
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe precise editing of surface sites on a molecular-like gold catalyst for modulating regioselectivity†
Shuohao
Li‡
,
Hongwei
Chen‡
,
Xu
Liu
 ,
Haoqi
Liu
,
Jing
Ma
* and
Yan
Zhu
*
,
Haoqi
Liu
,
Jing
Ma
* and
Yan
Zhu
*
School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China. E-mail: zhuyan@nju.edu.cn; majing@nju.edu.cn
First published on 21st July 2020
Abstract
It is extremely difficult to precisely edit a surface site on a typical nanoparticle catalyst without changing other parts of the catalyst. This precludes a full understanding of which site primarily determines the catalytic properties. Here, we couple experimental data collection with theoretical analysis to correlate rich structural information relating to atomically precise gold clusters with the catalytic performance for the click reaction of phenylacetylene and benzyl azide. We also identify a specific surface site that is capable of achieving high regioselectivity. We further conduct site-specific editing on a thiolate-protected gold cluster by peeling off two monomeric RS–Au–SR motifs and replacing them with two Ph2P–CH2–PPh2 staples. We demonstrate that the surface Au–Ph2P–CH2–PPh2–Au motifs enable extraordinary regioselectivity for the click reaction of alkyne and azide. The editing strategy for the surface motifs allows us to exploit previously inaccessible individual active sites and elucidate which site can explicitly govern the reaction outcome.
Introduction
The identification of catalytically active sites in particle catalysts has long been an important task in catalytic chemistry.1–3 In the sub-nanometer size regime, each site of a catalyst may be directly or indirectly involved in a reaction process.4–6 Hence, it is highly desirable to explicitly conclude the extents to which individual sites of a catalyst contribute to its overall catalytic performance. However, due to the complexity of conventional investigations, it remains an open challenge to directly observe the catalytic performance of each site in a typical catalyst; microscopy and spectroscopy techniques usually provide statistically averaged or sample-averaged information.7 Enlightened by organic chemists who can deliberately remove or replace any group in an organic molecule without changing other parts, tailoring a specific site without changing the other parts of a solid catalyst would allow us to unveil the contribution of the specific site to a given chemical reaction.Atomically precise metal clusters constitute a new generation of metal catalysts in catalysis science and exhibit unexpected catalytic properties.8–13 These metal clusters provide the potential to precisely correlate intricate structural information (such as aesthetic structural pattern, ligand effect, metal–metal bonding, metal–ligand interfacial bonding, and metal charge) with the catalytic properties, which can shed light on current mysteries in the field of catalysis.14–21 By solving their atomic structures, we can determine the active site structure and establish definitive structure–property relationships, which have previously been elusive.22 Such advances in these clusters inspire us to use them as proof-of-concept catalysts to pursue a fundamental understanding of how a specific site in a catalyst controls the catalytic properties.
In this work, the cycloaddition reaction of alkyne and azide by means of click chemistry, which has been widely applied to generate high-value organic compounds, was chosen as a probe reaction.23 A series of ligand-protected Aun (n = gold atom number) clusters with different atomic structures and surface motifs (see ESI for structural details, Fig. S1†) were screened to catalyze the click reaction. As shown in Fig. 1, these Aun clusters failed to provide regioselectivity for the 1,4- or 1,5-triazole products of the click reaction of phenylacetylene and benzyl azide. The above Aun clusters could not selectively drive the reaction process toward either of the two products. We thus questioned whether a purposeful editing on the surface motifs of the clusters might be used to manipulate the regioselectivity toward one product. If successful, this promising strategy could be developed to precisely modulate the surface active sites of catalysts to obtain an exclusive product through an operative process.
