
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ K-edge X-ray absorption spectroscopy of the ligand environment of single-site Au/C catalysts during acetylene hydrochlorination†
Grazia
Malta
 a,
Simon A.
Kondrat
b,
Simon J.
Freakley
c,
David J.
Morgan
a,
Emma K.
Gibson
de,
Peter P.
Wells
fg,
Matteo
Aramini
g,
Diego
Gianolio
g,
Paul B. J.
Thompson
hi,
Peter
Johnston
j and
Graham J.
Hutchings
*a
a,
Simon A.
Kondrat
b,
Simon J.
Freakley
c,
David J.
Morgan
a,
Emma K.
Gibson
de,
Peter P.
Wells
fg,
Matteo
Aramini
g,
Diego
Gianolio
g,
Paul B. J.
Thompson
hi,
Peter
Johnston
j and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: hutch@cardiff.ac.uk
bDepartment of Chemistry, Loughborough University, Loughborough, Leicestershire LE11 3TU, UK
cDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK
dSchool of Chemistry, University of Glasgow, Joseph Black Building, Glasgow G12 8QQ, UK
eUK Catalysis Hub, Research Complex at Harwell, RAL, Oxford, OX11 0FA, UK
fSchool of Chemistry, University of Southampton, Southampton, SO17 1BJ, UK
gDiamond Light Source, Harwell Science and Innovation Campus, Chilton, Didcot OX11 0DE, UK
hXMaS, UK CRG, ESRF, 71 Avenue des Martyrs, 38043 Grenoble, France
iDepartment of Physics, University of Liverpool, Oliver Lodge Laboratory, Liverpool L69 7ZE, UK
jProcess Technologies, Johnson Matthey PLC, Billingham, TS23 1LB, UK
First published on 24th June 2020
Abstract
The replacement of HgCl2/C with Au/C as a catalyst for acetylene hydrochlorination represents a significant reduction in the environmental impact of this industrial process. Under reaction conditions atomically dispersed cationic Au species are the catalytic active site, representing a large-scale application of heterogeneous single-site catalysts. While the metal nuclearity and oxidation state under operating conditions has been investigated in catalysts prepared from aqua regia and thiosulphate, limited studies have focused on the ligand environment surrounding the metal centre. We now report K-edge soft X-ray absorption spectroscopy of the Cl and S ligand species used to stabilise these isolated cationic Au centres in the harsh reaction conditions. We demonstrate the presence of three distinct Cl species in the materials; inorganic Cl−, Au–Cl, and C–Cl and how these species evolve during reaction. Direct evidence of Au–S interactions is confirmed in catalysts prepared using thiosulfate precursors which show high stability towards reduction to inactive metal nanoparticles. This stability was clear during gas switching experiments, where exposure to C2H2 alone did not dramatically alter the Au electronic structure and consequently did not deactivate the thiosulfate catalyst.
Introduction
The commercialisation of carbon-supported gold catalysts (Au/C), as a replacement for toxic mercuric chloride (HgCl2/C), represents a significant reduction in environmental impact of large scale vinyl chloride monomer (VCM) production via acetylene hydrochlorination.1–4 Since the prediction that Au would be an effective catalyst,5 intensive studies to understand and optimise these catalysts have been on-going.6–10 These catalysts were originally developed using strongly acidic and oxidising solvents, including impregnation of HAuCl4 from aqua regia (Au/C-AR).11,12 The preparation and use of such Au/C catalysts at an industrial scale represents an economic and technical challenge, which hindered the validation of this type of catalyst. Moreover, the high activity of these Au/C materials is often coupled with poor lifetimes under extended testing. The reduction of the active cationic Au to metallic nanoparticles and the development of acetylene-oligomers on the catalyst surface at strong acid sites introduced from the aqua regia are the primary deactivation mechanisms.1,13 In comparison, catalyst preparation from aqueous HAuCl4 results in the formation of large Au nanoparticles which have limited activity.14 Alternative solvents to aqua regia, such as “organic aqua regia (OAR)”, have been developed and utilised for the preparation of mono and bimetallic catalysts;15,16 however, despite the appreciable catalytic performances this still does not provide a solution to large scale catalyst synthesis.To obtain effective catalysts using aqueous metal-precursor solutions, strongly coordinating ligands are required to prevent nanoparticle formation.17,18 Johnston and collaborators,1,19 reported the use of soft donor ligands such as thiosulphate could produce a class of active and stable catalysts due to the increased stability constants of the Au–S species compared to the Au–Cl, avoiding the use of aggressive impregnation conditions. This catalyst (Au/C-S2O3) has been industrially validated as a replacement for the HgCl2/C. The synthesis is based on the in situ formation of a Au–thiosulfate complex before immobilisation onto carbon and the active form of the catalyst has been shown to consist of atomically dispersed cationic Au species under reaction conditions. We have previously demonstrated by operando X-ray Absorption Spectroscopy (XAS) that catalysts containing chloride or sulphur ligands had the same structure/activity correlation, with a Au(I) spectroscopic feature being proportional to VCM productivity and catalysts were shown to be comprise of atomically dispersed Au centres.13 The two catalysts (Au/C-AR and Au/C-S2O3) displayed differing induction periods; the Au/C-AR required 3 h to reach steady state activity, due to initial oxidation of Au(I) to Au(III) and subsequent re-equilibration of metal oxidation states, while Au/C-S2O3 achieved steady state immediately due to higher stability of the Au(I) species with a similar Au(I) to Au(III) ratio. In analogy with homogenous Au complexes,20 the choice of ligand and solvent, used during the catalyst preparation plays a major role in determining performance and stability.21,22
We now report a study using in situ K-edge S and Cl X-ray absorption spectroscopy, under relevant reaction conditions, to examine Cl and S speciation in the Au/C-AR and Au/C-S2O3 catalysts and correlate this to the observed catalytic activities. We show how the nature of the Cl and S evolve during the induction periods and at steady state to give information about the metal ligand environment and provide further information towards designing effective catalysts. The stability of the Au metal centre in the Au/C-S2O3 was studied in a operando Au L-edge XAS experiment, where reaction gases were switched to expose the catalyst to conditions that have already shown to deactivate the Au/C-AR catalyst.
Results and discussion
Initially, ex situ normalised Cl K-edge XANES spectra of fresh Au/C-AR and Au/C-S2O3 were compared (Fig. 1a). For comparative purposes a Au/C-H2O catalyst containing Au nanoparticles was also analysed (Fig. S1†). Three characteristic features, labelled as A, B and C, were observed in the XANES spectra of the catalysts with varying relative intensity. The absolute amount of Cl, determined from raw XANES spectra before data reduction23 differs in each sample (Fig. S2†); Au/C-AR has significantly more Cl (0.32) than Au/C-S2O3 (0.08) as expected from a preparation method using aqua regia compared to aqueous thiosulphate solution; the Au/C-H2O sample has lower amounts of Cl than either of the previous catalysts (0.03). These results are in accordance with Cl(2p) core-level XPS spectra (Fig. 1b and S3†), which verifies this difference in Cl concentration with Au/C-AR having 1.98 at% surface Cl and the Au/C-S2O3 0.25 at% (Table S1†).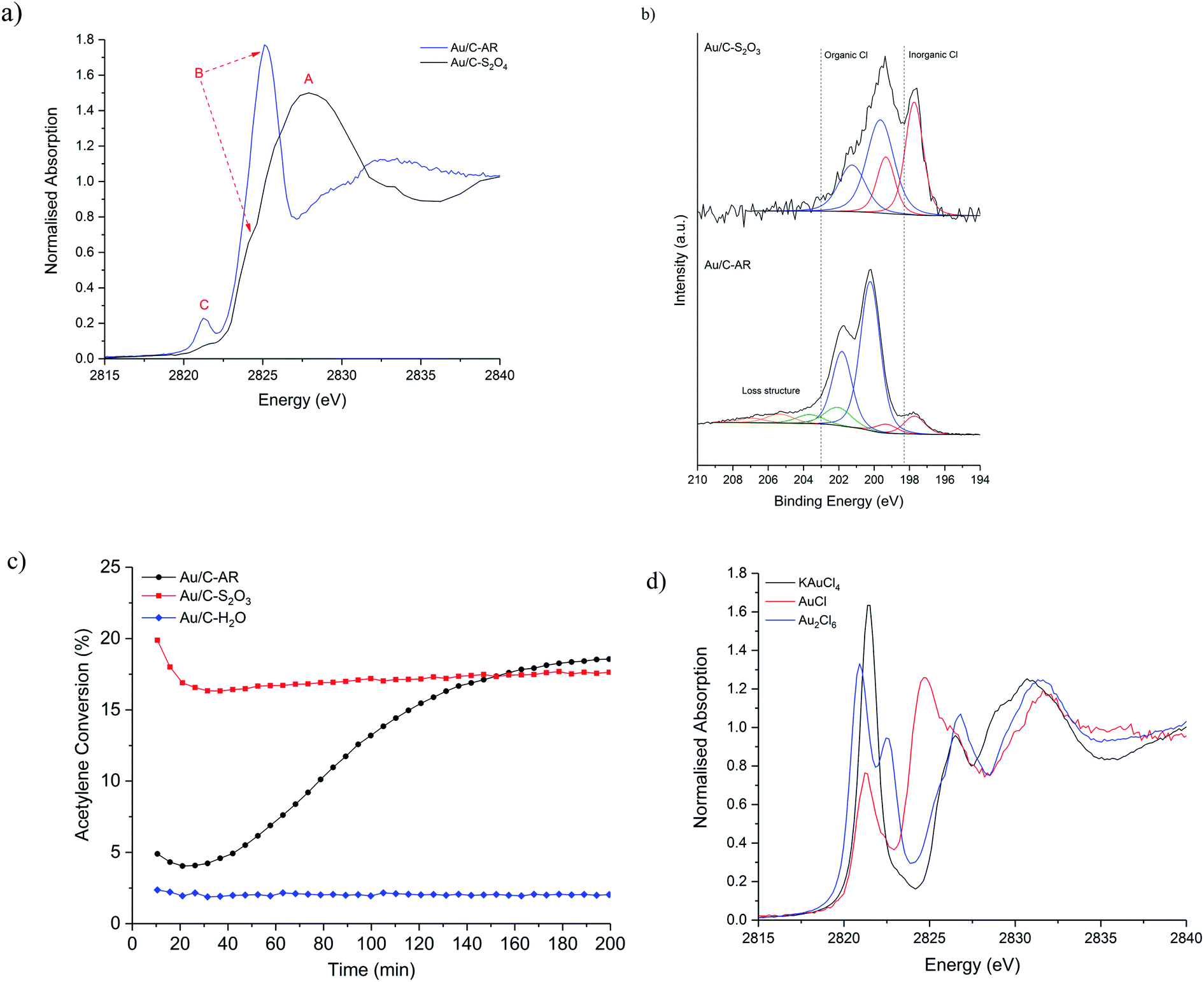 | ||
