
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmulsion-templated synthetic polypeptide scaffolds prepared by ring-opening polymerization of N-carboxyanhydrides†
Ozgun Can
Onder‡
 a,
Petra
Utroša‡
a,
Simon
Caserman
b,
Marjetka
Podobnik
b,
Magda Tušek
Žnidarič
c,
Jože
Grdadolnik
d,
Sebastijan
Kovačič
a,
Ema
Žagar
a and
David
Pahovnik
*a
a,
Petra
Utroša‡
a,
Simon
Caserman
b,
Marjetka
Podobnik
b,
Magda Tušek
Žnidarič
c,
Jože
Grdadolnik
d,
Sebastijan
Kovačič
a,
Ema
Žagar
a and
David
Pahovnik
*a
aDepartment of Polymer Chemistry and Technology, National Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia. E-mail: david.pahovnik@ki.si
bDepartment of Molecular Biology and Nanobiotechnology, National Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia
cDepartment of Biotechnology and Systems Biology, National Institute of Biology, Večna pot 111, 1000 Ljubljana, Slovenia
dTheory Department, National Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia
First published on 15th June 2020
Abstract
Unique morphology of scaffolds prepared by high internal phase emulsion (HIPE) templating is extremely attractive for tissue engineering purposes. However, the synthetic approaches in HIPE are mainly limited to the preparation of either nondegradable polymers or polyesters lacking the pendant functionality. In order to prepare functional scaffolds capable of providing a suitable micro-environment for cell growth, we have developed a method for the preparation of emulsion-templated polypeptide scaffolds, where ring-opening polymerization (ROP) of N-carboxyanhydrides (NCA) of amino acids is performed directly in the oil-in-oil HIPE. Such previously unreported combination of HIPE templating and ROP of NCA requires controlled experimental conditions to ensure gelation of the system before the emulsion phase separates. At the same time, foaming of the scaffold due to CO2 evolution during polymerization has been successfully prevented. The disclosed method was applied for the preparation of γ-benzyl-L-glutamate (BLG) and/or Nε-carbobenzyloxy-L-lysine-based polyHIPE scaffolds crosslinked with L-cystine. Cell culture studies reveal good viability, migration, and proliferation of cells on PBLG polyHIPE. The disclosed method thus represents an attractive approach for the preparation of polyHIPE scaffolds composed entirely of synthetic polypeptides, indicating the applicability of the synthetic method to various NCA monomers for tuning the chemical composition of the scaffolds.
Introduction
The ultimate goal of scaffolding for tissue engineering is to create functional and implantable scaffolds capable of providing suitable micro-environments for optimal cell growth, differentiation and function.1 In this respect, the scaffolds should possess an interconnected macroporous structure for cell penetration and transfer of metabolites and nutrients.2 Average pore size, pore-size distribution, porosity, pore interconnectivity, and permeability throughout the scaffold are therefore among the structural features that need to be adjusted to meet the tissue-specific demands.3 Such specific morphological features can be conveniently tuned by high internal phase emulsion (HIPE) templating.4–6 The scaffold material itself should also be biocompatible and biodegradable, and it should possess suitable functionality to promote cell adhesion. Until recently, HIPE-templating has been mainly limited to various polymers prepared by free-radical polymerization within the water-in-oil emulsions, resulting in the nondegradable scaffolds. Because polymerized HIPE (polyHIPE) morphology is extremely attractive for tissue engineering purposes,7 other approaches in HIPE have also been developed with a key goal to prepare the biodegradable and biocompatible scaffolds distinguished by the interconnected porous morphology, e.g. crosslinking of natural polymers such as gelatin in HIPE.8–10 Although natural polymers allow for good cell adhesion, their poorly defined structure, inter-batch variability, and risks of carrying the pathogens hamper their use in tissue engineering applications.11 Development of synthetic biodegradable and biocompatible polyHIPE scaffolds is currently spearheaded by the synthesis of polyester-based polyHIPEs in water-in-oil HIPEs, usually applying a thiol–ene click chemistry on the appropriately end-functionalized (macro)monomers bearing ester group in the structure already. Such polyHIPEs were found to be capable of supporting the growth of different cell types.12–16 Nevertheless, the main limitations of polyester-based polyHIPEs are their inherently high hydrophobicity and lack of pendant functionality, which can be alleviated by attaching the peptides and proteins on their surface.17,18 Synthetic polypeptides prepared by ring-opening polymerization (ROP) of N-carboxyanhydrides (NCAs) have been identified as one of the most promising candidates for the scaffold design, since they are distinguished by biodegradability, biocompatibility and cell adhesive properties.19–21 Moreover, their composition and thus the chemical and mechanical properties can be modulated by the monomer selection.22,23 So far, macroporous polypeptide-based materials have been produced by cryogelation,23,24 or porogen leaching25,26 which, unlike the HIPE templating, offer only a limited control over the pore structure and connectivity. Until now, ROP of NCA in combination with HIPE templating has been utilized only to tailor the surface properties of nondegradable amine-functionalized polystyrene-based polyHIPE monoliths. In this particular work, primary amine functional groups served as the initiating sites for ROP of γ-benzyl-L-glutamate (BLG) and Nε-carbobenzyloxy-L-lysine (ZLL) NCAs to form the polypeptide brushes on the polyHIPE surface. Subsequent deprotection of the grafted chains resulted in highly-dense and pH responsive functional coating on the polystyrene-based polyHIPE.27In this study, we disclose a previously unreported method for the preparation of emulsion-templated scaffolds composed entirely of synthetic polypeptides. Such scaffolds merge the morphological features of polyHIPEs with advantageous chemical and physical properties of synthetic polypeptides. For this purpose, ROP of BLG NCA or ZLL NCA, as well as their combination, was performed in the oil-in-oil HIPEs. For successful preparation of emulsion-templated synthetic polypeptide scaffolds, polymerization rate plays a very important role since the gelation of the system has to be ensured before the emulsion becomes phase separated. However, too fast polymerization rate of NCA leads to foaming of the scaffold due to the insufficient release of formed CO2 from the HIPE. Therefore, carefully controlled experimental conditions, disclosed in this study, are necessary to prepare well-defined synthetic polypeptide polyHIPEs. The synthesized polyHIPEs were evaluated by cell culture studies, the results of which are very promising for further investigations of emulsion-templated synthetic polypeptide scaffolds for tissue engineering purposes.
Experimental
Chemicals
BLG and ZLL were obtained from Iris Biotech GmbH. Di-N-tert-butyloxycarbonyl-L-cystine, triphosgene, N,N-diisopropylethylamine (DIPEA), Pluronic F-127, sodium hydride, iodomethane, sodium thiosulfate were purchased from Sigma Aldrich, and were used without further purification. Petroleum benzine (boiling range 100–140 °C), anhydrous N,N-dimethylformamide (DMF), 1,4-dioxane, anhydrous tetrahydrofuran (THF), anhydrous ethyl acetate, n-hexane, isopropanol, ethanol, chloroform, and glacial acetic acid were purchased from Merck, and were used as received.Synthesis of NCA monomers
BLG (10 g, 42.2 mmol) was suspended in anhydrous THF (90 mL). The suspension was heated to 50 °C in an oil bath. A solution of triphosgene (6.76 g, 22.8 mmol) in anhydrous THF (10 mL) was then added dropwise under argon. The mixture was stirred at 50 °C under argon for 2 hours to obtain clear solution. The mixture was concentrated by vacuo, the product was precipitated from n-hexane and placed in the freezer overnight. The filtered product was purified by recrystallization from the THF/n-hexane three more times. BLG NCA in the form of a white powder was obtained (9.57 g, 86% yield). 1H and 13C NMR spectra with signal assignation are presented in Fig. S1.† C13H13NO5: calcd C 59.3, H 5.0, N 5.3; found C 59.5, H 5.2, N 5.4.Di-N-tert-butyloxycarbonyl-L-cystine (1 g, 2.3 mmol) was suspended in anhydrous ethyl acetate (15 mL). Then, triphosgene (0.91 g, 3.1 mmol) was added to the suspension in one portion at room temperature. The mixture was heated to 60 °C in an oil bath and stirred at this temperature under argon for 12 hours. Afterwards, the mixture was filtered through the syringe filter (0.45 μm, PTFE, Aldrich) into the centrifuge tube (50 mL) containing 40 mL of n-hexane. After centrifugation at 8000 rpm for 7 minutes, the supernatant was decanted. The product was dried in a desiccator and purified further by repetition of the precipitation two more times. L-Cystine NCA in the form of a white powder was obtained (0.58 g, 83% yield). 1H and 13C NMR with signal assignation are presented in Fig. S2.† C15H18N2O5: calcd C 58.8, H 5.9, N 9.2; found C 58.9, H 5.7, N 9.4.