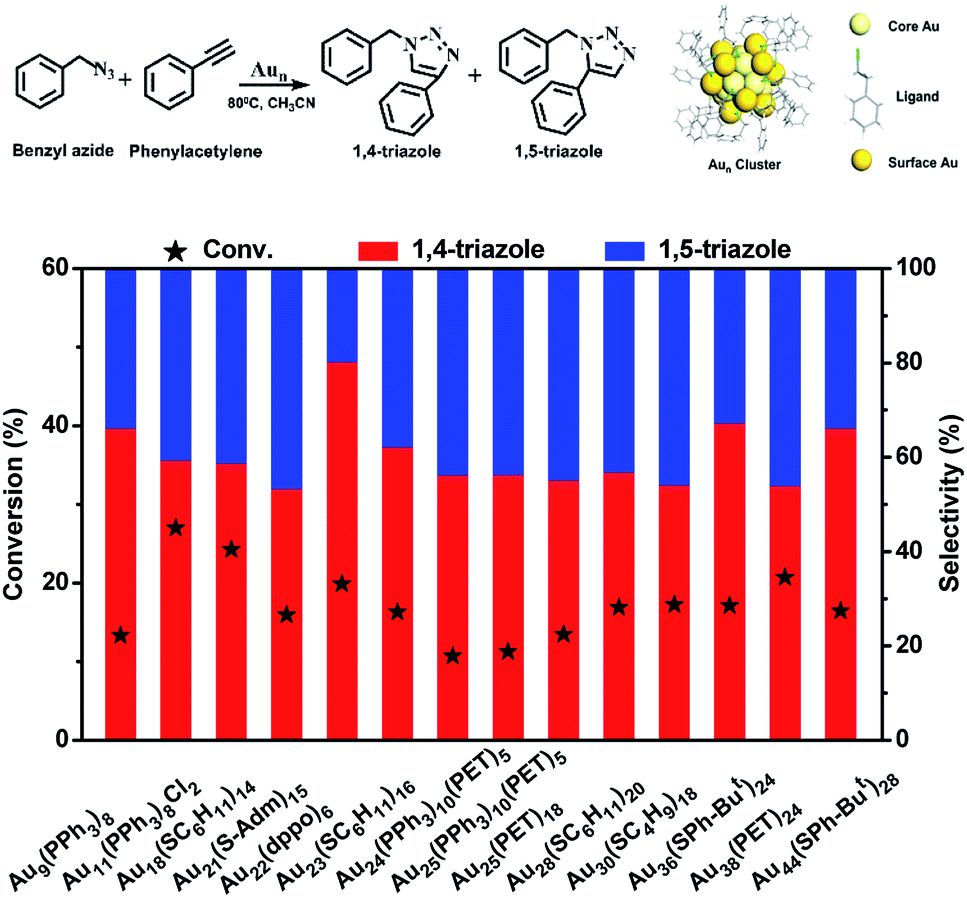 | ||
| Fig. 1 The catalytic performances of ligand-protected Aun catalysts for the click reaction of phenylacetylene and benzyl azide. Reaction conditions: 2 mg of Aun, phenylacetylene (0.15 mmol), benzyl azide (0.15 mmol), 2 ml of acetonitrile, 80 °C, 5 h. The upper panel shows the scheme for the click reaction of phenylacetylene and benzyl azide and a general model of Aun clusters. PET = 2-phenylethanethiol; S-Adm = 1-adamantanethiolate; dppo = 1,8-bis(diphenyl-phosphino)octane. | ||
Results and discussion
Given the complexity of the geometric structures and surface structures of the Aun clusters, it is difficult to rapidly identify the key structures in inducing high regioselectivity toward an exclusive product. Therefore, we analyzed the relationships between the structural information of the Aun clusters with the reaction activity and selectivity using existing reaction data (Fig. 1) to aid in establishing the structure–property relationship. Fig. 2a shows the effect of the surface gold atoms on the catalytic activity. In general, more surface gold atoms resulted in higher catalytic activity (more details of surface Au atoms are shown in Fig. S2†). It is interesting to consider the steric hindrance effect of the ligands on the regioselectivity of these Aun clusters protected by thiolates (Fig. 2b). Steric hindrance was qualitatively expressed by the surface area occupied by each ligand (i.e., the ratio of the total van der Waals surface area to the number of ligands of a cluster, referred to as the surface area per ligand). The steric hindrance effect of the ligands can also be illustrated by the electrostatic potentials of the Aun clusters determined by density functional theory (DFT) calculations,24 as shown in Fig. 2b and c (more details can be found in Fig. S3 and Table S1†). On the other hand, in the systems of the Aun clusters capped by PPh3 ligands, the steric hindrance is larger than in the thiolate-protected clusters. The Aun clusters capped by PPh3 ligands show larger surface areas per ligand and much narrower pockets for