
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManufacturing, characterization, and degradation of a poly(lactic acid) warp-knitted spacer fabric scaffold as a candidate for tissue engineering applications
Flavia
Caronna
 ab,
Nikola
Glimpel
c,
Georg-Philipp
Paar
c,
Thomas
Gries
c,
Andreas
Blaeser
d,
Khoa
Do
b,
Eimear B.
Dolan
a and
William
Ronan
*a
ab,
Nikola
Glimpel
c,
Georg-Philipp
Paar
c,
Thomas
Gries
c,
Andreas
Blaeser
d,
Khoa
Do
b,
Eimear B.
Dolan
a and
William
Ronan
*a
aBiomechanics Research Centre (BMEC), Biomedical Engineering, NUI Galway, Ireland. E-mail: william.ronan@nuigalway.ie
bITA GmbH, Aachen, Germany
cInstitut für Textiltechnik of RWTH Aachen University, Germany
dInstitute for BioMedical Printing Technology, Technical University of Darmstadt, Germany
First published on 24th May 2022
Abstract
Three-dimensional bioabsorbable textiles represent a novel technology for the manufacturing of tissue engineering scaffolds. In the present study, 3D bioabsorbable poly(lactic acid) (PLA) spacer fabric scaffolds are fabricated by warp-knitting and their potential for tissue engineering is explored in vitro. Changes in physical properties and mechanical performance with different heat setting treatments are assessed. To characterize the microenvironment experienced by cells in the scaffolds, yarn properties are investigated prior to, and during, hydrolytic degradation. The differences in yarn morphology, thermal properties, infrared spectra, and mechanical properties are investigated and monitored during temperature accelerated in vitro degradation tests in phosphate buffered saline (PBS) solution at 58 °C and pH 7.4 for 55 days. Yarn and textile cytocompatibility are tested to assess the effect of materials employed, manufacturing conditions, post processing and sterilization on cell viability, together with the cytocompatibility of the textile degradation products. Results show that the heat setting process can be used to modify scaffold properties, such as thickness, porosity, pore size and stiffness within the range useful for tissue regeneration. Scaffold degradation rate in physiological conditions is estimated by comparing yarn degradation data with PLA degradation data from literature. This will potentially allow the prediction of scaffold mechanical stability in the long term and thus its suitability for the remodelling of different tissues. Mouse calvaria preosteoblast MC3T3-E1 cells attachment and proliferation are observed on the scaffold over 12 days of in vitro culture by 4′,6-diamidino-2-phenylindole (DAPI) fluorescent staining and DNA quantification. The present work shows the potential of spacer fabric scaffolds as a versatile and scalable scaffold fabrication technique, having the ability to create a microenvironment with appropriate physical, mechanical, and degradation properties for 3D tissue engineering. The high control and tunability of spacer fabric properties makes it a promising candidate for the regeneration of different tissues in patient-specific applications.
Introduction
Traditional surgical approaches for the repair or replacement of severely damaged tissue (including autograft and allograft) are expensive and painful, providing the potential for infection and foreign body rejection. Furthermore, major reconstructive surgeries (e.g., following trauma or tumour resection) are limited by a paucity of tissue availability and donor site morbidity.1 Tissue engineering, or regenerative medicine, aims to develop new functional tissues by combining highly porous scaffold biomaterials with cells from the body and mechanobiological or biophysical cues, to regenerate damaged tissue in vitro or in vivo.2,3 The field of regenerative medicine is in continuous growth, with the global tissue engineering market size computed at USD 9.9 billion in 2019 and expected to witness a compound annual growth rate of 14.2% from 2020 to 2027.4 The ideal tissue engineering scaffold should act as a template for tissue formation. It should feature excellent biocompatibility and provide a 3D interconnected open porous architecture allowing cells infiltration, nutrient diffusion, and waste removal, with suitable pore size distribution to stimulate tissue ingrowth and promote vascularization.5 Physical and chemical scaffold surface properties should be optimized to stimulate the attachment, proliferation, differentiation of the cell type of interest (depending on the target tissue to regenerate) promoting fully functional new tissue ingrowth.6 A tissue engineering scaffold must also have adequate mechanical properties to be handled during surgical procedure. It must provide enough mechanical stability at the time of implantation, allowing for sufficient load transfer to the host tissue, which is necessary to promote tissue regeneration and integration. Scaffold constructs, should be able to degrade in the body at a suitable rate to allow for gradual load transfer to the newly formed tissue, until achieving complete scaffold replacement.2,7 The balance of suitable 3D architecture, surface characteristics, mechanical properties, and degradation kinetics becomes the main challenge for the development of a scaffold for each specific tissue engineering application. Additionally, scaffold target requirements vary, not only due to the structural and functional diversity between various tissues, but also within the same tissue, according to implant location, pathologic conditions, patient age and activity level.2 A suitable scaffold manufacturing method should then enable the tailoring of physical, biochemical, and mechanical properties of the construct to facilitate the manufacturing of tissue and patient specific implants. Additionally, scaffold fabrication should be cost effective and easily scalable to large volumes of production.8,9 In this context, combining textile technology and bioabsorbable polymers has the potential to fulfil the complex requirements of optimal scaffold manufacturing.Textile technology offers versatile tools for precise control of material, pore size, geometry, morphology, interconnectivity, and mechanical properties. It provides design freedom at the micro (fibre) and macro (fabric) scale, while allowing production at an industrial scale. Textile fibres can mimic fibrous biological structures of the extracellular matrix, such as collagen and elastin fibre.10,11 Several fibre-based structures, such as woven,12 braided,13 knitted14 and non-woven15 constructs, and their combination,16 have been studied as tissue engineering scaffolds. Among the wide and diverse range of textile technologies, knitting enables a high control over complex fabric architectures and provides highly flexible and extensible fabrics with optimal bursting properties. The qualities of knitted fabrics highlights such structures for the engineering of both non- and load-bearing tissues.17 Besides two-dimensional knits,14,18 3D knitted spacer fabrics (i.e., textiles composed of two flat cover areas knitted using multifilament yarns and interconnected by a stiff monofilament yarn) better resemble the natural 3D microenvironment offered by the extracellular matrix to cells during tissue formation.11 For example, Schäfer et al.19 developed poly(ethylene terephthalate) warp-knitted spacer fabric reinforced hydrogels for soft tissue engineering, whereas Ribeiro et al.20 demonstrated the potential of weft-knitted silk based spacer fabrics for flat bone regeneration, both in vitro and in vivo.
Bioabsorbable polyesters, i.e., polymers having the ability to disintegrate into smaller fragments metabolizable by the human body,21 have become very attractive for use in scaffold-based implants. Degradation of such polymers when implanted in vivo, occurs through chemical bond cleavage mostly by hydrolyzation of covalent bonds, with consequent decrease of their molecular weight and, in turn, mechanical integrity. Several factors influence degradation such as material properties, geometry, and microstructure.21 Among bioabsorbable polyesters, poly(lactic acid) (PLA) can be produced by renewable resources, and it is promising due to its excellent biocompatibility, good mechanical properties, and predictable degradation rate.22 The degradation of bioabsorbable polyesters has been extensively studied using both experimental23 and computational24,25 approaches. However, there is no good description of the degradation of bioabsorbable yarns, which is highly influenced by the characteristics of the base material and processing routes (e.g., spinning method, drawing ratio, spinneret geometry, filament number).21,26,27 Additionally, studying the evolution of mechanical properties at a yarn level can provide useful information on the microenvironment experienced by cells in the different parts of the fabric prior to and during degradation.
To the authors’ knowledge, no studies have previously investigated the use of plain PLA warp-knitted spacer fabric scaffolds for tissue engineering applications. It is unknown how warp-knitted spacer fabric properties, which are relevant for tissue regeneration, change with post processing treatments. Heat setting, which is a thermal treatment performed after manufacturing, is of particular interest; being necessary to stabilize a structure in the equilibrium form which a textile assumes.26
In the present study, novel warp-knitted spacer fabric scaffolds were designed for 3D tissue engineering applications using commercially available PLA monofilament and multifilament yarns. The effect of heat setting on scaffold morphology and 3D structure was evaluated using micro-computed tomography (micro-CT). Mechanical properties of heat set fabrics were assessed through compression tests. Temperature accelerated in vitro degradation tests were carried out on the same PLA yarns employed for scaffold manufacturing. With the aim to predict scaffold behaviour during long-term degradation at physiological temperature, the yarns’ mechanical properties, thermal response, and infrared spectra were investigated throughout hydrolytic degradation. The yarns employed for the fabrication of the textile, the best performing textile scaffold, and its degradation products were tested for in vitro cytocompatibility, to assess any potential cytotoxic effect of base materials, and manufacturing/sterilization processes in the short and long term. Considering the possible application of the scaffold for bone tissue regeneration, spontaneously immortalized murine calvarial preosteoblast MC3T3-E1 cell line attachment and proliferation on the scaffold were qualitatively and quantitatively assessed over 12 days of culture in vitro.