ZLL (10 g, 35.7 mmol) was suspended in anhydrous ethyl acetate (90 mL). The suspension was heated to 78 °C in an oil bath. A solution of triphosgene (4.70 g, 15.9 mmol) in anhydrous ethyl acetate (10 mL) was then added to the suspension dropwise under argon. The mixture was stirred at 78 °C under argon for 4 hours to obtain clear solution. The mixture was concentrated by vacuo, the product was precipitated from n-hexane and placed in the freezer overnight. The filtered product was purified by recrystallization from the THF/n-hexane three more times. ZLL NCA in the form of a white powder was obtained (7.65 g, 70% yield). 1H NMR and 13C NMR spectra with signal assignation are presented in Fig. S3.† C8H8N2O6S2: calcd C 32.9, H 2.8, N 9.6, S 21.9; found C 32.9, H 2.7, N 9.6, S 21.4.
End-group methylation of pluronic F-127
Pluronic F-127 was methylated as reported previously.28 To summarize, Pluronic F-127 (10 g, 0.8 mmol) and sodium hydride (0.63 g of 60% oil dispersion, 15.9 mmol) were suspended in 30 mL of THF. After cooling in an ice bath, 0.74 mL (15.9 mmol) of iodomethane was added. The mixture was left to warm up and stirred at room temperature for 48 hours. Afterwards, the mixture was cooled in an ice bath and quenched sequentially by dropwise addition of isopropanol (30 mL) and ethanol (2 mL). Then the mixture was neutralized by the addition of glacial acetic acid, whereupon the color changed from the transparent to orange. The solvent was evaporated and the residue was dissolved in chloroform. Subsequently, extraction with aqueous sodium thiosulfate solution (5 wt%) was performed repeatedly until discoloration to ensure the complete reduction and removal of iodine. The organic phase was then washed with water and dried over anhydrous magnesium sulfate. The organic phase was concentrated in vacuo and the product (F127-OMe) was isolated by precipitation from n-hexane to afford a white powder after drying (8.9 g, 89% yield). 1H NMR spectrum with signal assignation of the product is shown in Fig. S4.†General procedure for HIPE-templating of NCAs
Monoliths were prepared by HIPE-templating. The experimental conditions for the oil-in-oil emulsions are listed in Table S1.† The external phase was prepared by dissolving sequentially the surfactant (F127-OMe), the monomer (BLG NCA, ZLL NCA or their mixture), and the crosslinker (L-cystine NCA) in anhydrous DMF at room temperature in a three-necked round bottom flask equipped with an overhead stirrer, an addition funnel, and a rubber septum through which the argon gas was introduced. Under mechanical stirring of the external phase at 330 rpm, the internal phase (petroleum benzine) was added dropwise at a rate of 0.5 mL min−1. After the completion of the addition, the stirring was continued for another 10 minutes and then the catalyst solution (0.5 M DIPEA in DMF) was added in one portion. To ensure even dispersion of the catalyst, the stirring was maintained for further 30 seconds. The emulsion was thereafter aliquoted via an automatic pipette into the 2 mL polypropylene Eppendorf tubes and sealed with septum caps. A needle was then inserted into each cap to enable the release of the CO2 generated in situ during ROP. Emulsions were allowed to polymerize at room temperature for 24 hours. The resulting gels were purified by Soxhlet extraction with dioxane for 48 hours and then they were freeze-dried. Gel content of polyHIPE was determined gravimetrically according to eqn (1), where mpolyHIPE presents the total mass of purified and dried polyHIPE, mNCA presents the total mass of all NCAs used for HIPE, and mCO2 is the calculated mass of released CO2 assuming 100% monomer conversion.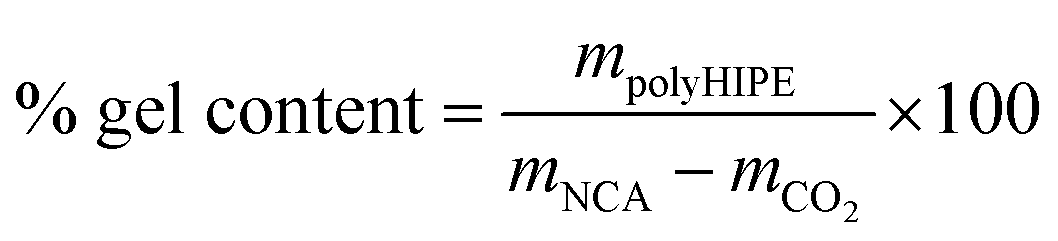 | (1) |
Total porosity was measured gravimetrically according to eqn (2),29 where the polyHIPE density (dpolyHIPE) was determined from the weight and volume of the dry polyHIPE. For polymer densities (dpolymer), the literature values of the PBLG (1.28 g cm−3) and PZLL (1.21 g cm−3) were used.30
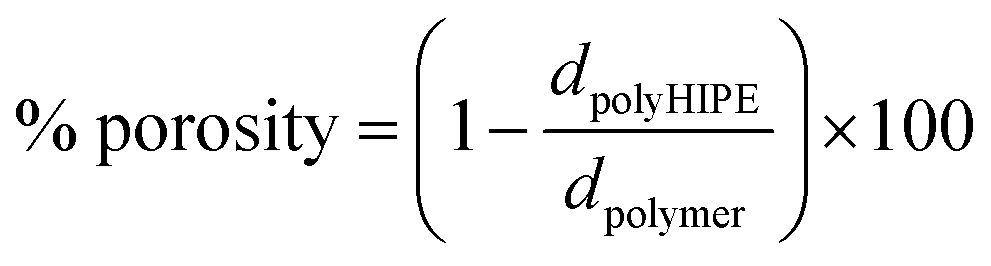 | (2) |
Characterization
1H NMR spectra were recorded at 25 °C in DMSO-d6 or CDCl3 on a Varian Unity Inova 300 MHz spectrometer (Varian, USA), operating in the pulse Fourier transform mode. Chemical shifts are expressed as δ, ppm relative to tetramethylsilane (TMS, δ = 0). The morphology of the monoliths was investigated by scanning electron microscopy (SEM) on a HR-SEM Zeiss Ultra plus instrument (Carl Zeiss, Germany). The samples were fractured with a razor after a liquid nitrogen dip. For charge dissipation during SEM analysis, the as-obtained cross-sections were coated with a 10 nm of gold layer using a Gatan PECS 682 (Gatan, USA). The pore size analyses were performed by using the Image J software, using a correction factor of 2/(30.5) to compensate for the statistical error.31 Infrared spectra were recorded by Bruker Vertex 80 spectrometer and collected in the attenuated total reflectance (ATR) absorbance mode with the nominal resolution of 2 cm−1 at temperature of 25 °C. Typically, 128 interferograms were averaged and apodized with the Gapp-Henzel function. MCT detector was used throughout. ATR spectra were collected by applying a temperature-controlled Specac Golden Gate ATR cell equipped with the diamond-bearing crystal. ATR spectra were analyzed as recorded. Raman spectra were recorded on a Bruker RAM II FT-Raman module coupled to the Vertex 80 infrared spectrometer using a NIR laser operating at 1064 nm as an excitation source. The Raman spectra were collected at room temperature with a nominal resolution of 4 cm−1. Raman scattered light was detected by the Ge-diode detector cooled by liquid nitrogen. Typically, laser power during the sample examination was 200 mW. 64 interferograms were averaged and apodized with the Blackman-Harris-4-term function. Mechanical tests were performed on a DMA Q800 (TA Instruments, USA) dynamic thermal analyzer using the compression discs with 15 mm in diameter. Uniaxial compressive stress–strain measurements were conducted at the temperature of 37 °C on the dry cylindrical specimens (∼8.0 mm diameter and ∼7.0 mm height) with a force rate of 1 N min−1 up to 18 N. For the “wet state” measurement, a solvent exchange from ethanol gradually to phosphate-buffered saline (PBS) was performed. PBS-swollen samples were compressed at 37 °C with a force rate of 1 N min−1 up to 6 N. The compressive Young's moduli (E) of the samples were determined from the slope of the initial linear region of the stress–strain curve. The average of three samples and their standard error are presented.Toxicity assay