reactant docking (compared Fig. 2b and c). More interestingly, the steric hindrance from PPh3 ligands is strongly correlated to the regioselectivity of the 1,4-product (Fig. 2c). Thus, the results suggest that the Aun clusters capped by phosphine ligands relative to thiolates are prone to driving the click reaction pathway toward the 1,4-product compared to Aun clusters capped by thiolates.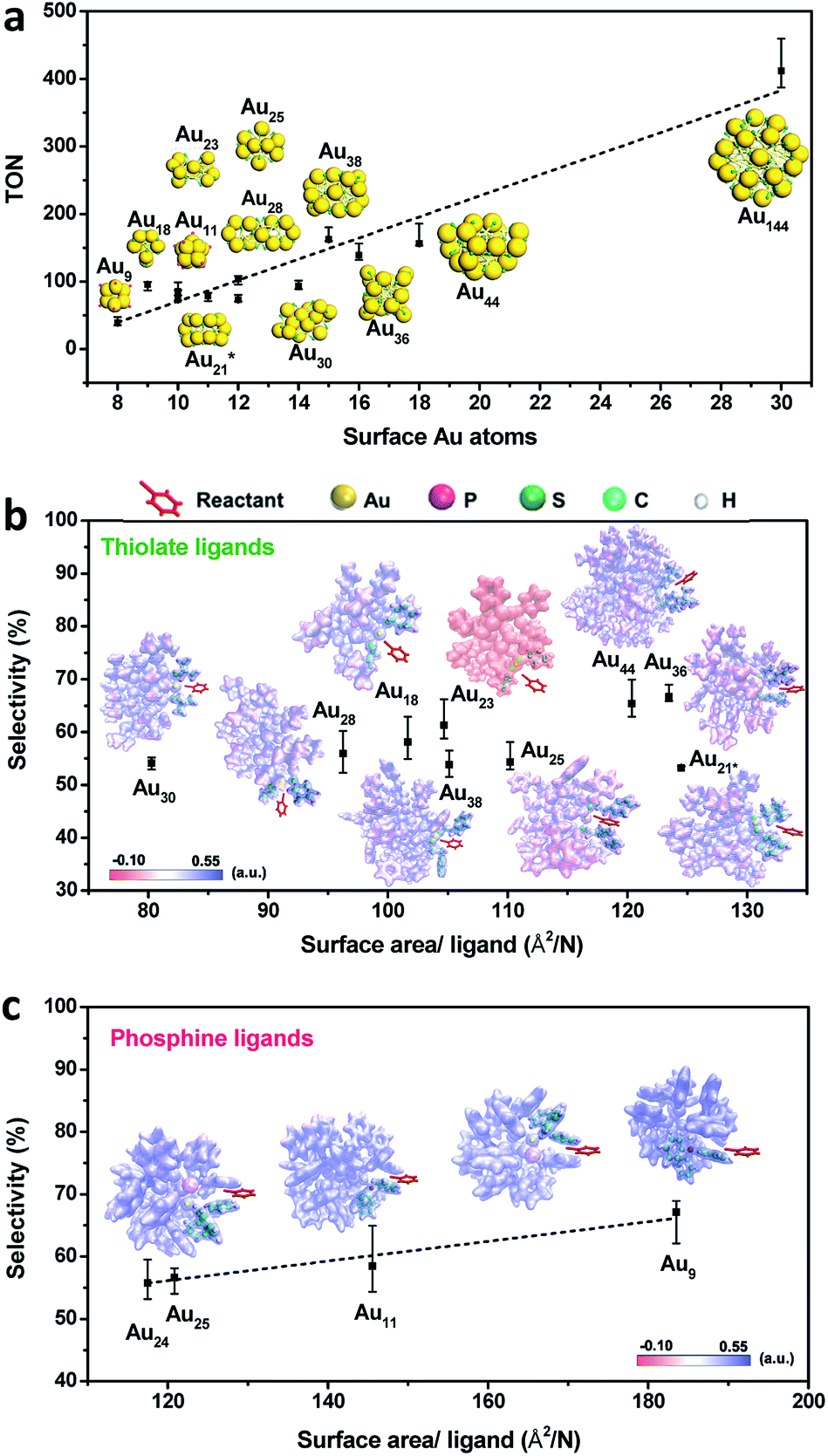 | ||
| Fig. 2 (a) Correlation between the catalytic activity and the surface Au atoms of Aun clusters. The insets show the atomic structures of the clusters. Color codes: yellow = Au; green = S; pink = P. (b) The relationship between the catalytic selectivity for 1,4-triazole and steric hindrance in thiolate-protected Aun clusters. Au21* = Au21(S-Adm)15. (c) The relationship between the catalytic selectivity for 1,4-triazole and the steric hindrance in PPh3-protected Aun clusters. The insets in (b) and (c) illustrate the electrostatic potentials of Aun clusters from DFT calculations. | ||
Inspired by the workable molecular tailoring of the Au23(SR)16 cluster,25 the specific surface site containing phosphine ligands (Au–Ph2P–CH2–PPh2–Au) was selected to enable regioselectivity in the click reaction. As illustrated in Fig. 3a, the Au23(SR)16 cluster possesses an Au13 cuboctahedron protected by two trimeric Au3(SR)4 staple motifs, two monomeric RS–Au–SR staples, and four bridging SR thiol ligands.26 By the resection of two monomeric RS–Au–SR motifs of the Au23(SR)16 cluster (abbreviated as Au23) and bandaging the gaps with two Ph2P–CH2–PPh2 staples, the Au21(SR)12(Ph2PCH2PPh2)2 cluster (abbreviated as Au21) was obtained (Fig. 3a). The two clusters showed similar UV-vis optical spectra (Fig. S4†) and hold similar atomic structures, with the exception that RS–Au–SR is replaced by Ph2P–CH2–PPh2. As shown in Fig. 3b, the peak corresponding to the mass/charge ratio (m/z) of 6371.09 was observed in the electrospray ionization mass spectrum (ESI-MS), corresponding to [Au23(SC6H11)16]−. The peak at m/z = 6285.3 in Fig. 3c was assigned to [Au21(SC6H11)12(Ph2PCH2PPh2)2]+.
