
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBNN-1,3-dipoles: isolation and intramolecular cycloaddition with unactivated arenes†
Rui
Guo
a,
Jingxing
Jiang
b,
Chenyang
Hu
c,
Liu Leo
Liu
 c,
Ping
Cui
a,
Meihua
Zhao
a,
Zhuofeng
Ke
*b,
Chen-Ho
Tung
a and
Lingbing
Kong
*ad
c,
Ping
Cui
a,
Meihua
Zhao
a,
Zhuofeng
Ke
*b,
Chen-Ho
Tung
a and
Lingbing
Kong
*ad
aSchool of Chemistry and Chemical Engineering, Key Laboratory of Colloid and Interface Chemistry of Ministry of Education, Shandong University, Jinan 250100, P. R. China. E-mail: konglb@sdu.edu.cn
bSchool of Materials Science & Engineering, PCFM Lab, Sun Yat-sen University, Guangzhou 510275, P. R. China. E-mail: kezhf3@mail.sysu.edu.cn
cShenzhen Grubbs Institute and Department of Chemistry, Southern University of Science and Technology, Shenzhen 518055, P. R. China
dState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China
First published on 19th June 2020
Abstract
The mono-base-stabilized 1,2-diboranylidenehydrazine derivatives featuring a 1,3-dipolar BNN skeleton are obtained by dehydrobromination of [ArB(Br)NH]2 (Ar = 2,6-diphenylphenyl (Dpp), Ar = 2,6-bis(2,4,6-trimethylphenyl)phenyl (Dmp) or Ar = 2,4,6-tri-tert-butylphenyl (Mes*)) with N-heterocyclic carbenes (NHCs). Depending on the Ar substituents, such species can be isolated as a crystalline solid (Ar = Mes*) or generated as reactive intermediates undergoing spontaneous intramolecular aminoboration of the proximal arene rings via [3 + 2] cycloaddition (Ar = Dpp or Dmp). The latter reactions showcase the 1,3-dipolar reactivity toward unactivated arenes at ambient temperature. In addition, double cycloaddition of the isolable BNN species with two CO2 molecules affords a bicyclic species consisting of two fused five-membered BN2CO rings. The electronic structures of these BNN species and the mechanisms of these cascade reactions are interrogated through density functional theory (DFT) calculations.
Introduction
The concept of 1,3-dipolar cycloaddition (1,3-DC), pioneered by Huisgen in the 1960s,1 has continued to influence a broad range of disciplines from medicine to materials sciences and even life sciences.2 1,3-DCs are reactions between a 1,3-dipole and a multiply bonded dipolarophile (e.g. alkenes, alkynes, carbonyls, and imines) to construct five-membered rings (Fig. 1a). This transformation represents one of the most effective approaches leading to synthetically and medicinally relevant five-membered heterocycles, and thus is pervasive in organic chemistry.3 However, the application of arenes as 2π-dipolarophiles in 1,3-DC reactions to produce fused heterocyclic scaffolds remains extremely challenging due to the substantial resonance stability of arenes. Although Huisgen showed that azomethine ylides are capable of undergoing 1,3-DC with dipolarophilic arenes as early as 1971,4 only activated arenes, such as heteroaromatic systems and nitro-substituted or fused arenes, are capable of engaging in such transformations.5 To date, only one set of examples describing intramolecular cycloadditions of azomethine ylides with unactivated arenes has been achieved, albeit under flash vacuum pyrolysis conditions.6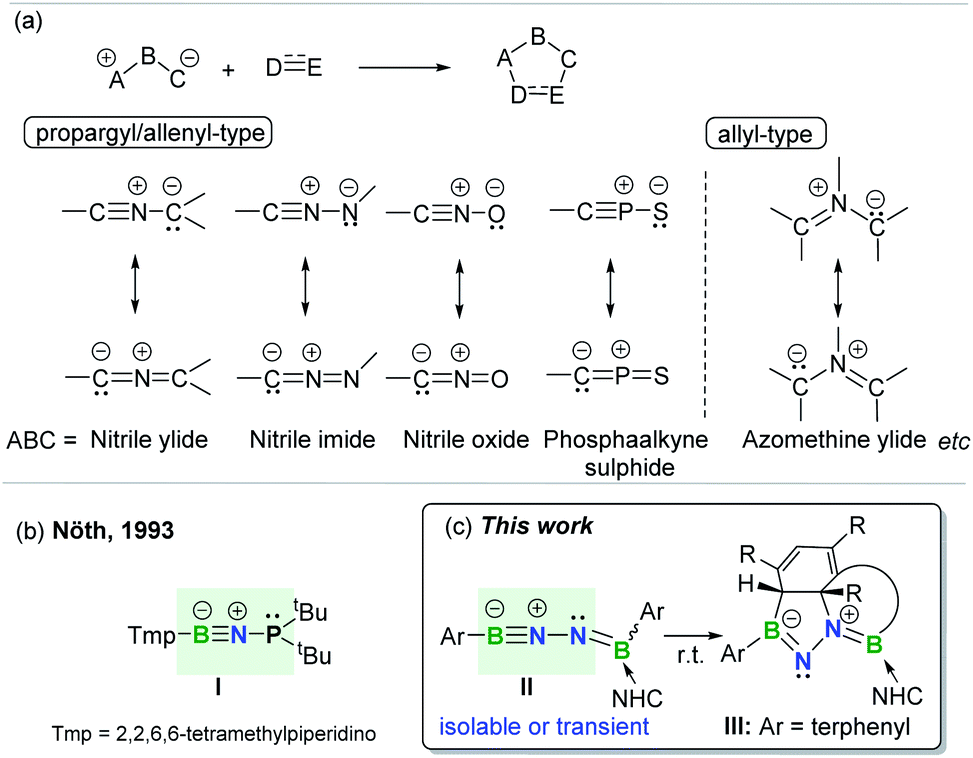 | ||
| Fig. 1 (a) 1,3-Dipolar cycloaddition and representative 1,3-dipoles. (b) BNP-1,3-dipole. (c) This work. | ||
1,3-Dipoles, which feature a delocalized three-center 4π-electron system with a separation of charge over the three atoms (Fig. 1a), can be classified into two broadly defined categories according to the geometry of the central atom: propargyl/allenyl and allyl 1,3-dipoles.7 The generation of a variety of transient or persistent 1,3-dipoles consisting of carbon, nitrogen, oxygen, sulfur and phosphorus as well as their cycloaddition reactivities have been extensively investigated over the past decades.7 However, boron-containing analogs of 1,3-dipoles are seldom encountered. To date, the only one example of a stable propargyl/allenyl-type BNP-1,3-dipole I was described by Nöth et al. in 1993 (Fig. 1b).8
Given the wide ranging applications of BN-containing heterocyclic compounds in organic synthesis, catalysis, materials science, and biomedical research, synthetic strategies toward a series of BN-scaffolds of azaborines, diazaboroles, diazadiborinines, triazadiboroles, and BN-polycyclic aromatic hydrocarbons etc. have been developed.9 For example, very recently, Kinjo and Ota described the synthesis of a neutral aromatic boron-rich inorganic benzene analog, namely 1,4-diaza-2,3,5,6-tetraborinine,10 while the group of Braunschweig synthesized complex BN-heterocycles via the reactions of organic azides with diaryl(dihalo)diboranes(4).11 However, synthesis of fused BN-heterocyclic skeletons via 1,3-DC with unactivated arenes is hitherto unknown.12 It is important to note that incorporation of heteroatoms into the 4π scaffold of 1,3-dipoles significantly effects the π-bond strength and frontier orbital energies,13 thereby modulating the ensuing reactivity. This prompted us to prepare and investigate reactions of an novel family of main group 1,3-dipoles containing a BNN-core. In the present work, we report the generation of mono-base-stabilized 1,2-diboranylidenehydrazines ArBNNB(NHC)Ar II (Fig. 1c). Depending on the Ar substituents, such species can be isolated and characterized or generated in situ as reactive intermediates that spontaneously engage with the proximal unactivated arenes via intramolecular 1,3-DC to afford fused BN-heterocycles III.