| Fig. 1 (a) Comparison of the normalised ex situ Cl K-edge XANES of the freshly prepared Au/C-AR (blue solid line) and Au/C-S2O3 (black solid line), (b) Cl(2p) core-level spectra for Au/C-AR and Au/C-S2O3 with associated binding energies; 197.7 eV – Au–Cl/NaCl (red), 200 and 202 eV – C–Cl functional groups (blue) and 203.4 and 205.2 eV loss structure associated with Cl species (orange). (c) Catalytic activity of the Au/C-AR (-●-, black), Au/C-S2O3 (-■-, red) and Au/C-H2O (-◆-, blue) (test conditions: 90 mg catalyst, 23.5 mL min−1 C2H2, 23.7 mL min−1 HCl and 2.7 mL min−1 Ar, 200 °C). (d) XANES of gold chloride standards. KAuCl4 (black solid line), Au2Cl6 (blue solid line) and AuCl (red solid line). | ||
The XANES feature at 2828 eV, labelled (A), is assigned to the Cl 1s → 4p dipole allowed transition of an inorganic Cl− species and it is comparable to the ex situ recorded reference material NaCl (Fig. S4†). The second feature at 2824–2826 eV, labelled (B), which is the predominant feature in Au/C-AR and a shoulder of the main edge for the Au/C-S2O3, can be assigned to a 1s → 3p* transition and associated with C–Cl functionality.24 This observation is in accordance with the assignments of XPS binding energies; Au–Cl/NaCl (197.7 eV), C–Cl groups (200 and 202 eV) and loss structure associated with high levels of carbon chlorination (203.4 and 205.2 eV)25 (Fig. 1b and S3†). Variation in the position of XANES C–Cl features is associated with the aliphatic/aromatic nature of the carbon species and the degree of chlorination;26 higher C–Cl energies, as seen in Au/C-AR, suggest a high degree of chlorination of the carbon surface. The high relative C–Cl content in Au/C-AR is rationalised as a result of chlorination from the highly acidic and oxidising aqua regia preparation. The XANES spectra of the Au/C-H2O catalyst is similar to Au/C-S2O3 however feature B (C–Cl) is more pronounced; aqueous impregnation of HAuCl4 on to the carbon support results is the deposition of metallic Au(0) nanoparticles and presumably consequent formation of a greater amount of C–Cl species.10,13
Bare activated carbon as received was also shown to contain both Cl− and C–Cl species, but in negligible amounts compared to the catalysts; both XANES and XPS results suggest that after washing the carbon support with water (C-H2O) the inorganic Cl− functionality was removed while treatment aqua regia (C-AR) was shown to be able to introduce C–Cl functionality (Fig. S5, S6 and Table S2†). The influence of water and aqua regia on the modification of the carbon surface, with and without the HAuCl4 metal precursor, has been also analysed via O 1s XPS. As expected, the aqua regia impregnation led to the highest oxygen content (Table S3†). However, the oxygen content does not seem to have direct influence on the catalysts' steady state activity when comparing the Au/C-AR and Au/C-S2O3 in our previous studies (catalytic data shown in Fig. 1c); this evidence also agrees with previous studies performed also on metal-free catalysts.27
The third feature in the Cl XANES at 2821 eV, labelled (C), is attributed to the Cl 1s → mixed Cl 3p and Au d orbital transition and is a pre-edge feature.28 The intensity of feature is dictated by the degree of covalency in the Au–Cl bond and is therefore sensitive to the electronic structure of the Au and its oxidation state. The hybridisation of Au 5d–6s orbitals, due to relativistic affects, allows the formally d10 electronic configuration of Au(I) to have molecular orbitals with Cl of sufficient character for electronic dipole allowed transitions. The feature is pronounced in Au/C-AR, while less intense in Au/C-S2O3 (Fig. 1a), showing that less Cl is associated with the Au and/or that the nature of the Au–Cl bonding is different, due to a change in bond covalency or lower oxidation state in the fresh Au/C-S2O3. Feature C is also negligible in the Au/C-H2O catalyst, in which the Au–Cl bond is almost absent (Fig. S1†).13 Given Au/C-S2O3 is prepared from a gold–thiosulfate complex made from HAuCl4, Au–Cl species could persist from the precursor. It is not possible to determine if the Au–Cl species is a discrete AuClx species or a partially chlorinated Au(S2O3)x species, raising the question if multiple Au speciation exists. However, the clear difference in catalytic stability and induction behaviour of this catalyst shows that most Au centres in Au/C-S2O3 are significantly different from the Au–Clx in Au/C-AR (Fig. 1c). Similar considerations can be made in the comparison between the Au/C-AR and Au/C-H2O. From analysis of the Au–Cl pre-edge for the catalysts it is possible to determine if the dimeric Au2Cl6 species are present. The dimeric Au2Cl6 structure has two bonding Cl environments and Au–Cl bond lengths, i.e. terminal and bridged. This is reflected in the XANES spectrum of the dimeric standard (Au2Cl6) as a splitting of the pre-edge (Fig. 1d). On inspection and consistent with monomeric standard (KAuCl4), the XANES of both catalysts showed a single feature (Fig. 1d), indicating no significant population of Au dimers where present, supporting the observations made previously that the Au is present as mono-dispersed cationic species.11
To understand the evolution of Cl species during the acetylene hydrochlorination reaction, in situ Cl K-edge XANES characterisation of Au/C-AR and Au/C-S2O3 was performed. On heating to reaction temperature (200 °C) under He (Fig. 2a–c), the position of feature A, associated with inorganic Cl− species, remains unchanged for both catalysts, with a slight decrease in normalised intensity in Au/C-S2O3. On the other hand, changes in feature C, associated with Au–Cl, were evident for both catalysts. In particular, the intensity of the pre-edge increased between 100–120 °C for the 2 catalysts, before reducing on further heating to 200 °C (Fig. 2c and S7†). This behaviour can be explained as following: on heating the catalysts the Au is oxidised to a higher oxidation state AuClx, through migration of Cl from the carbon to the Au. This causes an increase in intensity of feature C as there is an increased Cl coordination number (CN) and a lower occupancy of the 5d–3p hybridized orbital. The small decrease in the C–Cl band in Au/C-S2O3 concurrent with the increase in feature C supports this hypothesis; the absence of any notable C–Cl population change in Au/C-AR is attributed to the significantly higher initial signal intensity, masking any small change in signal from Cl migration. The reduced intensity of feature C above 110–130 °C is due to the decomposition of Au(III) Cl to Au(I) Cl, where fully occupied d orbitals limit the pre-edge feature,29 in agreement with Au L3-edge experiments.13 This observation suggests that the C–Cl species can be mobile and interact with the Au species at elevated temperatures by acting as a reservoir of Cl functionality. It is important to acknowledge that while changes between Cl environments can be inferred, the intensities of allowed 1s → 4d transitions cannot be proportionately compared with feature C, which is dependent on Cl 3p–Au 5d/6s hybridisation.