Materials and methods
Textile manufacturing and post processing
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m of yarn) (TREVIRA® 41053588, 32 filaments) supplied by Trevira Gmbh. The spacer was prepared using bar B10, according to the following chart: bar B10: 1-0/1-0/1-2/1-2//. Bar B10 was threaded with PLLA 99 dtex monofilament yarn from Luxilon Industries NV, to give thickness and stiffness to the three-dimensional structure. The manufactured textile featured a Tricot lapping and an X pile yarn configuration (Fig. 1A–C). Linear density of the yarns was measured prior to the experiments following DIN EN ISO 2060. A value of 8 effective arches per centimetre (EAC) (which is a machine parameter that regulates the drawing speed of the produced fabric, and thus the theoretical number of stitch courses per centimetre in production direction) was employed for fabric manufacturing.
000 m of yarn) (TREVIRA® 41053588, 32 filaments) supplied by Trevira Gmbh. The spacer was prepared using bar B10, according to the following chart: bar B10: 1-0/1-0/1-2/1-2//. Bar B10 was threaded with PLLA 99 dtex monofilament yarn from Luxilon Industries NV, to give thickness and stiffness to the three-dimensional structure. The manufactured textile featured a Tricot lapping and an X pile yarn configuration (Fig. 1A–C). Linear density of the yarns was measured prior to the experiments following DIN EN ISO 2060. A value of 8 effective arches per centimetre (EAC) (which is a machine parameter that regulates the drawing speed of the produced fabric, and thus the theoretical number of stitch courses per centimetre in production direction) was employed for fabric manufacturing.
 | ||
| Fig. 1 (A) Schematic diagram of warp-knitted pattern used on faces S1 and S2. (B) Simplified illustration of pile yarn geometry. (C) μCT image of PLA warp-knitted spacer fabric after manufacturing. (D) Custom made heat setting stenter frame with schematic diagram of stenter pin. Scale bar 1 cm. (E) Optical image of PLA multifilament yarn, 200 μm scale bar. (F) Detail of the cross section of multifilament yarn after embedding in epoxy resin. (G) Schematic diagram of samples for mechanical testing during degradation. | ||
Textile characterization
Yarn characterization
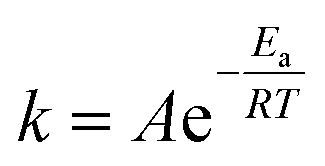 | (1) |
| F0 [cN] = 2 [cN tex−1] × LDMulti [tex] | (2) |
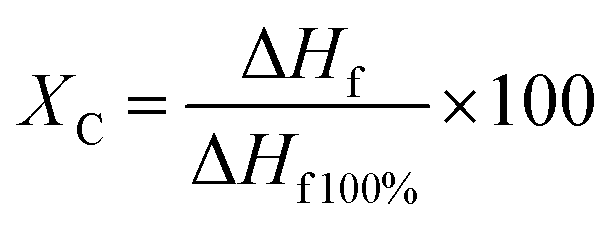 | (3) |
Glass transition temperature and melting point were also monitored during degradation. The cooling and second heating cycle were analysed to gain insights on crystalline morphology, material properties, and yarn manufacturing processes which may be of interest for degradation process characterization.
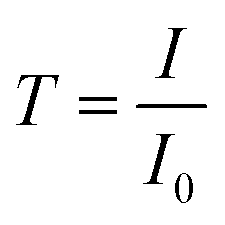 | (4) |
In vitro cell study
Prior to extraction, the PLA monofilament and multifilament yarns (i and ii) were autoclaved at 121 °C and the textile was sterilized via Ethylene Oxide (EtO) by Tepha Medical Inc. While steam sterilization has been shown to cause damage under autoclave conditions,21,44 PLA yarns were steam sterilized to assess their cytocompatibility in vitro. To closely represent the intended clinical use of the scaffold, the cytocompatibility of the final textile scaffold sterilized via EtO, and its degradation products (described in the next section), were also assessed.
Supplemented culture medium consisting of αMEM (Minimum Essential Eagle with Alpha modification) supplemented with 2 mM L-glutamine, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, and 10% foetal bovine serum (FBS) (all supplied by Sigma Aldrich) was used as both culture and extraction medium. Sample extracts of (i–iii) were prepared by immersing the test material in supplemented culture medium at sample mass per extract liquid volume ratio of 0.1 g mL−1, as the ISO standard suggests for irregularly shaped porous devices. Extraction was performed for 72 ± 2 h at 37 ± 1 °C. To account for the absorbency of test samples, as suggested by the ISO standard, the volume of extraction media absorbed per 0.1 g of material was measured beforehand and then, performing the extraction of each sample, this extra volume was added to each respective extraction mixture.
After 72 h incubation time, the liquid extracts from each sample type (i–iii) were diluted in fresh supplemented media to reach concentrations of 10, 50 and 100% (non-diluted) sample extracts. In parallel, osteoblast precursor cell line MC3T3-E1 from the American Type Culture Collection (ATCC, Manassas, VA, USA) (passage 24–25) were seeded at a density of 104 cells per well in a 96-well plate dispensing 100 μL cell suspension (1 × 105 cells per mL) in each well, using 12 wells per each condition studied (n = 12). The cells were then incubated for 24 h under standard conditions to allow attachment. After 24 h, the media was replaced with 100 μL of treatment medium containing either the appropriate concentration of sample extract (conditioned media), control media or cytotoxic media. Control media consisted of supplemented αMEM (100% viability), while the cytotoxic media consisted of 10% DMSO in complete culture medium (0% viability).45 After further 24 h, alamarBlue™ (Thermo Scientific™ Cell Viability Reagent) assay was used to determine metabolic activity of the cells according to manufacturer's instructions. After 4 h incubation with Alamar Blue solution, samples fluorescence was read at an excitation wavelength of 530 nm and emission wavelength of 590 nm, using black-walled 96-well plates and a Synergy™ HT multi-mode microplate reader (BioTek).
