
Stereocontrolled, multi-functional sequence-defined oligomers through automated synthesis†
Chiel
Mertens
 a,
Matthieu
Soete
a,
Marcin L.
Ślęczkowski
b,
Anja R. A.
Palmans
b,
E. W.
Meijer
b,
Nezha
Badi
a and
Filip E.
Du Prez
*a
a,
Matthieu
Soete
a,
Marcin L.
Ślęczkowski
b,
Anja R. A.
Palmans
b,
E. W.
Meijer
b,
Nezha
Badi
a and
Filip E.
Du Prez
*a
aPolymer Chemistry Research group, Centre of Macromolecular Chemistry (CMaC), Department of Organic and Macromolecular Chemistry, Faculty of sciences, Ghent University, Krijgslaan 281 S4, 9000 Ghent, Belgium. E-mail: Filip.DuPrez@UGent.be
bLaboratory of Macromolecular and Organic Chemistry, Institute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
First published on 9th June 2020
Abstract
In contrast to biomacromolecules, synthetic polymers generally lack a defined monomer sequence, therefore one of the challenges of polymer chemists these days is gaining more control over the primary structure of synthetic polymers and oligomers. In this work, stereocontrolled sequence-defined oligomers were synthesised using a thiolactone-based platform. Step-wise elongation of the oligomer occurs via ring-opening of the thiolactone, resulting in the formation of stereocenters along the backbone. These initial studies indicate remarkable differences in the strength of non-covalent interactions in isotactic and atactic oligomers. Different side-chain moieties were introduced using alkyl halide building blocks and the synthetic protocol was succesfully optimised and automated. Furthermore, the possible post-synthesis modification of the oligomers was demonstrated using ‘click’ chemistry.
Introduction
Inspired by the precision in the biosynthesis of macromolecules, the synthesis of non-natural polymers with a precise monomer sequence has become a new and rapidly expanding area in polymer chemistry.1–3 Recent advances at the interface between organic and polymer chemistry offer accessible ways to prepare this class of materials. To differentiate between the levels of control that is obtained, a new terminology was introduced.4 In this regard, it is important to distinguish between the terms ‘sequence-controlled’ and ‘sequence-defined’ polymers. While the first category includes polymers possessing a high degree of order in their monomer arrangement and a low dispersity, these polymers are still non-uniform. Only the second subclass of precision structures includes uniform macromolecules, having defined length and monomer sequence, i.e. every chain is identical.While many conceptually different strategies for the synthesis of sequence-defined macromolecules have been explored, the most versatile approach relies on the iterative coupling of building blocks.5–12 These coupling reactions can be performed in solution, which often require extensive purification by column chromatography after each step, or using a soluble support which allows purification via precipitation.13–15 Nevertheless, the purification process can be simplified by making use of a solid support.11,12 Each approach is widely used and the strengths and weaknesses have been compared recently.16 The possibility to automate the synthetic procedure by using a liquid handling robot is one of the significant advantages, as the purification after each coupling step is limited to a simple wash- and filtration-step.
Besides applications in the area of data-storage,17–21 it is envisioned that sequence-defined oligomers and polymers could mimic the properties exhibited by biomacromolecules, including molecular recognition and catalysis.22–28 However, these advanced applications require control over the folding and self-assembly behavior of these macromolecules. In this context, it has been shown that stereochemistry plays an important role with regard to the properties of peptides and peptidomimetics.29–32 For example, Li et al. showed that inverting the absolute configuration of a single amino acid in a short peptide disrupts the supramolecular interaction that drives its self-assembly.29 In another example by Hartley and co-workers, the influence of the stereochemistry of each stereocenter in a tripeptide was investigated.31 Only two out of eight diastereomers acted as gelators, while other diastereomers precipitated or crystallized instead. These examples underline that strict control of the stereochemistry is essential for assigning function to macromolecules, as it could dramatically influence their properties.
Despite the fact that in recent years many procedures for the synthesis of uniform oligomers and polymers with diverse backbone compositions have been published, in many cases, side-chain moieties are attached to a nitrogen atom in the backbone.8,27,33,34 However, some scarce examples where the backbone possesses carbon-stereocenters have been reported recently.35–37 Johnson and co-workers reported the synthesis of uniform macromolecules with a controlled, stereoregular triazole-ether backbone, where the stereochemistry results from the chiral epoxide building blocks that were used.38,39 Moreover, the synthesis of monodisperse, isotactic (L)-lactic acid oligomers was described by Hawker and co-workers.40 The synthesis was later further explored by one of us by coupling these oligomers with oligodimethylsiloxanes to obtain discrete block co-oligomers.41,42
We recently introduced various strategies for the synthesis of uniform macromolecules with tailored functionalisation, based on the inherent double modification properties of the thiolactone moiety.43–46 In the most successful approach, a racemic thiolactone was immobilised on a solid support and ring-opened with an amino-alcohol, thereby releasing a thiol moiety in situ.43 Subsequently, a side-chain functionality could be introduced by making use of any acrylate monomer. To complete this two-step iterative procedure, a new latent thiol was introduced at the end of the growing chain by means of a thiolactone bearing an isocyanate functionality. With this procedure, atactic oligomers up to a decamer were made and this process was also automated.43 While this approach can be used to synthesise sequence-defined oligomers from simple and readily available building blocks, it also suffers from some inherent disadvantages. First of all, the β-thioesters resulting from the Michael addition of the thiol to the acrylate, have been reported to be susceptible towards hydrolysis.47 As a result, long term applications in aqueous environments might be hampered. In addition, the distance between the backbone and the introduced functionality is always extended by the acrylate moiety, thereby increasing its mobility. This could diminish the effect of the sequence in certain applications for which the group placement is of importance. Finally, the stereochemistry of the backbone was not considered so far, and racemic building blocks were used.
Here we do not only report the use of thiolactone chemistry for the synthesis of stereocontrolled sequence-defined oligomers, but we also report a novel synthetic strategy using thiol-bromo chemistry,48 to bring the functionalities closer to the backbone. Simultaneously, the scope of the building blocks that can be used to introduce a side-chain functionality is extended from acrylates towards compounds with a halide leaving group (Fig. 1). In an automated fashion, oligomers up to an octadecamer (DP = 18) have been prepared. To study the effect of the backbone's tacticity, the individual enantiomeric building blocks were prepared on large scales and utilised to synthesise isotactic oligomers. By comparing the solution and bulk properties of an enantiomerically pure (isotactic) oligomer with the statistical mixture of diastereomers (atactic oligomer), the importance of stereochemistry was demonstrated.