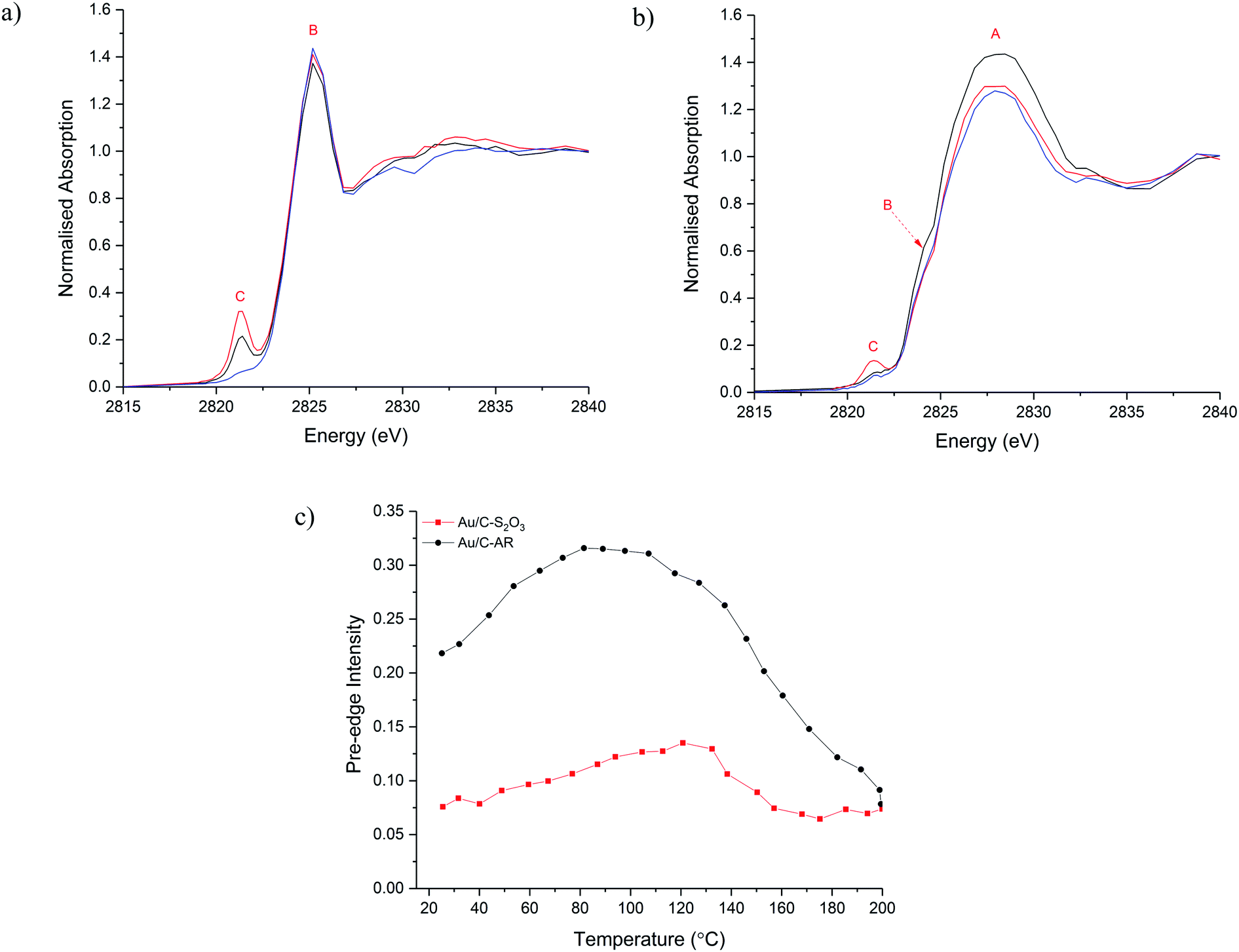 | ||
| Fig. 2 Cl K-edge XANES during the heating ramp of Au/C-AR (a) and Au/C-S2O3 (b) at start of heating ramp (black), maximum pre-edge intensity (red) and end of heating ramp (blue). (c) Pre-edge intensity against temperature for Au/C-AR (-●-, black) and Au/C-S2O3 (-■-, red). | ||
The catalysts were then characterised during the acetylene hydrochlorination reaction (Fig. 3a–c). Over a 100 minutes of reaction, the overall Cl concentration modestly increased for Au/C-S2O3 and was unchanged for Au/C-AR, which remained higher than the Au/C-S2O3 throughout (Fig. S8, S9 and Table S1†) which suggests that gaseous HCl cannot significantly chlorinate the carbon surface. Given the comparable VCM productivities between the catalysts (Fig. 1c),13 there seems to be no correlation between the total Cl concentration within the catalysts and catalytic activity. Moreover, it suggests that the Au complex and carbon surface within Au/C-S2O3 catalyst which is not highly chlorinated during the preparation is relatively resistant to possible chlorination phenomena directly from HCl and it remains significantly different to the highly chlorinated surface of the Au/C-AR. Cl(2p) XPS of the used catalysts (Fig. S10 and Table S1†) show a slight increase in Cl content in the Au/C-S2O3 catalyst after reaction, especially of the inorganic Cl species, which is attributable to a certain level of chlorination of the metal centre under reaction condition.
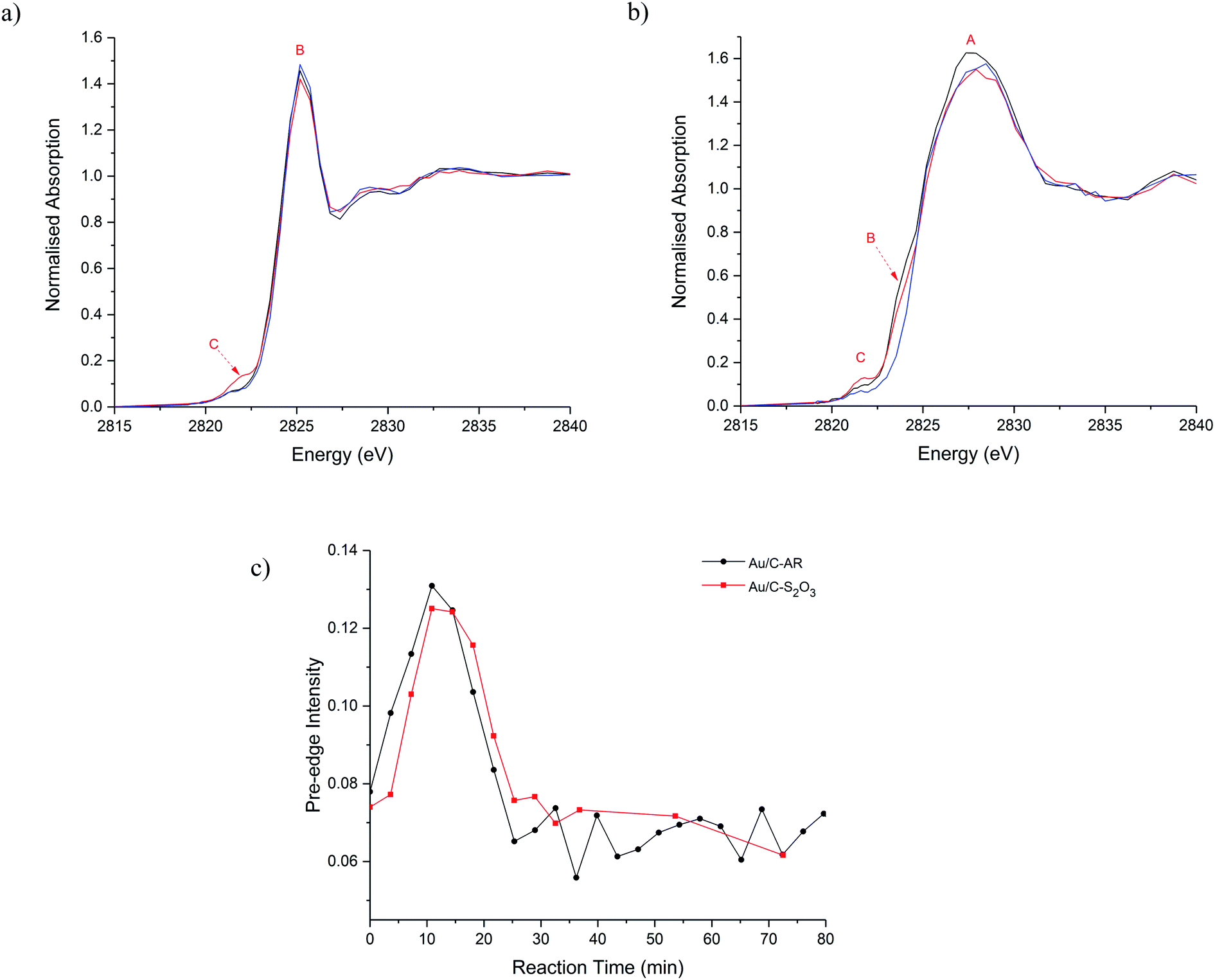 | ||
| Fig. 3 In situ Cl K-edge XANES spectra of (a) Au/C-AR and (b) Au/C-S2O3 catalysts during acetylene hydrochlorination at: beginning of the reaction (black line) max of pre-edge intensity (red line) and at steady state (blue line). (c) Change in pre-edge intensity with reaction time for Au/C-AR (-●-, black) and Au/C-S2O3 (-■-,red). | ||
Comparison of the normalised Cl-edge XANES spectra (Fig. 3 and S9†), show variation in feature C (Au–Cl) during reaction in both catalysts; shown as a function of reaction time-on-line in Fig. 3c. Feature C intensifies, broadens and shifts to higher energy during the first 20 min and then decreases, suggesting changes in the Au–Cl bonding, Au oxidation state and Au–Clx geometry during reaction.18 Broadening possibly indicates multiple Au–Cl bond lengths or Au–Cl species. These observations are in line with those observed from operando Au L3-edge XAFS.13 The comparable changes in feature C with respect to reaction time (Fig. 3c) suggest that the Au–Cl species are similar for both catalysts. However, the significantly lower Cl concentration in Au/C-S2O3 compared to Au/C-AR throughout the reaction suggests fewer Au–Cl species in Au/C-S2O3. Given all the Au within this catalyst is cationic and atomically dispersed, this lower Cl signal shows that most of the Au is coordinated to other non-chloride ligands, most probably S ligands, throughout the reaction and that the Au in this catalyst does not become excessively chlorinated or convert to a purely AuClx species while operating. Under operation Au/C-S2O3 contains multiple Au speciation, a robust Au–S complex and a minority Au–Cl species analogous to Au/C-AR.
Another difference between the two catalysts Cl speciation, under reaction conditions, is the change in C–Cl species, feature B. During heating to 200 °C and during reaction this gradually reduces in intensity for Au/C-S2O3 (Fig. 3a). This behaviour can be attributed to the migration of Cl from the carbon to the Au centre, confirming the Cl(2p) XPS results on the used sample (Fig. S10†). Although the corresponding feature appears to remain stable in Au/C-AR, this is again due to the high C–Cl concentration making small differences difficult to observe. The relatively high amount and possible mobility or evolution of C–Cl species in the Au/C-AR catalyst could also explain the induction period typically observed in this type of catalyst.13 On heating of the catalyst, we first observe an increase in the amount of Au(III), confirmed by both XAS of the Au L3-edge13 and Cl K-edge, before conversion to Au(I) beyond the thermal decomposition temperatures of AuCl3, which is approximately 130–160 °C. This observation of Au(III) is likely the result of oxidation of Au(I) rather than the disproportionation reaction (3AuCl → 2Au + AuCl3) as we do not observe metallic Au in these or our previous experiments.13 We have previously shown that exposure to HCl alone does not result in the oxidation of Au(I) to Au(III) species and that the induction period observed is related to the formation of Au(III) on exposure to both reactants with activity increasing as the ratio of Au(I) to Au(III) equilibrates. The presence of large amounts of surface chlorination could facilitate evolution of Cl2 from the surface at reaction temperature which is a strong enough oxidant to convert AuCl into AuCl3 in addition to residual NOx species remaining from the preparation. The evolution of these species could be accelerated by the large initial exotherm passing through the bed on introduction of the reactant gases and only when the evolution of oxidant has subsided can the catalyst equilibrate to a Au(I)/Au(III) ratio determined by catalytic turnover and steady state activity.