Cytocompatibility of textile degradation products
To further investigate cytocompatibility, PLA textile scaffold degradation products were assessed for in vitro cytotoxicity using an adaption of the testing method described in the previous section. The degradation products of the textile (iv) were obtained by immersing an EtO sterilized PLA textile samples in cell culture grade PBS at ratio of 0.004 g mL−1 (sample mass to volume of PBS, as recommended by the standard for in vitro degradation testing of PLA33) in an oven at 100 °C for 5 days. Almost complete textile degradation (>95%) was achieved during this process. The PBS containing the degradation products of the textile was then mixed with fresh PBS and culture media and used as conditioned media on cells. Since in a physiological environment, complete hydrolytic degradation of the textile would take place over several years, with a gradual release and metabolization of degradation products (mainly monomers, i.e., lactic acid), the conditions studied here represents an exaggerated release condition, suitable to study potential cytotoxicity effects of a substance in vitro. The immersed textile sample before and after degradation is shown in Fig. 2A and B, with the pH of the PBS solution, measured using pH stripes, dropping from 7.4 (Fig. 2A) to 3–3.5 (Fig. 2B) after degradation, confirming the presence of lactic acid in solution. Prior to the test, the effect of fresh PBS on cells in culture was investigated. It was observed that cells treated with 30% v/v PBS in complete culture media maintained the same metabolic activity of cells treated with media only. As a result, a concentration of 30% v/v PBS in culture media was employed for the present test.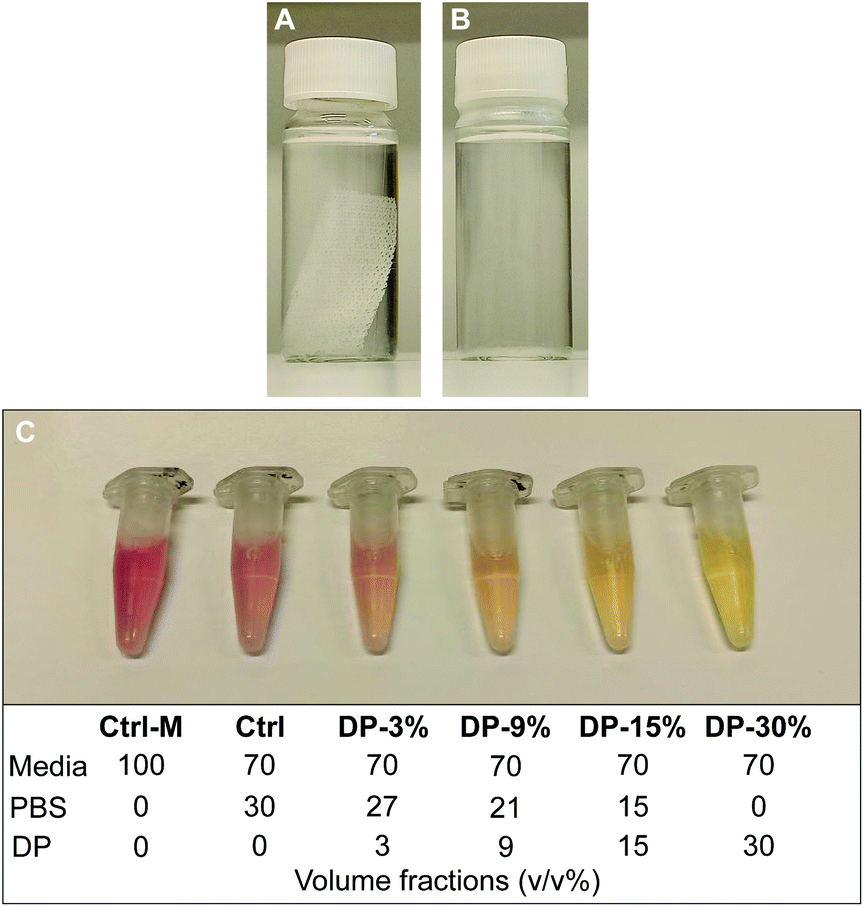 | ||
| Fig. 2 PLA textile immersed in PBS (0.004 g mL−1) (A) before and (B) after degradation at 100 °C for 5 days. (C) Aliquots of conditioned media. Ctrl-M = fresh media only, Ctrl = control with 30% fresh PBS in fresh media, DP-3% = 3% PBS from degraded sample in media. Samples compositions are also reported in volume fractions (v/v%). | ||
MC3T3-E1 cells (passage 22) were seeded at a density of 104 cells per well in a 96-well plate dispensing 100 μL cell suspension (1 × 105 cells per mL) in each well, using 6 wells per each condition studied (n = 6). 24 h after seeding, the media was replaced with 100 μL of treatment medium containing either the appropriate concentration of PBS mixture (PBS with degradation products, Fig. 2B), control media or cytotoxic media. Control media consisted of 30% v/v fresh PBS in supplemented αMEM (positive control), another control was used consisting of supplemented αMEM only (untreated healthy cells), while the cytotoxic media consisted of 10% DMSO in complete culture medium (negative control). The PBS from the degraded samples was mixed with fresh PBS in different ratios, reaching a final concentration of 30% total PBS in media. Cells were therefore exposed to conditioned media with a final concentration of 3, 9, 15 and 30% v/v of PBS from degraded samples. The composition of all conditioned and control media is reported in Fig. 2C. A change of the colour of conditioned media solutions, containing media with phenol red as pH indicator, was observed with increasing concentration of degradation products, with pH = 7.7–8 for Ctrl-M, Ctrl and DP-3% groups, pH = 7.4–7.7 for DP-9% group, and pH = 7–7.2 for DP-15% and DP-30%. After further 24 h, alamarBlue™ assay was used to determine metabolic activity of the cells as described in the previous section.
Results
Warp-knitted textiles properties
 | ||
| Fig. 3 (A) Optical images of fabric surfaces (S1 and S2) before (NS = not heat set) and after heat setting, 2 mm scale bar. (B) Fabric thickness and porosity with and without heat setting. (C) Pore size (diameter) distribution. (D) Stress–strain curves from uniaxial compression tests, for different heat setting conditions (±standard deviation). Significance (in B and C) refers to non-set textile. | ||
As shown in Fig. 3B, the average textile thickness (1.42 ± 0.08 mm before heat setting) significantly decreased with high setting temperatures (1.06 ± 0.9 mm for 120 °C and 1.05 ± 0.08 mm for 150 °C, p = 0.01), while not being significantly affected when setting at 90 °C (1.30 ± 0.18 mm, p = 0.36).
Textile shrinkage during heat setting causes the observed decrease in thickness and pore size. At low setting temperatures (90 °C), pore size decreases more than thickness, suggesting that yarn shrinkage in the cover areas is taking place without affecting the spacer yarn between the surfaces. When increasing the setting temperature, shrinkage of the pile yarn leads to a concurrent decrease of thickness and pore size. Overall porosity does not change considerably, as the decrease in textile thickness is accompanied by a drop of pore size. Pore definition is improved, being the structure in a position of lower minimum energy from the state prior to heat setting,26 thus providing a stable environment for cell attachment and growth.
Yarns degradation properties
 | ||
| Fig. 4 (A) Monofilament and (B) multifilament yarn samples at different days of accelerated degradation at 58 °C. Estimated equivalent duration at 37 °C is also shown. Note: samples in (A and B) are used for DSC and FTIR analysis. | ||
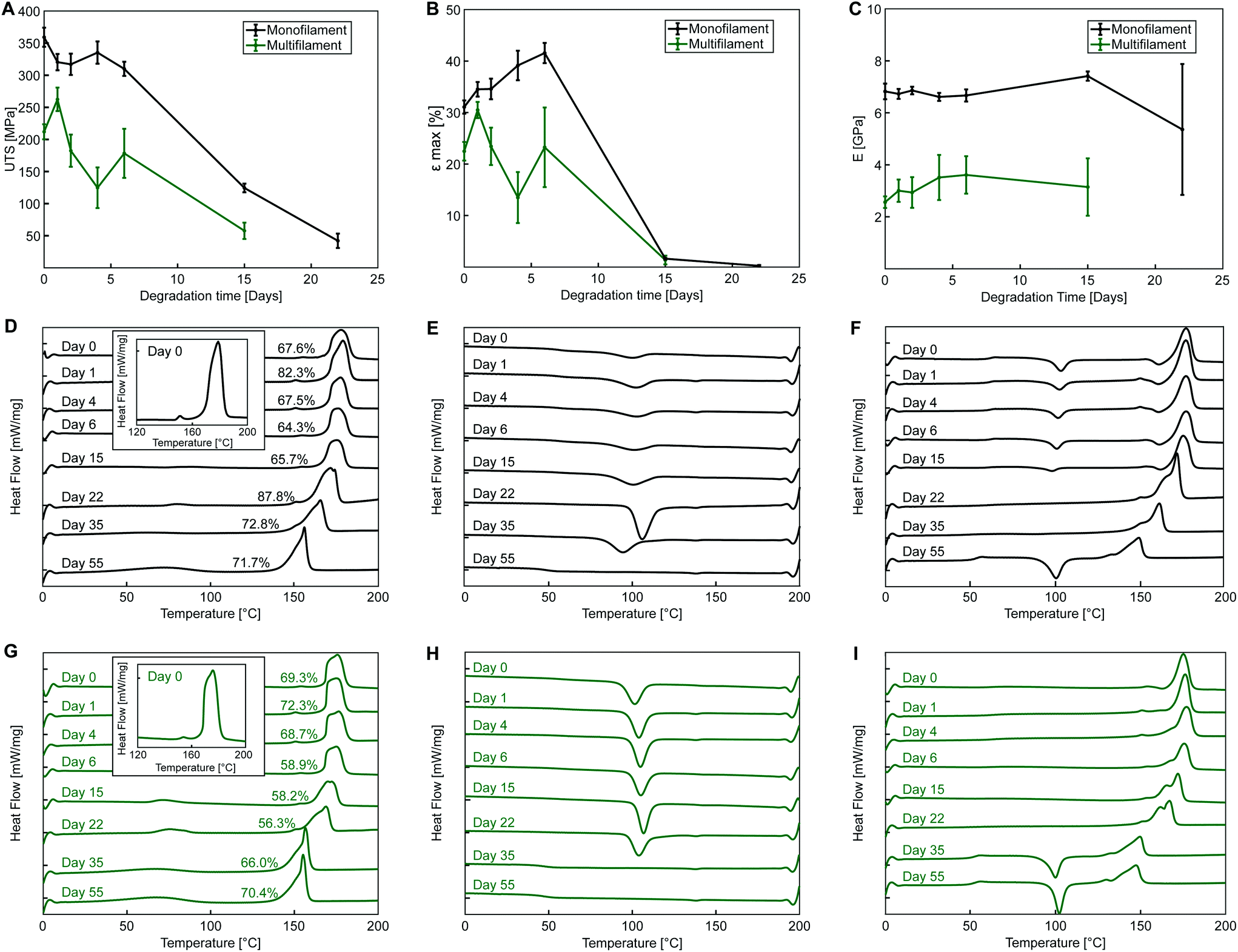 | ||