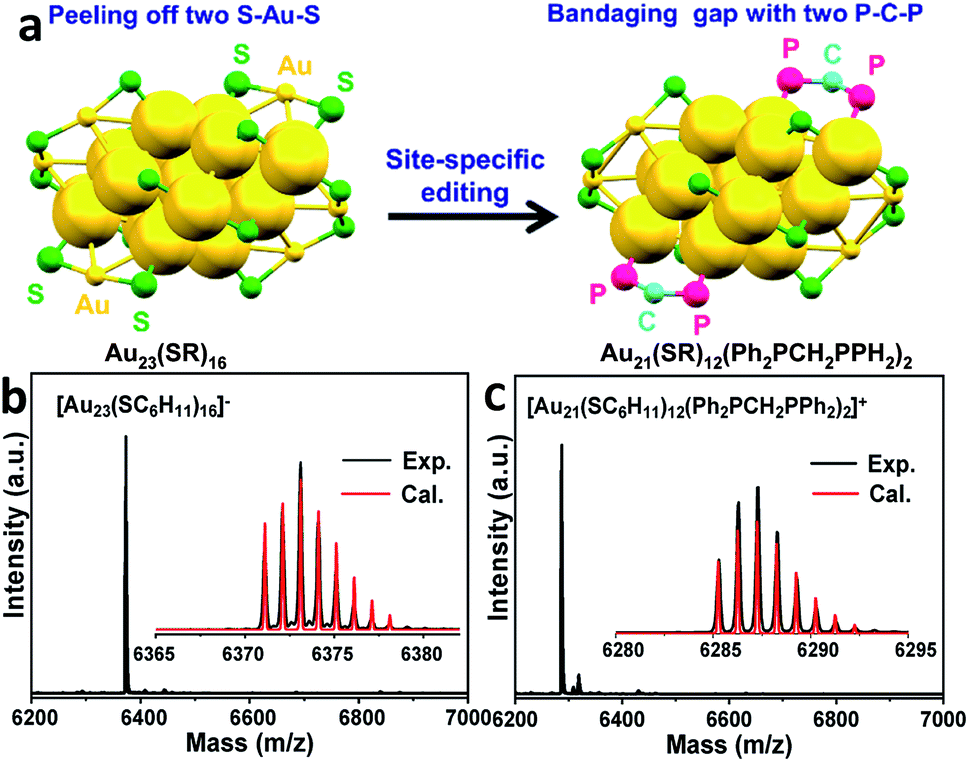 | ||
| Fig. 3 (a) A schematic diagram of the site-specific editing of Au23(SR)16 by resecting two RS–Au–SR motifs and bandaging the gaps with two Ph2P–CH2–PPh2 staples to obtain Au21(SR)12(Ph2PCH2PPh2)2. Color codes: yellow = Au; green = S; pink = P; blue = C. Carbon tails are omitted for clarity. SR = cyclohexanethiol. The ESI-MS spectra of (b) [Au23(SC6H11)16]− and (c) [Au21(SC6H11)12(Ph2PCH2PPh2)2]+. | ||
As shown in Fig. 4a, Au21(SC6H11)12(Ph2PCH2PPh2)2 exhibited a high selectivity for 1,4-triazoles. In contrast, the Au23(SC6H11)16 cluster showed no regioselectivity for 1,4- or 1,5-triazoles under identical reaction conditions. The replacement of two surface RS–Au–SR motifs by two Ph2P–CH2–PPh2 motifs successfully tuned the reaction pathway of the click reaction, leading to the transformation from non-regioselectivity to regioselectivity accompanied by the transformation from Au23(SC6H11)16 to Au21(SC6H11)12(Ph2PCH2PPh2)2. Additionally, Au21(SC6H11)12(Ph2PCH2PPh2)2 exhibited better activity than Au23(SC6H11)16 (Fig. 4a), which may be attributed to the stronger chemical adsorption capability of the former catalyst, as deduced from in situ time-resolved infrared Fourier transform infrared (FTIR) spectroscopic analysis of CO molecules adsorbed onto the two catalysts (Fig. S5†). The two clusters seemed robust during the reactions (Fig. S6 and S7†). Furthermore, the relationship between regioselectivity and steric hindrance in the Au21(SC6H11)12(Ph2PCH2PPh2)2 and Au23(SC6H11)16 systems was corroborated by the analysis of the surface area/ligand ratio. The steric hindrance from the organic ligands can be significantly increased by the site-specific replacement of SR–Au–SR with Ph2P–CH2–PPh2 (Fig. 4b), favoring an increase in regioselectivity toward 1,4-products.