This behaviour is observed to a much lesser extent for Au/C-S2O3, due to the C–Cl reservoir being quickly depleted. The increased C–Cl in Au/C-AR results in a more extensive oxidation of the Au(I) chloride like species to a Au(III) type chloride species and consequently the process of Au(I) reformation, which is correlated to activity, is considerably slower, resulting in a notable induction period as the species equilibrate.
Simple impregnation of the metal precursor, as in the aqua regia preparation, is however preferable to the in situ formation of the Au–thiosulphate complex followed by its impregnation onto the carbon support required in the preparation of Au/C-S2O3. Clearly the use of an alternative non-chlorinated/organic solvent to aqua regia for the dissolution of the HAuCl4 metal precursor could be an efficient solution for the preparation of active, stable and scalable Au/C catalysts for this reaction. Recently we have shown that catalysts can be prepared using organic solvents, such as acetone.30 It is important to note that these catalysts which are prepared using HAuCl4 and without aqua regia (and therefore without a high Cl content) did not have an induction period and behaved analogously to Au/C-S2O3 catalysts. The Cl K-edge XANES of a catalyst prepared using HAuCl4 and an acetone solvent is shown in Fig. 4. Critically, the C–Cl feature is notably less intense than that seen in Au/C-AR, confirming the relationship of this species to the catalytic induction period. Although the Au/C-acetone catalyst does not have a pronounced induction period and is similar in initial activity to the Au/C-S2O3 catalyst, a proportion of the Au within the catalyst was found to reduce to form Au(0) nanoparticles. This reduction was not observed for Au/C-S2O3 catalysts, suggesting that in addition to limiting C–Cl surface species the presence of sulphur species increases the stability of the Au(I) active site.
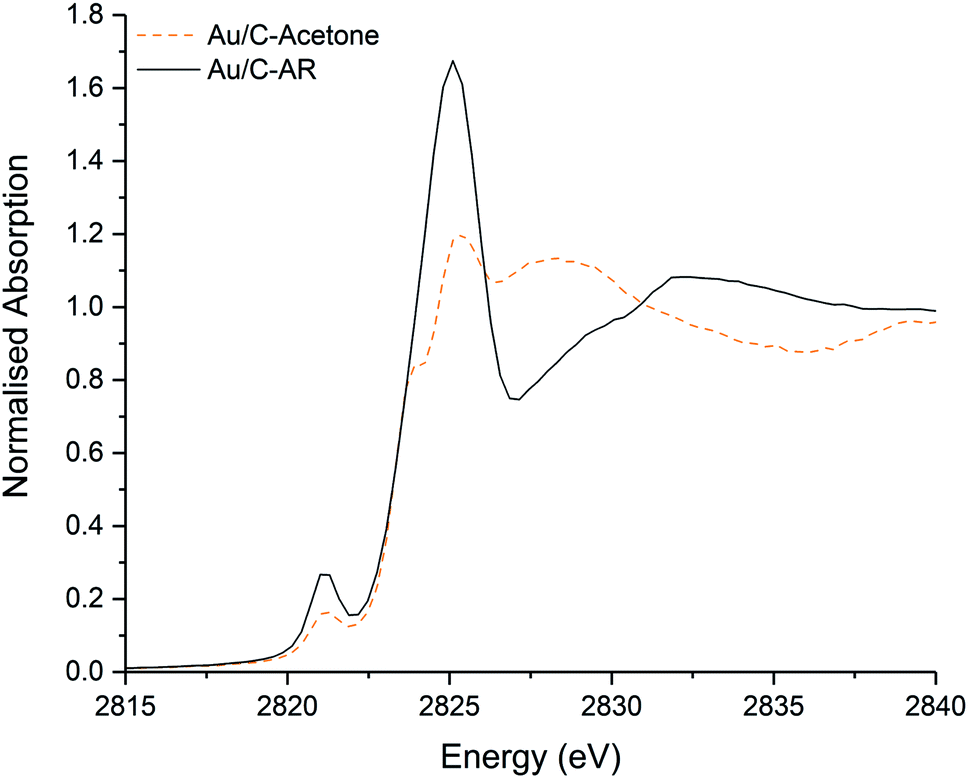 | ||
| Fig. 4 Comparison of the normalised ex situ Cl K-edge XANES of the freshly prepared Au/C-acetone (dashed orange line) and Au/C-AR (black solid line). | ||
To understand the role of sulphur-based ligands in Au/C-S2O3in situ S K-edge XANES experiments were also performed. The S1s → S3p electronic transitions are extremely sensitive to the sulphur oxidation state.31 Moreover, in the case of oxyanions, where the sulphur can be present in various oxidation states, the spectrum originated will show one feature (or edge) for each oxidation states of the element in the sample. Sulphur compounds have unique patterns of transitions on the absorption edge, making the sulphur K-edge XANES suitable for qualitative determination of sulphur species in samples with complex composition by using appropriate reference materials.32,33
Fig. 5a shows the S K-edge XANES of Au/C-S2O3 and Na3S2O3 (XANES spectra of other standards materials recorded are reported in Fig. S11†), where the sulphide can be differentiated from the more oxidised sulphate like species. While the formal oxidation state of sulphur in thiosulfate ions is debated,34–36 it is clear that species of comparable, but not identical, oxidation state are present in Na3S2O3 and Au/C-S2O3. Features in Na2S2O3 at 2472 and 2482 eV, associated with 1s → 3p transitions of the different sulphur environments, are shifted to higher energy in Au/C-S2O3 (2473 and 2483 eV) (Fig. 5a). However, features observed in Na2S2O3, between at 2477–2479 eV which are not satisfactorily explained in the literature, are absent in Au/C-S2O3. Abinitio DFT simulations of the XANES spectra of [S2O3]2− show that these undefined features can be assigned to transitions to excited states where atomic orbitals of the 2 S atoms and O 2p from thiosulfate are strongly hybridized (Fig. 6a). [S2O3]2− interaction with the carbon support (via C-π orbitals) only weakly affect these excited states (Fig. 6b). A relevant decrease of the intensity of the corresponding peaks could only be reproduced by including a strong chemical interaction in the models for the simulations (such as the addition of H covalently bonded to O of SO3, or the chemical binding of S2O3 to the C-based support, whose spectra are shown in Fig. 6b). The absence of these features in Au/C-S2O3 therefore suggests that the thiosulphate ligand has not remained intact on deposition onto the carbon and has potentially undergone a disproportionation reaction to S2− and SO32−. Furthermore, the XANES simulations of the model structures where a strong chemical interaction is consistently included, also successfully predict an increase in the energy of the first electronic transition to 2473 eV consistent with the shift of the first peak observed in experimental spectra shown in Fig. 5 between Na2S2O3 and Au/C-S2O3.