| Fig. 5 Yarns characterization during degradation. (A) Ultimate tensile strength (UTS) (B) elongation at ultimate strength (εmax) and (C) Young's modulus (E) of PLA monofilament and multifilament yarns during degradation. DSC heat flows for monofilament (D–F) and multifilament (G–I) yarns during degradation showing first heating curve (D and G), cooling curve (E and H) and second heating curve (F and I). In all DSC plots, the tick marks are at 1 mW mg−1 intervals with each sample offset vertically for clarity. Degree of crystallinity indicated for all curves during first heating. Inset in (D and G) shows additional peak at ∼150 °C. | ||
For both PLA yarns, during the first heating curve, no observable cold crystallization takes place (Fig. 5D and G) with a defined melting peak (onset temperature of 169 °C at day 0), and a small endothermic peak preceding the main one (measured at around 150 °C at day 0), which was included for enthalpy calculations and crystallinity measurements. This peak might indicate the presence of a conformationally disordered α′ polymorph together with a main ordered α phase, with the small peak being the result of the combination of an endothermic heat flow due to melting of α′-crystals at around 150 °C, superimposed to the exothermic formation of new α-crystals that melt at higher temperature.48,49 This phenomenon is even more evident in the second heating cycle of monofilament yarn (Fig. 5F) where an exothermic dip appears. Another possible explanations is that the two peaks are due to melting of crystals of different structural arrangement and directionality, not necessarily of α or α′ form.50 It is also possible that β polymorph was obtained during fibre manufacturing via stretching the α polymorph at high temperature to a high drawing ratio.51
The thermal plots show a decrease of yarn melting temperature during degradation, clearly observable from day 15 for both yarns, which may indicate an increase of the content of amorphous phase and molecular weight reduction with degradation,52 with the two melting peaks gradually merging into one.
The exothermic dip in the cooling cycles (Fig. 5E and H) indicates polymer crystallization from the melt. A more pronounced crystallization dip is observed for the multifilament yarn, suggesting a higher ability of sample molecules to rearrange and crystallize during cooling at the employed rate. The second heating curve (Fig. 5F and I) can be used to investigate the characteristics of the polymer once its thermal history has been erased by the first heating cycle.
Thermographs of monofilament samples during second heating shown in Fig. 5F feature the typical shape observed for semicrystalline polymers, with cold crystallization, i.e., exothermic transition where molecular rearrangement takes place in the amorphous phase at temperatures higher than TG, causing an increase of polymer crystallinity, followed by melting. Cold crystallization observed for the monofilament samples indicates that, differently from the multifilament yarn, not all the molecules were able to organize during cooling, which might suggest that a lower cooling rate was employed during manufacturing. In the second heating cycle shown in Fig. 5F, the glass transition is also more pronounced, most likely due to the lowered crystallinity of monofilament samples after cooling.
Fig. 5E and H show that the crystallization peak during cooling flattens for both yarns with increasing degradation time. This suggests a decrease in molecular chain length with degradation, leading towards an amorphous structure, with cooling rate of 10 K min−1 being unable to allow the complete crystallization of the melt. Flattening of the cooling curve is accompanied by cold crystallization during second heating and occurs at day 55 for the monofilament and at day 35 for the multifilament yarn. This might be an indication of a faster degradation rate of the multifilament compared to the monofilament yarn, with faster molecular weight loss. The reported difference could be related to a lower initial molecular weight or a different crystals arrangement and morphology, favouring degradation in multifilament yarn.
Glass transition temperature decreases with degradation with TG at the inflection point reaching values of 53.2 °C for the monofilament and 51.7 °C for the multifilament yarn after 55 days of accelerated degradation of monofilament.
In vitro assessment of yarns and textile
Results from the cytocompatibility study on PLA yarns and textile extracts, and textile degradation products are shown in Fig. 6. For all materials, Alamar Blue reduction, directly proportional to cells metabolic activity, is significantly different (p < 0.001) to the cytotoxic media control (DMSO treated cells), confirming the assay's capability to detect cytotoxic effects. No significant decrease of cells metabolic activity with respect to the control is observed for cells treated with extracts of all samples studied (PLA monofilament, multifilament, and textile) or with PLA textile degradation products.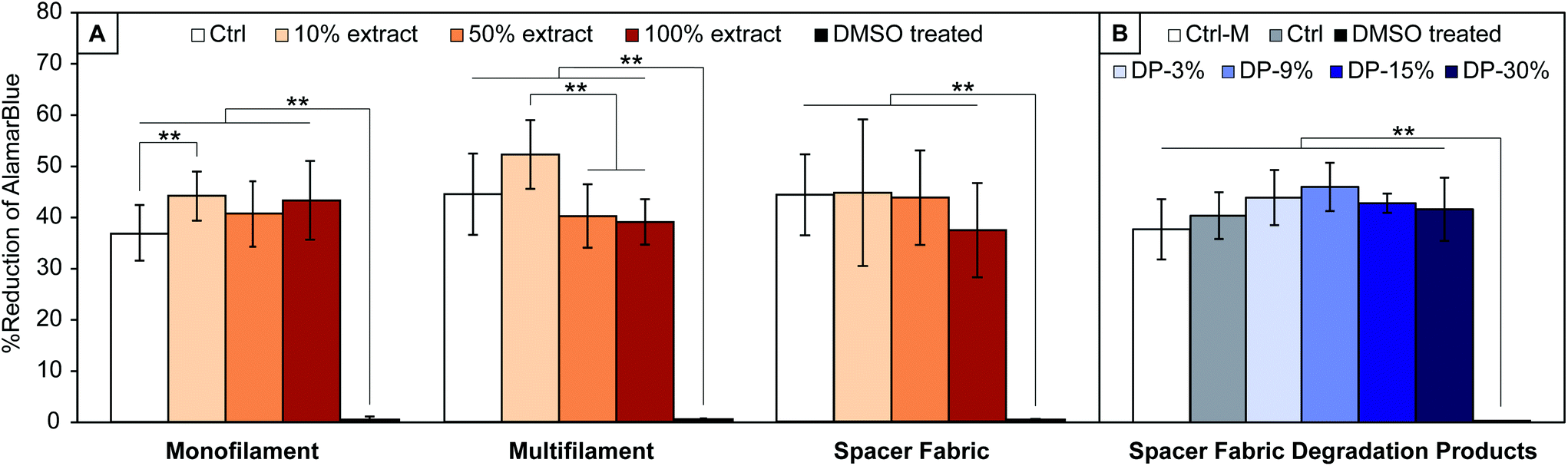 | ||
| Fig. 6 Results of cytotoxicity tests on extracts of (A) PLA monofilament yarn, multifilament yarn and spacer fabric heat set at 150 °C and sterilized via EtO (in orange scale), and (B) on degradation products of the latter (blue scale). Results are given in % of reduction of AlamarBlue which is proportional to MC3T3-E1 cells metabolic activity, with 100% extract being the most concentrated one, DP-30% = 30% v/v degradation products in media, Ctrl-M = media only, Ctrl = control (30% v/v fresh PBS in media). *p ≤ 0.05, **p ≤ 0.01. | ||
A significant difference with respect to the control media is observed for cells treated with 10% extract of PLA monofilament yarn, which show a higher metabolic activity (44.6 ± 4.0% vs. 36.8 ± 5.4% reduction of Alamar Blue, p = 0.009). No significant increase was observed with cells treated with 50 or 100% extracts of PLA monofilament.