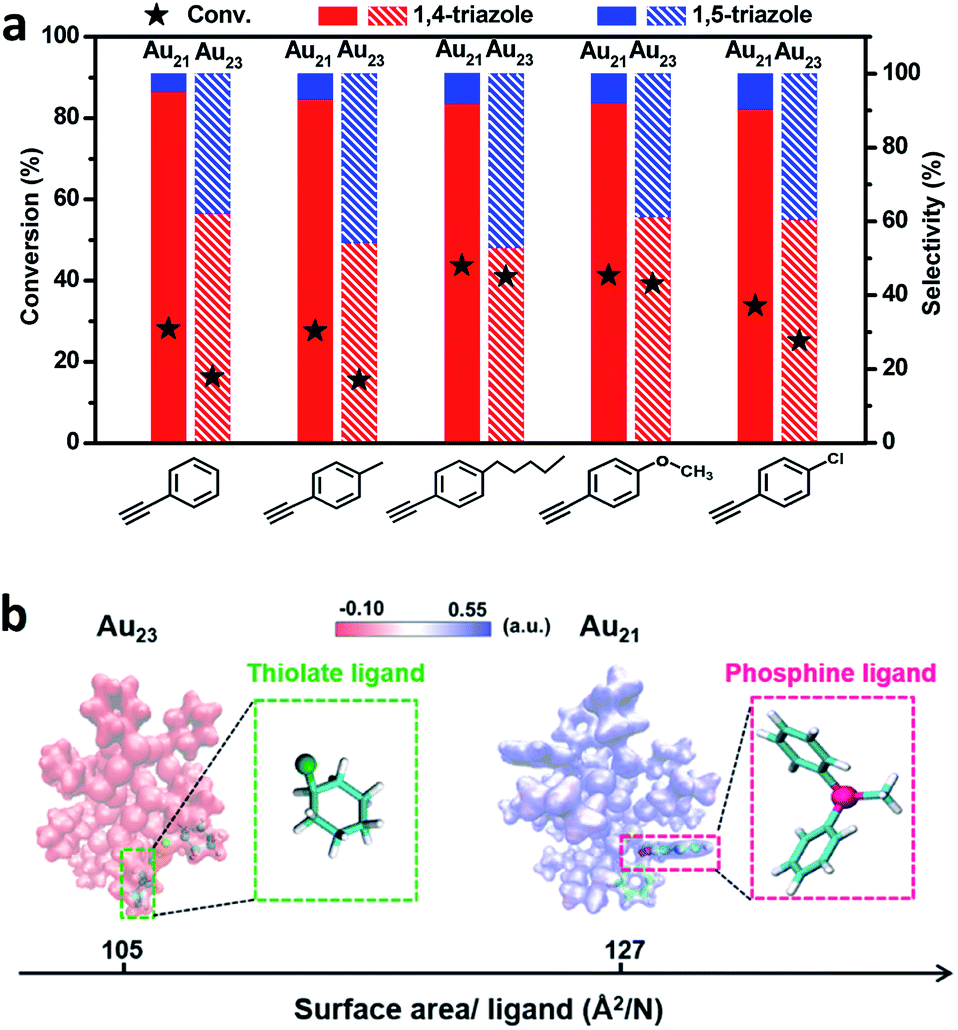 | ||
| Fig. 4 (a) The catalytic performances of Au21(SC6H11)12(Ph2PCH2PPh2)2 and Au23(SC6H11)16 for the click reactions of alkynes and benzyl azide. The catalytic data are averaged over three independent measurements. (b) Steric hindrance from the surface ligands in the Au21(SC6H11)12(Ph2PCH2PPh2)2 and Au23(SC6H11)16 clusters. Color codes: yellow = Au; pink = P; green = S. | ||
We now rationalize how Au21(SC6H11)12(Ph2PCH2PPh2)2 can achieve regioselectivity while Au23(SC6H11)16 cannot. First, the distinct selectivity cannot be attributed to the electronic structures of the two clusters because the exchange of surface motifs had little influence on the electronic properties of the clusters. As presented in Fig. 5a, the binding energies of the Au 4f X-ray photoelectron spectroscopy (XPS) peaks of Au21(SC6H11)12(Ph2PCH2PPh2)2 were not noticeably difference from those of Au23(SC6H11)16. The gold charge states were also monitored by X-ray absorption near-edge structure (XANES) studies, indicating that the gold charge state of Au21 was similar to that of Au23 based on liner combination fits (Fig. 5b and S8†). This means that tailoring the specific motifs did not cause a significant perturbation in the electronic structures of the two gold clusters.