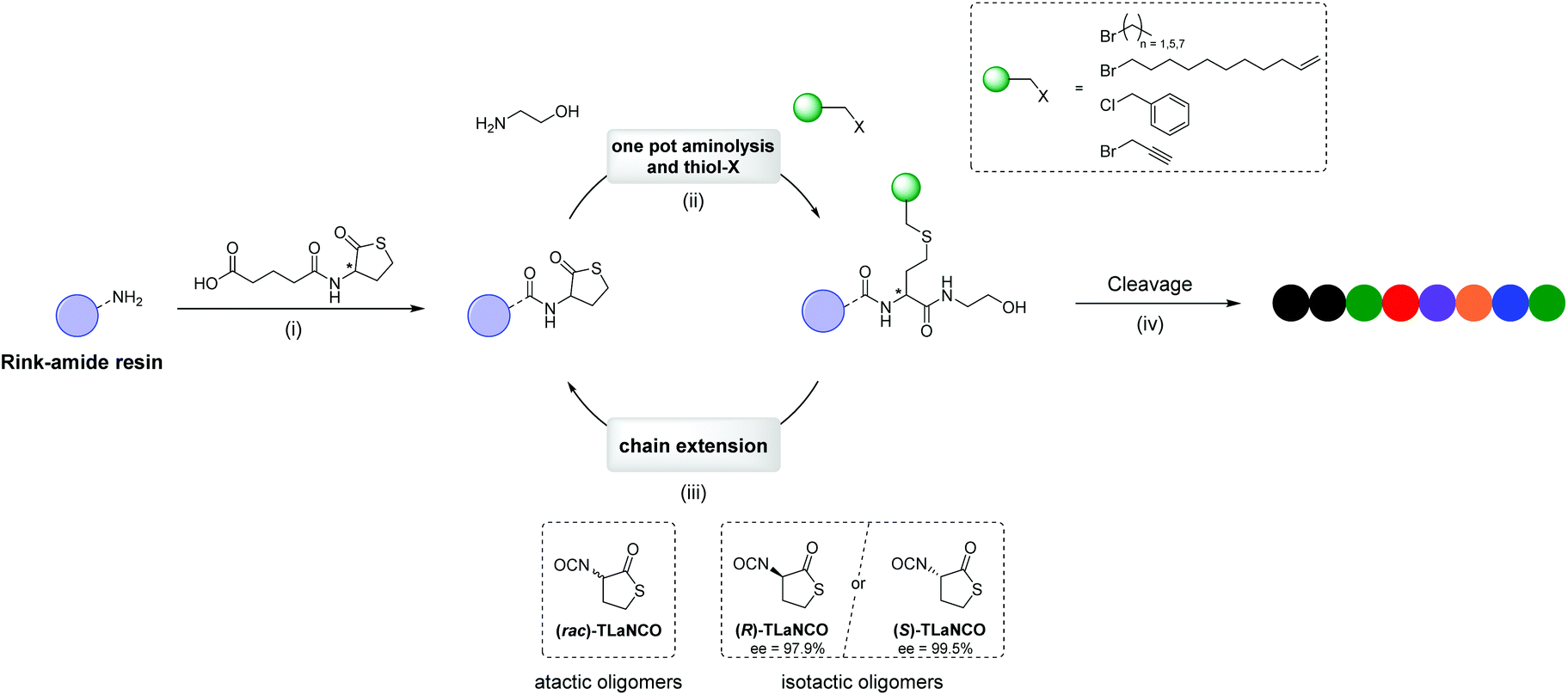 | ||
| Fig. 1 Solid-phase synthesis protocol for the preparation of multifunctional sequence-defined oligomers using thiolactone chemistry. (i) Rink amide functionalisation with a racemic or enantiomeric enriched thiolactone derivative containing a carboxylic acid moiety (TLaCOOH). (ii) One pot aminolysis and thiol-X reaction where the side-chain functionalities are introduced using alkyl halide species; DMPP and a drop of water were added to the reaction mixture as reducing agent. (iii) Chain extension with either racemic thiolactone isocyanate ((rac)-TLaNCO) to obtain atactic oligomers, or either enantiomeric enriched (R)- or (S)-TLaNCO for the synthesis of isotactic oligomers. Zirconium(IV) acetylacetonate was added as catalyst for urethane formation. (iv) Cleavage of the final oligomer from the resin using trifluoroacetic acid. | ||
Results and discussion
Development and automation of the synthetic protocol
The reaction conditions of the first step in the synthetic cycle, i.e. the amine-thiol-bromo coupling, was first optimised manually (Fig. S1†) with a racemic thiolactone connected to a Rink amide resin before it was automated using a liquid handling robot (Fig. S2†), usually utilised for peptide synthesis. To this end, 1-bromohexane was used as model to investigate the reaction conditions (Fig. 2a). A Rink amide resin with an immobilised thiolactone moiety (Fig. 1 (i)) was first swollen for 5 minutes and subsequently ethanolamine (15 eq.) and 1-bromohexane (20 eq.) were added simultaneously. Initial experiments revealed that besides the desired product, also a significant fraction of disulfide by-product was formed, which was drastically affected by the solvent that was used (see ESI page S4† for detailed investigation of the reaction conditions). Nevertheless, as already suggested in a previous study,45,49 the addition of dimethylphenylphosphine (DMPP) and a drop of water to the reaction mixture allowed for the elimination of this by-product. Eventually, dimethylformamide (DMF) was chosen as preferred solvent considering the desire to automate these reactions. As reagents and reaction vessels are placed in an open system in the robot set-up, a solvent with low volatility is desired. This allows longer reaction times and storage of the reagents, without significantly altering its concentration due to evaporation. In the next step, the reaction was performed using the synthesizer. The kinetics of the reaction were investigated by liquid chromatography mass spectrometry (LCMS) by monitoring the disappearance of the starting compound trace (t ≈ 3.19 min) and the appearance of the product trace (t ≈ 5.33 min) (Fig. 2b). After 5 minutes already most of the starting compound was transformed, while full conversion could be observed within 60 minutes. In another experiment the reaction mixture was refreshed after 15 minutes and shaking the reactor for another 15 minutes facilitated full conversion, thereby halving the reaction time (Fig. S4†). Nonetheless, to ensure full conversion each time, this step was performed twice with a reaction time of 1 hour in all the following automated syntheses, as a decrease in accessibility of the reactive end group is often observed with increasing chain length.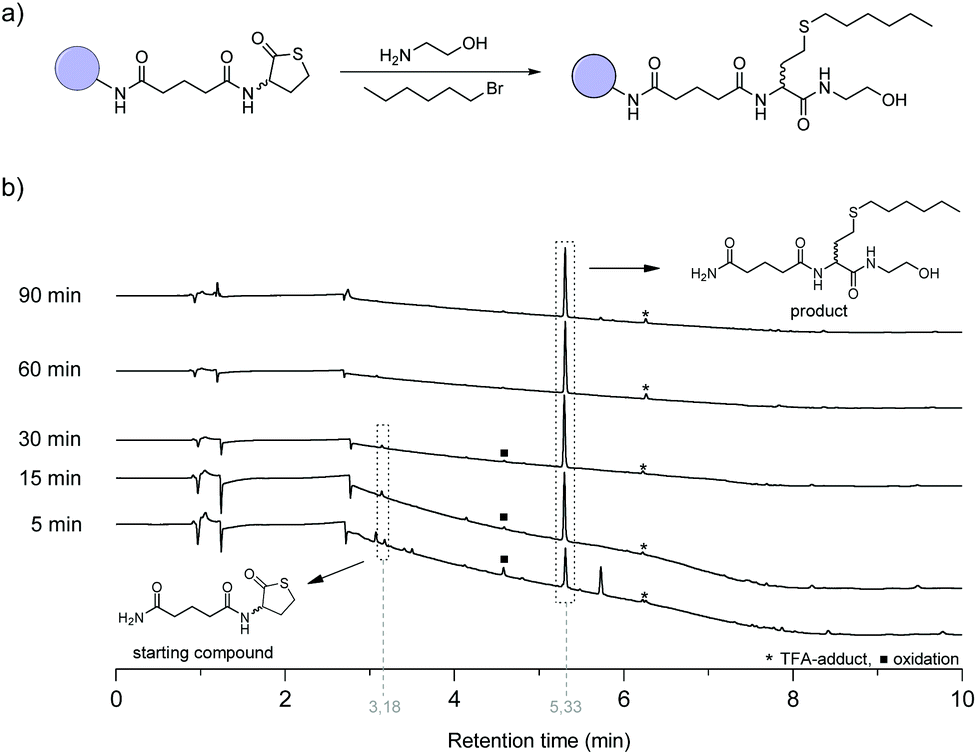 | ||