There was no significant increase in metabolic activity with cells treated with 10% extract of PLA multifilament yarn compared to the control (52.1 ± 6.7% vs. 44.4 ± 7.9%, p = 0.13), however a significantly higher metabolic activity was seen with cells treated with 10% compared to cells treated with 50 and 100% extracts (52.1 ± 6.7% vs. 40.1 ± 6.2% vs. 39 ± 4.4%, p < 0.001). These results (Fig. 6A) suggest that the yarns and textile are not cytotoxic, since cells exposed to all materials extracts showed comparable or higher metabolic activity than 2D controls.
When treating cells with PLA textile degradation products, no significant change of cells metabolic activity is observed for any concentration of degradation products with respect to the control (Ctrl) (Fig. 6B). The metabolic activity of cells treated with media only (Ctrl-M) was also not significantly different than those of other groups. The present results suggest that the degradation products of the manufactured PLA textile are not cytotoxic, since cells exposed to all conditioned media at any concentrations showed comparable metabolic activity to that of healthy controls. These conclusions are in agreement with the in vivo metabolization of lactic acid reported in literature,21,22 and show that no additional cytotoxic components are present in the yarns’ raw material, whose exact composition is unknown.
Histological staining of cells nuclei shows that PLA textile supports MC3T3-E1 cells attachment (Fig. 7A). Considerable cell infiltration occurred in the 3D fabric, with a higher portion of cells attached on the fabric top surface (seeding surface), with cell attachment also observed on both the bottom surface and spacer yarn. Cells proliferation occurs on the fabric, with a qualitative increase in the number of DAPI stained nuclei at day 12 (Fig. 7B). Good cell attachment is observed on monofilament yarns (Fig. 7C), with higher cell density observed on multifilament yarns (Fig. 7D), likely due to the higher surface area of the latter. DNA quantification results in Fig. 7E confirm that cell proliferation occurred on the scaffolds, with an approximate 5-fold increase of the average amount of double stranded DNA over 12 days of culture (70.4 ± 10.3 pg μL−1 at day 1 and 343.8 ± 93.7 pg μL−1 at day 12, p = 0.015).
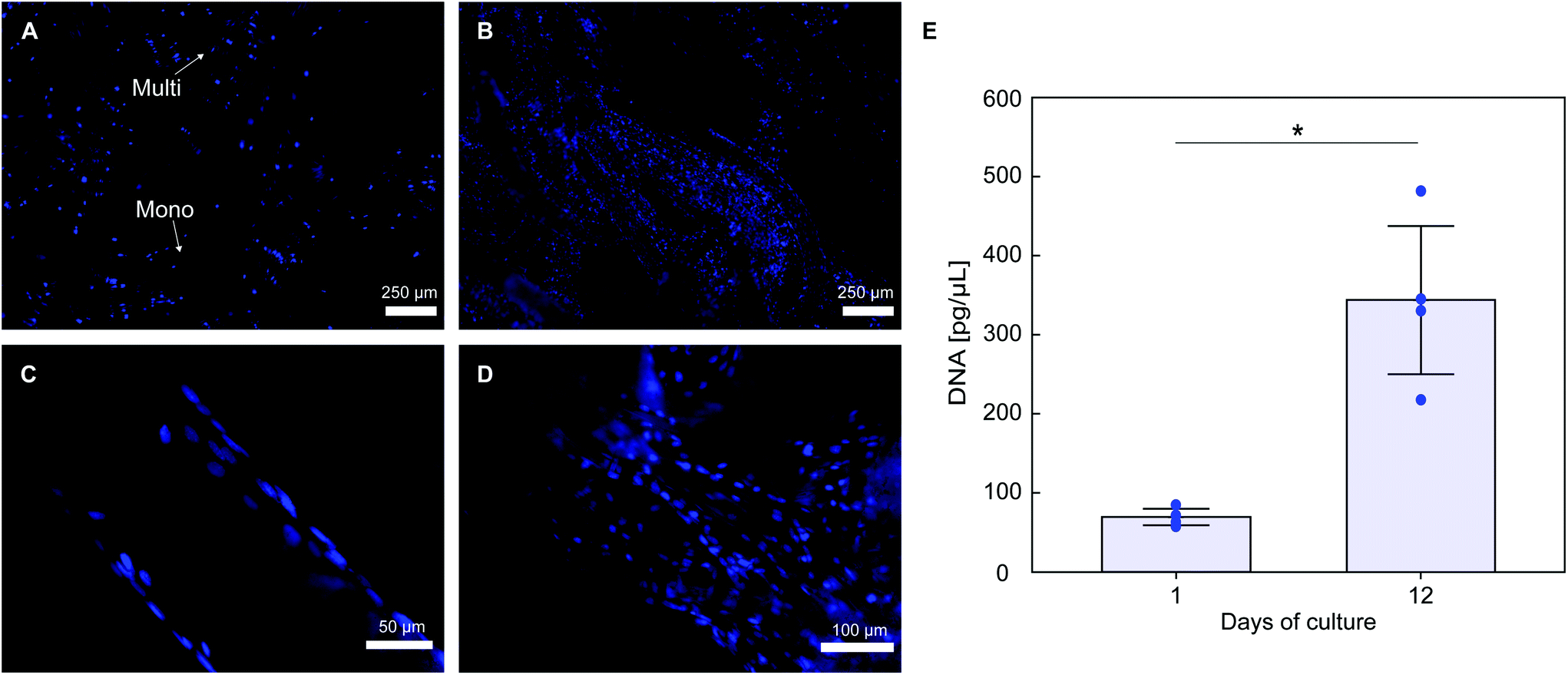 | ||
| Fig. 7 DAPI stained nuclei of MC3T3-E1 cells on the textile after (A) 1 day of culture and (B) 12 days of culture. Detail of cells attachment on (C) monofilament yarn and (D) multifilament yarn in the textile at day 12. (E) DNA content in scaffolds quantified using Pico488 assay at day 1 and 12 of culture. Individual data from 4 different samples (n = 4) for each time point are presented, together with average value (bar) ± standard deviation. | ||
Discussion
Medical textiles are widely used in clinical practice as medical implants for treating pelvic organ prolapse, hernia, and vascular diseases.17 Textiles and fibre-based structures are currently being investigated for a variety of tissue engineering applications in the cardiovascular and musculoskeletal areas.10 Fibre-based structures allow design control at a variety of scales, from yarn material, morphology, to fabric topography, pore shape and distribution, and structure mechanical properties; all of which are responsible for different pivotal phenomena occurring during the complex tissue regeneration process.10 Several technologies can also be combined to optimize scaffold properties for the regeneration of a specific tissue53 or imitate in vivo-like tissue development processes.54In the present work, three-dimensional bioabsorbable spacer fabric structures made of PLA monofilament and multifilament yarns are warp-knitted, heat set, and investigated as a possible strategy for tissue engineering applications. The characteristics of the manufactured spacer fabrics are described, and the effect of thermal post processing treatments assessed in regard to the main properties of scaffold-based implants. The heat setting process is performed to enhance the fabric's dimensional stability during use55 by causing a release of the stresses generated within the fibre structure during textile manufacturing. The present analysis of thickness, porosity, and pore size of the heat set PLA fabrics from CT scans confirms that the heat setting process led to an improved pore definition and textile structure homogeneity. These characteristics are pivotal to allow a stable environment for cell support, with scaffold porosity and pore size correlating well with those of tissue engineering scaffolds in literature.7,56–58 The presence of both macropores and micropores, the first within and between the cover areas, the second between yarns and filaments, allows nutrients and waste diffusion, while offering a wide available surface area for cell proliferation and tissue formation.