PBLG 80-3 polyHIPE discs were prepared manually by cutting-off the polymer blocks of cylindrical shape with 8 mm in diameter from the frozen samples in liquid nitrogen. PolyHIPE discs of about 15 mg were put in absolute ethanol overnight to soak and sterilize. The next day, ethanol was replaced by sterile Milli-Q water. A day before the cell toxicity experiment, polyHIPE discs were washed for 15 minutes in colorless test medium (DMEM D5921, supplemented with 10% v/v FBS and 1% v/v Na-Pyruvate S8636 all from Sigma and with 1% v/v Glutamax 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 050061 from Gibco). Then, polyHIPE discs were transferred to separate wells of 12-well cell culture plate in 1 mL of test medium and incubated overnight in cell culture incubator at normal conditions (5% CO2, 37 °C, close to 100% air humidity). A day before the experiment, the mice fibrosarcoma cell line L929 was seeded on a white 96-well microtiter plate (Costar 3610) at a density of 10000 cells per well in 100 μL of the test medium and incubated overnight in the cell culture incubator. The next day, 100 μL of the media incubated in the presence and absence of the polyHIPE sample were transferred to the cells on a microtiter plate as the treatment and control, respectively. Cells were incubated in the cell culture incubator for an additional day. Afterwards, 20 μL of a Presto blue cell viability reagent (Invitrogen) was added and incubated for an additional three hours to allow the living cells to form the fluorescent product. The signal was measured by a plate reader using the 570 nm excitation filter and 590 nm emission filter. Viability signal from the cells in a polymer-conditioned medium was expressed as a percentage of the control. The average of tests of three samples and their standard error are presented.
050061 from Gibco). Then, polyHIPE discs were transferred to separate wells of 12-well cell culture plate in 1 mL of test medium and incubated overnight in cell culture incubator at normal conditions (5% CO2, 37 °C, close to 100% air humidity). A day before the experiment, the mice fibrosarcoma cell line L929 was seeded on a white 96-well microtiter plate (Costar 3610) at a density of 10000 cells per well in 100 μL of the test medium and incubated overnight in the cell culture incubator. The next day, 100 μL of the media incubated in the presence and absence of the polyHIPE sample were transferred to the cells on a microtiter plate as the treatment and control, respectively. Cells were incubated in the cell culture incubator for an additional day. Afterwards, 20 μL of a Presto blue cell viability reagent (Invitrogen) was added and incubated for an additional three hours to allow the living cells to form the fluorescent product. The signal was measured by a plate reader using the 570 nm excitation filter and 590 nm emission filter. Viability signal from the cells in a polymer-conditioned medium was expressed as a percentage of the control. The average of tests of three samples and their standard error are presented.
Confocal microscopy
Cells of mice fibrosarcoma cell line L929 were seeded on the sterilized and culture medium-soaked PBLG 80-3 polyHIPE discs of 8 mm in diameter, sterilized in the same way as for the toxicity assay. 12 μL of the medium containing 30000 cells were seeded on the top of the polyHIPE disc partially dipped into the medium and cultured for one to seven days. During this period, polyHIPE discs were imaged by a Leica TCS SP5 inverted laser scanning microscope mounted on a Leica DMI 6000 CS module (Leica Microsystems, Germany) using the dry objective HCX PL Fluotar L (20x; NA, 0.4) at different digital zoom. Cell nuclei were labelled by a Hoechst 33342 nuclear stain at the final 4 μM concentration and nuclei of dead cells only were labeled with a SYTOX® Green Nucleic Acid stain (Molecular probes) at a final 1 μM concentration in the culture medium. Sequential excitation was performed using 405 nm diode and 476 nm line of the argon laser. The Hoechst fluorescence emission was detected at 440–480 nm and the emission of SYTOX Green at 520–550 nm.
Cell proliferation assay
Cells of mice fibrosarcoma cell line L929 were seeded on the sterilized and culture medium-soaked PBLG 80-3 polyHIPE discs of 8 mm in diameter in the same way as for the confocal and optical microscopy (∼30000 cells per scaffold). After 2, 4, and 6 days of culturing, the discs with seeded cells and unseeded controls were transferred to 900 μL of the fresh medium in 24 well microplate and after 30 minutes 100 μL of Presto blue cell viability reagent was added and incubated for 3 hours in cell culture incubator. Immediately after addition of the substrate and after 3 hours, 100 μL of medium was collected for the fluorescence measurements as described in the toxicity section to estimate the amount of living cells. Fluorescent signal values in the medium before incubation were subtracted from the values determined in medium after 3 hours of incubation. After incubation with the substrate, the discs were transferred to fresh medium for continuation of the culturing. Signals are presented as the relative fluorescent units to demonstrate the increase in cell number on discs with time.
Optical microscopy
Cells of mice fibrosarcoma cell line L929 were seeded on the sterilized and culture medium-soaked PBLG 80-3 polyHIPE discs of 8 mm in diameter in the same way as for the confocal microscopy (∼30000 cells per scaffold). The cells were cultured for 8 days. Then, the polyHIPE discs were put into a fixative solution (3% glutaraldehyde in 0.1 M phosphate buffer) and cut into 2–3 mm thick slices. Slices of sterilized polyHIPE discs without cells were prepared in the same way and served as a control. Samples were post-fixed with 1% OsO4 in 0.1 M phosphate buffer and embedded in an Agar 100 resin (Agar Scientific). Semithin slices (0.6 μm) were cut on an ultramicrotome (Reichert Ultracut S, Leica, Germany) and stained with the Azure II – Methylene Blue and examined with an AxioCam HR light microscope (Carl Zeiss, Germany).
