through flow-cytometry; Ms. Jamila Colica performed experiments related to the in vitro BBB model; Dr Attilio Marino performed confocal imaging; Dr Martina Bartolucci and Dr Andrea Petretto performed proteomic analysis; Dr Valentina Mollo and Dr Francesca Santoro performed FIB/SEM imaging; Dr Stefano Doccini and Prof. Filippo Maria Santorelli contributed to the discussion of the results concerning ARSACS disease and provided clinical insights; Prof. Gianni Ciofani designed and supervised the study and edited the manuscript.The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.
Ethics statement
The studies involving cells from human donors were reviewed and approved by Tuscany Pediatric Ethic Committee. The patients/participants provided their written informed consent to participate in this study.Conflicts of interest
Authors declare no conflicts of interest.Acknowledgements
This research was partially supported by the Italian Ministry of Health (grant no. RF-2016-02361610).References
- C. Martinelli, C. Pucci, M. Battaglini, A. Marino and G. Ciofani, Adv. Healthcare Mater., 2020, 9, 1901589 CrossRef CAS PubMed.
- V. Lobo, A. Patil, A. Phatak and N. Chandra, Pharmacogn. Rev., 2010, 4, 118–126 CrossRef CAS PubMed.
- G. Pizzino, N. Irrera, M. Cucinotta, G. Pallio, F. Mannino, V. Arcoraci, F. Squadrito, D. Altavilla and A. Bitto, Oxid. Med. Cell. Longevity, 2017, 2017, 8416763 Search PubMed.
- S. Kwiecien, K. Jasnos, M. Magierowski, Z. Sliwowski, R. Pajdo, B. Brzozowski, T. Mach, D. Wojcik and T. Brzozowski, J. Physiol. Pharmacol., 2014, 65, 613–622 CAS.
- M. Bélanger, I. Allaman and P. J. Magistretti, Cell Metab., 2011, 14, 724–738 CrossRef PubMed.
- P. R. Angelova and A. Y. Abramov, FEBS Lett., 2018, 592, 692–702 CrossRef CAS PubMed.
- I. Ricca, F. Morani, G. M. Bacci, C. Nesti, R. Caputo, A. Tessa and F. M. Santorelli, Neurogenetics, 2019, 20, 45–49 CrossRef CAS PubMed.
- M. Synofzik, A. S. Soehn, J. Gburek-Augustat, J. Schicks, K. N. Karle, R. Schüle, T. B. Haack, M. Schöning, S. Biskup, S. Rudnik-Schöneborn, J. Senderek, K.-T. Hoffmann, P. MacLeod, J. Schwarz, B. Bender, S. Krüger, F. Kreuz, P. Bauer and L. Schöls, Orphanet J. Rare Dis., 2013, 8, 41 CrossRef PubMed.
- S. Vermeer, R. P. P. Meijer, B. J. Pijl, J. Timmermans, J. R. M. Cruysberg, M. M. Bos, H. J. Schelhaas, B. P. C. van de Warrenburg, N. V. A. M. Knoers, H. Scheffer and B. Kremer, Neurogenetics, 2008, 9, 207–214 CrossRef PubMed.
- J. Baets, T. Deconinck, K. Smets, D. Goossens, P. Van den Bergh, K. Dahan, E. Schmedding, P. Santens, V. M. Rasic, P. Van Damme, W. Robberecht, L. De Meirleir, B. Michielsens, J. Del-Favero, A. Jordanova and P. De Jonghe, Neurology, 2010, 75, 1181–1188 CrossRef CAS PubMed.
- M. Girard, R. Larivier̀e, D. A. Parfitt, E. C. Deane, R. Gaudet, N. Nossova, F. Blondeau, G. Prenosil, E. G. M. Vermeulen, M. R. Duchen, A. Richter, E. A. Shoubridge, K. Gehring, R. A. McKinney, B. Brais, J. P. Chapple and P. S. McPherson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(5), 1661–1666 CrossRef CAS PubMed.
- T. Y. Bradshaw, L. E. L. Romano, E. J. Duncan, S. Nethisinghe, R. Abeti, G. J. Michael, P. Giunti, S. Vermeer and J. P. Chapple, Hum. Mol. Genet., 2016, 25, 3232–3244 CrossRef CAS PubMed.
- C. Criscuolo, C. Procaccini, M. C. Meschini, A. Cianflone, R. Carbone, S. Doccini, D. Devos, C. Nesti, I. Vuillaume, M. Pellegrino, A. Filla, G. De Michele, G. Matarese and F. M. Santorelli, J. Neurol., 2015, 262, 2755–2763 CrossRef CAS PubMed.
- C. Xian, Z. Gu, G. Liu and J. Wu, Chin. Chem. Lett., 2020, 31, 1612–1615 CrossRef CAS.