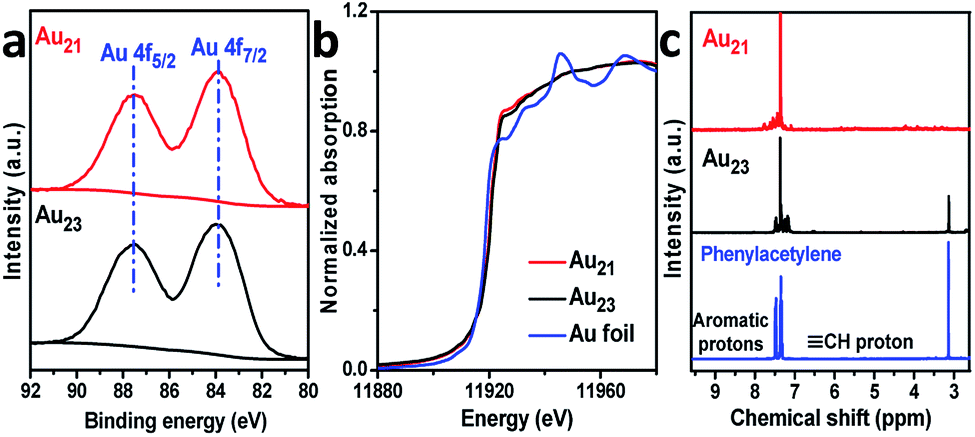 | ||
| Fig. 5 (a) Au 4f XPS and (b) Au L3-edge XANES profiles of Au21(SR)12(Ph2PCH2PPh2)2 and Au23(SR)16. (c) The 1H NMR spectra of Au-acetylide species. | ||
Going further, the Au-acetylide intermediates on the Au21(SC6H11)12(Ph2PCH2PPh2)2 and Au23(SC6H11)16 clusters were studied by 1H nuclear magnetic resonance (NMR) spectroscopy. Free phenylacetylene showed a singlet proton peak (δ = 3.1 ppm) and two sets of peaks (δ = 7.3 and 7.5 ppm) for the aromatic hydrogen atoms (Fig. 5c).27 Of note, the acetylenic proton peak of phenylacetylene adsorbed on the Au21(SC6H11)12(Ph2PCH2PPh2)2 cluster was nearly absent, excluding the binding mode of phenylacetylene in the intact form (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H) and supporting its binding mode in the dehydrogenated form (CC) (Fig. S9†). In the Au23(SC6H11)16 system, the peak of the C–H proton is observed (Fig. 5c), although the peak intensity is weak, implying that the deprotonation-induced activation process of phenylacetylene was more difficult for Au23(SC6H11)16 than for Au21(SC6H11)12(Ph2PCH2PPh2)2. Taken together, the results imply that two binding modes of alkyne on Au23(SC6H11)16 were obtained: terminal binding and internal binding (Fig. S9†).
C–H) and supporting its binding mode in the dehydrogenated form (CC) (Fig. S9†). In the Au23(SC6H11)16 system, the peak of the C–H proton is observed (Fig. 5c), although the peak intensity is weak, implying that the deprotonation-induced activation process of phenylacetylene was more difficult for Au23(SC6H11)16 than for Au21(SC6H11)12(Ph2PCH2PPh2)2. Taken together, the results imply that two binding modes of alkyne on Au23(SC6H11)16 were obtained: terminal binding and internal binding (Fig. S9†).