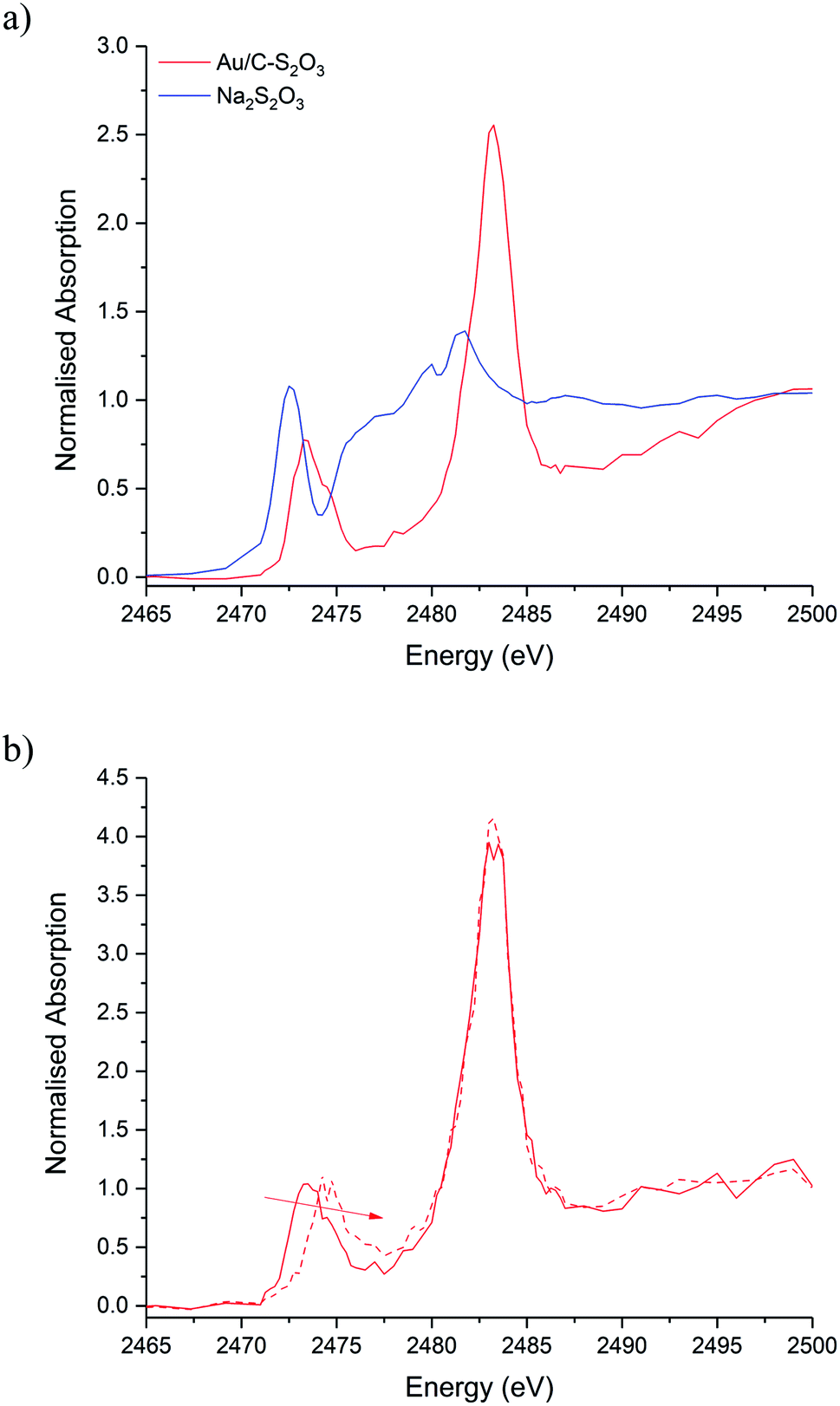 | ||
| Fig. 5 S K-edge XANES of the Au/C-S2O3. (a) Ex situ XANES normalised absorption spectra of Au/C-S2O3 (red solid line) with Na2S2O3 standard (blue solid line). (b) In situ XANES normalised absorption spectra during reaction time-on-line: start of reaction (red solid line) and after 20 minutes (red dashed line). | ||
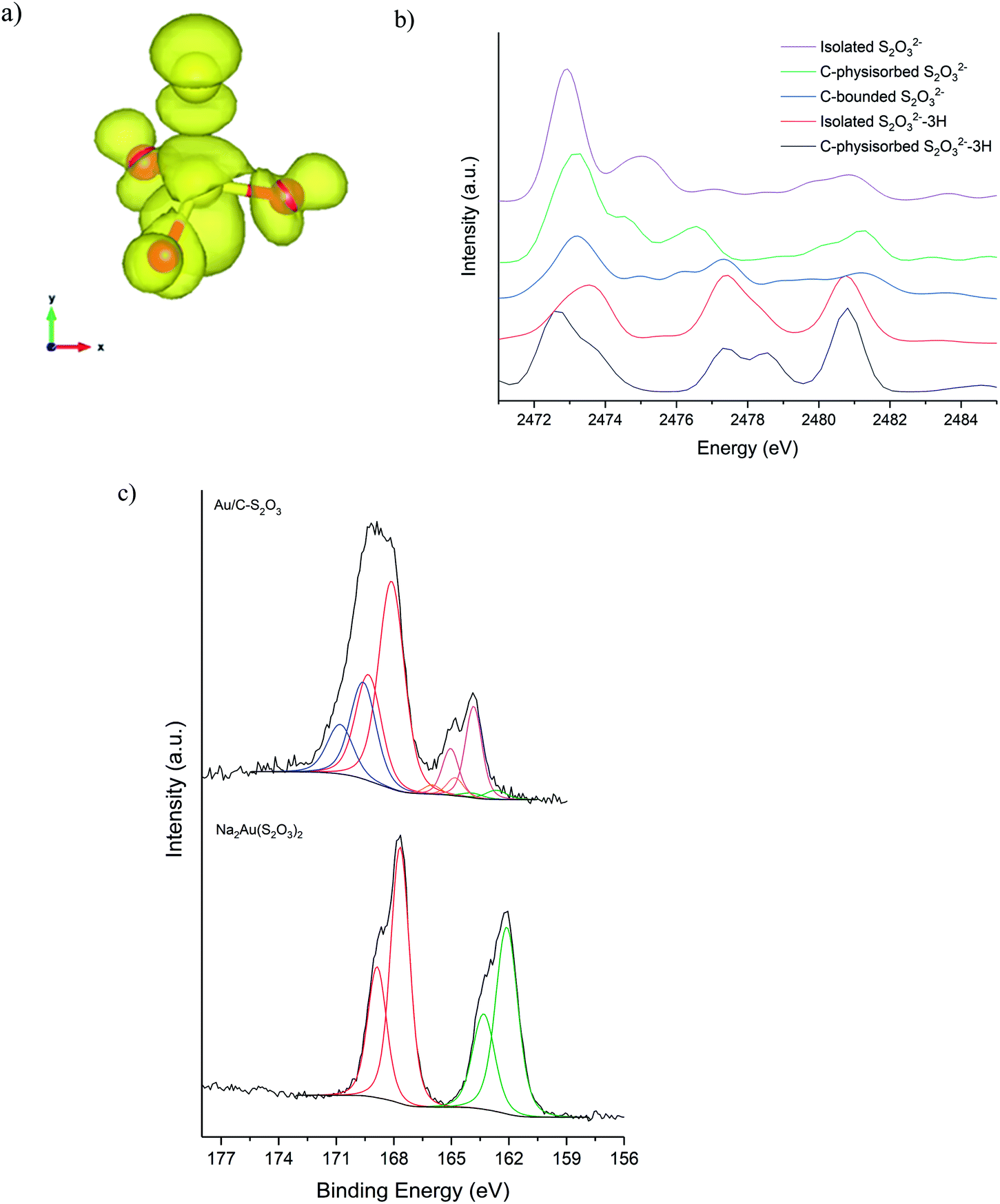 | ||
| Fig. 6 Sulphur selected orbital K-edge XANES obtained by DFT simulations. (a) Excited state responsible for transition at 2477 eV. (b) Comparison of simulated S K edge in thiosulfate S2O32− and model structures. (c) S(2p) core-level spectra for Na2Au(S2O3)2 and the Au/C-S2O3 catalyst with associated binding energy; two sulphur states in the former (S6+ and S2− at 166.7 and 162.1 eV) and several sulphur states in the catalyst, attributed to Au–S (162.6 eV), S–S (163.8 eV), S–H (164.8 eV), SO3 (168.1 eV) and SO4 (169.6 eV), consistent with disproportionation. | ||
XPS analysis of the Au/C-S2O3 catalyst supports this supposition. Fig. 6c shows the S(2p) region for Na3Au(S2O3)2 and Au/C-S2O3; with two sulphur states in the former (S6+ and S2− at 166.7 and 162.1 eV) and several additional sulphur states in the catalyst, attributed to Au–S (162.6 eV), S–S (163.8 eV), S–H (164.8 eV), SO3 (168.1 eV) and SO4 (169.6 eV), consistent with disproportionation. The presence of Au–S bonds in Au/C-S2O3 proposed based on XPS binding energy and splitting of the XANES S2− feature, as seen in Na3Au(S2O3)2 (Fig. S11†), confirms that the Au is predominantly bound to the sulphide species. To understand the dynamic nature of S within Au/C-S2O3, in situ S K-edge XANES studies were performed. During heating to reaction temperature under an inert atmosphere, the two main absorption edges did not change in intensity or position (Fig. S12†), in contrast with Cl speciation. Demonstrating that S speciation in Au/C-S2O3 was more thermally stable than the Cl species.
Introduction of reaction gases resulted in a shift in the absorption-edge of the S2− bound to Au, during the first 20 min of reaction, while the SO32− group remains stable (Fig. 5b and S13†). The shift to higher absorption-edge for the sulphide species shows a decrease in electron density and oxidation of this S species bound to Au. We suggest that Au(I) oxidation, caused by C–Cl surface species on the carbon is suppressed by the polarisable Au–S bond. Therefore, in addition to the significantly lower concentration of C–Cl species, the active Au(I) species in Au/C-S2O3 do not over oxidise and the catalyst is immediately active. This supports the proposed detrimental role of the Cl excess on the carbon surface. The S 2p XPS of the Au/C-S2O3 catalysts before and after reaction (Fig. S14 and Table S4†) show that multiple sulphur states can be observed as in Fig. 6c with identical speciation assignments. It should be noted however, binding energies for the species differ by ca. 0.2–0.6 eV, the smaller difference attributable to experimental errors and uncertainty in peak fits whilst the larger difference may again be attributed to agglomeration of sulphur species and/or higher uncertainty in the peak fits due to the signal to noise in the data.