- S. Zhang, J. Hou, Q. Yuan, P. Xin, H. Cheng, Z. Gu and J. Wu, Chem. Eng. J., 2020, 392, 123775 CrossRef CAS.
- V. S. Bollimpelli, P. Kumar, S. Kumari and A. K. Kondapi, Neurochem. Int., 2016, 95, 37–45 CrossRef CAS PubMed.
- A. Marino, M. Battaglini, A. Desii, C. Lavarello, G. Genchi, A. Petretto and G. Ciofani, Biomater. Sci., 2021, 9, 8171–8188 RSC.
- O. Binyamin, L. Larush, K. Frid, G. Keller, Y. Friedman-Levi, H. Ovadia, O. Abramsky, S. Magdassi and R. Gabizon, Int. J. Nanomed., 2015, 10, 7165–7174 CAS.
- S. Dolati, M. Ahmadi, L. Aghebti-Maleki, A. Nikmaram, F. Marofi, R. Rikhtegar, H. Ayromlou and M. Yousefi, Pharmacol. Rep., 2018, 70, 1158–1167 CrossRef CAS PubMed.
- Y. Kageyama, Z. Zhang, R. Roda, M. Fukaya, J. Wakabayashi, N. Wakabayashi, T. W. Kensler, P. H. Reddy, M. Iijima and H. Sesaki, J. Cell Biol., 2012, 197, 535–551 CrossRef CAS PubMed.
- M. Battaglini, A. Marino, A. Carmignani, C. Tapeinos, V. Cauda, A. Ancona, N. Garino, V. Vighetto, G. La Rosa, E. Sinibaldi and G. Ciofani, ACS Appl. Mater. Interfaces, 2020, 12(32), 35782–35798 CrossRef CAS PubMed.
- X. Bao, J. Zhao, J. Sun, M. Hu and X. Yang, ACS Nano, 2018, 12, 8882–8892 CrossRef CAS PubMed.
- H. Zhao, Z. Zeng, L. Liu, J. Chen, H. Zhou, L. Huang, J. Huang, H. Xu, Y. Xu, Z. Chen, Y. Wu, W. Guo, J. H. Wang, J. Wang and Z. Liu, Nanoscale, 2018, 10, 6981–6991 RSC.
- L. B. Vong, Y. Sato, P. Chonpathompikunlert, S. Tanasawet, P. Hutamekalin and Y. Nagasaki, Acta Biomater., 2020, 109, 220–228 CrossRef CAS PubMed.
- D. Maziukiewicz, B. F. Grześkowiak, E. Coy, S. Jurga and R. Mrówczyński, Biomimetics, 2019, 4(1), 3 CrossRef CAS PubMed.
- B. F. Grześkowiak, D. Maziukiewicz, A. Kozłowska, A. Kertmen, E. Coy and R. Mrówczyński, Int. J. Mol. Sci., 2021, 22(2), 738 CrossRef PubMed.
- R. S. Ambekar and B. Kandasubramanian, Biomater. Sci., 2019, 7, 1776–1793 RSC.
- H. Safari Yazd, Y. Yang, L. Li, L. Yang, X. Li, X. Pan, Z. Chen, J. Jiang, C. Cui and W. Tan, iScience, 2020, 23, 101750 CrossRef CAS PubMed.
- Ö. Şen, M. Emanet, A. Marino, M. Belenli Gümüş, M. Bartolucci, S. Doccini, F. Catalano, G. G. Genchi, F. M. Santorelli, A. Petretto and G. Ciofani, Mater. Des., 2021, 209, 110012 CrossRef.
- C. Martinelli, M. Battaglini, C. Pucci, S. Gioi, C. Caracci, G. Macaluso, S. Doccini, F. M. Santorelli and G. Ciofani, ACS Omega, 2020, 5, 12451–12466 CrossRef CAS PubMed.
- D. Iandolo, F. A. Pennacchio, V. Mollo, D. Rossi, D. Dannhauser, B. Cui, R. M. Owens and F. Santoro, Adv. Biosyst., 2019, 3, 1800103 CrossRef PubMed.
- F. A. Pennacchio, F. Caliendo, G. Iaccarino, A. Langella, V. Siciliano and F. Santoro, Nano Lett., 2019, 19, 5118–5123 CrossRef CAS PubMed.
- F. Santoro, W. Zhao, L.-M. Joubert, L. Duan, J. Schnitker, Y. van de Burgt, H.-Y. Lou, B. Liu, A. Salleo, L. Cui, Y. Cui and B. Cui, ACS Nano, 2017, 11, 8320–8328 CrossRef CAS PubMed.
- C. Pucci, C. Martinelli, D. De Pasquale, M. Battaglini, N. di Leo, A. Degl’Innocenti, M. Belenli Gümüş, F. Drago and G. Ciofani, ACS Appl. Mater. Interfaces, 2022, 14(14), 15927–15941 CrossRef CAS PubMed.