To further understand the distinct catalytic regioselectivity of the Au21(SC6H11)12(Ph2PCH2PPh2)2 and Au23(SC6H11)16 clusters for the click reaction of phenylacetylene and benzyl azide, we performed DFT calculations to explore the possible reaction pathways of 1,4- and 1,5-product formation on the two clusters. The overall pathways may include the adsorption, coupling, and reductive elimination of azide and phenylacetylene. Fig. 6 shows the calculated energy profiles for the possible reaction pathways of phenylacetylene and benzyl azide in the Au21(SC6H11)12(Ph2PCH2PPh2)2 model (the all-atom model, abbreviated as A-Au21) and the Au23(SC6H11)16 model (A-Au23) along with the calculated reaction barriers. For the A-Au21 catalyst (Fig. 6 and S10†), the energy difference between the activation barrier for the pathway toward the 1,4-product (ΔG1,4 = 12.2 kcal mol−1) and that for the pathway toward the 1,5-product (ΔG1,5 = 30.0 kcal mol−1) was large (ΔΔG = 17.8 kcal mol−1). Thus, the formation of 1,4-triazole on the Au21 catalyst is kinetically favored, resulting in regioselectivity on Au21. The larger the energy difference ΔΔG, the greater the selectivity for the 1,4-triazole. The large energy difference is related to the oxidative coupling of phenylacetylene and benzyl azide, which results in the generation of the six-membered intermediate IN1 (Fig. S11†). For A-Au21-IN11,4, the carbon atom on phenylacetylene is firmly bound by two gold atoms, resulting in a more stable intermediate, while the carbon atom is only captured by one gold atom in the intermediate A-Au21-IN11,5 (Fig. S11†). The click reaction occurs preferably on the Au–AuP–P–P–AuP–Au (AuP denotes the Au atom attached to the phosphine ligand) and Au–S–Au sites of the Au21 catalyst. In contrast, for the A-Au23 catalyst (Fig. 6 and S10†), the energy difference between the 1,4-product and 1,5-product is relatively small (ΔΔG = 8.6 kcal mol−1), leading to comparable selectivity for the formation of the 1,4- and 1,5-triazoles. This rationalizes the non-regioselectivity of the Au23 cluster. Similar trends were observed for simplified models of Au21 and Au23 (S-Au21 and S-Au23; Fig. S12 and S13†), for which the energy differences between the pathways toward the 1,4-product and the 1,5-product over the S-Au21 and S-Au23 catalysts were 19.2 and 3.1 kcal mol−1 (Fig. S14†), respectively, in good agreement with the experimental results.
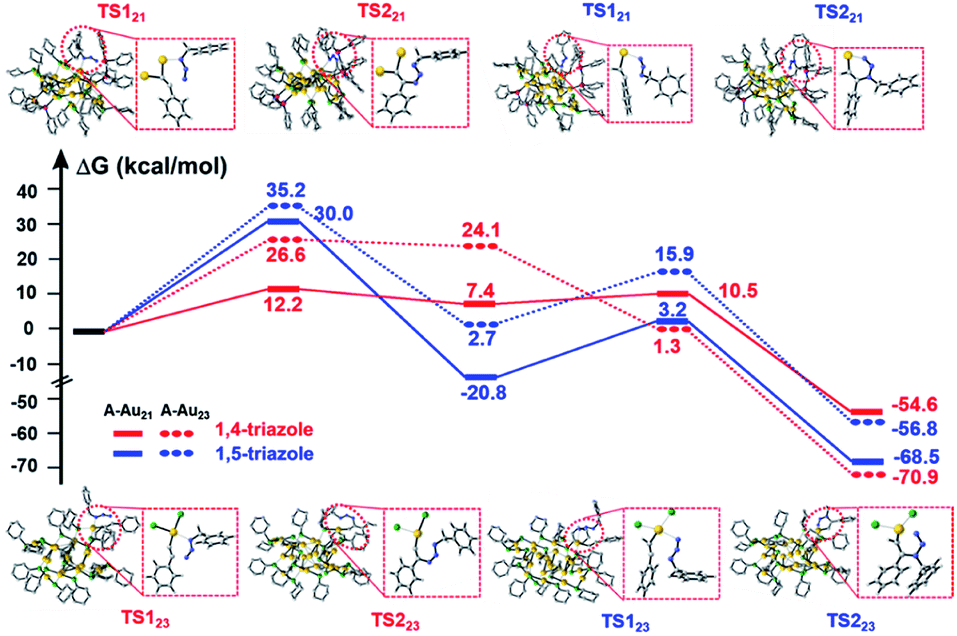 | ||
| Fig. 6 DFT-calculated energy profiles for the reaction pathways of Au21 and Au23 clusters with the all-atom models, termed A-Au21 and A-Au23. The insets indicate the reactive sites in the transition states. Color codes: yellow = Au, green = S, pink = P, gray = C, blue = N. To improve clarity, all the hydrogen atoms are hidden. TS = transition state. | ||
Conclusions
In summary, we have demonstrated that site-specific editing on an Au23(SC6H11)16 cluster via the exchange of the surface RS–Au–SR motifs with Ph2P–CH2–PPh2 staples can achieve a transformation from non-regioselectivity to regioselectivity for the click reaction of azide and alkyne. The editing strategy for surface motifs offers an exciting opportunity to elucidate how a surface site of a catalyst can precisely control the reaction pathway. This work not only improves the understanding of the contribution of a single site in a solid catalyst to the overall catalytic performance, but it can also guide the development of a feasible strategy to enable highly efficient chemical processes based on the editing of surface sites of catalysts with atomic precision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (21773109, 91845104, 21673111, 21873045).Notes and references
- M. Chen and D. W. Goodman, Chem. Soc. Rev., 2008, 37, 1860–1870 RSC.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zeleny, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- S. Xie, H. Tsunoyama, W. Kurashige, Y. Negishi and T. Tsukuda, ACS Catal., 2012, 2, 1519–1523 CrossRef CAS.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef PubMed.