The stability of the S containing Au/C-S2O3 catalyst was investigated at the L3-edge using an “accelerated deactivation test”, in which the catalyst is sequentially exposed to; (i) HCl + C2H2, (ii) HCl only, (iii) HCl + C2H2 (iv) C2H2 only and finally (v) HCl + C2H2 during one experiment. We have already reported that exposure to HCl only (i.e. in the absence of C2H2) resulted in Au/C-AR catalytic performance being perturbed on the re-introduction of C2H2, resulting in a second induction period to regain steady state activity.1,37 Also, Au/C-AR rapidly deactivates after treatment with only C2H2.37 This deactivation was attributed to an interaction with AuClx and C2H2 that on the re-introduction of HCl causes the formation of Au(0) nanoparticles. To clarify the influence of the individual reactants on the Au/C-S2O3, the VCM productivity (Fig. S15†) and the Au L3-edge XANES (Fig. 7) and EXAFS (Fig. 8 and Table 1) were correlated during an operando experiment in a fixed bed reactor. XANES and EXAFS spectra of the Au/C-AR catalyst, reported previously,37 are also shown for comparison. These previous studies show that under steady state reaction conditions a combination of Au(I) and Au(III) chloride/sulphur compounds are present, with EXAFS determined coordination numbers matching those determined from XANES analysis, by white line height or linear combination fitting.13,37
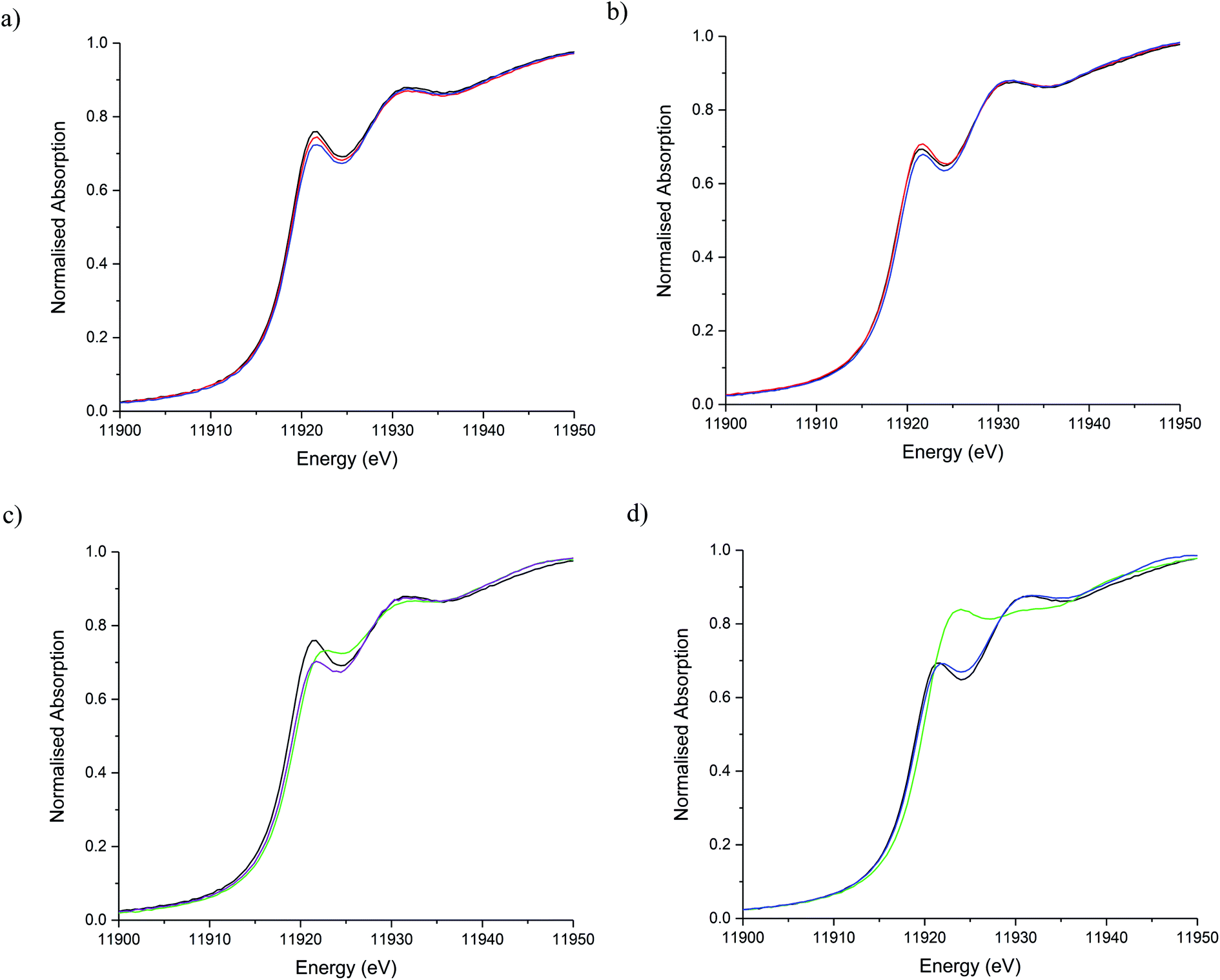 | ||
| Fig. 7 Au L3-edge XANES of Au/C-S2O3 and Au/C-AR during the sequential gas exposure experiments. (a) Au/C-S2O3 and (b) Au/C-AR at steady state for steps i (red line), steps ii (blue line) and steps iii (black line). (c) Au/C-S2O3 and (d) Au/C-AR at steady state for steps iii (black line), steps iv (green line) and steps v (purple line). | ||
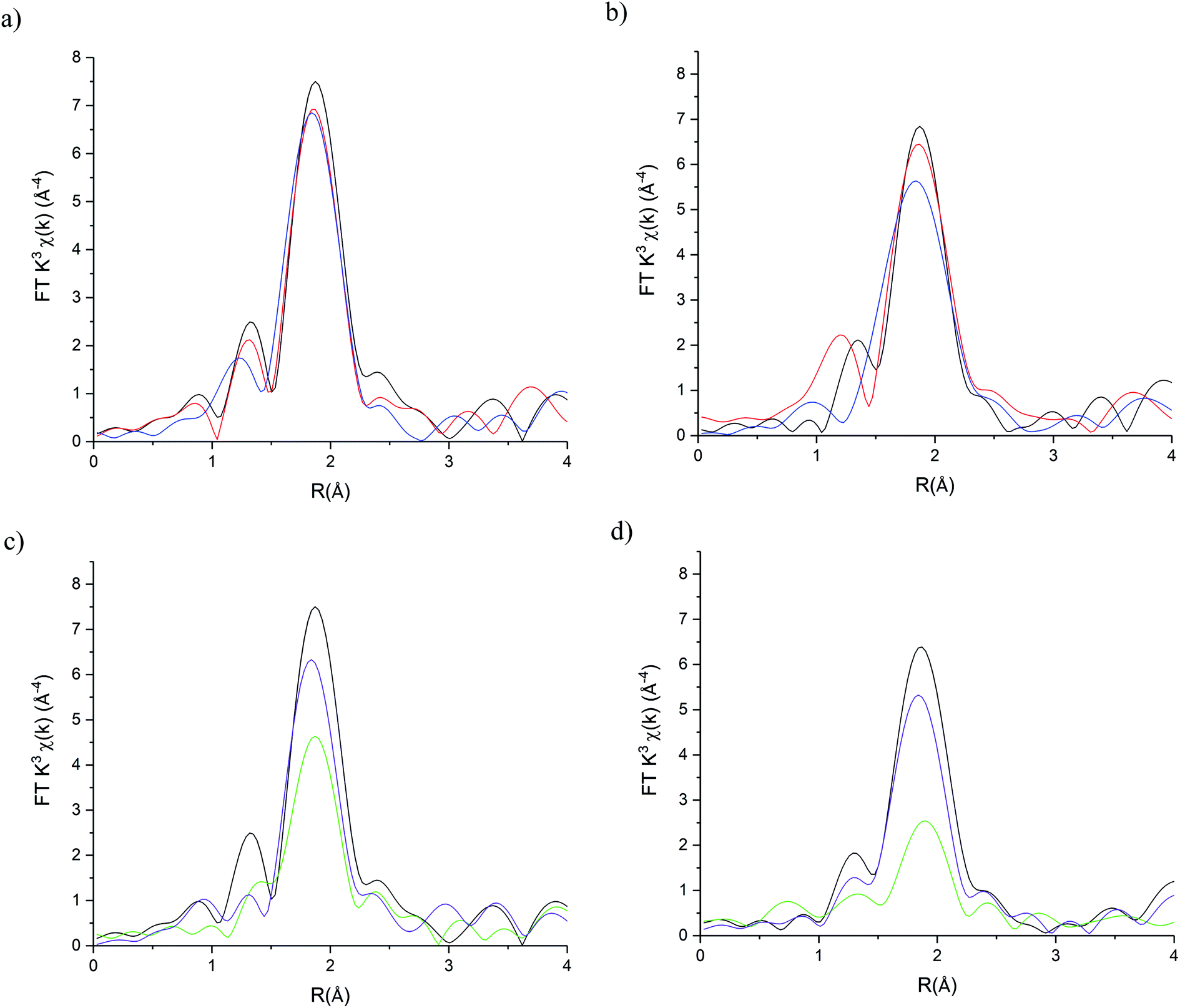 | ||
| Fig. 8 Au L3 edge EXAFS Fourier Transform magnitudes of Au/C-S2O3 and Au/C-AR during the sequential gas exposure experiments. (a) Au/C-S2O3 and (b) Au/C-AR at steady state for steps i (red line), steps ii (blue line) and steps iii (black line). (c) Au/C-S2O3 and (d) Au/C-AR at steady state for steps iii (black line), steps iv (green line) and steps v (purple line). | ||
| Reaction stepa | Scattering pathb | Coordination number | 2σ2 (Å2)e | R (Å) | E f (eV) | R factor | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Au/C-S2O3 | Au/C-ARc | Au/C-S2O3 | Au/C-AR | Au/C-S2O3 | Au/C-ARc | Au/C-S2O3 | Au/C-ARc | Au/C-S2O3 | Au/C-ARc | ||
| a EXAFS data taken under steady state conditions. b Fitting of multiple Au–Cl paths was attempted for all spectra. Only is step iv did this provide realistic fits. c Au/C-AR data published in ref. 37. d Au–C CN fixed at 1. e Debye–Waller and amplitude reduction factors were determined from fitting of KAuCl4 with a fixed CN of 4 or Au(0) with fixed CN of 12. f Debye–Waller fitted for model 1 (step iv). | |||||||||||
| i | Au–Cl | 2.5(1) | 2.5(1) | 0.0037 | 2.27(1) | 2.27(1) | 2(1) | 1(1) | 0.011 | 0.012 | |
| ii | Au–Cl | 2.5(1) | 2.4(1) | 2.27(1) | 2.27(1) | 1(1) | 3(1) | 0.012 | 0.07 | ||
| iii | Au–Cl | 2.6(2) | 2.5(1) | 2.28(1) | 2.27(1) | 2(1) | 3(1) | 0.022 | 0.04 | ||
| iv (model 1)f | Au–Cl | 2.2(3) | 2.0(4) | 0.006(2) | 0.011(2) | 2.28(1) | 2.29(2) | 1(2) | 1(2) | 0.031 | |
| iv (model 2) | Au–Cl (1) | 1.6(1) | 0.9(2) | 0.0037 | 2.28(2) | 2.30(3) | 1(1) | 1(4) | 0.020 | 0.037 | |
| Au–Cd | 1 | 1 | 0.0090 | 2.28(1) | 2.04(7) | ||||||
| v | Au–Cl | 2.2(1) | 1.7(3) | 0.0037 | 2.27(1) | 2.26(1) | 1(1) | 2(2) | 0.019 | 0.017 | |
| Au–Aud | 1.0(4) | 0.6(4) | 0.007 | 2.88(2) | 2.84(5) | ||||||
| Fixed parameters: So2 = 0.75, 2σ2 (Å2) = 0.0037 (Au–Cl) and 0.007 (Au–Au) | |||||||||||
After a small drop in productivity over the first 20 min time-on-line Au/C-S2O3 remained stable for the complete test duration with minimal deactivation despite periods of being exposed to only HCl or C2H2. The XANES spectra of Au/C-S2O3 showed a slight decrease in white-line height, as seen for Au/C-AR, under the HCl environment. It is clear that for both catalysts the Au(III/I) centres are not strongly influenced by the dilute HCl stream and that neither catalyst is oxidised to a higher degree of chlorination under these conditions. This is corroborated by the EXAFS which shows no change in the Au–Cl/S (indistinguishable by EXAFS) coordination number of 2.5 or bond lengths between reaction (i) and under HCl (ii).