Results and discussion
Cycloaddition reactivity of transient BNN-1,3-dipoles
In targeting a 1,2-diboranylidenehydrazine, we sought to incorporate abundant steric protection around boron to disfavor cyclooligomerization.14 Accordingly, we chose ArBBr2 with a sterically encumbering 2,6-diphenylphenyl (Dpp) or 2,6-bis(2,4,6-trimethylphenyl)phenyl (Dmp) substituent (Scheme 1). The borylated hydrazines 1 (11B NMR: 36.3 ppm) and 2 (11B NMR: 32.4 ppm) were prepared in moderate yields via reactions of ArBBr2 with bis(trimethylsilyl)hydrazine. The formulations of 1 and 2 as [DppB(Br)NH]2 and [DmpB(Br)NH]2, respectively, were unambiguously validated by high resolution mass spectrometry and X-ray diffraction studies (Fig. 2a and b). In the solid-state structures, the BN-involved-butadiene framework of 2 appears to be a E,E-configuration with a nearly coplanar B2Br2C2N2H2 structure, whereas that of 1 is a formal Z,Z-configuration.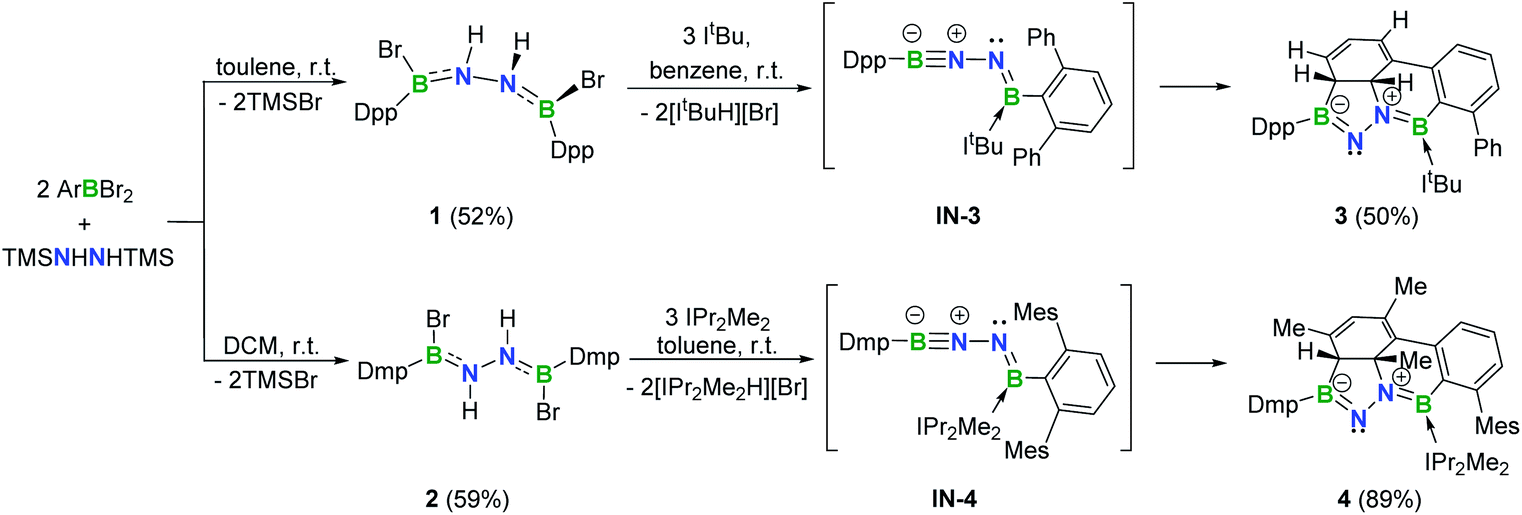 | ||
| Scheme 1 Synthesis of 1–4 (Dpp = 2,6-diphenylphenyl; Dmp = 2,6-bis(2,4,6-trimethylphenyl)phenyl; Mes = 2,4,6-trimethylphenyl). | ||
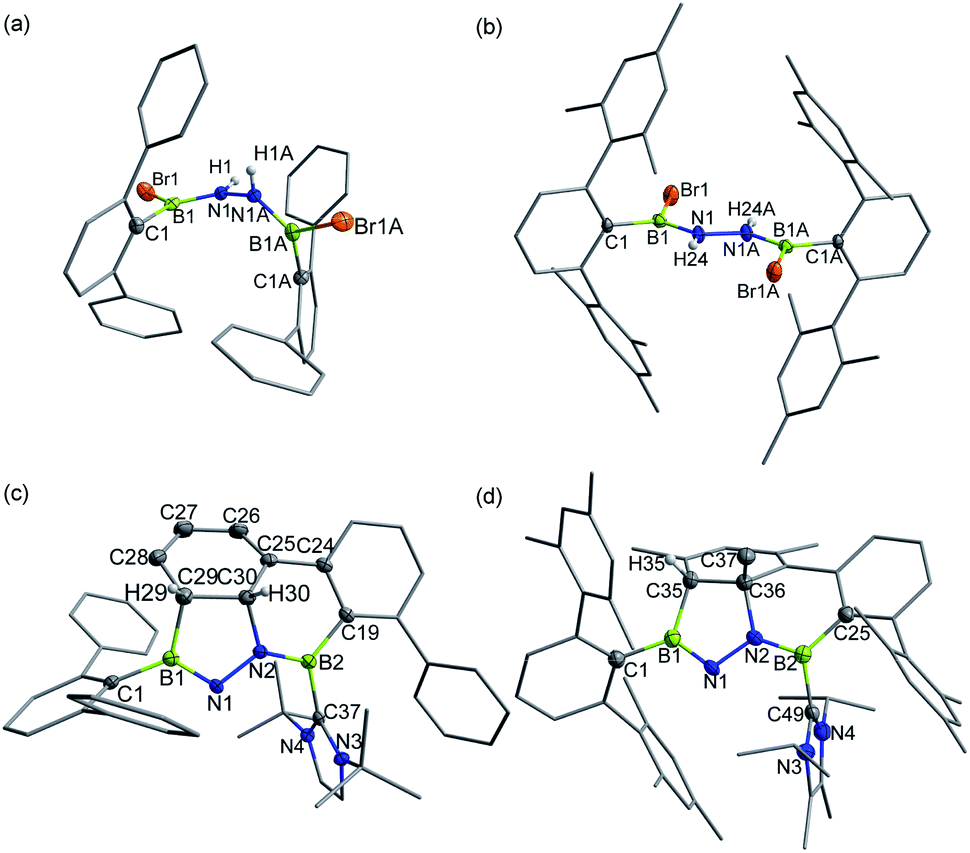 | ||
| Fig. 2 Solid-state structures of 1 (a), 2 (b), 3 (c) and 4 (d). Hydrogen atoms except for N–H, C29–H, C30–H and C35–H are omitted for clarity. Thermal ellipsoids are set at the 30% probability level. | ||
Noting that N-heterocyclic carbenes (NHCs) are potent Brønsted bases15 and ancillary ligands exhibiting excellent capability for stabilization of highly reactive main group species,16 we reacted 1 with 3 equivalents of 1,3-di-tert-butylimidazol-2-ylidene (ItBu) in benzene at room temperature (Scheme 1). The dehydrobromination proceeded immediately with precipitation of [ItBuH][Br]. After recrystallization, a new species 3 was obtained as a yellow crystalline solid in 50% yield. The 11B NMR spectrum of 3 revealed two broad singlets at 43.0 and 23.0 ppm, indicating the presence of two inequivalent tricoordinate boron centers. The 1H NMR spectrum displayed a multiplet resonance at 2.68 ppm and a doublet at 4.42 ppm, integrating for one proton each.
Single crystal X-ray diffraction revealed 3 to be a tetracyclic BN-embedded 3aH-3a1H-acephenanthrylene derivative, resulting from the apparent [3 + 2] cycloaddition of a transient BNN-1,3-dipole IN-3 across the proximal flanking phenyl substituent of Dpp moiety (Fig. 2c). The bond lengths of C25–C26 (1.350(3) Å) and C27–C28 (1.339(4) Å) are in the typical range of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, whereas those of C25–C30 (1.486(3) Å), C26–C27 (1.452(3) Å), C28–C29 (1.480(3) Å) and C29–C30 (1.543(3) Å) appear to be C–C single bonds. These structural parameters are in line with the formation of a 1,3-cyclohexadiene unit from an arene. Both the cyclohexadiene and BC2N2 rings are essentially planar (the sum of internal angles = 719.86° and 539.54°, respectively), and the torsion angle of B1–C29–C30–C25 is 123.43(17)°. The B1–N1 and B2–N2 bond lengths are 1.387(3) and 1.375(3) Å, respectively, are significantly shorter than those in 1 (1.444(7) Å, av.) and fall within the typical range of BN double bonds in aminoboranes.17 Additionally, the N1–N2 bond distance of 1.4243(19) Å is marginally longer than that in 1 (1.391 (8) Å) and close to that reported for [Mes2B(H)N]2 (1.411(4) Å).18 These structural parameters reveal a B
C double bonds, whereas those of C25–C30 (1.486(3) Å), C26–C27 (1.452(3) Å), C28–C29 (1.480(3) Å) and C29–C30 (1.543(3) Å) appear to be C–C single bonds. These structural parameters are in line with the formation of a 1,3-cyclohexadiene unit from an arene. Both the cyclohexadiene and BC2N2 rings are essentially planar (the sum of internal angles = 719.86° and 539.54°, respectively), and the torsion angle of B1–C29–C30–C25 is 123.43(17)°. The B1–N1 and B2–N2 bond lengths are 1.387(3) and 1.375(3) Å, respectively, are significantly shorter than those in 1 (1.444(7) Å, av.) and fall within the typical range of BN double bonds in aminoboranes.17 Additionally, the N1–N2 bond distance of 1.4243(19) Å is marginally longer than that in 1 (1.391 (8) Å) and close to that reported for [Mes2B(H)N]2 (1.411(4) Å).18 These structural parameters reveal a B![[partial double bond, top dashed]](https://www.rsc.org/images/entities/char_e12f.gif) N–NB skeleton. The B1–N1–N2–B2 torsion angle of −171.37(18)° suggests a near coplanarity. Atoms B1 and B2 bear sp2 hybridization character, judging from the angular sums of 359.95° and 359.93°, respectively.
N–NB skeleton. The B1–N1–N2–B2 torsion angle of −171.37(18)° suggests a near coplanarity. Atoms B1 and B2 bear sp2 hybridization character, judging from the angular sums of 359.95° and 359.93°, respectively.
In an analogous fashion, treatment of 2 with 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene (IPr2Me2) in toluene led to a species 4 (11B NMR: 41.3 and 22.3 ppm) (Scheme 1) with a fused tetracyclic framework (Fig. 2d). It is noteworthy that compounds 3 and 4 could also be prepared in the dark, which rules out a light-induced radical mechanism for this transformation.
Mechanistic studies
To gain more insight into the mechanism leading to 3, density functional theory (DFT) calculations were carried out at the SMD-M06-2X/def2-TZVP//M06-2X/def2-SVP level of theory (Fig. 3a). While the double dehydrobromination of 1 with ItBu followed by mono-ligation to generate IN-3 is endergonic by 11.8 kcal mol−1, the precipitation of [ItBuH][Br] over the course of the reaction promotes this conversion. Subsequently, the approach of the proximal phenyl group toward the BNN moiety in IN-3 gives rise to TS (27.7 kcal mol−1) in a concerted manner. Intrinsic bond orbital (IBO) calculations,19 which have been demonstrated to give an exact representation of any Kohn–Sham DFT wave function, showcase that the redistribution of π-electrons over the phenyl and BNN fragments forms B1–C29 and C30–N2 σ-bonds to furnish 3 (−2.9 kcal mol−1) (Fig. 3a). The additional linkage (i.e., C24–C25 or B2–C19 bonding) of 3 plays an essential role in stabilizing the dearomatized framework. In the absence of either the C24–C25 or B2–C19 linkage (Fig. 3b), the corresponding intermolecular 1,3-DC using model molecules is calculated to be endergonic by 14.1 or 8.1 kcal mol−1, respectively. Collectively, these results demonstrate that the intramolecular 1,3-DC is exergonic and favorable whereas the formation of 3a or 3b in an intermolecular fashion is endergonic and unfavorable.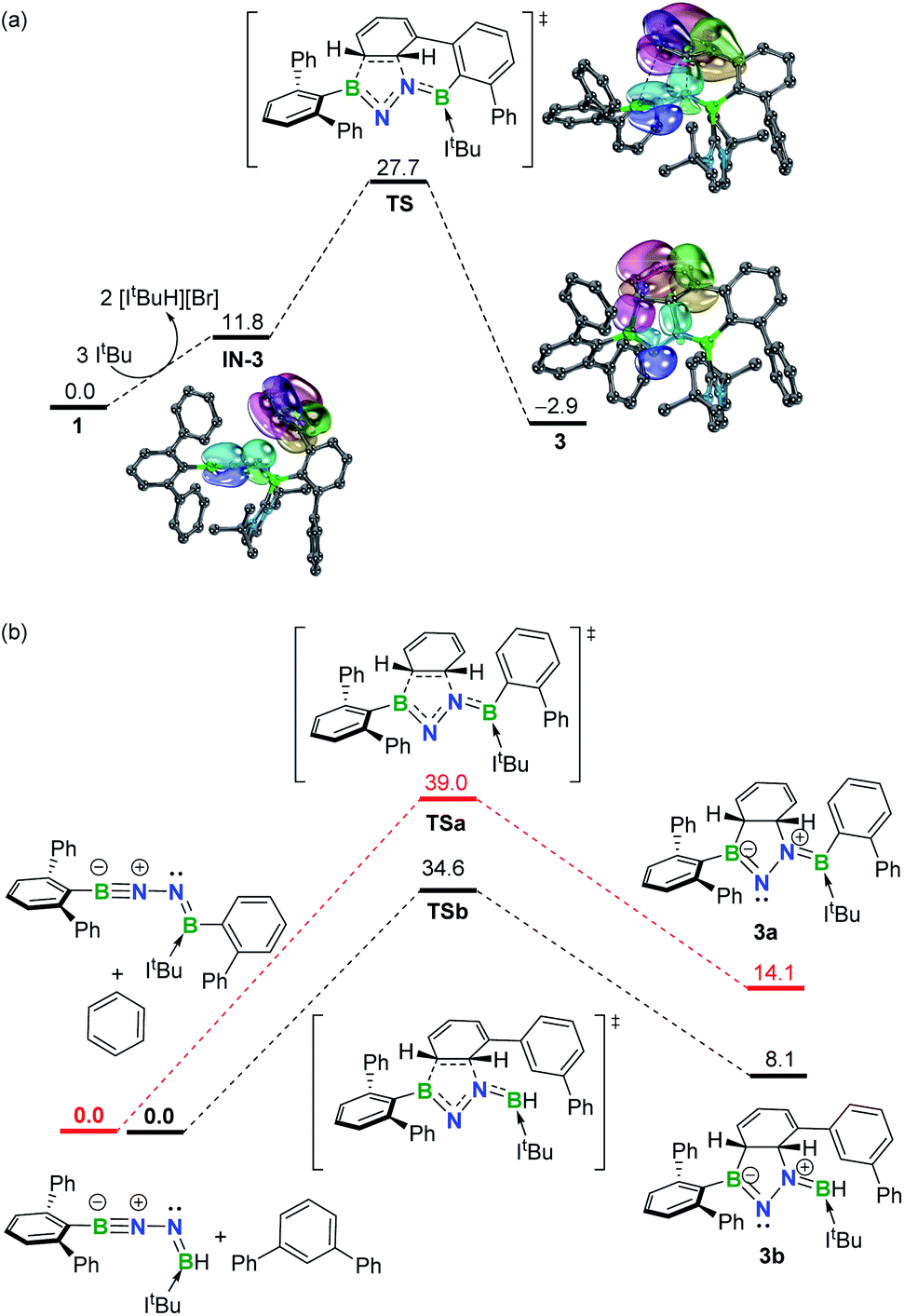 | ||
| Fig. 3 (a) Free energy profile for formation of 3 with selected IBOs (75% of the orbital electron's density). (b) Free energy profiles for formation of 3a and 3b. Energies are given in kcal mol−1. | ||
The NHC coordination in 3 is also crucial for stabilization of the fused tetracycle, probably due to the compensation of the intrinsic electron deficiency of B2 and the enhancement of the nucleophilicity of N2. The absence of ItBu is spontaneously destructive to the tetracyclic framework leading to the base-free 1,2-diboranylidenehydrazine DppBN–NBDpp upon an unrestricted geometry optimization.
It is important to note that the incorporation of heteroatoms into unsaturated organic skeletons forms multiply bonded main group species have proven particularly effective in enabling cycloadditions with arenes in the ground state.20 For example, the Power and Tokitoh groups independently demonstrated intermolecular [4 + 2] cycloadditions of transient dialumenes with benzene/toluene.21 Stephan and co-workers described a reversible intramolecular [4 + 2] cycloaddition of a phosphaalkene to an arene ring.22 More recently, the group of Kinjo showed the diboration of a series of polycyclic aromatic hydrocarbons by a 1,3,2,5-diazadiborinine derivative.23 The formation of 3 and 4 represents the first examples of dearomative bifunctionalization of arenes via 1,3-dipolar cycloaddition under mild conditions, thus providing another avenue to access novel fused BN-heterocyclic scaffolds. In addition, such transformations are rare examples of unactivated arenes behaving as 2π components in cycloaddition reactions.6,20h,23
A stable BNN-1,3-dipole
Efforts to prevent the reaction of the BNN moiety toward sterically accessible arene substituents were undertaken. To this end, we prepared [Mes*B(Br)NH]2 (5, Mes* = 2,4,6-tri-tert-butylphenyl) analogously to 1 and 2 (Fig. 4a). Reaction of 5 with 3 equivalents of IPr2Me2 in toluene afforded a yellow solution, from which species 6 was isolated in 31% yield after work-up (Scheme 2). In the 11B NMR spectrum of 6, two broad resonances at 33.7 and 2.8 ppm were observed. The former is considerably downfield shifted with respect to those of iminoborane–carbene adducts (12.6–24.0 ppm),24 while the latter is comparable to the chemical shift seen for iminoborane, Mes*B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) NtBu (4.3 ppm).25 The 1H NMR spectrum of 6 in C6D6 showed one set of peaks assignable to the IPr2Me2 ligand and no evidence of N–H resonances.
NtBu (4.3 ppm).25 The 1H NMR spectrum of 6 in C6D6 showed one set of peaks assignable to the IPr2Me2 ligand and no evidence of N–H resonances.
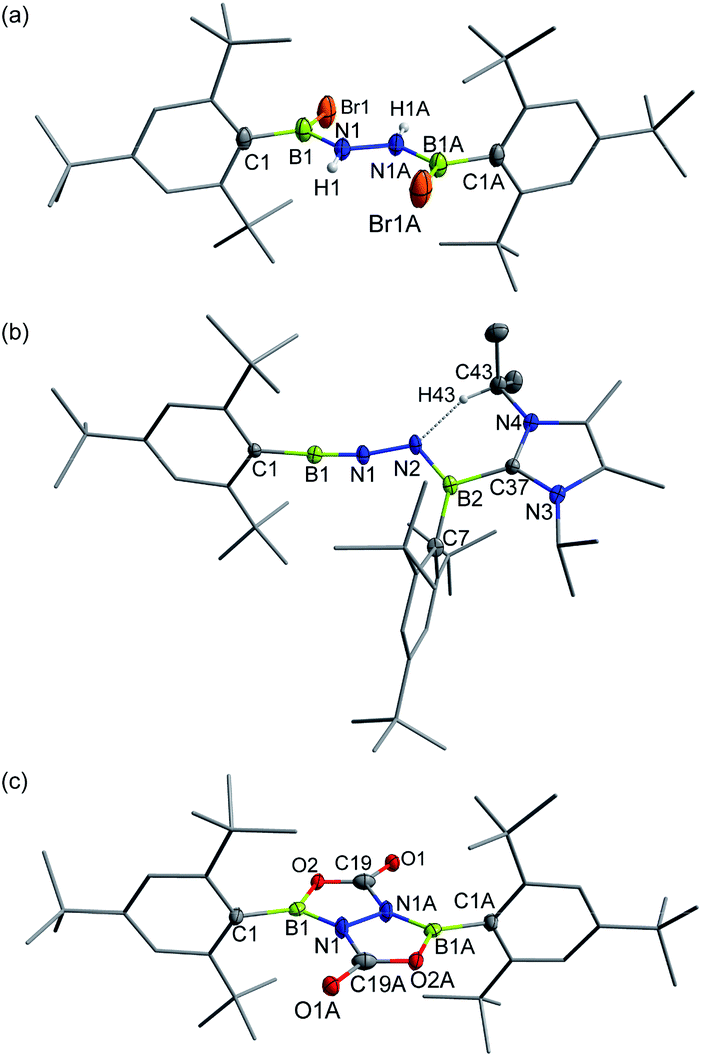 | ||
| Fig. 4 Solid-state structures of 5 (a), 6 (b) and 7 (c). Hydrogen atoms except for N–H and C43–H are omitted for clarity. Thermal ellipsoids are set at the 30% probability level. | ||
 | ||
| Scheme 2 Synthesis of 5–7 (Mes* = 2,4,6-tri-tert-butylphenyl). | ||
Single crystals of 6 suitable for X-ray diffraction were obtained by slow evaporation of a concentrated n-hexane solution at −25 °C within 24 h. X-ray crystallography confirmed the structure as Mes*BN–NB(IPr2Me2)Mes* (Fig. 4b). While such a linear BNN moiety is reminiscent of recent works of main group diazo and N2 chemistry by the Stephan26 and Braunschweig27 groups, respectively, compound 6 contains a BNNB motif in which one of the boron atoms is only dicoordinate. In a related work, Klingebiel et al. disclosed that the iminoborane supported by two bis(trimethylsilyl)amido groups could easily self-cyclize through C–H addition across the BN bond.28 The most prominent structural feature is the nearly linear C1–B1–N1–N2 chain (∠C1–B1–N1 = 176.0(2)°; ∠B1–N1–N2 = 169.24(19)°) with the B1–N1 bond length of 1.236(4) Å that is almost same to that observed for Mes*BNtBu (1.232(6) Å).25 The N1, N2, B2, C7, C37 atoms are in a planar arrangement (mean deviation form plane = 0.0081 Å). The B2–N2 bond (1.368(3) Å) appears at lengths seen for carbene-stabilized iminoborane complexes (1.304(3)–1.360(5) Å).24 The N1–N2 bond distance of 1.351(2) Å is significantly longer in comparison with that in the dinitrogen bis(borylene) compound {[(CAAC)DurB]2(μ2-N2)} (1.248(4) Å).27 Nevertheless, this N1–N2 bond length is markedly shorter than that in 5 (1.396(4) Å), indicating the enhanced interaction between these two unsaturated BN fragments in 6. The geometry of the B2N2 bond adopts a formal E-configuration, similar to iminoborane-CAAC adducts (CAAC = cyclic (alkyl)(amino)carbene), reported by Braunschweig, Bertrand, Stephan et al.24b–d The presence of a hydrogen bonding interaction of N(2)⋯H(43)–C(43) (D = 2.9482(1) Å, θ = 137.947(4)°) is identified in the solid state. Collectively, the dipolar BNN fragment features a propargyl/allenyl-like structure. Compound 6 represents the first example of a room-temperature-stable BNN-1,3-dipole.
Bonding
The electronic properties of 6 were further investigated based on DFT calculations using the M06-2X/def2-SVP optimized structure. The LUMO and LUMO + 1 predominantly involves the π*-antibonding interaction, which is delocalized over one of the Mes* rings, B1–N1 and B2–N2 bonds, and the imidazole ring (Fig. 5a and b). The HOMO is the out-of-plane B1–N1 and B2–N2 π orbitals (Fig. 5c), whereas the in-plane B1–N1 π orbital as well as a lone pair of electrons on N2 are represented by the HOMO − 1 (Fig. 5d). Furthermore, the Wiberg bond indices (WBIs) of the B1–N1, N1–N2 and B2–N2 bonds from the natural bond orbital (NBO) calculations (M06-2X/TZVP) are 1.87, 1.14 and 1.35, respectively (Table S2†), suggesting that the NHC ligation on B2 remarkably diminishes the π-donation from N2 to B2. To gain more insight into the interaction between two unsaturated BN fragments, the second-order perturbation theory of the NBO method was applied, which shows a strong hyperconjugative delocalization from the p-type N2 lone pair to the B1–N1 π*-antibonding orbital with a donor–acceptor stabilization energy of 20.2 kcal mol−1 (Fig. S33†). The partial double bond character of N1–N2 is further evidenced by natural localized molecular orbital (NLMO) analysis that reveals a highly delocalized N2 lone pair toward N1 (Fig. S33†). The B1 (0.82) and B2 (0.73) atoms are positively charged, while N1 (−0.65) and N2 (−0.72) atoms carry negative charges (Table S3†). Bending the B–N–N angle up to 155° from 172° in 6 has negligible energetic cost (2.7 kcal mol−1), indicating a flexibility of the molecule along this coordinate.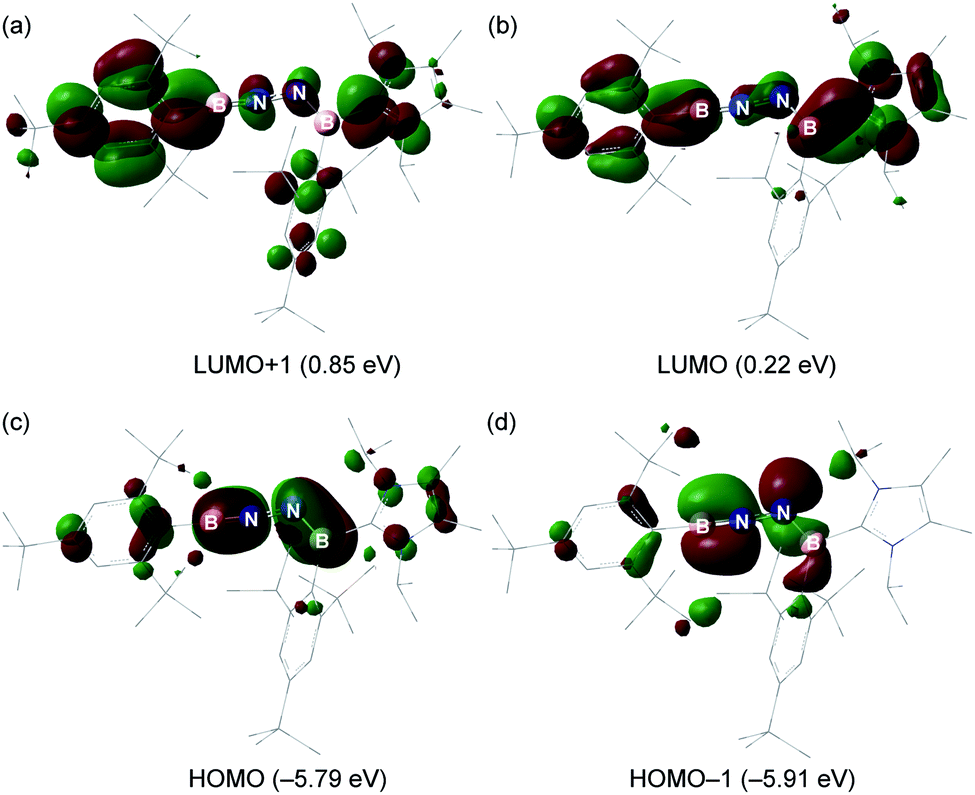 | ||
| Fig. 5 (a–d) Key molecular orbitals of 6 (isovalue = 0.03, calculated at the M06-2X/Def2-SVP level of theory). | ||
Compound 6 exhibits an absorption maximum in the UV/Vis spectrum in n-hexane at 372.3 nm (f = 0.7601) (Fig. S28†). This absorption was primarily attributed to the π–π* transitions (HOMO − 1 → LUMO and HOMO → LUMO) according to TD-DFT calculations (Fig. S34†).
Natural resonance theory (NRT) calculations (M06-2X/TZVP) on the simplified model 6′ shows that two leading propargyl-type forms 6′a (24.4%) and 6′b (22.5%) (Fig. 6a), coupled with several other propargyl-type forms (weights <5%; Fig. S35†), predominately contribute to the electronic structure of this BNN-1,3-dipole. Two allenyl-type forms 6′c (2.2%) and 6′d (2.1%) are identified with low weights (Fig. 6a). It is noteworthy that NHCs have been shown to feature a carbon–nitrogen multiple bond based on NRT analysis.29 In contrast, the nitrile ylide CH3CNC(CH3)2 shows an opposite trend with propargyl-type form weight of 16.5% and allenyl-type form weight of 66.1% (Fig. 6b). This finding provides hints that substitution of C atoms with heteroatoms is capable of shifting electronic properties.
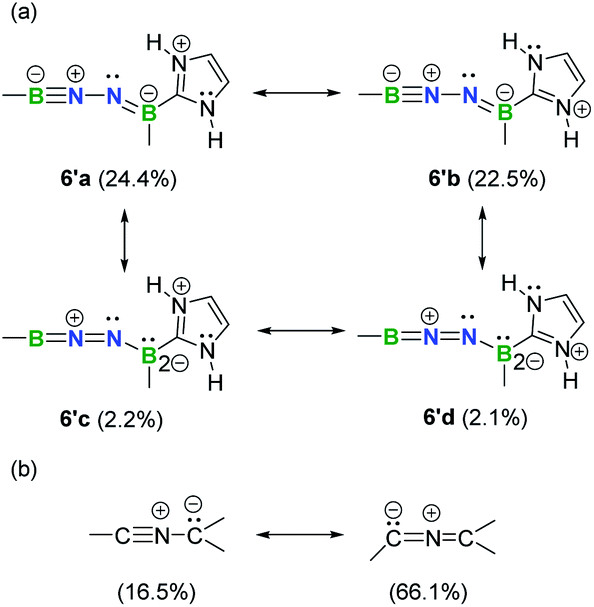 | ||
| Fig. 6 Selected predominant resonance structures and their weights for 6′ (a) and nitrile ylide CH3CNC(CH3)2 (b). | ||
Reactivity of 6 toward CO2
As a further probe of the 1,3-dipole ability of 6, we examined its reactivity toward carbon dioxide (CO2) (Scheme 2). Exposure of a toluene solution of 6 to 1 atm dry CO2 at room temperature for 5 min resulted in a slight yellow cloudy solution. After work-up, colorless crystals of 7 (11B NMR: 36.0 ppm) were isolated in 32% yield. The presence of a carbonyl carbon is evidenced by a resonance at 155.5 ppm in the 13C NMR spectrum. An unambiguous elucidation of the structure of 7 was performed based on single crystal X-ray diffraction (Fig. 4c), revealing a system containing a fused bicyclic B,N,O-involved heterocycle. Interestingly, two CO2 molecules were fixed via a double 1,3-DC, concurrent with the loss of IPr2Me2. It is important to mention that sequential CO2 cycloaddition by BN species to construct a fused bicyclic compound is hitherto unknown, although I ([3 + 2] cycloaddition),8 iminoboranes ([2 + 2] cycloaddition)14b,24b and a series of N/B based frustrated Lewis pairs (FLPs)30,31 for CO2 activation have been explored.Conclusions
More than six decades after Huisgen's classification of 1,3-dipoles, this work demonstrates that a BNN-1,3-dipole, namely a mono-base-stabilized 1,2-diboraneylidenehydrazine 6, is synthetically achievable. Single crystal X-ray diffraction in combination with DFT calculations provide insights into the most plausible electronic structure of 6, which features a conjugated BN chain with a formal B1N1 triple bond and a B2N2 double bond. Remarkably, in the presence of a sterically accessible flanking arene substituent, the corresponding BNN-1,3-dipoles are highly fleeting and susceptible to [3 + 2] cycloaddition reactions, affording BN-containing fused tetracycles 3 and 4. Such 1,3-dipolar reactivity is also reflected by the 1,3-DC of 6 with CO2 to give 7. Further reactivity investigations of such BNN-1,3-dipoles, as well as the synthesis of their constitutional isomers, are currently underway in our laboratory.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We gratefully acknowledge financial support from the Natural Science Foundation of China (Grants 21971144, 21702228 and 21673301), the Natural Science Foundation of Shandong Province (Grants ZR2019ZD46 and ZR2017MB021), the Key R&D Program of Shandong Province (Grant 2019GGX102032), the Guangdong Natural Science Funds for Distinguished Young Scholars (Grant 2015A030306027), the Qilu Youth Scholar Funding of Shandong University (Grant 11190088963021), the Multidisciplinary Research and Innovation Team of Young Scholars of Shandong University (Grant 2020QNQT007) and the Fundamental Research Funds for the Central Universities. L. L. L. gratefully acknowledge the start-up fund of SUSTech. The DFT calculations were made possible by the facilities of the Shared Hierarchical Academic Research Computing Network (SHARCNET: http://www.Sharcnet.ca) and Compute Canada. L. L. L. thanks Prof. D. W. Stephan (U of T) and Prof. J. Zhu (XMU) for kindly providing access to computational facilities. L. L. L thanks Dr E. A. Romero at the University of California, Berkeley and Dr R. Wei (SINAP) for valuable discussion. We thank Prof. D. Sun (SDU) and Dr Y. Li (NTU) for assistance with the X-ray crystal structure analysis. We thank the reviewers for their valuable suggestions.Notes and references
- (a) R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 633–645 CrossRef; (b) R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef; (c) R. Huisgen, Proc. Chem. Soc., 1961, 357–396 Search PubMed; (d) R. Huisgen, Festschrift der Zehnjahresfeier des Fonds der Chemischen Industrie, Düsseldorf, 1960, p. 73 Search PubMed.
- (a) N. Nishiwaki, Methods and Applications of Cycloaddition Reactions in Organic Synthesis, Wiley-VCH, Weinheim, Germany, 2014 CrossRef; (b) Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, in The Chemistry of Heterocyclic Compounds, ed. A. Padwa and W. H. Pearson, Wiley, New York, NY, USA, 2003, vol. 59 Search PubMed.
- (a) L.-J. Wang and Y. Tang, Intermolecular 1,3-dipolar cycloadditions of alkenes, alkynes and allenes, in Comprehensive Organic Synthesis, ed. P. Knochel and G. A. Molander, Elsevier, Amsterdam, The Netherlands, 2nd edn, 2014, vol. 4, pp. 1342–1383 Search PubMed; (b) R. S. Menon and V. Nair, Intramolecular 1,3-dipolar cycloadditions of alkenes, alkynes and allenes, in Comprehensive Organic Synthesis, ed. P. Knochel and G. A. Molander, Elsevier, Amsterdam, The Netherlands, 2nd edn, 2014, vol. 4, pp. 1281–1341 Search PubMed.
- R. Huisgen and W. Scheer, Tetrahedron Lett., 1971, 12, 481–484 CrossRef.
- J. H. Ryan, Arkivoc, 2015, 1, 160–183 Search PubMed.
- B. R. Henke, A. J. Kouklis and C. H. Heathcock, J. Org. Chem., 1992, 57, 7056–7066 CrossRef CAS.
- R. Huisgen, J. Org. Chem., 1976, 41, 403–419 CrossRef CAS.
- J. Kroner, H. Nöth, K. Polborn, H. Stolpmann, M. Tacke and M. Thomann, Chem. Ber., 1993, 126, 1995–2002 CrossRef CAS.
- (a) C. R. McConnell and S.-Y. Liu, Chem. Soc. Rev., 2019, 48, 3436–3453 RSC; (b) Z. X. Giustra and S.-Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef CAS PubMed; (c) G. Bélanger-Chabot, H. Braunschweig and D. K. Roy, Eur. J. Inorg. Chem., 2017, 4353–4368 CrossRef; (d) B. Su and R. Kinjo, Synthesis, 2017, 49, 2985–3034 CrossRef CAS; (e) P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed; (f) M. J. D. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8–29 CrossRef.
- K. Ota and R. Kinjo, Angew. Chem., Int. Ed., 2020, 59, 6572–6575 CrossRef CAS PubMed.
- D. Prieschl, G. Bélanger-Chabot, X. Guo, M. Dietz, M. Müller, I. Krummenacher, Z. Lin and H. Braunschweig, J. Am. Chem. Soc., 2020, 142, 1065–1076 CrossRef CAS PubMed.
- (a) M. Schäfer, J. Schäfer, R. D. Dewhurst, W. C. Ewing, M. Krahfuß, M. W. Kuntze-Fechner, M. Wehner, C. Lambert and H. Braunschweig, Chem.–Eur. J., 2016, 22, 8603–8609 CrossRef PubMed; (b) H. Braunschweig, K. Geetharani, J. O. C. Jimenez-Halla and M. Schäfer, Angew. Chem., Int. Ed., 2014, 53, 3500–3504 CrossRef CAS PubMed; (c) H. Braunschweig, A. Damme, J. O. C. Jimenez-Halla, B. Pfaffinger, K. Radacki and J. Wolf, Angew. Chem., Int. Ed., 2012, 51, 10034–10037 CrossRef CAS PubMed.
- (a) L. Zhao, S. Pan, N. Holzmann, P. Schwerdtfeger and G. Frenking, Chem. Rev., 2019, 119, 8781–8845 CrossRef CAS PubMed; (b) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- (a) M. Nutz, B. Borthakur, R. D. Dewhurst, A. Deißenberger, T. Dellermann, M. Schäfer, I. Krummenacher, A. K. Phukan and H. Braunschweig, Angew. Chem., Int. Ed., 2017, 56, 7975–7979 CrossRef CAS PubMed; (b) L. Xie, J. Zhang, H. Hu and C. Cui, Organometallics, 2013, 32, 6875–6878 CrossRef CAS; (c) B. Kröckert, K.-H. van Bonn and P. Paetzold, Z. Anorg. Allg. Chem., 2005, 631, 866–868 CrossRef; (d) M. Geschwentner, G. Elter and A. Meller, Z. Anorg. Allg. Chem., 1993, 619, 1474–1478 CrossRef CAS; (e) P. Paetzold, Adv. Inorg. Chem., 1987, 31, 123–170 CrossRef CAS.
- (a) P. Cui, R. Guo, L. Kong and C. Cui, Inorg. Chem., 2020, 59, 5261–5265 CrossRef CAS PubMed; (b) R. Guo, X. Huang, M. Zhao, Y. Lei, Z. Ke and L. Kong, Inorg. Chem., 2019, 58, 13370–13375 CrossRef CAS PubMed; (c) Y. Wang, H. Hu, J. Zhang and C. Cui, Angew. Chem., Int. Ed., 2011, 50, 2816–2819 CrossRef CAS PubMed; (d) H. Cui and C. Cui, Chem.–Asian J., 2011, 6, 1138–1141 CrossRef CAS PubMed; (e) Y. Gao, J. Zhang, H. Hu and C. Cui, Organometallics, 2010, 29, 3063–3065 CrossRef CAS; (f) H. Cui, Y. Shao, X. Li, L. Kong and C. Cui, Organometallics, 2009, 28, 5191–5195 CrossRef CAS; (g) R. S. Ghadwal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 5683–5686 CrossRef CAS PubMed.
- (a) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed; (b) T. W. Hudnall, R. A. Ugarte and T. A. Perera, N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools (2), The Royal Society of Chemistry, UK, 2017, pp. 178–237 Search PubMed; (c) L. J. Murphy, K. N. Robertson, J. D. Masuda and J. A. C. Clyburne, N-Heterocyclic Carbenes, Wiley-VCH Verlag GmbH & Co. KGaA, New York, 2014, pp. 427–498 Search PubMed; (d) E. Rivard, in Comprehensive Inorganic Chemistry II, ed. K. Poeppelmeier, Elsevier, Amsterdam, 2013, pp. 457–484 Search PubMed.
- K.-A. Østby, G. Gundersen, A. Haaland and H. Nöth, Dalton Trans., 2005, 2284–2291 RSC.
- D. C. Pestana and P. P. Power, Inorg. Chem., 1991, 30, 528–535 CrossRef CAS.
- (a) G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2015, 54, 5518–5522 CrossRef CAS PubMed; (b) G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- For low-valent main group species for arene activation, see: (a) A. Hermann, M. Arrowsmith, D. E. Trujillo-Gonzalez, J. O. C. Jimenez-Halla, A. Vargas and H. Braunschweig, J. Am. Chem. Soc., 2020, 142, 5562–5567 CrossRef CAS PubMed; (b) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000–11003 CrossRef CAS PubMed; (c) L. Zhu, J. Zhang and C. Cui, Inorg. Chem., 2019, 58, 12007–12010 CrossRef CAS PubMed; (d) L. L. Liu, L. L. Cao, J. Zhou and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 273–277 CrossRef CAS PubMed; (e) L. L. Liu, J. Zhou, L. L. Cao, R. Andrews, R. L. Falconer, C. A. Russell and D. W. Stephan, J. Am. Chem. Soc., 2018, 140, 147–150 CrossRef CAS PubMed; (f) D. Wendel, A. Porzelt, F. A. D. Herz, D. Sarkar, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 8134–8137 CrossRef CAS PubMed; (g) P. Bissinger, H. Braunschweig, K. Kraft and T. Kupfer, Angew. Chem., Int. Ed., 2011, 50, 4704–4707 CrossRef CAS PubMed. For cyclobutadiene for arene cycloaddition, see: (h) Y. Inagaki, M. Nakamoto and A. Sekiguchi, Nat. Commun., 2014, 5, 3018–3023 CrossRef PubMed. For carboryne for arene cycloaddition, see: (i) D. Zhao, J. Zhang and Z. Xie, Angew. Chem., Int. Ed., 2014, 53, 8488–8491 CrossRef CAS PubMed.
- (a) R. J. Wright, A. D. Phillips and P. P. Power, J. Am. Chem. Soc., 2003, 125, 10784–10785 CrossRef CAS PubMed; (b) T. Agou, K. Nagata and N. Tokitoh, Angew. Chem., Int. Ed., 2013, 52, 10818–10821 CrossRef CAS PubMed.
- L. L. Liu, J. Zhou, L. L. Cao, Y. Kim and D. W. Stephan, J. Am. Chem. Soc., 2019, 141, 8083–8087 CAS.
- Y. Su, D. C. H. Do, Y. Li and R. Kinjo, J. Am. Chem. Soc., 2019, 141, 13729–13733 CrossRef CAS PubMed.
- (a) L. Winner, A. Hermann, G. Bélanger-Chabot, O. F. González-Belman, J. O. C. Jiménez-Halla, H. Kelch and H. Braunschweig, Chem. Commun., 2018, 54, 8210–8213 RSC; (b) F. Dahcheh, D. W. Stephan and G. Bertrand, Chem.–Eur. J., 2015, 21, 199–204 CrossRef CAS PubMed; (c) F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159–13163 CrossRef CAS PubMed; (d) H. Braunschweig, W. C. Ewing, K. Geetharani and M. Schäfer, Angew. Chem., Int. Ed., 2015, 54, 1662–1665 CrossRef CAS PubMed.
- G. Elter, M. Neuhaus, A. Meller and D. Schmidt-Bäse, J. Organomet. Chem., 1990, 381, 299–313 CrossRef CAS.
- (a) D. Zhu, Z.-W. Qu and D. W. Stephan, Dalton Trans., 2020, 49, 901–910 RSC; (b) L. L. Cao, J. Zhou, Z.-W. Qu and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 18487–18491 CrossRef CAS PubMed; (c) C. Tang, Q. Liang, A. R. Jupp, T. C. Johnstone, R. C. Neu, D. Song, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2017, 56, 16588–16592 CrossRef CAS PubMed. See also: (d) R. L. Melen and D. W. Stephan, Dalton Trans., 2015, 44, 5045–5048 RSC.
- (a) M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed; (b) M.-A. Légaré, M. Rang, G. Bélanger-Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329–1332 CrossRef PubMed.
- K. Bode, U. Klingebiel, M. Noltemeyer and H. Witte-Abel, Z. Anorg. Allg. Chem., 1995, 621, 500–505 CrossRef CAS.
- L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CAS.
- (a) A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1, 35–48 CrossRef; (b) D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- (a) B. R. Barnett, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Chem. Commun., 2015, 51, 541–544 RSC; (b) Z. Lu, Y. Wang, J. Liu, Y. Lin, Z. H. Li and H. Wang, Organometallics, 2013, 32, 6753–6758 CrossRef CAS; (c) T. Voss, T. Mahdi, E. Otten, R. Fröhlich, G. Kehr, D. W. Stephan and G. Erker, Organometallics, 2012, 31, 2367–2378 CrossRef CAS; (d) M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 13559–13568 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1977878–1977884. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02162h |
| This journal is © The Royal Society of Chemistry 2020 |