Fig. 2 (a) Model reaction for the one pot amine-thiol-bromo coupling. (b) Reaction progress followed by LCMS analysis, using the optimised conditions (see ESI pS4;† DMPP and a drop of water were added to the reaction mixture as reducing agent). *![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Esterification of the hydroxyl group with trifluoroacetic acid during the cleavage from the solid support. ■Acid catalysed oxidation of the thioether to a sulfoxide during cleavage from the solid support that can be avoided by eliminating oxygen during this process. Esterification of the hydroxyl group with trifluoroacetic acid during the cleavage from the solid support. ■Acid catalysed oxidation of the thioether to a sulfoxide during cleavage from the solid support that can be avoided by eliminating oxygen during this process. | ||
In the second step, a new thiolactone moiety is introduced, completing the iterative synthetic cycle (Fig. 1 (iii)). This chain extension was performed by attaching a new thiolactone to the hydroxyl chain end, making use of the aforementioned racemic thiolactone isocyanate ((rac)-TLaNCO) and zirconium(IV) acetylacetonate as catalyst for the urethane formation. This (rac)-TLaNCO has already been reported as an efficient compound to functionalise various substrates.17,43,50 Again, this reaction was performed twice for 1 hour to ensure full conversion.
To prove the effectiveness of this protocol, two atactic (at) undecamers, i.e. at-undeca-C6 and at-undeca-C8, were synthesised in an automated fashion using 1-bromohexane (C6) and 1-bromooctane (C8) as alkyl halide, respectively. After cleavage from the resin (Fig. 1 (iv)), LCMS analysis confirmed that at-undeca-C6 (Fig. S5†) was successfully formed. However, the more apolar at-undeca-C8 was difficult to analyse by LCMS. Thus, this oligomer was analysed by matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF) and size exclusion chromatography (SEC) (Fig. 3). Only a single peak was found in the mass spectrum, which corresponds to the sodium adduct. In addition, SEC confirmed that a uniform oligomer was obtained. The number-average molecular weight obtained was in good accordance with the actual molecular weight, although polystyrene standards were used for calibration.
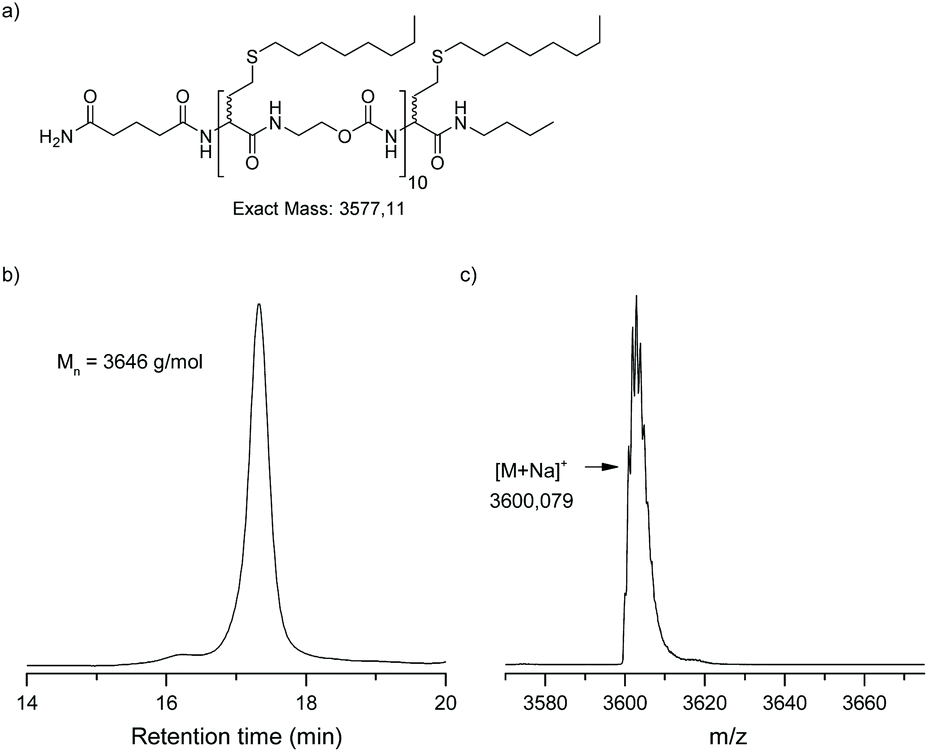 | ||
| Fig. 3 Undecamer, at-undeca-C8, synthesised in an automated fashion: (a) molecular structure, (b) SEC chromatogram in THF, (c) MALDI-TOF spectrum. | ||
In order to determine the limits of the automated procedure, a (homo)oligomer was synthesised in a single run with hexyl side-chains. Small samples of resin (∼5 mg) were taken from the reactor every two cycles, starting from the 10th cycle. The product was cleaved from the resin and analysed by MALDI-TOF and SEC (Fig. 4 and S6†). Again, the mass spectrum of the at-decamer showed a single peak corresponding to the sodium adduct, indicating the high reproducibility of the synthetic protocol. However, from the at-dodecamer onwards, additional peaks were observed, resulting from incomplete reaction cycles. For instance, the at-dodecamer showed the presence of a small fraction of at-undecamer, which could be the result of non-quantitative conversion at this stage, as for example also observed for other high molar mass uniform structures such as dendrimers with high generation number.51 Remarkably, these truncated molecules are still active and can continue to grow during the subsequent cycles. This indicates that this chain length cannot be considered as the limit of this chemistry, but rather of the reaction conditions used. Indeed, the latter could be further optimised after the decamer stage, if needed. For instance, reaction times could be prolonged or reaction steps could be repeated multiple times to ensure full conversion. In addition, a resin with a lower loading could be used to reduce steric crowding around the chain-end and thereby improve the synthesis. Here lessons can be learned from the synthesis of dendrimers, oligopeptides and oligonucleotides.
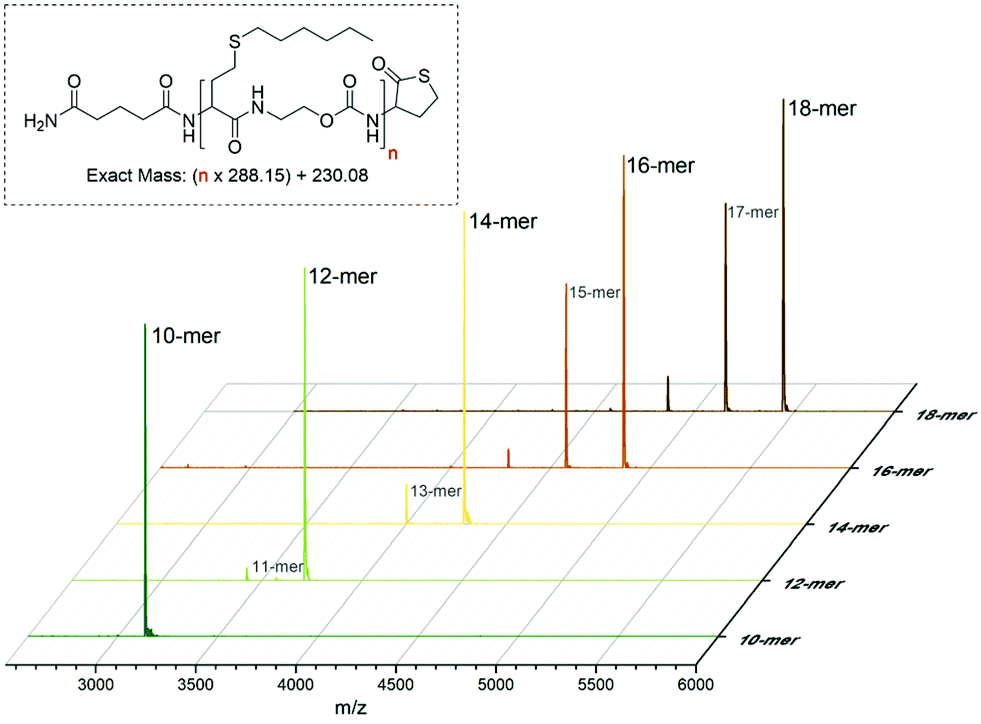 | ||
| Fig. 4 MALDI-TOF spectrum of oligomer samples during the synthesis of an at-octadecamer in a single continuous run after the 10, 12, 14, 16 and 18th reaction cycle. The peaks corresponds to the sodium adduct ([M + Na]+) of the oligomers. | ||
While these results show that (homo)oligomers up to an undecamer can be successfully obtained in high purities by this procedure, other halide-building blocks were also investigated (see Fig. 1). Initially a single monomeric cycle was performed in order to check the compatibility of those building blocks with the reaction conditions, before the synthesis of an oligomer with different side-chain functionalities was undertaken. Only in the case of propargyl bromide, the conditions had to be adapted, as reactions carried out with this substrate suffered from incomplete conversion of the thiolactone. It was suspected that this could be the result of the higher reactivity of propargyl bromide, enabling a side-reaction with ethanolamine.52 Nevertheless, when the solvent was changed from DMF to chloroform, only the desired product was obtained (Fig. S7†). It should be noted that this side-reaction can also occur to a certain extent with the other bromides used in this work. Because no issues were observed for those, it is assumed that this side-reaction is negligible and only becomes dominant when more activated bromides are used. Nevertheless, the formed by-product being not attached to the resin, it can be washed away after reaction.
Secondary alkyl bromides (e.g. 2-bromobutane) were not sufficiently reactive to be compatible with this protocol. After overnight reaction, only trace amounts of the desired product could be found with the ring-opened thiolactone as main product. In principle higher temperatures could be used, but this was not further investigated due to hardware limitation of the automated synthesiser.
Using the automated procedure, with adapted conditions for propargyl bromide, a sequence-defined at-hexamer was synthesised containing a double and triple bond available for post-synthesis modification (Fig. 5). The hexamer was modified using a Cu(I)-catalysed azide alkyne ‘click’ reaction (CuAAC) with 1-azidodecane. After overnight reaction full conversion was observed as demonstrated both by mass spectrometry and SEC (Fig. S8†). The presence of a terminal olefin in the first functional side-group, could also be considered for a radical thiol–ene reaction or a hydrosilylation reaction (not done), thus increasing the scope of orthogonal modification that can be performed on those multifunctional oligomers.
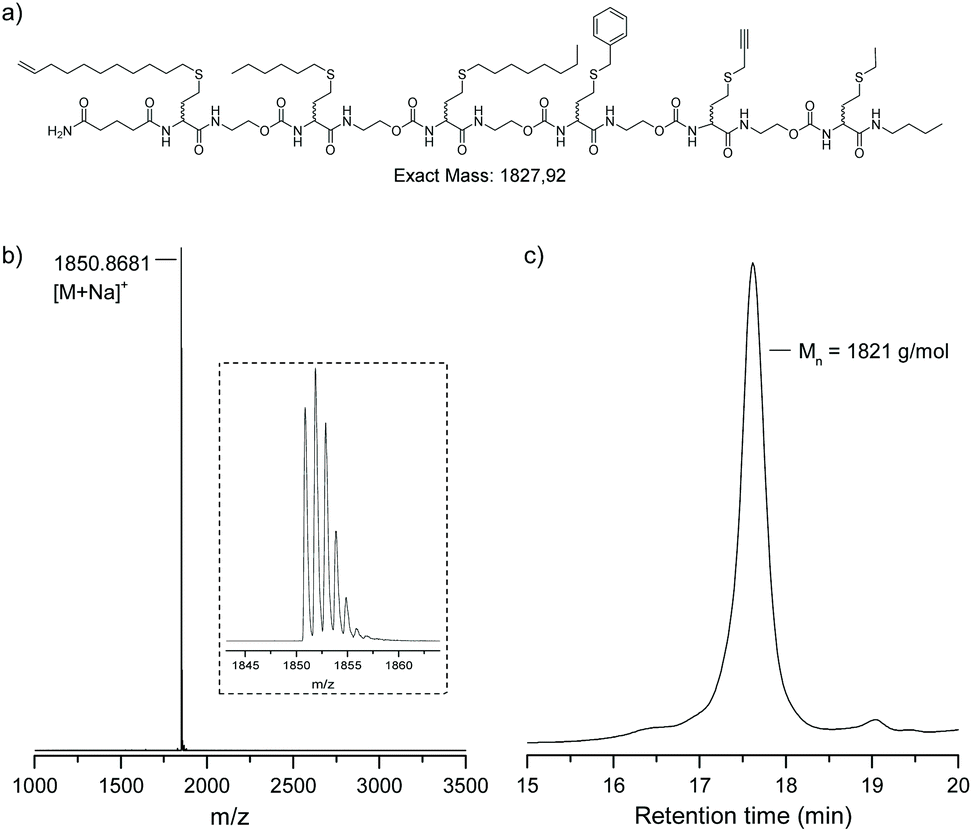 | ||
| Fig. 5 (a) Structure of the atactic sequence-defined hexamer obtained using multiple bromine building blocks. (b) MALDI-TOF spectrum. (c) SEC chromatogram in THF. | ||
All the results presented above demonstrate the viability of this new protocol. Moreover, the oligomers obtained were found to be thermally more stable than the ones resulting from the protocol previously reported where ester side-chains were present43 (see TGA results in Fig. S10†).
Stereocontrolled oligomers
The oligomers obtained in the previous section are mixtures of diastereomers, which is the result of using the racemic thiolactone building blocks during the synthesis (Fig. 1). For applications such as macromolecular data storage or encryption, it can be essential to control the stereochemistry by assigning the R- and S-enantiomeric building block as a 0 and 1. However, then other read-outs than tandem mass spectrometry techniques have to be developed, as usually no difference in fragmentation is observed.17,53 Nevertheless, the absolute configuration of the stereocenters will affect the physical properties of the oligomers formed, also in their interactions with other optically active molecules, as typically found in biological environments.In the previous part, (rac)-TLaNCO was used for the development of the protocol, which was synthesised from (rac)-homocysteine thiolactone. While this starting compound is also available as a single enantiomer, the high cost considerably limits its use. Nevertheless, we isolated both enantiomers by means of a chiral resolution strategy that was adapted from a patent procedure (Scheme S2†) in scales up to 500 g.54 After isolation, both enantiomers were further transformed into the corresponding isocyanate by treatment with triphosgene. Chiral liquid chromatography was then performed to assess the enantiopurity of the products obtained. However, prior to that, the isocyanates were transformed into their carbamate counterparts by reaction with methanol to avoid side-reactions with the solvent used during the analysis. The analysis confirmed that both enantiomers were obtained in enantiomeric excess (ee) of 97.9% and 99.5% for the R- and S-enantiomer, respectively. The purity of the S-enantiomer was slightly higher, which was a direct result from higher purity of the (R)-mandelic acid that was used to selectively precipitate the enantiomer.
Subsequently, oligomers were synthesised using the two enantiomeric building blocks, as well as the racemic mixture. An isotactic oligomer iso(R)-nona-C8, built with only (R)-TLa-NCO and (R)-TLaCOOH for Rink amide functionalisation (Fig. 1(i)) and an atactic oligomer at-nona-C8, using the racemic thiolactone derivatives, have been made. During the iterative synthesis, no difference in reactivity was observed and, in a previous study, it was already shown that racemisation does not occur during the thiolactone ring opening.55 After the cleavage of the oligomers with 30% trifluoroacetic acid (TFA) in dichloromethane (DCM) and filtration of the resin, the solvent was evaporated under a nitrogen flow and residual TFA was removed by freeze-drying. Subsequently, it was attempted to re-dissolve the oligomers in dichloromethane. Surprisingly, it was impossible to solubilise the isotactic oligomer again in this solvent, while its atactic counterpart could be readily dissolved. Even after refluxing for up to 2 hours and prolonged sonication, no clear solution could be obtained. Only when a drop of TFA was added, the isotactic oligomer could be solubilised again. It was later observed that TFA effectively competes for hydrogen bonds, thereby increasing the solubility (vide infra). Also in other solvents, a clear difference in solubility could be observed between both oligomers (see Table S2†). Contrary to iso(R)-nona-C8, at-nona-C8 was soluble in the majority of solvents investigated. For instance, iso(R)-nona-C8 was insoluble in solvents like decaline and methylcyclohexane, but soluble after reflux in dipolar solvents such as tetrahydrofuran and 1,4-dioxane. Similarly, protic solvents such as ethanol were not capable of solvating the iso(R)-nona-C8, whereas they readily dissolved the atactic counterpart. This difference in solubility indicates that control of the stereochemistry reinforces supramolecular structures resulting from multiple hydrogen bonds.
To gain more insight into the molecular origin of this difference, infrared spectra of both oligomers were recorded in tetrahydrofuran at the same concentration. After solvent subtraction, two distinct N–H absorption bands could be detected (Fig. 6a). The band around 3500 cm−1 is associated with N–H in a molecularly dissolved state, while the band around 3300 cm−1 corresponds to a N–H stretch involved in hydrogen bonding. Not surprisingly, for the isotactic oligomer, the hydrogen bonded state is the most abundant, whereas the molecularly dissolved state is the most abundant for the atactic oligomer. Nevertheless, the atactic oligomer also displayed a fraction of hydrogen bonded state, ascribed to the high density of amide and carbamate moieties present in the structure.