Results and discussion
Experimental conditions for ROP of NCA in HIPE
To develop a synthetic approach for the preparation of emulsion-templated synthetic polypeptides, the BLG with carboxylic and ZLL with amine protected pendant functional groups were selected as suitable candidates. BLG and ZLL amino acids were converted to their NCA derivatives by reaction with triphosgene. Natural di-amino acid L-cystine was used as a crosslinker (Fig. 1). The method for preparation of L-cystine NCA was optimized since direct triphosgenation of the unprotected and in organic solvents poorly soluble L-cystine resulted in a very low yield of the L-cystine NCA (<25%), which was in addition difficult to purify. Therefore, L-cystine NCA was synthesized in higher yield (83%) and purity from the di-N-tert-butyloxycarbonyl-L-cystine, which is well-soluble in ethyl acetate.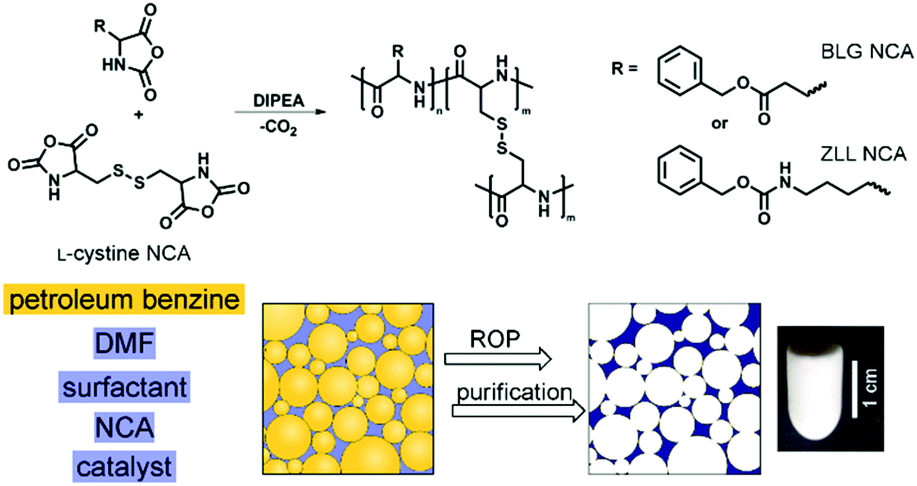 | ||
| Fig. 1 Schematic representation for the preparation of emulsion-templated synthetic polypeptide scaffolds by performing ROP of NCA in the oil-in-oil HIPE. | ||
Recently, NCA polymerization was performed in water-based emulsions, where NCA monomers had been dissolved in chloroform or dichloromethane in order to either prepare polypeptide nanoparticles32 or to achieve fast and controlled ROP of even nonpurified NCA monomers.33,34 In our case, DMF proved to be the best choice as the polymerization medium and continuous phase of HIPE since it is able to dissolve the NCA monomers in very high concentrations (≥2.0 M), which was found to be of utmost importance for the preparation of well-defined polyHIPEs. Because DMF is miscible with water, the polymerization has to be performed in an oil-in-oil emulsion in the presence of petroleum benzine as an internal phase. The oil-in-oil HIPEs were stabilized by a polymeric surfactant (Pluronic F-127).28,35 In order to prevent any chance of hydroxyl groups of Pluronic F-127 to react with NCA,36,37 they were first methylated by iodomethane.
ROP of NCA is usually initiated by the primary amines since they enable good control over the polypeptide molecular weight characteristics. However, the propagation rate of ROP of NCA initiated by the primary amines proved to be too slow to be successfully used in HIPE. Namely, the oil-in-oil HIPEs are delicate to stabilize as they are more prone to phase separation compared to the water-in-oil HIPEs. The HIPEs in this work are stable under stirring, however, after stirring stops, partial phase separation of emulsion is observed within ∼15 min. Therefore, the gelation of the system to lock-in the polyHIPE morphology has to be accomplished before the emulsion undergoes extensive phase separation (Fig. 2a). Fast chain propagation of ROP of NCA can be achieved by the use of tertiary amine initiators. In such cases, polymerization is governed by an activated monomer polymerization mechanism, characterized by the fast propagation and slow initiation, which results in the polypeptides of high molecular weight but broad molecular weight distribution.38 Nevertheless, broad molecular weight distribution does not present a major issue in our case since the resulting polypeptide chains are covalently crosslinked. DIPEA as the tertiary amine catalyst was therefore used to ensure the gelation of the system within a time-frame of emulsion stability.
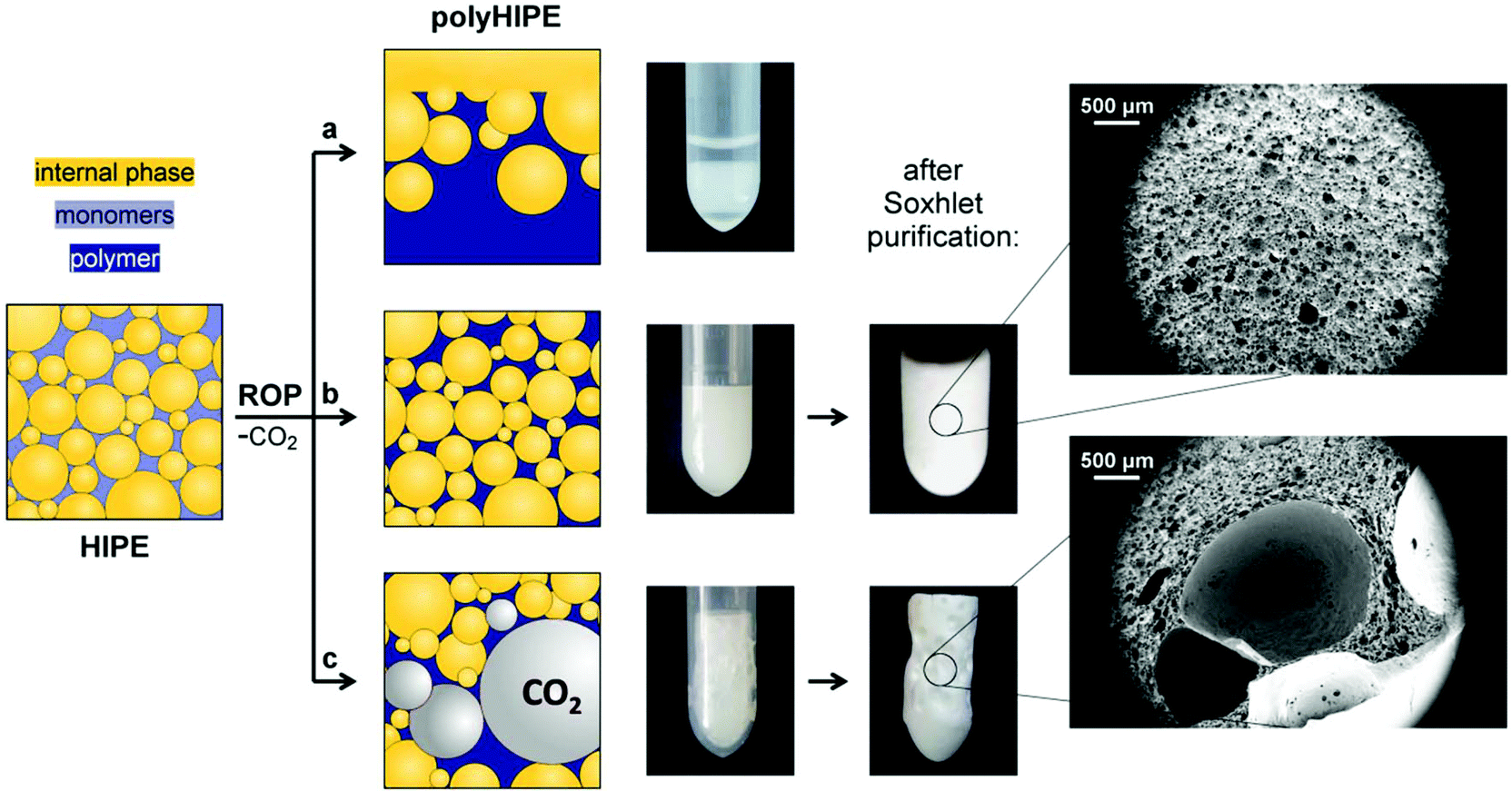 | ||
| Fig. 2 Graphical representation and sample images taken before purification of (a) phase separated polyHIPE obtained when gelation of the system was too slow (PBLG 80-3 with 0.05% catalyst), (b) polyHIPE with optimal morphology (PBLG 80-3 with 0.5% catalyst), and (c) foamed polyHIPE as a result of too fast polymerization (PBLG 80-3 with 2.0% catalyst) or excessive stirring of the emulsion after catalyst addition. A difference between the foamed and non-foamed polypeptide polyHIPEs obtained after Soxhlet purification can be clearly seen from the shape of the sample as well as from the SEM images. | ||
Another major obstacle of NCA polymerization in HIPE is the formation of CO2 since gaseous by-products can cause foaming of the polyHIPE gel. Foaming has to be prevented in order to obtain scaffolds with well-defined polyHIPE morphology (Fig. 2b). The issue of foaming has already been reported by David et al.39 in the study related to the preparation of polyurethane polyHIPEs, where they were not able to avoid the formation of large cavities due to the nucleation and growth of CO2 bubbles. The problem of foaming occurs when the release rate of CO2 exceeds its diffusion rate from the saturated solution, causing the system to become supersaturated.40 This leads to the nucleation of bubbles and foaming, which results in the formation of large cavities and deteriorated control over the porous structure (Fig. 2c). For successful implementation of ROP of NCA in HIPE, the polymerization has to be fast enough to induce the gelation of the system within the time-frame of emulsion stability, but at the same time it should be slow enough to prevent foaming. Therefore, having a control over the polymerization rate is of utmost importance and can be simply tuned by experimental conditions, mainly by the amount of added tertiary amine catalyst and concentration of NCA monomers. Since polymerization takes place at room temperature, the catalyst solution in DMF was added as the last component to the already formed emulsion, which was then further stirred for approximately 30 seconds. Importantly, at prolonged stirring (2–3 minutes) the foamed polyHIPEs were obtained even if the optimal catalyst amount had been used. This is most likely associated with the entrapment of air bubbles into the HIPE during stirring as a consequence of increasing HIPE viscosity. Air bubbles may act as the potential nucleation sites for the released CO2 during polymerization. For the same reason, it is important that the mold is filled in a continuous manner without creating any gaps to avoid CO2 or air entrapment within the emulsion.