Heat setting the PLA textile at the highest studied temperature (150 °C), lead to minimum fabric thickness (1.05 ± 0.08 mm) and maximum stiffness (16.1 ± 4.4 kPa) due to textile shrinkage upon heating. There is no standard consensus about the mechanical testing of spacer fabric textiles;32 they are commonly tested for compression properties since often used as energy absorbing structures.46 Testing other mechanical properties, e.g., burst strength, commonly measured for 2D surgical meshes,14 or uniaxial/biaxial strength retention,19 may be more relevant according to the specific mechanical requirements of the tissue to regenerate. Nevertheless, the mechanical properties of a scaffold do not necessarily need to match those of the native tissue to regenerate20 and the modulus is used primarily to quantitatively differentiate between treatments. Internal or external fixation systems can be used to shield the tissue engineering construct from part of the load until sufficient tissue maturation.59 However, the scaffold must resist flow in vivo and maintain cell physical integrity while transferring adequate biomechanical forces to stimulate initial tissue formation. In the present study, we have manufactured a bioabsorbable 3D construct able to fold and be adjusted to complex shapes, featuring sufficient mechanical integrity to support cells and withstand handling and fluid flow in vitro.
As an essential preliminary step for the development of tissue engineering scaffolds, yarns, textile, and degradation products cytocompatibility was assessed and confirmed in vitro, based on the ISO standard for in vitro cytotoxicity testing. An adaption of the standard method to test the cytotoxicity of degradation products is reported here as a fast and effective method. It is suitable for hydrolytically degradable polymers with a glass transition temperature which allows fast and complete degradation in temperature accelerated conditions. In addition, it was shown that the manufactured scaffold supports cell infiltration, attachment, and proliferation over 12 days of culture in vitro, being a basic property necessary for all tissue engineering applications.
Polylactide, or poly(lactic acid), is a biodegradable polyester which has been widely employed in drug delivery systems, sutures, vascular grafts, surgical implants together with porous scaffolds for soft and hard tissue regeneration.22 The mechanism of bulk degradation of bioabsorbable polyesters is well known, with a number of numerical models currently under investigation for predicting the degradation of 3D structures with different geometries25,60 as well as their mechanical properties evolution during hydrolysis.
In the present work, the activation energy for the in vitro hydrolytic degradation of PLLA calculated by Weir et al.35 was used to estimate the time range for yarn degradation in physiological conditions. The results presented here are consistent with the PLA degradation time scale from literature;23,35 however, care must be taken when comparing the hydrolysis of constructs of different shapes and crystalline fractions, since both device geometry and material crystallinity are known to affect the degradation rate of bioabsorbable polymeric devices.41,52 In the present study, hydrolytic degradation has limited effect on the mechanical properties of PLA yarns up to 6 days of accelerated degradation (72 days at physiological temperature). Therefore, the stability of yarn mechanical properties in the initial stage is expected to favour cells attachment, proliferation, and differentiation, together with initial tissue formation. The higher cell density observed on multifilament yarns suggests that substantial tissue formation is more likely to happen on the cover areas. The latter, based on hydrolysis results in vitro, are expected to experience a faster loss of mechanical properties with respect to the pile yarn, with multifilament strength dropping after approximately 6 months at 37 °C, which implies subsequent gradual load transfer to the newly formed tissue. The monofilament yarn, primarily responsible for the compressive properties of the scaffold, is expected to retain its strength for a longer time (8–9 months at 37 °C), thus offering mechanical support to the entire developing tissue construct. Full structure resorption at physiological temperature is expected to occur after more than 660 days. The results obtained for PLA multifilament yarn demonstrate its high cytocompatibility, together with the increased MC3T3-E1 cell attachment and proliferation, and are in a countertendency compared with other studies, showing a higher susceptibility of multifilament yarns to infections.14
The faster degradation of the studied PLA yarns with respect to some other PLA devices24,47 might be due to the high initial degree of crystallinity of the studied yarns.41 In the present work, crystallinity increases only slightly during degradation (consistent with PLA yarns degradation data from literature61). These results suggest that during the studied time frame, polymer chains in the amorphous region have limited mobility, with hydrolysis induced crystallization not contributing to slow down the yarn's degradation rate, as is likely to happen for constructs with lower initial crystallinity.24
The high regularity of the developed structure together with its high porosity will likely result in a homogeneous degradation of the overall construct. Macroporosities likely favour acid diffusion outside the textile, with little autocatalysis happening at yarn and filament geometrical scale, as observed for other bioabsorbable polyesters.25 Taken together, these results demonstrate the potential of 3D bioabsorbable textile scaffolds.
Although technologies such as 3D printing or additive manufacturing allow for quick innovation and testing of new designs and production of patient specific scaffolds, they are slow, with limited resolution and usable materials, and difficult to scale up in manufacturing.62 Additionally, the act of thermal processing of bioabsorbable polymers in presence of moisture introduces an additional degradation mechanism, occurring rapidly due to the elevated processing temperatures.44 Textile production has continuously developed starting from the industrial era to fulfil the growing demand for textiles at a reasonable cost.63 Fibre-based scaffolds benefit from the easy scalability of textile production, its cost effectiveness and high structural control, together with the existing expertise on a wide range of technical textiles. In concert with novel materials, textile technology represent a multidisciplinary design strategy with the textile industry having the potential to accelerate the fabrication, translation, and commercialization of tissue engineering scaffolds.11
The limitations of the current study also need to be discussed. It must be noted that in vitro degradation tests only partially model the complex environment surrounding a tissue in vivo. When a device is implanted, mechanical load and fluid flow contribute to its degradation,64 surface erosion occurs due to enzymatic attack,65 and acid release from the degrading implant can cause inflammation of neighbouring tissue.21 Additionally, yarn glass transition temperature during the later stages of degradation decreased to values below the immersion temperature (58 °C). However, Weir et al.,35 comparing in vitro degradation results for PLLA degraded at 37 and 70 °C, demonstrated that the activation energy of the hydrolytic reaction remained unchanged. This confirms the assumption that no major alteration of the material degradation kinetic occurred in the present study with respect to physiological temperature. MC3T3-E1 pre-osteoblast cell line was chosen in the present work, being a reliable alternative to primary human osteoblast for testing the in vitro cytocompatibility and osteogenicity of novel biomaterials.66 While it is necessary to show that a scaffold is cytocompatible at all stages and can support cell invasion, attachment, and proliferation; a tissue specific investigation at both in vitro and in vivo level is required to demonstrate the suitability of the presented scaffold for each specific tissue engineering application. Polymeric synthetic three-dimensional scaffolds which can incorporate cells or growth factors are crucial for the development of a tissue engineered construct to induce the growth of normal tissue. For investigating the use of cell-loaded scaffolds, depending on the repair strategy chosen, in vivo or ex vivo bioreactor studies would be needed to confirm the growth of tissue with functional and mechanical properties similar to the native target tissue. For vascularized native tissues, this includes investigating the formation of interconnected, functional, and perfused vascular networks in the construct, which is a major challenge. Furthermore, studying the host immune response is of critical importance to prevent the risk of infection after implantation.2
Future outlooks of this research include the stacking and sewing of multiple layers of the manufactured PLA spacer fabric textile, to produce modular scaffolds with adaptable size, with fixed overall porosity and pore size distribution. Moreover, the work of Entezari et al.,67 who demonstrated numerically the possibility of using weak biomaterials in modular scaffolds for repairing large bone defects, with only few percent of strain being sufficient to promote osteogenesis, supports our proposed use of the present scaffold for the repair of non and load-bearing tissues. The bioabsorbable scaffold developed here can also be used in bottom-up tissue engineering strategies,54,68 culturing different cell types on different fabric layers, which assembled could better mimic the native tissue environment and improve tissue healing. Even though some scaffold requirements are specific to each tissue and a single scaffold design is unlikely to meet the requirements of all human tissues; we propose bioabsorbable warp-knitted spacer fabrics as versatile platform for scaffold manufacturing. These structures offer design freedom in terms of yarns material, size and shape, cover area structure, pile yarn arrangement, scaffold thickness and mechanical properties, and have the potential to be used directly, in modules, or in combination with other materials or constructs, thus meriting further exploration in the tissue engineering field. All structural, biological and degradation features of the scaffold make it a potential candidate for further investigation in the fields of hard and soft tissue regeneration. Scaffold porosity and pore size fall in the range of those used for bone3 and cartilage69 tissue engineering, where scaffold biodegradation is desired. Bioabsorbable 2D and 3D knitted structures are also being investigated for the repair of large complex abdominal wall defects,14,70 as either an alternative to permanent meshes when contraindicated (e.g., repair of infected defects), or to improve tissue healing. The drapability of warp-knitted spacer fabric scaffolds also makes them attractive candidates in complex tissue engineering applications such as muscle-tendon junctions.71
With only few engineered tissues successfully used for clinical purposes,54 the present work describes the design and manufacturing of a 3D textile scaffold able to support cell attachment and growth, and gives insights on the properties of bioabsorbable polymeric yarns during degradation and their potential effects on cell behaviour, thus contributing to the further development of bioabsorbable tissue engineering scaffolds.