The difference in catalyst performance on re-introduction of both reactants following HCl treatment (step iii) between Au/C-S2O3 and Au/C-AR is therefore due to the C–Cl content on the two respective support structures. Namely, the absence of significant C–Cl content in Au/C-S2O3 resulting in an immediate return to reactivity, without an induction period. Under a C2H2 flow, the energy of the absorption edge increased for Au/C-S2O3, with the observed white-line shifting from 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 921.6 eV to 11922.7 eV. Comparison with the change in XANES seen for Au/C-AR under identical C2H2 conditions showed similar trends to that observed for Au/C-S2O3. The change in white-line for both catalysts is attributed to an uncharacterised Au–acetylene interaction.37 However, the extent of the edge shift was significantly less for Au/C-S2O3 than that seen for Au/C-AR. The Fourier transformed Au L3-edge EXAFS (Fig. 8) showed a decrease in the magnitude of the signal at 1.8 Å for both catalysts. Again, the dampening of the signal was notably less for Au/C-S2O3 under C2H2 relative to Au/C-AR.
921.6 eV to 11922.7 eV. Comparison with the change in XANES seen for Au/C-AR under identical C2H2 conditions showed similar trends to that observed for Au/C-S2O3. The change in white-line for both catalysts is attributed to an uncharacterised Au–acetylene interaction.37 However, the extent of the edge shift was significantly less for Au/C-S2O3 than that seen for Au/C-AR. The Fourier transformed Au L3-edge EXAFS (Fig. 8) showed a decrease in the magnitude of the signal at 1.8 Å for both catalysts. Again, the dampening of the signal was notably less for Au/C-S2O3 under C2H2 relative to Au/C-AR.
The damping of oscillations seen for Au/C-AR was previously attributed to out of phase multiple Au–Cl path lengths.37 The catalyst was considered to be Au(I) based on the edge position but with a significant change in geometry. The rational for this interpretation being that χ space data showed a damping of oscillations across the k range under C2H2, with no change in the phasing between a catalyst under C2H2 (step iv) or under steady state reaction conditions (step i–iii) (Fig. S16†), suggestive of no new scatter being present and an increase in disorder. However, an unresolved question of the cause of this affect, in addition to the significant change in the XANES of both the Au/C-AR and Au/C-S2O3 catalysts under C2H2, prompts consideration of an alternative interpretation of the data. A compelling hypothesis being that in addition to Au–Cl paths there is a Au–C path attributable to Au–C2H2 bonding. Therefore, the fitting of the EXAFS data of Au/C-AR and Au/C-S2O3 catalysts under C2H2 was performed using two models; (1) a single Au–Cl path with a floated 2σ2 parameter to account for an increased structural disorder due to multiple Au–Cl paths or (2) fitting Au–Cl and Au–C paths.
Both models provided reasonable fits of the data. Model 1 showed a significant increase in 2σ2 from 0.0037 Å2 under steady state (step i) to values under C2H2 of 0.006(2) Å2 and 0.011(2) for Au/C-AR and Au/C-S2O3 respectively. Model 2 provides a viable Au–C path length of 2.04(7) Å which, despite the significant error, fits with distances seen for Au(I) σ-bonded alkyne complexes at 1.98 Å.38 Freeing of the Au–Cl/S coordination number showed that this path was more significant in Au/C-S2O3 and suggest that more free Au(I) is present in this catalyst than Au/C-AR. Comparison of the fits shows, however, no statistical improvement on the addition of a Au–C path, further this model significantly misrepresented the first oscillation of the χ space data where a soft scatterer would be best represented (low k distances, Fig. S17†). It is concluded, that whist compelling to evoke Au–alkyne complex formation it could not be definitively proven with the current data. Regardless of the fitting model used it is clear that C2H2 has a similar influence on Au/C-S2O3, although it is far less dramatic than that seen for Au/C-AR.
The effect of reintroduction of both reactant gases in step v on Au speciation was notably different for the two catalysts. While Au/C-AR was found to deactivate after the re-introduction of HCl, Au/C-S2O3 remained relatively stable with only minimal deactivation. As stated previously, the growth of Au(0) responsible for deactivation in Au/C-AR, occurred only after re-introduction of HCl (step iv) and not under C2H2.37 Au–Au paths could be fitted for both catalysts at the end of reaction step iv, at all other points during the reaction no acceptable fit of this path was found. The proportion of Au(0) could not accurately be determined by EXAFS due to the large error in Au–Au coordination numbers.
Conclusion
S and Cl K-edge XANES have been used to gain further insight into the structure–activity relationship of cationic Au catalysts in acetylene hydrochlorination. Three Cl species; unreactive inorganic Cl, covalently bound C–Cl and Au–Cl species are found on Au/C-AR and Au/C-S2O3. The concentration of these species was significantly higher in Au/C-AR. Au–Cl species are dynamic during heating and under reaction conditions, with changes in Au–Cl bond hybridisation and bond length. In addition, surface bound C–Cl is found to be titrated from the carbon surface during reaction suggesting the potential importance of these surface groups in influencing Au–Cl speciation. High concentrations of C–Cl, as seen in Au/C-AR, therefore result in a catalytic induction period. The presence of Au–Cl in Au/C-S2O3 was concluded to be from a minority phase that coexists with Au–S species. The presence of intact [S2O3]2− ligands could not be confirmed on deposition of the Au complex onto the carbon support. However, the S species present were found to be stable under reaction conditions and stabilises Au(I) to produce a robust and commercially viable acetylene hydrochlorination catalyst.Experimental
Catalysts preparation
1 wt% gold supported on activated carbon catalysts were prepared by wet impregnation of the HAuCl4 precursor dissolved in aqua regia or water (denoted as Au/C-AR and Au/C-H2O respectively). Activated carbon was initially ground to obtain a 100–140 mesh powder. The gold precursor, HAuCl4·xH2O (Alfa Aesar, 99.9% (metals basis), Au 49%) was dissolved in aqua regia (3 parts by volume HCl [(Fisher, 32 wt%)]: 1 part by volume HNO3 [(Fisher, 70 wt%)]) or water. The gold precursor solution was then added drop-wise with stirring to the acid washed, steam activated wood carbon. Stirring was continued at ambient temperature for 1 hour or until NOx production subsided. The product was then dried for 16 h at 140 °C under an inert flow of nitrogen. A different procedure was employed for the preparation of the 1 wt% Au/C-S2O3 catalyst: an aqueous HAuCl4·3H2O solution was mixed with an aqueous solution of sodium thiosulfate, Na2S2O3, in order to obtain a NaAuS2O3 complex. The mixture obtained was added slowly in aliquots to the support while stirring. The product was then dried at 110 °C overnight under a nitrogen flow.Catalysts testing
Within this setup the dedicated space for the catalyst bed is very limited and the amount of catalyst used was ca. 10 mg. For this reason, the total flow has been reduced to 5 mL min−1: C2H2/He (2.5 mL min−1) HCl/He (2.5 mL min−1). Dilute gas mixtures were used C2H2/He (4.97% balanced in He, Air Liquide) and HCl/He (5.00% balanced in He, Air Liquide). The gases were dried, using moisture traps, prior to introduction to the reactor setup. In all cases, the reactor was purged with He (99.99%, Air Liquide), heated to 200 °C at a ramp rate of 2.5 °C min−1 and held at temperature for 30 min, all under a flow of He (5 mL min−1), prior to admitting the hydrochlorination reaction mixture. The outlet gas line was connected to a mass spectrometer (Hiden QGA), to detect VCM during reactions and showed that the catalysts were functioning.
000 h−1. When both reactants were present, the C2H2:HCl ratio was kept at a constant value of 1:1.02.
The reaction mixture was analysed on-line by mass spectrometry (Hiden QGA), and Professional Edition software was used for both qualitative and quantitative analyses. The catalyst activity presented is shown in terms of productivity toward vinyl chloride monomer (VCM). The response factor of the mass spectrometer toward VCM was correlated with the productivity (mol kgcat−1 h−1) obtained by using a Varian 450 gas chromatograph equipped with a flame ionization detector (FID). Chromatographic separation and identification of the products was carried out using a Porapak N packed column (6 ft × 1/8′′ stainless steel).