Synthesis and characterization of PBLG-based polyHIPEs
For successful emulsion-templating, gelation of the external phase is required to resist the buoyancy forces in order to prevent a coalescence of the internal phase droplets that leads to the phase separation.41 Physical gelation of the PBLG homopolymer in various solvents,42,43 including DMF,44 has already been reported. The creation of the network structure is believed to stem from the aggregation of PBLG helices.45,46 Therefore, the gelation of the PBLG-based polyHIPE did not depend exclusively on the presence of L-cystine NCA crosslinker, since PBLG polyHIPE gelled without the L-cystine NCA as confirmed by the vial inversion test (Fig. S5†). The physical crosslinking of growing PBLG chains importantly contributes to the emulsion stabilization, however, the covalent crosslinking viaL-cystine NCA is essential to obtain well-defined polyHIPEs, which retain network structure and polyHIPE morphology after the purification step. The composition of HIPE for the preparation of PBLG-based polyHIPEs is listed in the Table S1.† The total monomer concentration (2.0 M in DMF) and the content of the crosslinking monomer (5 mol% relative to the total NCA monomers) were kept constant. The samples are coded as 80-10, 80-3, and 85-3, where the numbers represent the volume percentage of the internal phase and the weight percentage of the surfactant relative to the monomer solution, respectively. 0.5 mol% of DIPEA proved to be the appropriate amount of catalyst in all three cases. The gels were left to stand at room temperature for 24 hours to ensure complete polymerization. The consumption of NCA monomers was followed by 1H NMR by taking emulsion aliquots during the polymerization. The aliquots were soaked in DMSO-d6 to monitor the –COCH(R)NH– signal of the NCA monomer at the chemical shift of 4.44 ppm. The obtained monoliths were purified by Soxhlet extraction with dioxane for 2 days, followed by freeze-drying. Complete removal of the surfactant and by-products was confirmed by 1H NMR after soaking the powdered polyHIPEs in CDCl3.Experimental and theoretical studies have shown that several infrared bands are sensitive to changes in polypeptide conformation.47–50 Among others, the most indicative bands are in the amide I (1620–1690 cm−1) and amide III (1220–1330 cm−1) regions. Amide I mode represents a predominant contribution of the backbone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration. Its frequency and band-shape depend on the backbone conformation, reflecting the type of polypeptide secondary structure. Although the amide III region due to the N–H bending and C–N stretching vibrations is the most sensitive indicator for the changes in polypeptide conformation, it may suffer from strong overlapping with the bands originating from the side chain vibrations, which therefore complicate inference on the polypeptide conformation. For this reason, the FTIR spectrum of PBLG 80-3 polyHIPE was compared to that of its linear PBLG analogue, which is well-known to form the helical secondary structure.42 The bands due to the amide I (1649 cm−1) and amide III (1250–1310 cm−1) vibrations in the FTIR spectra of both samples match in the frequency and band shape (Fig. S6†). In both cases, the frequency and narrow symmetrical band shape of the amide I band reveal typical helical conformation of polypeptide chains.50 These results confirm our expectation that introduction of 5% of L-cystine crosslinker does not disrupt the helicity of the PBLG chains significantly. The gel content of PBLG 80-3 was determined to be 85%, thus indicating successful incorporation of the chemical crosslinks throughout the polypeptide network. The presence of crosslinker in PBLG structure was further confirmed by the band at the 503 cm−1 shift in its Raman spectrum, corresponding to the stretching vibration of the disulfide bond of the L-cystine crosslinker (Fig. S7†).
O stretching vibration. Its frequency and band-shape depend on the backbone conformation, reflecting the type of polypeptide secondary structure. Although the amide III region due to the N–H bending and C–N stretching vibrations is the most sensitive indicator for the changes in polypeptide conformation, it may suffer from strong overlapping with the bands originating from the side chain vibrations, which therefore complicate inference on the polypeptide conformation. For this reason, the FTIR spectrum of PBLG 80-3 polyHIPE was compared to that of its linear PBLG analogue, which is well-known to form the helical secondary structure.42 The bands due to the amide I (1649 cm−1) and amide III (1250–1310 cm−1) vibrations in the FTIR spectra of both samples match in the frequency and band shape (Fig. S6†). In both cases, the frequency and narrow symmetrical band shape of the amide I band reveal typical helical conformation of polypeptide chains.50 These results confirm our expectation that introduction of 5% of L-cystine crosslinker does not disrupt the helicity of the PBLG chains significantly. The gel content of PBLG 80-3 was determined to be 85%, thus indicating successful incorporation of the chemical crosslinks throughout the polypeptide network. The presence of crosslinker in PBLG structure was further confirmed by the band at the 503 cm−1 shift in its Raman spectrum, corresponding to the stretching vibration of the disulfide bond of the L-cystine crosslinker (Fig. S7†).