Conclusions
In this study, a highly cytocompatible 3D bioabsorbable warp-knitted spacer fabric scaffold featuring a regular and highly interconnected open porous structure was manufactured employing commercially available monofilament and multifilament PLA yarns, through a highly automated manufacturing method. It was demonstrated that the heat setting process can be used to tune the physical and mechanical properties of the manufactured PLA spacer fabric scaffolds. All manufactured fabrics presented suitable porosity and pore size to allow cell infiltration, nutrients, and waste diffusion, with sufficient mechanical strength to withstand handling and flow in vivo, providing a stable environment for cell attachment and growth. The cytocompatibility of degradation products of the manufactured PLA textile is also assessed. From yarn mechanical data obtained during temperature-accelerated in vitro degradation tests, scaffold degradation in the long term is predicted together with cell–scaffold interactions in the different parts of the constructs. Complete loss of scaffold mechanical integrity is predicted to occur after 6–8 months of hydrolysis at physiological temperature. The developed bioabsorbable PLA textile (heat set at 150 °C) was able to support MC3T3-E1 cells attachment and proliferation, up to a 5-fold DNA increase on the scaffold, during 12 days of culture in vitro. The employed 3D-knitting technology allows high structural control while permitting the manufacturing of high volumes of production, which is a necessary requirement for the clinical development of tissue engineering. An easily mass-manufactured PLA textile scaffold is proposed here as potential candidate for 3D tissue engineering research due to its biological properties, ability to adapt to complex shapes, tunability of its physical and mechanical properties, and predictable degradation rate. Superimposition of multiple textile layers is proposed for future investigation for bottom-up tissue regeneration approaches.Author contributions
Flavia Caronna: conceptualization, data curation, formal analysis, methodology, investigation, writing – original draft, visualization. Nikola Glimpel: methodology, writing – review & editing. Georg-Philipp Paar: methodology, writing – review & editing. Thomas Gries: resources, supervision. Andreas Blaeser: conceptualization, writing – review & editing. Khoa Do: supervision, funding acquisition. Eimear B. Dolan: conceptualization, resources, writing – review & editing, supervision. William Ronan: conceptualization, funding acquisition, resources, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 813869. This presentation reflects only the author's view, and the REA is not responsible for any use that may be made of the information it contains.The authors would like to thank: ITA-RWTH technicians involved in the present project and C. Emonts for their support in textile manufacturing and characterization; A. Holzapfel and Y. Zhou for their help on yarns characterization during degradation; L. McNamara for kindly providing the cells, T. Marangon for the scientific contribution and D. Connolly for his help with DSC analyses.
The authors acknowledge Tepha Medical Inc. (now part of Becton & Dickinson) and their support for the sterilization of the PLA textile, and very much appreciate the technical and scientific contribution of S. Limem and S. Rizik from Tepha Medical Inc. to the present work.
References
- J. Henkel, M. A. Woodruff, D. R. Epari, R. Steck, V. Glatt, I. C. Dickinson, P. F. Choong, M. A. Schuetz and D. W. Hutmacher, Bone Res., 2013, 1, 216–248 CrossRef CAS PubMed.
- F. J. O'Brien, Mater. Today, 2011, 14, 88–95 CrossRef.
- M. N. Collins, G. Ren, K. Young, S. Pina, R. L. Reis and J. M. Oliveira, Adv. Funct. Mater., 2021, 31, 1–22 CrossRef.
- G. V. Research, Tissue Engineering Market Size, Share & Trends Analysis Report By Application (Cord Blood & Cell Banking, Cancer, GI & Gynecology, Dental, Orthopedics, Musculoskeletal, & Spine), By Region, And Segment Forecasts, 2020–2027.
- N. Abbasi, S. Hamlet, R. M. Love and N.-T. Nguyen, J. Sci.: Adv. Mater. Devices, 2020, 5, 1–9 Search PubMed.
- M. A. Fernandez-Yague, S. A. Abbah, L. McNamara, D. I. Zeugolis, A. Pandit and M. J. Biggs, Adv. Drug Delivery Rev., 2015, 84, 1–29 CrossRef CAS PubMed.
- J. Henkel and D. W. Hutmacher, BioNanoMaterials, 2013, 14, 171–193 Search PubMed.
- Z. Hao, Z. Song, J. Huang, K. Huang, A. Panetta, Z. Gu and J. Wu, Biomater. Sci., 2017, 5, 1382–1392 RSC.
- M. I. Echeverria Molina, K. G. Malollari and K. Komvopoulos, Front. Bioeng. Biotechnol., 2021, 9, 617141 CrossRef PubMed.
- Y. Jiao, C. Li, L. Liu, F. Wang, X. Liu, J. Mao and L. Wang, Biomater. Sci., 2020, 8, 3574–3600 RSC.
- H. G. Senel-Ayaz, Y.-E. Har-El, H. Ayaz and P. I. Lelkes, Functional 3D Tissue Engineering Scaffolds, 2018, pp. 175–201. DOI:10.1016/b978-0-08-100979-6.00008-2.
- M. Persson, P. P. Lehenkari, L. Berglin, S. Turunen, M. A. J. Finnila, J. Risteli, M. Skrifvars and J. Tuukkanen, Sci. Rep., 2018, 8, 10457 CrossRef PubMed.
- A. Pagan, S. D. Aznar-Cervantes, J. Perez-Rigueiro, L. Meseguer-Olmo and J. L. Cenis, J. Biomed. Mater. Res., Part B, 2019, 107, 2209–2215 CrossRef CAS PubMed.
- D. P. Martin, A. Badhwar, D. V. Shah, S. Rizk, S. N. Eldridge, D. H. Gagne, A. Ganatra, R. E. Darois, S. F. Williams, H. C. Tai and J. R. Scott, J. Surg. Res., 2013, 184, 766–773 CrossRef CAS PubMed.
- M. A. Brennan, A. Renaud, A. L. Gamblin, C. D'Arros, S. Nedellec, V. Trichet and P. Layrolle, Biomed. Mater., 2015, 10, 045019 CrossRef PubMed.
- Y. Gao, W. Shao, W. Qian, J. He, Y. Zhou, K. Qi, L. Wang, S. Cui and R. Wang, Mater. Sci. Eng., C, 2018, 84, 195–207 CrossRef CAS PubMed.
- M. Akbari, A. Tamayol, S. Bagherifard, L. Serex, P. Mostafalu, N. Faramarzi, M. H. Mohammadi and A. Khademhosseini, Adv. Healthcare Mater., 2016, 5, 751–766 CrossRef CAS PubMed.
- X. Wang, C. Han, X. Hu, H. Sun, C. You, C. Gao and Y. Haiyang, J. Mech. Behav. Biomed. Mater., 2011, 4, 922–932 CrossRef CAS PubMed.
- B. Schäfer, C. Emonts, N. Glimpel, T. Ruhl, A. S. Obrecht, S. Jockenhoevel, T. Gries, J. P. Beier and A. Blaeser, Materials, 2020, 13, 1–16 CrossRef PubMed.
- V. P. Ribeiro, J. Silva-Correia, A. I. Nascimento, A. da Silva Morais, A. P. Marques, A. S. Ribeiro, C. J. Silva, G. Bonifacio, R. A. Sousa, J. M. Oliveira, A. L. Oliveira and R. L. Reis, Biomaterials, 2017, 123, 92–106 CrossRef CAS PubMed.
- F. Buchanan, Degradation rate of bioresorbable materials: Prediction and evaluation, Woodhead Publishing Limited, 2008 Search PubMed.
- M. A. Ghalia and Y. Dahman, J. Polym. Res., 2017, 24, 1–22 CrossRef CAS.