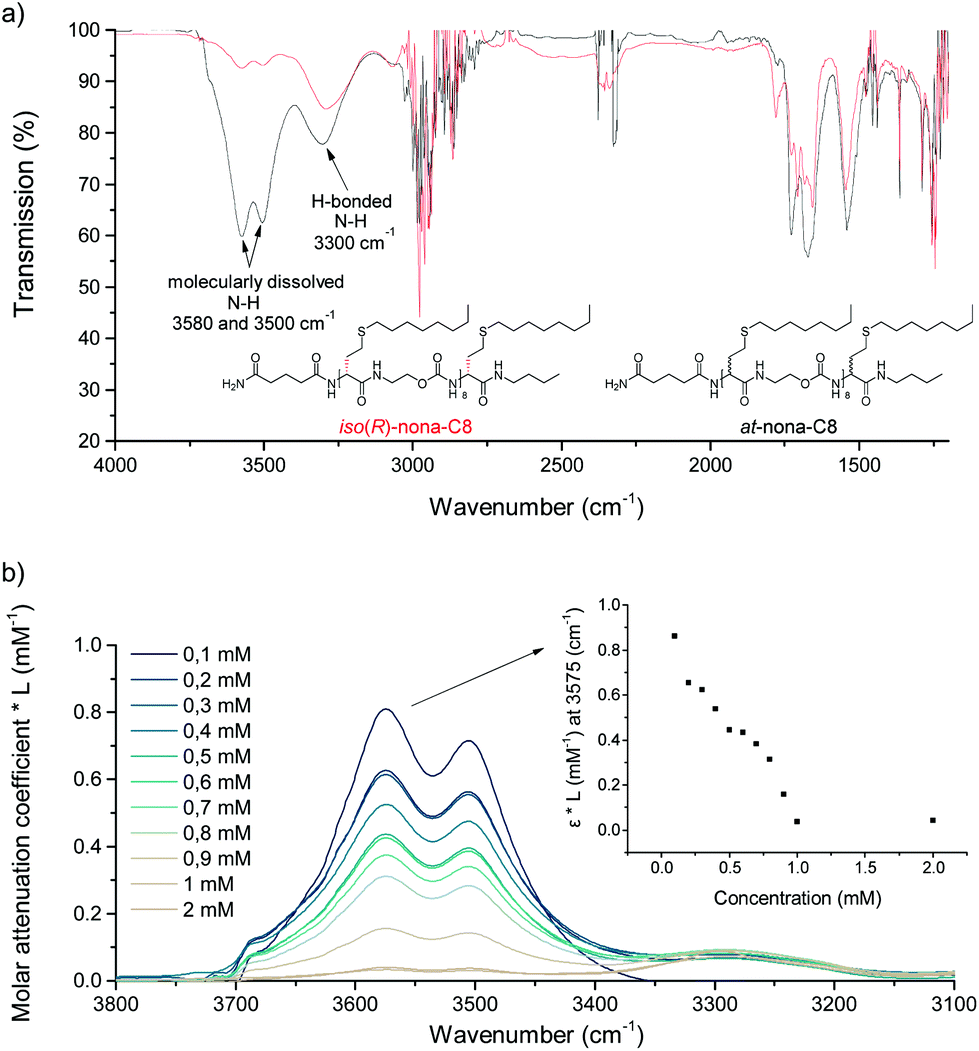 | ||
| Fig. 6 (a) Comparison of IR-absorption iso(R)-nona-C8 and at-nona-C8, whereby two different N–H stretches are indicated. (b) Molar attenuation coefficient of iso(R)-nona-C8 at different concentration and at 3575 cm−1. | ||
Dilution experiments were performed with iso(R)-nona-C8 to investigate whether these hydrogen bond interactions occur intramolecularly, as a result of folding, or intermolecularly (Fig. 6b). In the latter, the oligomer concentration should have an influence on the equilibrium between free and hydrogen bonded N–H. Since the infrared absorbance is directly correlated to the concentration, the molar attenuation coefficient was calculated to allow a comparison of the data. As the sample was further diluted, the value increased, indicating a shift towards the molecularly dissolved N–H state. Therefore, hydrogen bonds in iso(R)-nona-C8 primarily occur between different oligomers in aggregated structures.
As the previous experiments indicated that control over the stereochemistry has a significant influence on the properties of the oligomers in solution, the (thermal) bulk properties have also been compared. Thermogravimetric analysis (TGA) showed that the oligomers are stable up to 200 °C. Differential scanning calorimetry (DSC) analysis (second heating curve) showed that the atactic oligomer exhibited only a single glass transition temperature (Tg) around 25 °C, suggesting an amorphous structure for this material. Interestingly, in the case of the isotactic oligomer, an additional reversible endothermic transition upon heating was observed around 140 °C (Fig. S11†). However, this transition in DSC was not observed when studying an undecamer prepared with 1-bromohexane (C6) instead of 1-bromooctane (C8) (Fig. S12†), thus suggesting a link with the alkyl side-chain. Further investigations are needed to confirm the origin of this effect.
Next, iso(R)-nona-C8 was also studied by variable temperature optical microscopy. In the case of the atactic oligomer, no birefringence was detected under cross-polarised light. In contrast, the isotactic oligomer showed a multidomain birefringent structure (Fig. S13†), which indicates the absence of any long-range order because of the multiple conformations that the backbone can adopt. When the temperature was raised no visual change was observed, until the sample started to degrade around 200 °C.
Conclusions
A new automated platform based on thiolactone chemistry was developed for the synthesis of stereocontrolled and sequence-defined oligomers, using alkyl halide compounds for the introduction of side-chain functionalities. Before translating the synthetic procedure towards a liquid handling robot, it was important that the procedure was developed manually. In this way the reaction parameters for the amine-thiol-bromo coupling could be optimized. Using these optimized conditions, the limits of the automated procedure in terms of chain length were determined, whereby oligomers up to the undecamer were obtained in high purities. Going beyond this length, small defects were obtained as a result of incomplete conversion in each step. The stereocenters in these oligomers directly result from the thiolactone building blocks used and both isotactic and the atactic oligomers were synthesised. For those oligomers, remarkable differences in solution and bulk properties were observed. Controlling the stereochemistry in sequence-defined oligomers is thus envisioned to be a powerful tool to address different properties and functions of these complex molecular systems.Experimental
Materials
All chemicals were used as supplied, unless otherwise stated. Deionised water was used in the procedures. Tetrahydrofuran was purified by filtration over activated basic aluminium oxide to remove organic peroxides. DMSO-d6, MeCN-d3 (≥99.5%) and CDCl3 (≥99.8%) were purchased from Eurisotop. (rac)-Homocysteine thiolactone hydrochloride (99%) was purchased from Haihang Industry (Jinan City, China). Magnesium sulphate (dried ≥99%), sodium hydrogen carbonate (99%) and sodium chloride (99%) were purchased from Carl Roth. Hydrochloric acid (36 wt%) was purchased from Chem Lab NV. Trifluoroacetic acid (peptide grade) and Fmoc-protected rink-amide resin (100–200 mesh, 1% DVB, 0.4 mmol g−1) were purchased from Iris Biotech GmbH. Dichloromethane (HPLC grade, 99.5%), anhydrous chloroform (99.8%), N,N′-dimethylformamide (HPLC grade, 99.8%), piperidine (anhydrous, 99%), 1,4-dioxane (HPLC grade, 99%), pyridine (anhydrous, 99%), triethylamine (99%) and methanol (HPLC grade, 99.5%) were purchased from Acros Organics. 1,2-Dichloroethane (spectroscopy grade, 99.8%), 1-bromohexane (98%), 1-bromooctane (98%), bromoethane (99%), dimethylphenylphosphine (99%), N,N′-dicyclohexylcarbodiimide (99%), glutaric anhydride (95%), zirconium(IV) acetylacetonate (97%), 4-dimethylaminopyridine (99%), sodium Iodide (99.9%), propargyl bromide solution (80 wt% in toluene), 11-bromo-1-undecene (95%), 2-bromoethanol (95%), copper sulfate pentahydrate (>98%), 11-azido-3,6,9-trioxaundecan-1-amine (>90%), (+)-sodium L-ascorbate (>98%), decalin (99%), isopropanol (99.7%), ethyl acetate (HPLC grade, 99.7%), chloroform (HPLC grade, 99.9%) and methanol (HPLC grade, 99.5%) were also purchased from Sigma Aldrich. Triphosgene (98.0%), methylcyclohexane (99.8%), n-butylamine (99%) and dibutyltin dilaurate (95%) were purchased from Tokyo Chemical Industry. Acetone (HPLC grade, 99.5%), ethanol (absolute HPLC grade), acetonitrile (HPLC grade, 99.9%), diethyl ether (HPLC grade, 99%) and N,N′-dimethylformamide (HPLC grade, 99.7%) were purchased from Fischer Scientific. Salicylaldehyde (99%), (S)-(+)-mandelic acid (99%) and (R)-(−)-mandelic acid (98%) were purchased from ABCR.Instrumentation
640 Da. To each sample, 2 μl of toluene was added as internal standard to check the accuracy of the calibrations.
Differential scanning calorimetry (DSC) analysis were performed with a Mettler-Toledo 1/700 under nitrogen atmosphere. The samples were analysed in aluminium sample pans which contained ∼2.5 mg of the sample. Glass transition temperatures were determined in the second heating run. Measurements were performed in a temperature range of 25–180 °C with a rate of 10 °C min−1.
Polarised optical microscopy images were obtained with an Olympus BH-2, equipped with a Canon EOS 1100D camera, using 20× and 40× objectives. The sample was put between two glass slides, placed on a Linkam scientific TMS 91 hot stage.
Synthesis of the building blocks
(S)-Homocysteine thiolactone hydrochloride was obtained in a similar fashion by using (S)-mandelic acid, instead of the other enantiomer, as chiral derivation agent. Yield: 287.6 g, 1.872 mol, 54.5%.
(S)-, (R)- and (rac)-thiolactone carboxylic acid was synthesised according to a previously reported procedure,46 whereby respectively (S)-, (R)- or (rac)-homocysteine thiolactone hydrochloride was used as starting compound.
General protocol for the manual synthesis of the oligomers
:H2O mixture (6:1, 0.7 mL for 20 mg resin) and after 5 minutes copper sulfate pentahydrate (0.75 eq.) and sodium ascorbate (5 eq.) was added. Next, the azide compound (10 eq.) was added and the reaction was shaken for 36 h in a closed solid-phase reactor. Afterwards the resin was washed by a DMF:H2O (6:1) mixture, followed by the regular washing steps.
Automated synthesis
Automated synthesis was performed in a similar way as the manual synthesis. The reader is referred to the ESI† for more information.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. M. would like to acknowledge funding of his Ph.D. scholarship from the Special Research Fund (BOF) of Ghent University. M. S. would like to acknowledge the Research Foundation Flanders (FWO) for the funding of his Ph.D. scholarship under application number 1SA3820N. F. D. P. would like to acknowledge financial support from the FWO under EOS-project 30650939. The authors would like to thank Dr Steven Martens for the help with the chiral resolution, Jos Van Den Begin for the assistance with the liquid handling robot used in this work, Jan Goeman for the LCMS measurements and Bernhard De Meyer.References
- Sequence-Controlled Polymers, ed. J.-F. Lutz, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2018 Search PubMed.
- N. Badi and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383 RSC.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS.
- J.-F. Lutz, Macromol. Rapid Commun., 2017, 38, 1700582 CrossRef PubMed.
- M. L. McKee, P. J. Milnes, J. Bath, E. Stulz, A. J. Turberfield and R. K. O'Reilly, Angew. Chem., Int. Ed., 2010, 49, 7948–7951 CrossRef CAS PubMed.
- J. J. Haven, J. A. De Neve and T. Junkers, ACS Macro Lett., 2017, 6, 743–747 CrossRef CAS.
- D. Oh, M. Sawamoto and M. Ouchi, Polym. Chem., 2019, 10, 1998–2003 RSC.
- M. Porel and C. A. Alabi, J. Am. Chem. Soc., 2014, 136, 13162–13165 CrossRef CAS PubMed.
- S. C. Solleder and M. A. R. Meier, Angew. Chem., Int. Ed., 2014, 53, 711–714 CrossRef CAS PubMed.
- E. A. Hoff, G. X. De Hoe, C. M. Mulvaney, M. A. Hillmyer and C. A. Alabi, J. Am. Chem. Soc., 2020, 142, 6729–6736 CrossRef CAS PubMed.
- S. C. Solleder, R. V. Schneider, K. S. Wetzel, A. C. Boukis and M. A. R. Meier, Macromol. Rapid Commun., 2017, 38, 1600711 CrossRef PubMed.
- S. Martens, J. O. Holloway and F. E. Du Prez, Macromol. Rapid Commun., 2017, 38, 1700469 CrossRef PubMed.
- T. J. Dickerson, N. N. Reed and K. D. Janda, Chem. Rev., 2002, 102, 3325–3344 CrossRef CAS PubMed.
- M. Landa, M. Kotera, J.-S. Remy and N. Badi, Eur. Polym. J., 2016, 84, 338–344 CrossRef CAS.
- A. Meszynska, N. Badi, H. G. Börner and J.-F. Lutz, Chem. Commun., 2012, 48, 3887 RSC.
- J. O. Holloway, K. S. Wetzel, S. Martens, F. E. Du Prez and M. A. R. Meier, Polym. Chem., 2019, 10, 3859–3867 RSC.
- S. Martens, A. Landuyt, P. Espeel, B. Devreese, P. Dawyndt and F. Du Prez, Nat. Commun., 2018, 9, 4451 CrossRef PubMed.
- J. O. Holloway, F. Van Lijsebetten, N. Badi, H. A. Houck and F. E. Du Prez, Adv. Sci., 2020, 1903698 CrossRef PubMed.
- R. K. Roy, A. Meszynska, C. Laure, L. Charles, C. Verchin and J.-F. Lutz, Nat. Commun., 2015, 6, 7237 CrossRef CAS PubMed.
- A. C. Boukis and M. A. R. Meier, Eur. Polym. J., 2018, 104, 32–38 CrossRef CAS.
- Z. Huang, Q. Shi, J. Guo, F. Meng, Y. Zhang, Y. Lu, Z. Qian, X. Li, N. Zhou, Z. Zhang and X. Zhu, Nat. Commun., 2019, 10, 1918 CrossRef PubMed.
- M. A. R. Meier and C. Barner-Kowollik, Adv. Mater., 2019, 31, 1806027 CrossRef PubMed.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- K. Kirshenbaum, R. Zuckermann and K. Dill, Curr. Opin. Struct. Biol., 1999, 9, 530–535 CrossRef CAS PubMed.
- J.-F. Lutz, ACS Macro Lett., 2020, 185–189 CrossRef CAS.
- M. Porel, D. N. Thornlow, N. N. Phan and C. A. Alabi, Nat. Chem., 2016, 8, 590–596 CrossRef CAS PubMed.
- A. E. Stross, G. Iadevaia, D. Núñez-Villanueva and C. A. Hunter, J. Am. Chem. Soc., 2017, 139, 12655–12663 CrossRef CAS PubMed.
- S. C. Leguizamon and T. F. Scott, Nat. Commun., 2020, 11, 784 CrossRef CAS PubMed.
- X. Li, X. Du, J. Li, Y. Gao, Y. Pan, J. Shi, N. Zhou and B. Xu, Langmuir, 2012, 28, 13512–13517 CrossRef CAS PubMed.
- Z. Luo, S. Wang and S. Zhang, Biomaterials, 2011, 32, 2013–2020 CrossRef CAS PubMed.
- S. Marchesan, C. D. Easton, K. E. Styan, L. J. Waddington, F. Kushkaki, L. Goodall, K. M. McLean, J. S. Forsythe and P. G. Hartley, Nanoscale, 2014, 6, 5172–5180 RSC.
- K. J. Nagy, M. C. Giano, A. Jin, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2011, 133, 14975–14977 CrossRef CAS PubMed.
- D. Ponader, F. Wojcik, F. Beceren-Braun, J. Dernedde and L. Hartmann, Biomacromolecules, 2012, 13, 1845–1852 CrossRef CAS PubMed.
- J. Sun and R. N. Zuckermann, ACS Nano, 2013, 7, 4715–4732 CrossRef CAS PubMed.
- S. Mosca, F. Wojcik and L. Hartmann, Macromol. Rapid Commun., 2011, 32, 197–202 CrossRef CAS PubMed.
- N. Franz, L. Menin and H.-A. Klok, Eur. J. Org. Chem., 2009, 5390–5405 CrossRef CAS.
- D. Núñez-Villanueva and C. A. Hunter, Chem. Sci., 2017, 8, 206–213 RSC.
- J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–815 CrossRef CAS PubMed.
- Y. Jiang, M. R. Golder, H. V.-T. Nguyen, Y. Wang, M. Zhong, J. C. Barnes, D. J. C. Ehrlich and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 9369–9372 CrossRef CAS PubMed.
- K. Takizawa, H. Nulwala, J. Hu, K. Yoshinaga and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5977–5990 CrossRef CAS.
- B. van Genabeek, B. A. G. Lamers, B. F. M. de Waal, M. H. C. van Son, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2017, 139, 14869–14872 CrossRef CAS PubMed.
- B. A. G. Lamers, B. van Genabeek, J. Hennissen, B. F. M. de Waal, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2019, 52, 1200–1209 CrossRef PubMed.
- S. Martens, J. Van den Begin, A. Madder, F. E. Du Prez and P. Espeel, J. Am. Chem. Soc., 2016, 138, 14182–14185 CrossRef CAS PubMed.
- J. O. Holloway, C. Mertens, F. E. Du Prez and N. Badi, Macromol. Rapid Commun., 2019, 40, 1800685 CrossRef PubMed.
- P. Espeel, L. L. G. Carrette, K. Bury, S. Capenberghs, J. C. Martins, F. E. Du Prez and A. Madder, Angew. Chem., Int. Ed., 2013, 52, 13261–13264 CrossRef CAS PubMed.
- J. O. Holloway, S. Aksakal, F. E. Du Prez and C. R. Becer, Macromol. Rapid Commun., 2017, 38, 1700500 CrossRef PubMed.
- A. Rydholm, K. Anseth and C. Bowman, Acta Biomater., 2007, 3, 449–455 CrossRef CAS PubMed.
- V. Beyer, B. Cattoz, A. Strong, D. Phillips, A. Schwarz and C. R. Becer, Polym. Chem., 2019, 10, 4259–4270 RSC.
- G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196 RSC.
- K. Bruycker, C. Mertens and F. E. Prez, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 322–333 CrossRef.
- M. V. Walter and M. Malkoch, Chem. Soc. Rev., 2012, 41, 4593–4609 RSC.
- V. J. Shiner and J. S. Humphrey, J. Am. Chem. Soc., 1967, 89, 622–630 CrossRef CAS.
- A. C. Boukis, K. Reiter, M. Frölich, D. Hofheinz and M. A. R. Meier, Nat. Commun., 2018, 9, 1439 CrossRef PubMed.
- European Patent Office, EP1964838A1, 2009, 10.
- D. Van Lysebetten, S. Felissati, E. Antonatou, L. L. G. Carrette, P. Espeel, E. Focquet, F. E. Du Prez and A. Madder, ChemBioChem, 2018, 19, 641–646 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00645a |
| This journal is © The Royal Society of Chemistry 2020 |