Cross-section SEM images of the PBLG-based polyHIPEs reveal highly interconnected porous structure composed of voids and interconnecting windows typical for the polyHIPE morphology (Fig. 3). The monoliths exhibit well-defined polyHIPE morphology throughout the whole samples. By changing the surfactant amount, the average void size of the final polyHIPE foams can be adjusted.51 Lower surfactant amount resulted in larger average void size and broader void size distribution as determined from SEM images. The average void size together with the porosity and density values are collected in Table 1. At the internal phase volume fraction of 80%, the average void size increases from 35 to 74 μm by decreasing the surfactant concentration from 10 to 3%. At the same surfactant amount of 3%, the increase in the volume of the internal phase to 85% provides more open polyHIPE morphology with larger voids (average size of 123 μm), however a somewhat lower porosity is observed probably due to a slightly more pronounced shrinkage. Pore size distribution of the scaffold most likely plays a very important role in tissue engineering since it is known that different processes involved in tissue development such as cell adhesion, migration, proliferation, and differentiation demand different optimal pore sizes,52 with large pores being especially important since they allow improved infiltration and migration of cells into the scaffold.53
 | ||
| Fig. 3 Cross-section SEM images of polypeptide polyHIPEs revealing the typical polyHIPE morphology composed of large voids, which are interconnected with smaller windows. Samples are coded by X–Y, where X and Y represent the volume percentage of the internal phase and the weight percentage of the surfactant relative to the monomer solution, respectively. | ||
| PolyHIPE | Average void size (μm) | Porosity (%) | Density (g cm−3) | Catalyst amount (mol%) | NCA concentration (M) |
|---|---|---|---|---|---|
| PBLG 80-10 | 34.8 ± 13.9 | 91.3 ± 0.9 | 0.11 ± 0.01 | 0.5 | 2.0 |
| PBLG 80-3 | 74.4 ± 52.0 | 92.0 ± 0.5 | 0.10 ± 0.01 | 0.5 | 2.0 |
| PBLG 85-3 | 123.4 ± 99.0 | 91.3 ± 0.9 | 0.11 ± 0.01 | 0.5 | 2.0 |
| PZLL 80-5.5 | 25.6 ± 10.1 | 91.4 ± 0.4 | 0.10 ± 0.01 | 1.5 | 2.4 |
| P(BLG-co-ZLL) 80-5 | 46.3 ± 33.7 | 90.3 ± 0.6 | 0.12 ± 0.01 | 1.0 | 2.0 |
Synthesis and characterization of PZLL- and P(BLG-co-ZLL)-based polyHIPEs
On the contrary to PBLG, the gelation of PZLL in HIPE or solution can be induced solely via the covalent crosslinking. Namely, PZLL does not form a gel in the absence of crosslinker even at a full monomer conversion, as confirmed by vial inversion test (Fig. S5†), indicating the lack of substantial physical crosslinking via association of polypeptide secondary structures. PZLL helices are reported to be more flexible than PBLG helices54,55 since PZLL bears longer pendant group in the structure, which could be a reason for the absence of physical crosslinking. Under the experimental conditions used for the synthesis of PBLG-based polyHIPE (2.0 M total NCA monomers, 5.0 mol% crosslinking monomer, 3.0 wt% surfactant, and 0.5 mol% catalyst), the gelation of the ZLL system was not observed within the emulsion stability period, despite the use of a crosslinker. It is known that reactivity of ZLL NCA is slower than that of BLG NCA during ROP through a normal amine mechanism.56 To compare the reactivity of both monomers during ROP via the activated monomer mechanism catalyzed by the tertiary amine, ROP of BLG NCA and ZLL NCA (2.0 M monomer) was monitored in DMF in the presence of 0.5 mol% DIPEA catalyst. Since the consumption of ZLL NCA was found to be ∼3 times slower than that of BLG NCA, the optimal HIPE composition has to be appropriately adjusted. In order to speed up the polymerization, the catalyst amount was increased from 0.5 to 1.5 mol%, and the total NCA concentration was increased from 2.0 to 2.4 M. To enhance the stability of the emulsion, the surfactant concentration was also increased from 3.0 to 5.5%. In this way, the PZLL polyHIPE of well-defined interconnected porous morphology was synthesized as indicated by its cross-section SEM images and structural parameters in Fig. 3 and Table 1. FTIR (Fig. S6†) and Raman (Fig. S7†) analyses revealed no significant changes in helical conformation of crosslinked PZLL polyHIPE as compared to its linear PZLL counterpart, similar as it was observed for the PBLG polyHIPEs.To further confirm the applicability of the reported method, the copolypeptide polyHIPE was also synthesized from a mixture of BLG NCA and ZLL NCA monomers in a mole ratio of 1:1. For this purpose, the synthetic experimental conditions were adjusted (2.0 M total monomer concentration, 5.0% surfactant, 5.0 mol% crosslinker, and 1.0 mol% catalyst) in a way to induce system gelation before emulsion phase separation. Similar as PBLG- and PZLL-based polyHIPEs, also the SEM images of the P(BLG-co-ZLL) polyHIPE show typical interconnected porous structure as evident in Fig. 3. The average void size, density, and porosity values of P(BLG-co-ZLL) polyHIPE are listed in Table 1. The presented results thus reveal the applicability of the disclosed synthetic method to a wide variety of NCA monomers and their combinations.
Cell culture studies
Since polypeptide polyHIPEs are potential candidates for tissue engineering applications, the toxicity and cell proliferation properties were assessed using the PBLG 80-3 as a model polypeptide polyHIPE. It is worth noting that PBLG polyHIPEs are hydrophobic and do not swell in the water-based mediums, instead they keep the original shape and size. Young's modulus, measured at 37 °C, decreases from 1.34 ± 0.15 MPa in dry state to 0.98 ± 0.19 MPa when soaked in PBS (Fig. S8†). The absence of toxic residues in the material was confirmed by cell viability experiments, where the PBLG 80-3 samples were soaked in a medium for 24 hours, followed by the transfer of the medium to the L929 mice fibrosarcoma cell culture. The results show no reduction in cell viability as indicated by the comparable viability signals of the treated cultures and control (Fig. 4a).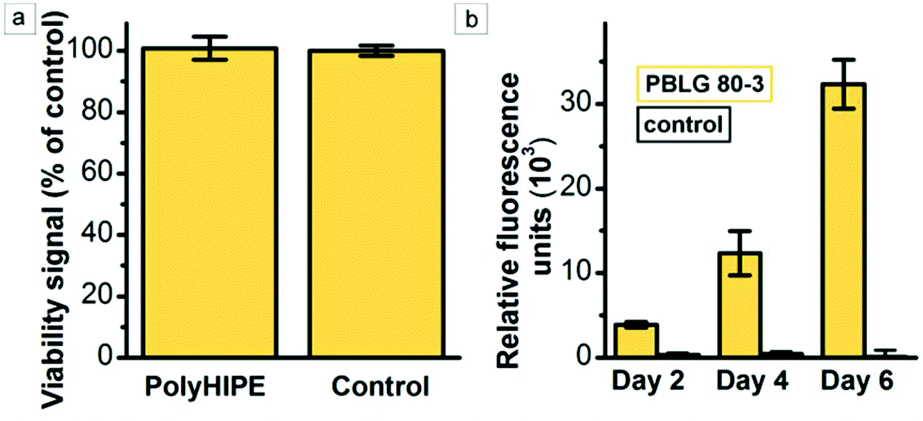 | ||
| Fig. 4 (a) Viability test of L929 cells treated with medium incubated overnight with polyHIPE or without it (control). The average of three tests and their standard error are presented. (b) Relative fluorescence signal of PBLG 80-3 scaffold discs at 2, 4, and 6 days after seeding with L929 cells or without cells (control). The average of five tests and their standard error are presented. | ||
For evaluation of the cell adherence and proliferation, the mice fibrosarcoma cell line L929 was seeded on the PBLG 80-3 polyHIPE scaffold. The extent of cell proliferation in the scaffold was evaluated by the PrestoBlue assay, allowing for quantification of a relative change of the living cell number present in the scaffold. Since PrestoBlue is metabolized by the living cells to produce a fluorescent product,57 the intensity of the fluorescence signal correlates with the number of the living cells in the scaffold. The cell population in the scaffold was estimated at 2, 4, and 6 days after seeding. Compared to day 2, the signal intensity increased by almost 3 (p = 1.78 × 10−3) and 8 times (p = 2.09 × 10−5) after 4 and 6 days, respectively (Fig. 4b). In contrast, the signal of the control without seeded cells did not change significantly (p-values 0.61 and 0.53 for days 4 and 6 respectively). Qualitative evaluation of cell proliferation was done by confocal microscopy. The images show the increasing number of viable L929 cells (blue) with culturing time and only a few dead cells (green) were observed (Fig. 5a–f and Videos S1–S3†). After seven days, the surface of the deeper voids was densely populated by the cells as observed through the windows of the polyHIPE structure. Since the scaffolds are not transparent there is a limitation on how deep can we observe cell migration using confocal microscopy, therefore, we have decided to perform optical microscopy of semithin (0.6 μm) slices of the scaffold which were cut parallel (Fig. 5g) and perpendicular (Fig. 5h) to the surface. Light microscopy images show that the cells spread over the surface of scaffold as well as penetrated into the scaffold and successfully inhabited the surface of the voids deep into the polyHIPE structure, confirming cell proliferation throughout the polyHIPE structure.
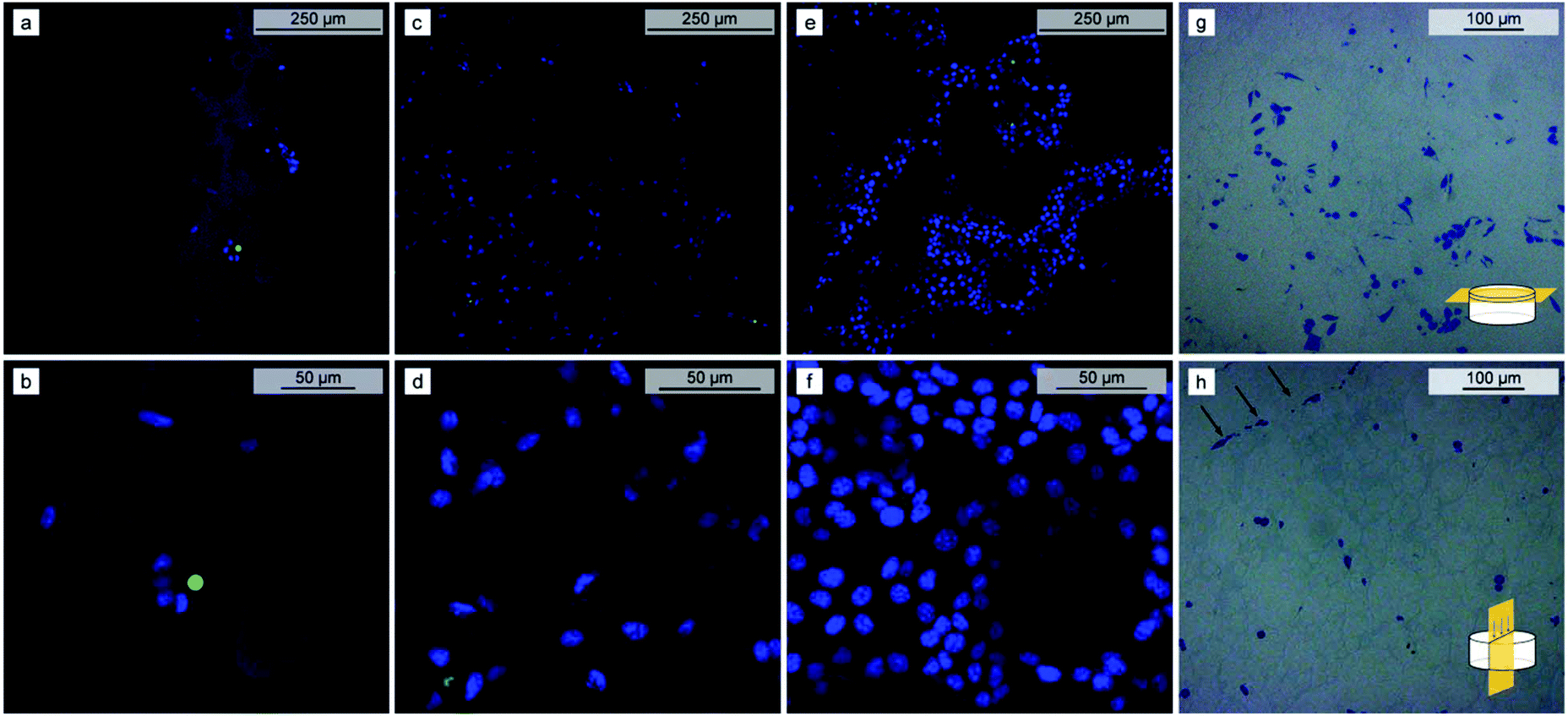 | ||
| Fig. 5 Confocal microscopy images indicating the increase in L929 cell number on the PBLG 80-3 scaffold at 2 (a and b), 4 (c and d) and 7 (e and f) days after seeding. Living cell nuclei are blue and dead cell nuclei are green. Images b, d and f are magnified area from a, c and e, respectively. Optical microscopy images of PBLG 80-3 scaffold slices cut parallel (g) to the surface (∼5 μm below the surface) and perpendicular (h) (black arrows mark the direction of top side) after 8 days of seeding. | ||
Conclusion
Here we report a versatile synthetic strategy for the preparation of highly interconnected macroporous synthetic polypeptide scaffolds by performing ROP of NCA in the oil-in-oil HIPE. To combine NCA chemistry and HIPE-templating it is necessary to achieve gelation before phase separation of oil-in-oil emulsion as well as to overcome the issue of HIPE foaming due to the CO2 evolution during NCA polymerization. In the case of PBLG polyHIPEs, the gelation is enhanced by the physical crosslinking through self-association of the growing polypeptide chains. However, such non-covalent crosslinking is not mandatory to obtain well-defined polypeptide polyHIPEs as it was shown for the PZLL-based polyHIPEs. Important parameter that has to be taken into account is the rate of polymerization since too fast ROP of NCA results in deteriorated control over the porous structure as a consequence of HIPE foaming. Foaming occurs when the release rate of CO2, formed as a by-product, exceeds its diffusion rate from the HIPE. Since the polymerization rate and thus the rate of CO2 release depends on the NCA reactivity and polymerization conditions used, it can be easily adjusted by the catalyst amount and monomer concentration with the aim to hit the optimal window, wherein the scaffolds with the typical polyHIPE morphology throughout the whole structure are obtained. Moreover, the pore size and pore size distribution of emulsion-templated synthetic polypeptides can be adjusted by changing the surfactant amount and/or increasing the internal phase volume fraction. Cell culture study confirms advantageous combination of polyHIPE morphology and polypeptide structure as revealed by excellent viability, migration, and proliferation of cells on the PBLG scaffold.The method described here thus opens up a way for the preparation of polyHIPE scaffolds composed entirely of synthetic polypeptides, where the chemical composition can be tuned by employing a large variety of available amino acids allowing for direct incorporation of various functionalities into the polyHIPE scaffold. Since the scaffolds are covalently crosslinked and therefore insoluble, countless possible post-polymerization modifications/functionalizations can also be performed to further fine-tune the scaffolds’ properties. Due to these reasons, the disclosed synthetic method represents an important basis for preparation of a new generation of functional scaffolds distinguished by highly interconnected macroporous morphology and tailored physical and chemical properties.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the financial support from the Slovenian Research Agency (Research Core Funding No. P2-0145 and Project No. J2-9214 and N2-0131). P.U. is a Ph.D. student at the Faculty of Chemistry and Chemical Engineering, University of Maribor, Slovenia.References
- J. Lee, M. J. Cuddihy and N. A. Kotov, Tissue Eng., Part B, 2008, 14, 61–86 CrossRef CAS PubMed.
- A. Khademhosseini and R. Langer, Nat. Protoc., 2016, 11, 1775–1781 CrossRef CAS PubMed.
- D. Griffon, M. Sedighi, D. Schaeffer, J. Eurell and A. Johnson, Acta Biomater., 2006, 2, 313–320 CrossRef PubMed.
- M. S. Silverstein, Polymer, 2014, 55, 304–320 CrossRef CAS.
- M. S. Silverstein, Prog. Polym. Sci., 2014, 39, 199–234 CrossRef CAS.
- R. J. Carnachan, M. Bokhari, S. A. Przyborski and N. R. Cameron, Soft Matter, 2006, 2, 608–616 RSC.
- W. Busby, N. R. Cameron and C. A. B. Jahoda, Biomacromolecules, 2001, 2, 154–164 CrossRef CAS PubMed.
- H. Tan, Z. Tu, H. Jia, X. Gou and T. Ngai, Langmuir, 2018, 34, 4820–4829 CrossRef CAS PubMed.
- A. Barbetta, M. Massimi, L. Conti Devirgiliis and M. Dentini, Biomacromolecules, 2006, 7, 3059–3068 CrossRef CAS PubMed.
- A. Barbetta, M. Dentini, E. M. Zannoni and M. E. De Stefano, Langmuir, 2005, 21, 12333–12341 CrossRef CAS PubMed.
- S. Venkatraman, F. Boey and L. L. Lao, Prog. Polym. Sci., 2008, 33, 853–874 CrossRef CAS.