- Y. Zou, X. Chen, P. Yang, G. Liang, Y. Yang, Z. Gu and Y. Li, Sci. Adv., 2020, 6, 36 Search PubMed.
- N. A. Kulak, G. Pichler, I. Paron, N. Nagaraj and M. Mann, Nat. Methods, 2014, 11, 319–324 CrossRef CAS PubMed.
- J. Cox and M. Mann, Nat. Biotechnol., 2008, 26, 1367–1372 CrossRef CAS PubMed.
- S. Tyanova, T. Temu, P. Sinitcyn, A. Carlson, M. Y. Hein, T. Geiger, M. Mann and J. Cox, Nat. Methods, 2016, 13, 731–740 CrossRef CAS PubMed.
- C. S. Greene, A. Krishnan, A. K. Wong, E. Ricciotti, R. A. Zelaya, D. S. Himmelstein, R. Zhang, B. M. Hartmann, E. Zaslavsky, S. C. Sealfon, D. I. Chasman, G. A. FitzGerald, K. Dolinski, T. Grosser and O. G. Troyanskaya, Nat. Genet., 2015, 47, 569–576 CrossRef CAS PubMed.
- Y. Perez-Riverol, J. Bai, C. Bandla, D. García-Seisdedos, S. Hewapathirana, S. Kamatchinathan, D. J. Kundu, A. Prakash, A. Frericks-Zipper, M. Eisenacher, M. Walzer, S. Wang, A. Brazma and J. A. Vizcaíno, Nucleic Acids Res., 2022, 50, D543–D552 CrossRef CAS PubMed.
- R. Pascua-Maestro, S. Diez-Hermano, C. Lillo, M. D. Ganfornina and D. Sanchez, PLOS Genet., 2017, 13, e1006603 CrossRef PubMed.
- I. Pezzini, A. Marino, S. Del Turco, C. Nesti, S. Doccini, V. Cappello, M. Gemmi, P. Parlanti, F. M. Santorelli, V. Mattoli and G. Ciofani, Nanomedicine, 2016, 12, 403–416 CrossRef PubMed.
- M. Battaglini, C. Tapeinos, I. Cavaliere, A. Marino, A. Ancona, N. Garino, V. Cauda, F. Palazon, D. Debellis and G. Ciofani, ACS Biomater. Sci. Eng., 2019, 5, 670–682 CrossRef CAS PubMed.
- S. Yuan, Z. Liu, Z. Xu, J. Liu and J. Zhang, J. Hematol. Oncol., 2020, 13, 91 CrossRef PubMed.
- D. Tang, M. T. Loze, H. J. Zeh and R. Kang, Autophagy, 2010, 6, 1181–1183 CrossRef CAS PubMed.
- S. A. McCallum, J. F. Bazan, M. Merchant, J. Yin, B. Pan, F. J. de Sauvage and W. J. Fairbrother, Biochemistry, 2006, 45, 11974–11982 CrossRef CAS PubMed.
- A. H. Crosby, H. Patel, B. A. Chioza, C. Proukakis, K. Gurtz, M. A. Patton, R. Sharifi, G. Harlalka, M. A. Simpson, K. Dick, J. A. Reed, A. Al-Memar, Z. M. A. Chrzanowska-Lightowlers, H. E. Cross and R. N. Lightowlers, Am. J. Hum. Genet., 2010, 87, 655–660 CrossRef CAS PubMed.
- F. Longo, D. De Ritis, A. Miluzio, D. Fraticelli, J. Baets, M. Scarlato, F. M. Santorelli, S. Biffo and F. Maltecca, Neurology, 2021, 97, e2315–e2327 CrossRef CAS PubMed.
- B. L. Fogel and S. Perlman, Lancet Neurol., 2007, 6, 245–257 CrossRef CAS PubMed.
- M. S. Cooke, M. D. Evans, M. Dizdaroglu and J. Lunec, FASEB J., 2003, 17, 1195–1214 CrossRef CAS PubMed.
- E. J. Duncan, R. Larivière, T. Y. Bradshaw, F. Longo, N. Sgarioto, M. J. Hayes, L. E. L. Romano, S. Nethisinghe, P. Giunti, M. B. Bruntraeger, H. D. Durham, B. Brais, F. Maltecca, B. J. Gentil and J. P. Chapple, Hum. Mol. Genet., 2017, 26, 3130–3143 CAS.
- A. Martelli and H. Puccio, Front. Pharmacol., 2014, 5, 130 Search PubMed.
- R. Abeti, A. F. Brown, M. Maiolino, S. Patel and P. Giunti, Front. Cell. Neurosci., 2018, 12, 264 CrossRef CAS PubMed.
- J. Paprocka, A. Jezela-Stanek, A. Tylki-Szymańska and S. Grunewald, Brain Sci., 2021, 11, 88 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm00729k |
| This journal is © The Royal Society of Chemistry 2022 |