The sequential flow experiment was performed simultaneously monitoring the Au L3-edge XAS and catalytic activity. Reaction sequence employed the following gas compositions: step i = HCl/C2H2/Ar, step ii = HCl/Ar, step iii = HCl/C2H2/Ar, step iv = C2H2/Ar, and step v = HCl/C2H2/Ar. The duration of each step in the sequence was not the same. The gas composition during the experiment was altered only when no further change in the XAS spectra was observed.
Catalysts characterisation
In situ and ex situ XAFS at the Au L3-edge and Cl K-edge have been also acquired at the B18. The Cl K-edge X-ray absorption spectra (XAS) have been measured to probe chloride-gold bonding. Spectra for the Au/C-AR samples at different time-on-line were recorded ex situ at the Cl K absorption edge in fluorescence mode, using beamline B18 of the Diamond Light Source, Harwell, UK. The measurements were performed using a QEXAFS setup with a fast-scanning Si (111) double crystal monochromator and a 36 element Ge fluorescence detector. The K-edge absorption spectrum of Cl when bound to a transition metal shows a pre-edge feature due to the forbidden 1s → 3d transition. This transition becomes partially allowed and therefore observed when the Cl p-orbitals mix with the metal d-orbitals. The position of the pre-edge feature is dependent on several factors, namely (i) the Cl 1s energy, related to the charge on the chloride, and (ii) the metal d-orbital energy which is itself determined by both the oxidation state of the metal and the coordination number. The intensity of the pre-edge feature is dependent on the mixing of the Cl orbitals and metal d-orbitals and so the bonding characteristic of the Cl to the metal.
We tested the consistency of the results obtained with 6-311G*, triple-ζ split-valence plus polarization and double-ζ split-valence basis set for both S and O atoms. Here we show the results obtained with def2-TZVP basis set. Geometries of the molecules were relaxed within an energy tolerance of 5.0 × 10−6 a.u. and a maximum displacement of 4.0 × 10−3 a.u. The electronic properties of the ground state were converged to an energy threshold of 1.0 × 10−7 a.u. Sulphur K edge spectra were then calculated within the TD-DFT formalism for the excited states.43 A rigid shift has been applied consistently among all the spectra shown in Fig. SIM1† to align them at the energy of the experimental S K absorption spectra.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge Diamond Light Source for time on Beamline B18 under Proposal SP15214-1. The K-edge experiments were performed on beamline BM28 (XMaS – UK CRG Beamline) at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. We are grateful to Dr Paul Thompson, Dr Laurence Bouchenoire and Dr Didier Wermeille at the ESRF for providing assistance in using beamline BM28 (XMaS – UK CRG Beamline). We thank S. Morris for technical support.References
- P. Johnston, N. Carthey and G. J. Hutchings, J. Am. Chem. Soc., 2015, 137, 14548–14557 CrossRef CAS PubMed.
- R. Ciriminna, E. Falletta, C. Della Pina, J. H. Teles and M. Pagliaro, Angew. Chem., Int. Ed., 2016, 55, 14210–14217 CrossRef CAS PubMed.
- J. Zhang, N. Liu, W. Li and B. Dai, Front. Chem. Sci. Eng., 2011, 5, 514–520 CrossRef CAS.
- http://www.mercuryconvention.org/ .
- G. J. Hutchings, J. Catal., 1985, 96, 292 CrossRef CAS.
- C. J. Davies, P. J. Miedziak, G. L. Brett and G. J. Hutchings, Chin. J. Catal., 2016, 37, 1600–1607 CrossRef CAS.
- G. Malta, S. J. Freakley, S. A. Kondrat and G. J. Hutchings, Chem. Commun., 2017, 53, 11733–11746 RSC.
- M. Zhu, Q. Wang, K. Chen, Y. Wang, C. Huang, H. Dai, F. Yu, L. Kang and B. Dai, ACS Catal., 2015, 5, 5306–5316 CrossRef CAS.
- M. Conte, A. F. Carley, C. Heirene, D. Willock, P. Johnston, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2007, 250, 231–239 CrossRef CAS.
- X. Liu, M. Conte, D. Elias, L. Lu, D. J. Morgan, S. J. Freakley, P. Johnston, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2016, 6, 5144–5153 RSC.
- B. Nkosi, N. J. Coville and G. J. Hutchings, J. Chem. Soc., Chem. Commun., 1988, 1, 71–72 RSC.
- B. Nkosi, N. J. Coville and G. J. Hutchings, Appl. Catal., 1988, 43, 33 CrossRef CAS.
- G. Malta, S. A. Kondrat, S. J. Freakley, C. J. Davies, L. Lu, S. Dawson, A. Thetford, E. K. Gibson, D. J. Morgan, W. Jones, P. P. Wells, P. Johnston, C. R. A. Catlow, C. J. Kiely and G. J. Hutchings, Science, 2017, 355, 1399–1403 CrossRef CAS PubMed.
- X. Liu, M. Conte, D. Elias, L. Lu, D. J. Morgan, S. J. Freakley, P. Johnston, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2016, 6, 5144 RSC.
- J. Zhao, B. Wang, X. Xu, Y. Yu, S. Di, H. Xu, Y. Zhai, H. He, L. Guo and Z. Pan, J. Catal., 2017, 350, 149–158 CrossRef CAS.
- H. He, J. Zhao, B. Wang, Y. Yue, G. Sheng, Q. Wang, L. Yu, Z.-T. Hu and X. Li, Materials, 2019, 12, 1310 CrossRef CAS PubMed.
- C. Huang, M. Zhu, L. Kang and B. Dai, Catal. Commun., 2014, 54, 61–65 CrossRef CAS.
- K. Zhou, J. Jia, C. Li, H. Xu, J. Zhou, G. Luo and F. Wei, Green Chem., 2014, 17, 356–364 RSC.
- P. T. Bishop, N. A. Carthey and P. Johnston, WO 2013/008004A3, May 16, 2013.
- M. Pernpointner and A. S. K. Hashmi, J. Chem. Theory Comput., 2009, 5, 2717–2725 CrossRef CAS PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kuhl, M. Havecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. L. Kniep, M. Tovar, R. W. Fischer, J. K. Norskov and R. Schlogl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- M. Flytzani-Stephanopoulos and B. C. Gates, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 545–574 CrossRef CAS PubMed.
- M. Newville, Rev. Mineral. Geochem., 2014, 78, 33–74 CrossRef CAS.
- (a) T. A. Smith, J. G. DeWitt, B. Hedman and K. O. Hodgson, J. Am. Chem. Soc., 1994, 116, 3836–3847 CrossRef CAS; (b) A. C. Leri, M. A. Marcus and S. C. B. Myneni, Geochim. Cosmochim. Acta, 2007, 71, 5834–5846 CrossRef CAS.
- E. Papirer, R. Lacroix, J.-B. Donnet, G. Nansé and P. Fioux, Carbon, 1995, 33, 63–72 CrossRef CAS.
- S. C. B. Myneni, Science, 2002, 295, 1039–1041 CrossRef CAS PubMed.
- T. Zhang, J. Zhao, J. Xu, J. Xu, X. Di and X. Li, Chin. J. Chem. Eng., 2016, 24, 484–490 CrossRef CAS.
- (a) S. E. Shadle, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1995, 117(8), 2259–2272 CrossRef CAS; (b) T. Glaser, B. Hedman, K. O. Hodgson and E. I. Solomon, Acc. Chem. Res., 2000, 33, 859–868 CrossRef CAS PubMed.
- L. S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433–6442 CrossRef CAS.
- X. Sun, S. R. Dawson, T. E. Parmentier, G. Malta, T. E. Davies, Q. He, L. Lu, D. J. Morgan, N. Carthey, P. Johnston, S. A. Kondrat, S. J. Freakley, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2020, 12, 560–567 CrossRef CAS PubMed.
- B. Hedman, P. Frank, J. E. Penner-Hahn, A. L. Roe, K. O. Hodgson, R. M. K. Carlson, G. Brown, J. Cerino, R. Hettel, T. Troxel, H. Winick and J. Yang, Nucl. Instrum. Methods Phys. Res., Sect. A, 1986, 246, 797–800 CrossRef.
- A. Vairavamurthy, B. Manowitz, G. W. Luther III and Y. Jeon, Geochim. Cosmochim. Acta, 1993, 57, 1619–1623 CrossRef CAS.
- F. Jalilehvand, Chem. Soc. Rev., 2006, 35, 1256–1268 RSC.
- A. Vairavamurthy, Spectrochim. Acta, Part A, 1998, 54, 2009–2017 CrossRef.
- H.-P. Loock, J. Chem. Educ., 2011, 88, 282–283 CrossRef CAS.
- P. Karen, P. McArdle and J. Takats, Pure Appl. Chem., 2014, 86, 1017–1108 CAS.
- G. Malta, S. A. Kondrat, S. J. Freakley, C. J. Davies, S. Dawson, X. Liu, L. Lu, K. Dymkowski, F. Fernandez-Alonso, S. Mukhopadhyay, E. K. Gibson, P. P. Wells, S. F. Parker, C. J. Kiely and G. J. Hutchings, ACS Catal., 2018, 8, 8493–8505 CrossRef CAS.
- J. V. Halliday and J. M. Lynam, Dalton Trans., 2016, 45, 12611–12626 RSC.
- P. B. J. Thompson, B. N. Nguyen, R. Nicholls, R. A. Bourne, J. B. Brazier, K. R. J. Lovelock, S. D. Brown, D. Wermeille, O. Bikondoa, C. A. Lucas, T. P. A. Hasee and M. A. Newton, J. Synchrotron Radiat., 2015, 22, 1426–1439 CrossRef PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, 1327 Search PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- F. Neese, Coord. Chem. Rev., 2009, 253, 526 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02152k |
| This journal is © The Royal Society of Chemistry 2020 |