- N. A. Weir, F. J. Buchanan, J. F. Orr and G. R. Dickson, Proc. Inst. Mech. Eng., Part H, 2004, 218, 307–319 CrossRef CAS PubMed.
- K. Polak-Krasna, A. R. Abaei, R. N. Shirazi, E. Parle, O. Carroll, W. Ronan and T. J. Vaughan, J. Mech. Behav. Biomed. Mater., 2021, 118, 104409 CrossRef CAS PubMed.
- R. N. Shirazi, W. Ronan, Y. Rochev and P. McHugh, J. Mech. Behav. Biomed. Mater., 2016, 54, 48–59 CrossRef PubMed.
- V. B. Gupta and V. K. Kothari, Manufactured Fibre Technology, Springer Science+Business Media, 1997 Search PubMed.
- O. Avinc and A. Khoddami, Fibre Chem., 2010, 42, 68–78 CrossRef CAS.
- G. Ertekin and A. Marmarali, J. Eng. Fibers Fabr., 2016, 11, 64–71 Search PubMed.
- C. Emonts, M.Sc., RWTH Aachen University, 2018.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- DIN:EN:ISO-139, Textiles – Standard atmospheres for conditioning and testing, 2011.
- Y. Liu and H. Hu, J. Eng. Fibers Fabr., 2014, 9, 61–69 Search PubMed.
- ISO13781, Implants for surgery – Homopolymers, copolymers and blends on poly(lactide) – In vitro degradation testing, 2017.
- C. M. Agrawal, D. Huang, J. P. Schmitz and K. A. Athanasiou, Tissue Eng., 1997, 3, 345–352 CrossRef CAS.
- N. A. Weir, F. J. Buchanan, J. F. Orr, D. F. Farrar and G. R. Dickson, Proc. Inst. Mech. Eng., Part H, 2004, 218, 321–330 CrossRef CAS PubMed.
- ISO2062:2009, Textiles – Yarns from packages – Determination of single-end breaking force and elongation at break using constant rate of extension (CRE) tester (ISO 2062:2009), German version EN ISO 2062:2009, 2009.
- ISO11357-3:2018, Plastics – Differential scanning calorimetry (DSC) – Part 3: Determination of temperature and enthalpy of melting and crystallization, 2013.
- E. W. Fischer, H. J. Sterzel and G. Wegner, Kolloid Z. Z. Polym., 1973, 251, 980–990 CrossRef CAS.
- G. Kister, G. Cassanas and M. Vert, Polymer, 1998, 39, 267–273 CrossRef CAS.
- A. Gleadall, J. Pan and H. Atkinson, Polym. Degrad. Stab., 2012, 97, 1616–1620 CrossRef CAS.
- H. Tsuji, A. Mizuno and Y. Ikada, J. Appl. Polym. Sci., 2000, 77, 1452–1464 CrossRef CAS.
- ISO10993-5, Biological evaluation of medical devices – Part 5: Tests for in vitro cytotoxicity, 2009.
- ISO10993-12, Biological evaluation of medical devices – Part 12: Sample preparation and reference materials, 2012.
- ASTM-F2902, ASTM Standard-F2902, Assessment of Absorbable Polymeric Implants, 2016.
- S. H. Bae, J. H. Che, J. M. Seo, J. Jeong, E. T. Kim, S. W. Lee, K. I. Koo, G. J. Suaning, N. H. Lovell, D. I. Cho, S. J. Kim and H. Chung, Invest. Ophthalmol. Visual Sci., 2012, 53, 2653–2657 CrossRef CAS PubMed.
- Y. Liu, H. Hu, L. Zhao and H. Long, Text. Res. J., 2011, 82, 11–20 CrossRef.
- C. Fiuza, K. Polak-Krasna, L. Antonini, L. Petrini, O. Carroll, W. Ronan and T. J. Vaughan, J. Mech. Behav. Biomed. Mater., 2021, 125, 104955 CrossRef PubMed.
- M. L. Di Lorenzo and R. Androsch, Polym. Int., 2019, 68, 320–334 CrossRef CAS.
- R. C. Zhang, D. Sun, A. Lu, M. Zhong, G. Xiong and Y. Wan, Polymers, 2017, 9, 1–16 CrossRef PubMed.
- R. Xu, J. Xie and C. Lei, RSC Adv., 2017, 7, 39914–39921 RSC.
- P. Pan, J. Yang, G. Shan, Y. Bao, Z. Weng, A. Cao, K. Yazawa and Y. Inoue, Macromolecules, 2011, 45, 189–197 CrossRef.
- H. Tsuji and Y. Ikada, Polym. Degrad. Stab., 2000, 67, 179–189 CrossRef CAS.
- H. G. Senel Ayaz, A. Perets, H. Ayaz, K. D. Gilroy, M. Govindaraj, D. Brookstein and P. I. Lelkes, Biomaterials, 2014, 35, 8540–8552 CrossRef CAS PubMed.
- T. Schmidt, Y. Xiang, X. Bao and T. Sun, Processes, 2021, 9, 1–17 Search PubMed.
- R. H. Yang and C. W. Kan, Fibers Polym., 2013, 14, 1347–1353 CrossRef CAS.
- Q. L. Loh and C. Choong, Tissue Engineering Part B: Reviews, 2013, 19, 485–502 CrossRef CAS PubMed.
- X. Wang, T. Lou, W. Zhao, G. Song, C. Li and G. Cui, J. Biomater. Appl., 2016, 30, 1545–1551 CrossRef CAS PubMed.
- G. Narayanan, V. N. Vernekar, E. L. Kuyinu and C. T. Laurencin, Adv. Drug Delivery Rev., 2016, 107, 247–276 CrossRef CAS PubMed.
- T. Winkler, F. A. Sass, G. N. Duda and K. Schmidt-Bleek, Bone Joint Res., 2018, 7, 232–243 CrossRef CAS PubMed.
- R. Shine, R. Neghabat Shirazi, W. Ronan, C. A. Sweeney, N. Kelly, Y. A. Rochev and P. E. McHugh, J. Med. Devices, 2017, 11, 1–12 CrossRef PubMed.
- X. Yuan, A. F. T. Mak and K. Yao, J. Appl. Polym. Sci., 2002, 85, 936–943 CrossRef CAS.
- D. G. Tamay, T. Dursun Usal, A. S. Alagoz, D. Yucel, N. Hasirci and V. Hasirci, Front. Bioeng. Biotechnol., 2019, 7, 164 CrossRef PubMed.
- B. Wulfhorst, T. Gries and D. Veit, Textile Technology, Hanser, 2006 Search PubMed.
- Y. Li, Z. Chu, X. Li, X. Ding, M. Guo, H. Zhao, J. Yao, L. Wang, Q. Cai and Y. Fan, Regener. Biomater., 2017, 4, 179–190 CrossRef CAS PubMed.
- H. Azevedo and R. L. Reis, in Biodegradable Systems in Tissue Engineering and Regenerative Medicine, ed. J. S. R. Rui and L. Reis, CRC Press, 2004, ch. 12 Search PubMed.
- E. M. Czekanska, M. J. Stoddart, R. G. Richards and J. S. Hayes, Eur. Cells Mater., 2012, 24, 1–17 CrossRef CAS PubMed.
- A. Entezari, M. V. Swain, J. J. Gooding, I. Roohani and Q. Li, Acta Biomater., 2020, 118, 100–112 CrossRef CAS PubMed.
- C. Simitzi, M. Vlahovic, A. Georgiou, Z. Keskin-Erdogan, J. Miller and R. M. Day, Front. Bioeng. Biotechnol., 2020, 8, 816 CrossRef PubMed.
- M. Wasyleczko, W. Sikorska and A. Chwojnowski, Membranes, 2020, 10, 1–28 CrossRef PubMed.
- F. Pu, N. P. Rhodes, Y. Bayon, R. Chen, G. Brans, R. Benne and J. A. Hunt, Biomaterials, 2010, 31, 4330–4340 CrossRef CAS PubMed.
- M. W. King, J. Chen, M. Deshpande, T. He, H. Ramakrishna, Y. Xie, F. Zhang and F. Zhao, Biotechnology and Bioengineering, 2019. DOI:10.5772/intechopen.84643.
| This journal is © The Royal Society of Chemistry 2022 |