- Y. Liu, X. Chai, X. Cai, M. Chen, R. Jin, W. Ding and Y. Zhu, Angew. Chem., Int. Ed., 2018, 57, 9775–9779 CrossRef CAS PubMed.
- Y. Zhang, P. Song, T. Chen, X. Liu, T. Chen, Z. Wu, Y. Wang, J. Xie and W. Xu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10588–10593 CrossRef CAS PubMed.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981–983 CrossRef CAS PubMed.
- E. C. Tyo and S. Vajda, Nat. Nanotechnol., 2015, 10, 577–588 CrossRef CAS PubMed.
- X. Wan, J. Wang, Z. Nan and Q. Wang, Sci. Adv., 2017, 3, e1701823 CrossRef PubMed.
- Y. Tan, X. Liu, L. Zhang, A. Wang, L. Li, X. Pan, S. Miao, M. Haruta, H. Wei, H. Wang, F. Wang, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2017, 56, 2709–2713 CrossRef CAS PubMed.
- H. Chen, C. Liu, M. Wang, C. Zhang, N. Luo, Y. Wang, H. Abroshan, G. Li and F. Wang, ACS Catal., 2017, 7, 3632–3638 CrossRef CAS.
- H. Zhang, H. Liu, Z. Tian, D. Lu, Y. Yu, S. Cestellos-Blanco, K. K. Sakimoto and P. Yang, Nat. Nanotechnol., 2018, 13, 900–905 CrossRef CAS PubMed.
- Y. Wang, X. Wan, L. Ren, H. Su, G. Li, S. Malola, S. Lin, Z. Tang, H. Häkkinen, B. K. Teo, Q. Wang and N. Zheng, J. Am. Chem. Soc., 2016, 138, 3278–3281 CrossRef CAS PubMed.
- Y. Zhu, H. Qian and R. Jin, J. Mater. Chem., 2011, 21, 6793–6799 RSC.
- T. Imaoka, H. Kitazawa, W. Chun, S. Omura, K. Albrecht and K. Yamamoto, J. Am. Chem. Soc., 2013, 135, 13089–13095 CrossRef CAS PubMed.
- W. Chen and S. Chen, Angew. Chem., Int. Ed., 2009, 48, 4386–4389 CrossRef CAS PubMed.
- C. Liu, X. Ren, F. Lin, X. Fu, X. Lin, T. Li, K. Sun and J. Huang, Angew. Chem., Int. Ed., 2019, 58, 11335–11339 CrossRef CAS PubMed.
- Q. Zhang, X. Chen and L. Wang, Acc. Chem. Res., 2018, 51, 2159–2168 CrossRef CAS PubMed.
- T. Imaoka, S. Tsuchiya and K. Yamamoto, Chem. Lett., 2016, 45, 1450–1452 CrossRef CAS.
- Y. Zhang, P. Song, T. Chen, X. Liu, T. Chen, Z. Wu, Y. Wang, J. Xie and W. Xu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10588–10593 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- W. Wang, X. Sun, J. Qu, X. Xie, Z. Qi, D. Hong, S. Jing, D. Zheng, Y. Tian, H. Ma, S. Yu and J. Ma, Phys. Chem. Chem. Phys., 2017, 19, 31443–31451 RSC.
- Q. Li, J. C. Russell, T. Luo, X. Roy, N. L. Rosi, Y. Zhu and R. Jin, Nat. Commun., 2018, 9, 3871 CrossRef PubMed.
- Q. Li, T. Luo, M. G. Taylor, S. Wand, X. Zhu, Y. Song, G. Mpourmpakis, N. L. Rosi and R. Jin, Sci. Adv., 2017, 3, e1603193 CrossRef PubMed.
- W. Chen, J. R. Davies, D. Ghosh, M. Tong, J. P. Konopelski and S. Chen, Chem. Mater., 2006, 18, 5253–5259 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02207a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |