
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regiodivergent construction of medium-sized heterocycles from vinylethylene carbonates and allylidenemalononitriles†
Xiang
Zhang
acd,
Xiang
Li
b,
Jun-Long
Li
 d,
Qi-Wei
Wang
cd,
Wen-Lin
Zou
d,
Yan-Qing
Liu
bd,
Zhi-Qiang
Jia
d,
Fu
Peng
*a and
Bo
Han
*b
d,
Qi-Wei
Wang
cd,
Wen-Lin
Zou
d,
Yan-Qing
Liu
bd,
Zhi-Qiang
Jia
d,
Fu
Peng
*a and
Bo
Han
*b
aWest China School of Pharmacy, Sichuan University, Chengdu 610041, China. E-mail: pengf@scu.edu.cn
bState Key Laboratory of Southwestern Chinese Medicine Resources, School of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China. E-mail: hanbo@cdutcm.edu.cn
cChengdu Institute of Organic Chemistry, Chinese Academy of Sciences, Chengdu 610041, China
dAntibiotics Research and Re-evaluation Key Laboratory of Sichuan Province, Sichuan Industrial Institute of Antibiotics, Chengdu University, Chengdu 610052, China
First published on 10th February 2020
Abstract
Medium-sized heterocycles exist in a broad spectrum of biologically active natural products and medicinally important synthetic compounds. The construction of medium-sized rings remains challenging, particularly the assembly of different ring sizes from the same type of substrate. Here we report palladium-catalyzed, regiodivergent [5 + 4] and [5 + 2] annulations of vinylethylene carbonates and allylidenemalononitriles. We describe the production of over 50 examples of nine- and seven-membered heterocycles in high isolated yields and excellent regioselectivities. We demonstrate the synthetic utility of this approach by converting a nine-membered ring product to an interesting polycyclic caged molecule via a [2 + 2] transannulation. Mechanistic studies suggest that the [5 + 2] annulation proceeds through palladium-catalyzed ring-opening/re-cyclization from the [5 + 4] adducts.
Introduction
Cyclic molecular frameworks have special importance in chemical research and industry.1 Medium-sized rings (MSR, 7–11 members),2 particularly hetero-rings, exist in a large number of biologically active natural products and medicinally important synthetic molecules3 (Fig. 1). However, MSRs are challenging to prepare because of their inherent entropic factors and transannular interactions. Most established methods to generate MSRs are based on a fixed reaction site and suitable only for rings of the same size;4 changing the size of the ring usually requires changing the substrate design.5 Such a substrate-controlled strategy can be quite costly and inefficient because of the need to prepare the necessary substrate variants and optimize them in the ring-forming reactions. It could be much more efficient to develop a way to generate medium-sized rings of various sizes from the same set of substrates, simply by altering the reaction conditions. However, to our knowledge, controlling the regioselectivity of medium-sized ring cyclization is notoriously difficult and remains underdeveloped6 (Scheme 1a).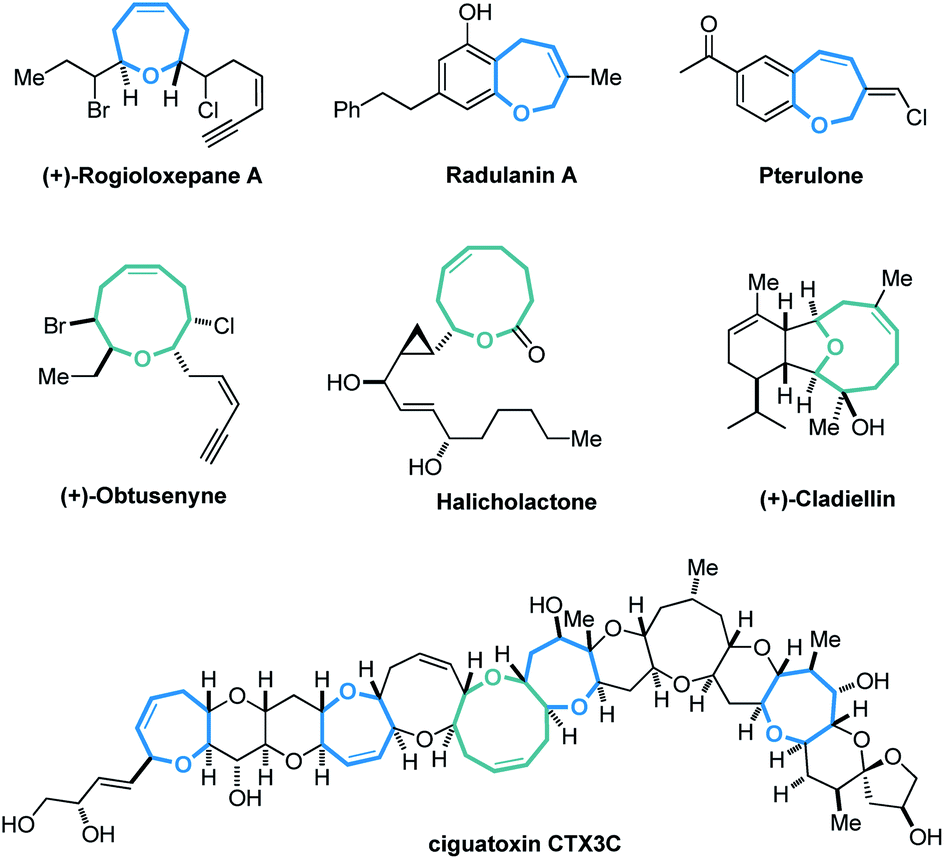 | ||
| Fig. 1 Selected natural products and synthetic bioactive compounds containing medium-sized oxo-heterocycles. | ||
 | ||
| Scheme 1 Divergent construction of medium-sized rings. | ||
Vinylethylene carbonates (VECs) have recently emerged as versatile building blocks for various cyclizations, because of their inherent ability to undergo decarboxylation in the presence of a palladium catalyst to generate highly reactive zwitterionic π-allyl palladium intermediates.7,8 Recently, Zhao and co-workers disclosed that π-allyl palladium species can serve as 1,5-dipoles in a highly efficient [5 + 4] annulation with 1,3-azadienes to construct nine-membered hetero-rings.9 Since then, palladium-catalyzed [5 + n] annulations involving vinylethylene carbonates have been described for generating various medium-sized heterocycles10 (Scheme 1b). However, rarely have vinylethylene carbonates been used for divergent annulation,10h,11 and to our knowledge, they have never been applied to regioselective [5 + n] cyclization, which could generate multiple ring sizes.
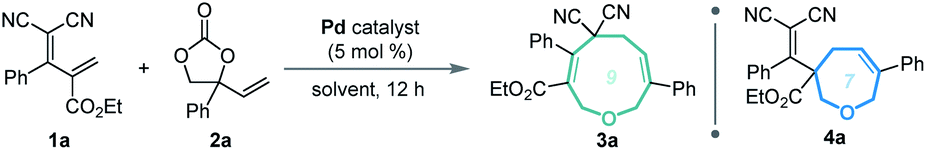
Given our experience with the assembly of biologically interesting heterocycles by exploring novel catalytic reactions,12 we aimed to develop a convenient strategy for ring size-divergent construction of medium-sized rings. We found that by using the versatile, electron-deficient diene substrate allylidenemalononitriles,13 we could achieve smooth [5 + 4] annulation with vinylethylene carbonates in MeCN in the presence of a palladium catalyst at room temperature, generating a nine-membered product. More importantly, we could completely shift the regioselectivity to [5 + 2] cyclization by changing the solvent to THF and increasing the reaction temperature, generating a seven-membered product. In both cases, the regioselectivity was nearly perfect (Scheme 1c). In addition, the nine-membered cyclic ether adducts were able to undergo intramolecular transannular [2 + 2] cycloaddition14 to build a structurally interesting caged polycycle.
Results and discussion
Our investigations began with a reaction between the easily accessible diene 1a and vinylethylene carbonate 2a. Different solvents were evaluated in the presence of Pd(PPh3)4 at 20 °C, and MeCN afforded the [5 + 4] adduct 3a with a high yield and regioselectivity, while other solvents provided a mixture of nine- and seven-membered products (Table 1, entries 1–5) or 3a in low yield (entry 6). The reaction in THF gave the highest ratio of [5 + 2] product 4a, which encouraged us to screen the reaction conditions further in order to switch the regioselectivity. With THF as the solvent, phosphine ligands L1–L7 were screened, but all reacted inefficiently (entry 7). To our gratification, conducting the reaction at 40 °C improved the relative amount of seven-membered cyclic ether 4a, and increasing the temperature to 60 °C afforded 4a as a single regioisomer in high yield (entries 8 and 9). Further increasing the temperature maintained the high regioselectivity but lowered the yield slightly (entry 10). Using other solvents at 60 °C did not improve the results in terms of yield and regioselectivity (entries 11–16).15|
|
|||||
|---|---|---|---|---|---|
|
|
|||||
| Entry | Catalyst | Solvent | Temp. (°C) | Yieldb (%) |
3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4ac :4ac |
|
a Unless noted otherwise, the reactions were carried out with 1a (0.10 mmol), 2a (0.15 mmol) and the Pd catalyst (5 mol%) in solvent (1 mL) for 12 h.
b Yield was determined by 1H-NMR analysis with CH2Br2 as the internal standard; the data in parentheses refer to isolated yields.
c The ratio of 3a:4a was determined by 1H-NMR analysis of the crude reaction mixture.
d For 48 h.
e For 24 h.
f The Pd/ligand complex was pre-prepared with Pd2(dba)3·CHCl3 and a ligand in THF at rt for 1 h.
|
|||||
| 1d | Pd(PPh3)4 | Toluene | 20 | 72 | 3.5:1 |
| 2 | Pd(PPh 3 ) 4 | MeCN | 20 | 96(90) |
>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
| 3d | Pd(PPh3)4 | DCM | 20 | 68 | 3.6:1 |
| 4d | Pd(PPh3)4 | CHCl3 | 20 | 85 | 2.6:1 |
| 5 | Pd(PPh3)4 | THF | 20 | 85 | 1.4:1 |
| 6e | Pd(PPh3)4 | DMF | 20 | 16 | >20:1 |
| 7f | Pd/L1–L7 | THF | 20 | <5 | — |
| 8 | Pd(PPh3)4 | THF | 40 | 98 | 1:4.6 |
| 9 | Pd(PPh 3 ) 4 | THF | 60 | 91(84) |
<1:20
|
| 10 | Pd(PPh3)4 | THF | 80 | 89 | <1:20 |
| 11 | Pd(PPh3)4 | 1,4-Dioxane | 60 | 83 | 16.0:1 |
| 12 | Pd(PPh3)4 | Toluene | 60 | 88 | 1:1.3 |
| 13 | Pd(PPh3)4 | MeCN | 60 | 87 | 8.6:1 |
| 14 | Pd(PPh3)4 | DMF | 60 | 81 | 14.8:1 |
| 15 | Pd(PPh3)4 | DCM | 60 | 76 | 1:1.4 |
| 16 | Pd(PPh3)4 | CHCl3 | 60 | 80 | 5.3:1 |
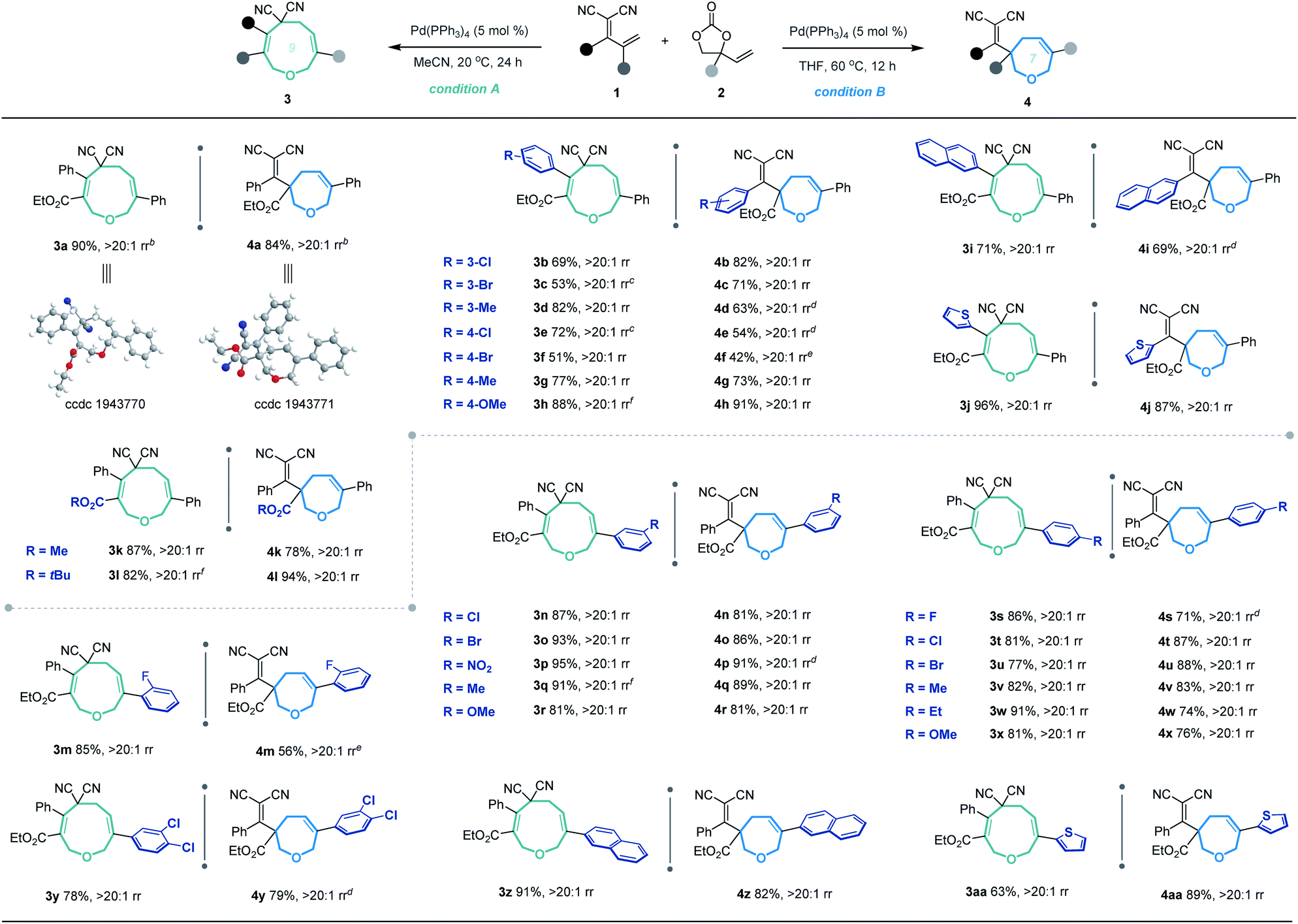
Based on the optimized conditions for generating the seven- and nine-membered rings, we explored the generality of our method with various substituted allylidenemalononitriles 1 and vinylethylene carbonates 2. Each substrate combination was tested under conditions A or B to generate, respectively, nine-membered products 3 or seven-membered products 4 (Table 2). First, we tested a range of electrophiles 1 with various aryl groups bearing different electronic and steric substituents, delivering the [5 + 4] adducts 3a–3h or [5 + 2] adducts 4a–4h in reasonable yields with excellent regioselectivities. Divergent annulations proceeded smoothly with a diene electrophile bearing a 2-naphthyl moiety, selectively affording the medium-sized rings 3i and 4i with satisfactory results. The reactions also worked well for thienyl-substituted 1, generating the products 3j and 4j with impressive yields and regioselectivities. Different ester groups on 1 did not harm the reaction (3k–3l and 4k–4l). We also tested three types of allylidenemalononitril substrates changing the ester group to hydrogen, but none of them could offer the desired products (see the ESI† for detailed experimental procedure). Next, we examined the reaction of 1a with vinylethylene carbonates 2 featuring either an electron-donating or -withdrawing group on the benzene ring. The corresponding nine-membered products 3m–3y and seven-membered products 4m–4y were obtained with high isolated yields and regioselectivities. Naphthyl- and heteroarene-substituted 2 also performed well in the regiodivergent cyclizations (3z–3aa and 4z–4aa). Moreover, this methodology is not tolerant to the VECs bearing aliphatic substituents (see the ESI† for more details).
|
a Unless noted otherwise, the [5 + 4] annulation was performed under conditions A: 1 (0.1 mmol), 2 (0.15 mmol) and Pd(PPh3)4 (5 mol%) in MeCN (1.0 mL) at 20 °C for 24 h, and the rr (regioisomeric ratio) refers to the ratio of 3:4; the [5 + 2] annulation was performed under conditions B: 1 (0.1 mmol), 2 (0.15 mmol) and Pd(PPh3)4 (5 mol%) in THF (1.0 mL) at 60 °C for 12 h, and the rr refers to the ratio of 4:3; yield of the isolated product; rr was determined by 1H-NMR analysis of the crude reaction mixture.
b The structures of 3a and 4a were determined by X-ray diffraction analysis, and the structures of other products were assigned by analogy.
c For 48 h.
d At 80 °C.
e At 100 °C.
f With 0.3 mmol of 2.
|
|---|
|
Subsequently, several experiments were performed to demonstrate the robustness and practicality of this synthetic method. Firstly, both [5 + 4] and [5 + 2] annulation of diene 1a and vinylethylene carbonate 2a could be scaled up to the 1 gram scale without drastic loss of yield (Scheme 2a). Then, the synthetic utility of our approach was explored, and we found that one of the two cyano groups on 3a could be selectively hydrolyzed in formic acid in the presence of a Pd(OAc)2 catalyst, delivering 5 in 81% yield (Scheme 2b). Treating 3a with L-selectride triggered reductive C–O bond cleavage that opened the nine-membered ring, offering linear 1,4-diene alcohol 6 in moderate yield. The product 4a could undergo a retro-Knoevenagel reaction under aqueous basic conditions to release the malononitrile moiety and give the ketone-containing derivative 7 in 52% yield. It could also undergo sequential retro-Knoevenagel and retro-Claisen condensation in the presence of Et3N, iPrOH and water to afford product 8 in excellent yield. In addition, we extended this divergent cyclization strategy to a reaction between 1a and vinyloxazolidinone 9, assembling the nine- and seven-membered azacycles 10 and 11 in satisfying yields with excellent regioselectivities (Scheme 2c).
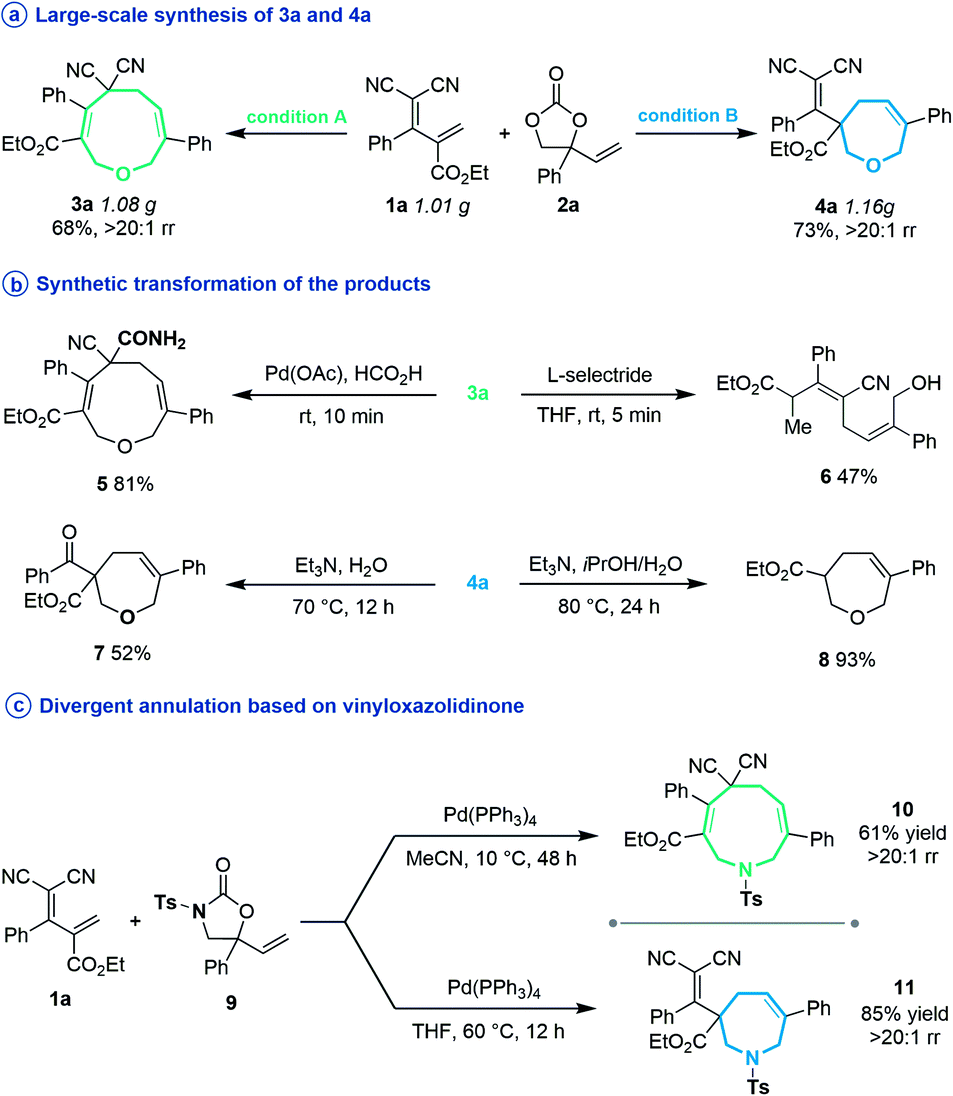 | ||
| Scheme 2 Large-scale reactions of regiodivergent cyclizations and further synthetic applications. | ||
Unexpectedly, heating the [5 + 4] adduct 3a without the Pd catalyst in toluene generated a cage-like molecule 12a in high yield. The structure of 12a was confirmed by X-ray diffraction analysis. We attribute the formation of this product to heat-induced isomerization of the styrene moiety from the E- to Z-configuration, followed by transannular [2 + 2] cycloaddition (for the preliminary mechanism investigation, see the ESI†). This reaction proved tolerant of various functional groups, allowing the rapid synthesis of caged compounds 12a–12j (Scheme 3a). With a series of synthesized molecule fused pharmacologically privileged frameworks in hand and motivated by the pharmaceutical properties of nitrile,16a–c oxygen heterocycles1f and caged-skeletons,16d–h we preliminarily evaluated their ability to inhibit the proliferation of a panel of cancer cell lines (Scheme 3b). In these experiments, the concentrations of tested compounds and paclitaxel (PTX) were 20 μM and 5 μM, respectively. Compounds 12c/j, 12j, 12a and 12d showed promising cytotoxicity against A549, PC12, SH-SY5Y and A375 cells, respectively (for the details, see ESI, Table S3†).
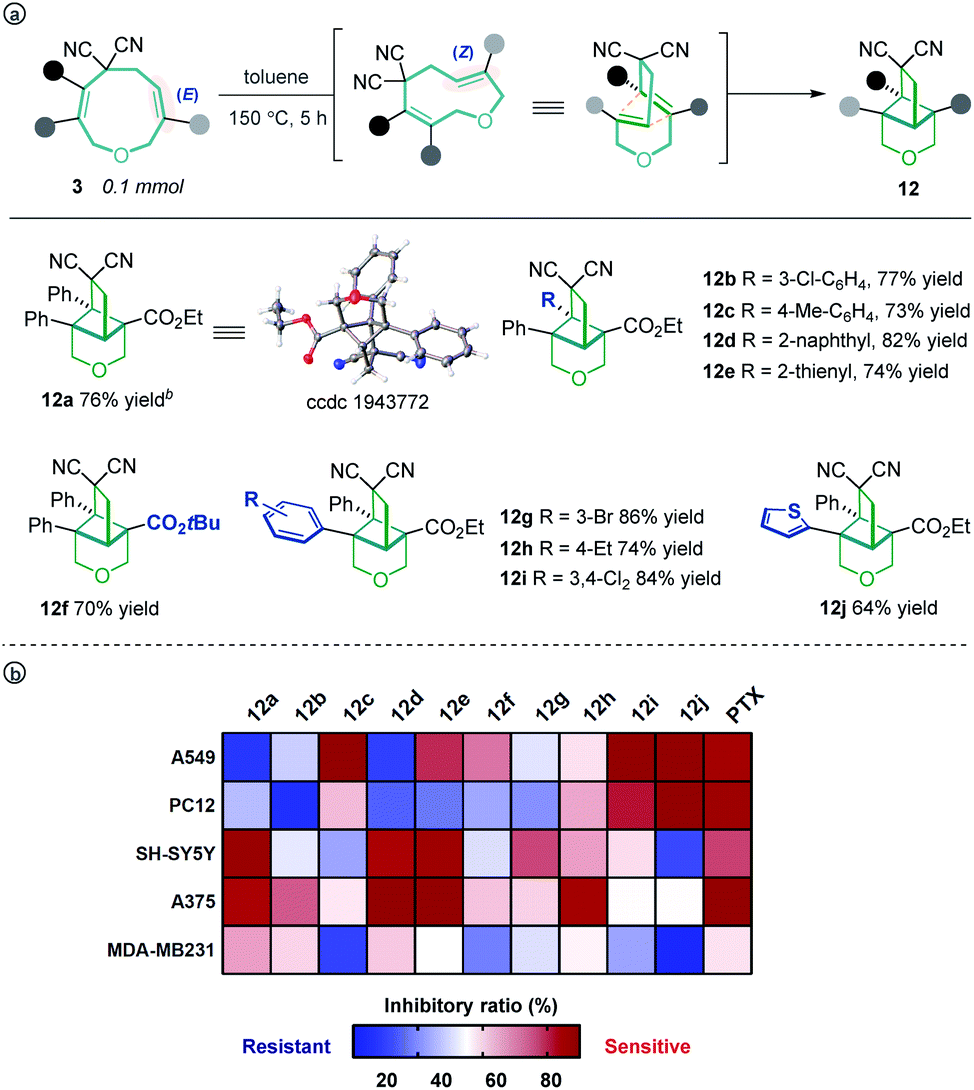 | ||
| Scheme 3 The transannular [2 + 2] cycloaddition of 3 (a) and heat map of the mean inhibitory ratio of compounds 12a–12j against a panel of cancer cell lines (b). | ||
In order to investigate the reaction mechanism, we performed several control experiments based on the reaction of allylidenemalononitril 1a and vinylethylene carbonate 2a. Firstly, the reaction progress was monitored by NMR analysis. As shown in Scheme 4a, under the [5 + 4] annulation reaction conditions, the nine-membered product 3a formed gradually, without concomitant emergence of the [5 + 2] seven-membered product 4a. In contrast, in the reaction meant to produce 4a, the starting material 1a was rapidly consumed and 3a was initially generated in high NMR yield, together with trace amounts of 4a. Subsequently, the ratio of 3a/4a slowly decreased until 4a was obtained as the sole regioisomer (Scheme 4b). Follow-up experiments showed that in the presence of a palladium catalyst in THF at 60 °C, 3a converted to 4a, but not vice versa (Scheme 4c). These results suggest that the nine-membered 3a undergoes palladium-catalyzed ring-opening/re-cyclization to produce 4a. In addition, we found that using excess vinylethylene carbonate inhibited the transformation from 3a into 4a under heating conditions in THF (Scheme 4d), probably because the palladium catalyst prefers to coordinate with a higher concentration of vinylethylene carbonate which blocks the palladium activation of 3a.17
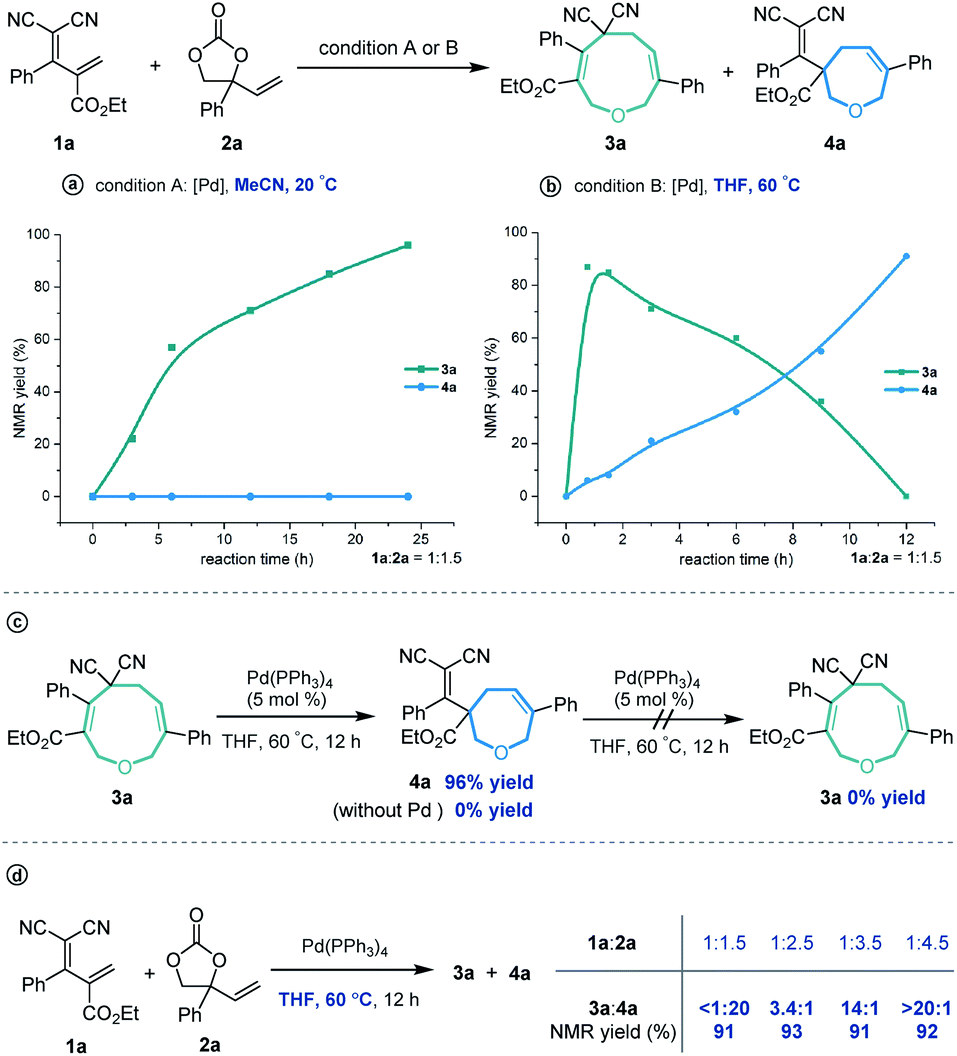 | ||
| Scheme 4 Control experiments. (a) Reaction progress was monitored in MeCN at 20 °C; (b) Reaction progress was monitored in THF at 60 °C; (c) Transformation from 3a to 4a; (d) Effect of the loading of VEC on the regioisomeric ratio. | ||
These experimental results suggest the following mechanism to rationalize the regioselectivity of the [5 + 4] and [5 + 2] annulations (Fig. 2). The palladium-catalyzed decarboxylation of vinylethylene carbonate 2a generates an ambiphilic π-allyl palladium intermediate I, which undergoes vinylogous Michael addition with allylidenemalononitril 1a to form intermediate II. At lower temperature and in MeCN solvent, the π-allylic anion is stabilized by dicyano electron-withdrawing groups, so the corresponding α terminal carbon attacks the electrophilic π-allyl palladium moiety to deliver 3ain a kinetically controlled manner. At higher temperature and in THF solvent, the same pathway generates 3a, which can revert to intermediate IIvia palladium-catalyzed ring-opening, but en route it can undergo a different ring-closing reaction between an internal γ-carbon and the π-allyl palladium moiety, delivering 4a in a thermodynamically controlled reaction.
 | ||
| Fig. 2 Proposed mechanism. | ||
Conclusions
In summary, we have developed a regiodivergent cyclization of vinylethylene carbonates and allylidenemalononitriles for the synthesis of medium-sized heterocycles. [5 + 4] annulation proceeds smoothly in MeCN at lower temperature, delivering nine-membered oxo-heterocycles in high yields. Changing the solvent to THF and raising the temperature completely reverse the regioselectivity of the ring-closing step, giving rise to [5 + 2] annulation that generates seven-membered heterocycles. In this way, our strategy allows the selective assembly of two heterocycle sizes from the same set of substrates through simple manipulation of reaction conditions. The nine-membered products efficiently undergo a transannular [2 + 2] cycloaddition to afford intriguing caged ring systems. Mechanistic studies suggest that [5 + 2] cyclization may occur via palladium-catalyzed ring-opening/cyclization from [5 + 4] adducts. Further biological studies of these novel cyclic molecules are currently underway in our laboratory, and the results will be reported in due course.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Financial support from the NSFC (21871031, 21702021 and 81573588), Science & Technology Department of Sichuan Province (2017JQ0032, 2017JZYD0001, and 2017JY0323), “Thousand Talents Program” of Sichuan Province, “Chengdu Talents Program” and Start-up Fund of Chengdu University is gratefully acknowledged.Notes and references
- (a) W. Carruthers, Cycloaddition reactions in organic synthesis, Pergamon, Oxford, 1990 Search PubMed; (b) Cycloaddition reactions in organic synthesis, ed. S. Kobayashi and K. A. Jorgensen, Wiley-VCH, New York, 2002 Search PubMed; (c) Handbook of cyclization reactions, ed. S.-M. Ma, Wiley-VCH, New York, 2010 Search PubMed; (d) Methods and applications of cycloaddition reactions in organic syntheses, ed. N. Nishiwaki, John Wiley & Sons, New York, 2014 Search PubMed; (e) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed; (f) M. D. Delost, D. T. Smith, B. J. Anderson and J. T. Njardarson, J. Med. Chem., 2018, 61, 10996 CrossRef CAS PubMed.
- For selected reviews on medium-sized rings, see: (a) L. Yet, Chem. Rev., 2000, 100, 2963 CrossRef CAS PubMed; (b) A. S. Kleinke, D. Webb and T. F. Jamison, Tetrahedron, 2012, 68, 6999 CrossRef CAS; (c) M. E. Maier, Angew. Chem., Int. Ed., 2000, 39, 2073 CrossRef CAS; (d) I. Shiina, Chem. Rev., 2007, 107, 239 CrossRef CAS PubMed; (e) J. R. Donald and W. P. Unsworth, Chem.–Eur. J., 2017, 23, 8780 CrossRef CAS PubMed; (f) A. Hussain, S. K. Yousuf and D. Mukherjee, RSC Adv., 2014, 4, 43241 RSC.
- (a) G. Guella, I. Mancini, G. Chiasera and F. Pietra, Helv. Chim. Acta, 1992, 75, 310 CrossRef CAS; (b) M. Yoshida, K. Nakatani and K. Shishido, Tetrahedron, 2009, 65, 5702 CrossRef CAS; (c) J. B. P. A. Wijinberg, A. van Veldhuizen, H. J. Swarts, J. C. Frankland and J. A. Field, Tetrahedron Lett., 1999, 40, 5767 CrossRef; (d) T. J. King, S. Imre, A. Öztunc and R. H. Thomson, Tetrahedron Lett., 1979, 20, 1453 CrossRef; (e) H. Niwa, K. Wakamatsu and K. Yamada, Tetrahedron Lett., 1989, 30, 4543 CrossRef CAS; (f) B. F. Bowden, J. C. Coll and M. C. Dai, Aust. J. Chem., 1989, 42, 665 CrossRef CAS; (g) M. Satake, M. Murata and T. Yasumoto, Tetrahedron Lett., 1993, 34, 1975 CrossRef CAS.
- For selected recent examples for the synthesis of medium-sized rings, see: (a) C. R. Kennedy, H. Zhong, R. L. Macaulay and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 8557 CrossRef CAS PubMed; (b) C. Zhu, B. Yang, B. K. Mai, S. Palazzotto, Y. Qiu, A. Gudmundsson, A. Ricke, F. Himo and J.-E. Bäckvall, J. Am. Chem. Soc., 2018, 140, 14324 CrossRef CAS PubMed; (c) L. Zhang, Y. Wang, Z.-J. Yao, S. Wang and Z.-X. Yu, J. Am. Chem. Soc., 2015, 137, 13290 CrossRef CAS PubMed; (d) X. Hong, M. C. Stevens, P. Liu, P. A. Wender and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 17273 CrossRef CAS PubMed; (e) S. Saito, K. Maeda, R. Yamasaki, T. Kitamura, M. Nakagawa, K. Kato, I. Azumaya and H. Masu, Angew. Chem., Int. Ed., 2010, 49, 1830 CrossRef CAS PubMed.
- For selected examples, see: (a) Y. A. Cheng, T. Chen, C. K. Tan, J. J. Heng and Y.-Y. Yeung, J. Am. Chem. Soc., 2012, 134, 16492 CrossRef CAS PubMed; (b) Y. Hu and H. Huang, Org. Lett., 2017, 19, 5070 CrossRef CAS PubMed; (c) F. Medina, C. Besnard and J. Lacour, Org. Lett., 2014, 16, 3232 CrossRef CAS PubMed; (d) G. Prado, A. X. Veiga, F. Fernández-Nieto, M. R. Paleo and F. J. Sardina, Org. Lett., 2015, 17, 2054 CrossRef CAS PubMed; (e) N. P. Tsvetkov, A. Bayir, S. Schneider and M. Brewer, Org. Lett., 2012, 14, 264 CrossRef CAS PubMed; (f) Z. Wang, S. Chen, J. Ren and Z. Wang, Org. Lett., 2015, 17, 4184 CrossRef CAS PubMed; (g) I. D. G. Watson, S. Ritter and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 2056 CrossRef CAS PubMed; (h) Z. Wu and J. Wang, ACS Catal., 2017, 7, 7647 CrossRef CAS.
- There is a report on the ligand controlled divergent synthesis of medium-sized rings; however, only one example achieved the regioselective switch. For details, see: M. M. Coulter, P. K. Dornan and V. M. Dong, J. Am. Chem. Soc., 2019, 131, 6932 CrossRef PubMed.
- For selected recent reviews on VECs, see: (a) J. E. Gómez and A. W. Kleij, Adv. Organomet. Chem., 2019, 71, 175 CrossRef; (b) W. Guo, J. E. Gómez, À. Cristòfol, J. Xie and A. W. Kleij, Angew. Chem., Int. Ed., 2018, 57, 13735 CrossRef CAS PubMed; (c) A. Khan and Y. J. Zhang, Synlett, 2015, 26, 853 CrossRef CAS.
- For selected examples on the transitional chemistry of VECs, see: (a) A. Khan, R. Zheng, Y. Kan, J. Ye, J. Xing and Y. J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 6439 CrossRef CAS PubMed; (b) A. Khan, L. Yang, J. Xu, L. Y. Jin and Y. J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 11257 CrossRef CAS PubMed; (c) A. Cai, W. Guo, L. Martínez-Rodríguez and A. W. Kleij, J. Am. Chem. Soc., 2016, 138, 14194 CrossRef CAS PubMed; (d) A. Khan, S. Khan, I. Khan, C. Zhao, Y. Mao, Y. Chen and Y. J. Zhang, J. Am. Chem. Soc., 2017, 139, 10733 CrossRef CAS PubMed; (e) W. Guo, L. Martínez-Rodríguez, R. Kuniyil, E. Martin, E. C. Escudero-Adán, F. Maseras and A. W. Kleij, J. Am. Chem. Soc., 2016, 138, 11970 CrossRef CAS PubMed; (f) W. Guo, L. Martínez-Rodríguez, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Angew. Chem., Int. Ed., 2016, 55, 11037 CrossRef CAS PubMed; (g) W. Guo, R. Kuniyil, J. E. Gómez, F. Maseras and A. W. Kleij, J. Am. Chem. Soc., 2018, 140, 3981 CrossRef CAS PubMed; (h) R. Zeng, J.-L. Li, X. Zhang, Y.-Q. Liu, Z.-Q. Jia, H.-J. Leng, Q.-W. Huang, Y. Liu and Q.-Z. Li, ACS Catal., 2019, 9, 8256 CrossRef CAS; (i) Y. Liu, Q.-W. Huang, Q.-Z. Li, H.-J. Leng, Q.-S. Dai, R. Zeng, Y.-Q. Liu, X. Zhang, B. Han and J.-L. Li, Org. Lett., 2019, 21, 7478 CrossRef CAS PubMed.
- (a) L.-C. Yang, Z.-Q. Rong, Y.-N. Wang, Z. Y. Tan, M. Wang and Y. Zhao, Angew. Chem., Int. Ed., 2017, 56, 2927 CrossRef CAS PubMed; (b) Z.-Q. Rong, L.-C. Yang, S. Liu, Z. Yu, Y.-N. Wang, Z. Y. Tan, R.-Z. Huang, Y. Lan and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 15304 CrossRef CAS PubMed.
- For selected examples on the [5 + n] annulation of VECs, see: (a) P. Das, S. Gondo, P. Nagender, H. Uno, E. Tokunaga and N. Shibata, Chem. Sci., 2018, 9, 3276 RSC; (b) X. Gao, M. Xia, C. Yuan, L. Zhou, W. Sun, C. Li, B. Wu, D. Zhu, C. Zhang, B. Zheng, D. Wang and H. Guo, ACS Catal., 2019, 9, 1645 CrossRef CAS; (c) C. Yuan, Y. Wu, D. Wang, Z. Zhang, C. Wang, L. Zhou, C. Zhang, B. Song and H. Guo, Adv. Synth. Catal., 2018, 360, 652 CrossRef CAS; (d) B. Niu, X.-Y. Wu, Y. Wei and M. Shi, Org. Lett., 2019, 21, 4859 CrossRef CAS PubMed; (e) S. Singha, T. Patra, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2018, 140, 3551 CrossRef CAS PubMed; (f) Y. Wei, S. Liu, M.-M. Li, Y. Li, Y. Lan, L.-Q. Lu and W.-J. Xiao, J. Am. Chem. Soc., 2019, 141, 133 CrossRef CAS PubMed; (g) H.-W. Zhao, J. Du, J.-M. Guo, N.-N. Feng, L.-R. Wang, W.-Q. Ding and X.-Q. Song, Chem. Commun., 2018, 54, 9178 RSC; (h) Y. Yang and W. Yang, Chem. Commun., 2018, 54, 12182 RSC . For Pd-catalyzed [5 + n] annulations with vinyloxiranes, see:; (i) Y. Wu, C. Yuan, C. Wang, B. Mao, H. Jia, X. Gao, J. Liao, F. Jiang, L. Zhou, Q. Wang and H. Guo, Org. Lett., 2017, 19, 6268 CrossRef CAS PubMed; (j) J.-J. Feng and J. Zhang, J. Am. Chem. Soc., 2011, 133, 7304 CrossRef CAS PubMed; (k) J.-J. Feng and J. Zhang, ACS Catal., 2017, 7, 1533 CrossRef CAS.
- For divergent annulations with VECs, see: (a) L.-C. Yang, Z. Y. Tan, Z.-Q. Rong, R. Liu, Y.-N. Wang and Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 7860 CrossRef CAS PubMed; (b) Y. Xia, Q.-F. Bao, Y. Li, L.-J. Wang, B.-S. Zhang, H.-C. Liu and Y.-M. Liang, Chem. Commun., 2019, 55, 4675 RSC.
- (a) Q. Li, L. Zhou, X.-D. Shen, K.-C. Yang, X. Zhang, Q.-S. Dai, H.-J. Leng, Q.-Z. Li and J.-L. Li, Angew. Chem., Int. Ed., 2018, 57, 1913 CrossRef CAS PubMed; (b) M.-C. Yang, C. Peng, H. Huang, L. Yang, X.-H. He, W. Huang, H.-L. Cui, G. He and B. Han, Org. Lett., 2017, 19, 6752 CrossRef CAS PubMed; (c) J.-L. Li, L. Fu, J. Wu, K.-C. Yang, Q.-Z. Li, X.-J. Gou, C. Peng, B. Han and X.-D. Shen, Chem. Commun., 2017, 53, 6875 RSC; (d) Q.-Z. Li, X. Zhang, R. Zeng, Q.-S. Dai, Y. Liu, X.-D. Shen, H.-J. Leng, K.-C. Yang and J.-L. Li, Org. Lett., 2018, 20, 3700 CrossRef CAS PubMed; (e) K.-C. Yang, Q.-Z. Li, Y. Liu, Q.-Q. He, Y. Liu, H.-J. Leng, A.-Q. Jia, S. Ramachandran and J.-L. Li, Org. Lett., 2018, 20, 7518 CrossRef CAS PubMed.
- (a) X.-N. Zhang, G.-Q. Chen, X.-Y. Tang, Y. Wei and M. Shi, Angew. Chem., Int. Ed., 2014, 53, 10768 CrossRef CAS PubMed; (b) L. Zhang, H. Lu, G.-Q. Xu, Z.-Y. Wang and P.-F. Xu, J. Org. Chem., 2017, 82, 5782 CrossRef CAS PubMed; (c) X. Zhang, Q.-F. Huang, W.-L. Zou, Q.-Z. Li, X. Feng, Z.-Q. Jia, Y. Liu, J.-L. Li and Q.-W. Wang, Org. Chem. Front., 2019, 6, 3321 RSC.
- For selected reviews on transannular reactions, see: (a) E. Reyes, U. Uria, L. Carrillo and J. L. Vicario, Tetrahedron, 2014, 70, 9461 CrossRef CAS; (b) A. Rizzo and S. R. Harutyunyan, Org. Biomol. Chem., 2014, 12, 6570 RSC; (c) S. Handa and G. Pattenden, Contemp. Org. Synth., 1997, 4, 196 RSC. For a recent example, see: (d) R. Mato, R. Manzano, E. Reyes, L. Carrillo, U. Uria and J. L. Vicario, J. Am. Chem. Soc., 2019, 141, 9495 CrossRef CAS PubMed.
- For more studies on condition screening, see the ESI.†.
- For selected reviews and examples, see: (a) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902 CrossRef CAS PubMed; (b) B. A. Klein, I. M. Robertson, B. Reiz, T. Kampourakis, L. Li and B. D. Sykes, ACS Med. Chem. Lett., 2019, 10, 1007 CrossRef CAS; (c) M. A. Cinelli, H. Li, G. Chreifi, T. L. Poulos and R. B. Silverman, J. Med. Chem., 2017, 60, 3958 CrossRef CAS PubMed; (d) Q.-B. Han and H.-X. Xu, Curr. Med. Chem., 2019, 16, 3775 CrossRef PubMed; (e) Z. Zheng, M. Wu, J. Zhang, W. Fu, N. Xu, Y. Lao, L. Lin and H. Xu, Frontiers in Oncology, 2019, 9, 654 CrossRef PubMed; (f) N. Anantachoke, P. Tuchinda, C. Kuhakarn, M. Pohmakotr and V. Reutrakul, Pharm. Biol., 2012, 50, 78 CrossRef CAS PubMed; (g) S. Alam and F. Khan, Sci. Rep., 2018, 8, 5524 CrossRef PubMed; (h) C. Bao, M. Jin, B. Li, Y. Xu, J. Jin and L. Zhu, Org. Biomol. Chem., 2012, 10, 5238 RSC.
- The regioselectivity of the annulation can be influenced by multiple factors, such as the solvent, temperature, the loading of ligand, etc. (for more details, see the ESI†).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data for new compounds and crystallographic data in CIF or other electronic formats. CCDC 1943770–1943772. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06377c |
| This journal is © The Royal Society of Chemistry 2020 |