- S. Caldwell, D. W. Johnson, M. P. Didsbury, B. A. Murray, J. J. Wu, S. A. Przyborski and N. R. Cameron, Soft Matter, 2012, 8, 10344–10351 RSC.
- D. W. Johnson, C. R. Langford, M. P. Didsbury, B. Lipp, S. A. Przyborski and N. R. Cameron, Polym. Chem., 2015, 6, 7256–7263 RSC.
- A. R. Murphy, I. Ghobrial, P. Jamshidi, A. Laslett, C. M. O'Brien and N. R. Cameron, Polym. Chem., 2017, 8, 6617–6627 RSC.
- A. M. Eissa, F. S. V. Barros, P. Vrljicak, J. J. Brosens and N. R. Cameron, Biomacromolecules, 2018, 19, 3343–3350 CrossRef PubMed.
- M. Whitely, S. Cereceres, P. Dhavalikar, K. Salhadar, T. Wilems, B. Smith, A. Mikos and E. Cosgriff-Hernandez, Biomaterials, 2018, 185, 194–204 CrossRef CAS PubMed.
- S. A. Richardson, T. M. Rawlings, J. Muter, M. Walker, J. J. Brosens, N. R. Cameron and A. M. Eissa, Macromol. Biosci., 2019, 19, 1800351 CrossRef PubMed.
- J. L. Ratcliffe, M. Walker, A. M. Eissa, S. Du, S. A. Przyborski, A. L. Laslett and N. R. Cameron, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1974–1981 CrossRef CAS.
- T. J. Deming, Prog. Polym. Sci., 2007, 32, 858–875 CrossRef CAS.
- C. Deng, J. Wu, R. Cheng, F. Meng, H.-A. Klok and Z. Zhong, Prog. Polym. Sci., 2014, 39, 330–364 CrossRef CAS.
- X. Zhou and Z. Li, Adv. Healthcare Mater., 2018, 7, 1800020 CrossRef PubMed.
- X.-Q. Dou and C.-L. Feng, Adv. Mater., 2017, 29, 1604062 CrossRef PubMed.
- S. J. Shirbin, F. Karimi, N. J.-A. Chan, D. E. Heath and G. G. Qiao, Biomacromolecules, 2016, 17, 2981–2991 CrossRef CAS PubMed.
- T. Sedlačík, V. Proks, M. Šlouf, M. Dušková-Smrčková, H. Studenovská and F. Rypáček, Biomacromolecules, 2015, 16, 3455–3465 CrossRef PubMed.
- J. Qian, X. Yong, W. Xu and X. Jin, Mater. Sci. Eng., C, 2013, 33, 4587–4593 CrossRef CAS PubMed.
- J. Fang, Q. Yong, K. Zhang, W. Sun, S. Yan, L. Cui and J. Yin, J. Mater. Chem. B, 2015, 3, 1020–1031 RSC.
- F. Audouin, M. Fox, R. Larragy, P. Clarke, J. Huang, B. O'Connor and A. Heise, Macromolecules, 2012, 45, 6127–6135 CrossRef CAS.
- P. Utroša, O. C. Onder, E. Žagar, S. Kovačič and D. Pahovnik, Macromolecules, 2019, 52, 9291–9298 CrossRef.
- V. Karageorgiou and D. Kaplan, Biomaterials, 2005, 26, 5474–5491 CrossRef CAS PubMed.
- C. G. Sridhar, W. A. Hines and E. T. Samulski, J. Chem. Phys., 1974, 61, 947–953 CrossRef CAS.
- A. Barbetta and N. R. Cameron, Macromolecules, 2004, 37, 3188–3201 CrossRef CAS.
- J. Jacobs, D. Pavlović, H. Prydderch, M.-A. Moradi, E. Ibarboure, J. P. A. Heuts, S. Lecommandoux and A. Heise, J. Am. Chem. Soc., 2019, 141, 12522–12526 CrossRef CAS PubMed.
- X. Wang, Z. Song, Z. Tan, L. Zhu, T. Xue, S. Lv, Z. Fu, X. Zheng, J. Ren and J. Cheng, ACS Macro Lett., 2019, 8, 1517–1521 CrossRef CAS.
- Z. Song, H. Fu, J. Wang, J. Hui, T. Xue, L. A. Pacheco, H. Yan, R. Baumgartner, Z. Wang, Y. Xia, X. Wang, L. Yin, C. Chen, J. Rodríguez-López, A. L. Ferguson, Y. Lin and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10658–10663 CrossRef CAS PubMed.
- N. R. Cameron and D. C. Sherrington, J. Chem. Soc., Faraday Trans., 1996, 92, 1543–1547 RSC.
- W. Zhao, Y. Gnanou and N. Hadjichristidis, Polym. Chem., 2015, 6, 6193–6201 RSC.
- Š. Gradišar, E. Žagar and D. Pahovnik, ACS Macro Lett., 2017, 6, 637–640 CrossRef.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528–5578 CrossRef CAS PubMed.
- D. David and M. S. Silverstein, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5806–5814 CrossRef CAS.
- V. Kumar and N. P. Suh, Polym. Eng. Sci., 1990, 30, 1323–1329 CrossRef CAS.
- A. Parker, P. A. Gunning, K. Ng and M. M. Robins, Food Hydrocolloids, 1995, 9, 333–342 CrossRef CAS.
- A. Niehoff, A. Mantion, R. McAloney, A. Huber, J. Falkenhagen, C. M. Goh, A. F. Thünemann, M. A. Winnik and H. Menzel, Colloid Polym. Sci., 2013, 291, 1353–1363 CrossRef CAS PubMed.
- S. Sasaki, M. Hikata, C. Shiraki and I. Uematsu, Polym. J., 1982, 14, 205–213 CrossRef CAS.
- B. Ginzburg, T. Siromyatnikova and S. Frenkel, Polym. Bull., 1985, 13, 139–144 CAS.
- C. D. Vacogne, S. M. Brosnan, A. Masic and H. Schlaad, Polym. Chem., 2015, 6, 5040–5052 RSC.
- Y. Izumi, H. Takezawa, N. Kikuta, S. Uemura and A. Tsutsumi, Macromolecules, 1998, 31, 430–435 CrossRef CAS.
- J. Bandekar, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1992, 1120, 123–143 CrossRef CAS.
- R. Schweitzer-Stenner, F. Eker, Q. Huang, K. Griebenow, P. A. Mroz and P. M. Kozlowski, J. Phys. Chem. B, 2002, 106, 4294–4304 CrossRef CAS.
- J. Grdadolnik, V. Mohacek-Grosev, R. L. Baldwin and F. Avbelj, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1794–1798 CrossRef PubMed.
- A. Mirtič and J. Grdadolnik, Biophys. Chem., 2013, 175–176 Search PubMed , 47–53.
- L. L. C. Wong, P. M. Baiz Villafranca, A. Menner and A. Bismarck, Langmuir, 2013, 29, 5952–5961 CrossRef CAS.
- I. Bružauskaitė, D. Bironaitė, E. Bagdonas and E. Bernotienė, Cytotechnology, 2016, 68, 355–369 CrossRef PubMed.
- C. M. Murphy, M. G. Haugh and F. J. O'Brien, Biomaterials, 2010, 31, 461–466 CrossRef CAS PubMed.
- S. Itou, N. Nishioka, T. Norisuye and A. Teramoto, Macromolecules, 1981, 14, 904–909 CrossRef CAS.
- G. Spach, L. Freund, M. Daune and H. Benoît, J. Mol. Biol., 1963, 7, 468–482 CrossRef CAS PubMed.
- M. Zelzer and A. Heise, Polym. Chem., 2013, 4, 3896–3904 RSC.
- X. Ren, L. F. Tapias, B. J. Jank, D. J. Mathisen, M. Lanuti and H. C. Ott, Biomaterials, 2015, 52, 103–112 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR, FTIR, and Raman spectra, stress–strain curves, and additional experimental details. See DOI: 10.1039/d0py00387e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |