
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new sol–gel route towards Nd3+-doped SiO2–LaF3 glass-ceramics for photonic applications
María Eugenia
Cruz
 *a,
Alicia
Durán
a,
Rolindes
Balda
bc,
Joaquín
Fernández
d,
Glenn C.
Mather
a and
Yolanda
Castro
a
*a,
Alicia
Durán
a,
Rolindes
Balda
bc,
Joaquín
Fernández
d,
Glenn C.
Mather
a and
Yolanda
Castro
a
aInstituto de Cerámica y Vidrio (CSIC), Campus de Cantoblanco, 28049 Madrid, Spain. E-mail: mariaeugenia@icv.csic.es
bDept. Física Aplicada I, Escuela Superior de Ingeniería, Universidad del País Vasco (UPV-EHU), 48013 Bilbao, Spain
cCentro de Física de Materiales, (UPV/EHU-CSIC), 20018 San Sebastian, Spain
dDonostia International Physics Center DIPC, 20018 San Sebastian, Spain
First published on 20th November 2020
Abstract
Glass-ceramic materials with composition 0.9Nd3+–80SiO2–20LaF3 were successfully obtained and further heat-treated at 450 °C for 6 h. Stable and homogeneous LaF3 nanoparticle suspensions with and without Nd3+ were first prepared by a chemical route, incorporating polyvinylpyrrolidone (PVP) as dispersant. The suspensions were then concentrated and characterised by XRD, HRTEM and zeta potential, confirming that LaF3 crystallises as the only phase, with particle size around 16 nm. The suspensions were incorporated in a silica sol to obtain a 0.9Nd3+–20LaF3–80SiO2 particulate sol, xerogel and glass-ceramic. HRTEM confirmed the homogeneous incorporation of the doped nanocrystals into the SiO2 matrix without modification of the nanoparticle structure. Rietveld refinement was used to determine the crystallinity and quantity of LaF3 nanoparticles present in the glass-ceramic after treatment of the particulate sol at 450 °C for 6 h. Luminescence measurements of near infrared Nd3+ ion emissions in the lanthanum fluoride nanoparticles and SiO2–LaF3 glass-ceramic showed well-structured emission spectra with lifetimes similar to those of theoretical Nd+3 in LaF3 crystals.
Introduction
Glass is a key material with a wide range of applications, ranging from traditional glassware products to advanced optical materials. Optical transparency in an extended spectrum, chemical durability, and manufacturability render glass optimal for producing materials of different shapes. In addition, significant improvements in certain properties may be achieved on preparation of glass ceramics (GCs) through controlled crystallisation.1S. D. Stookey first developed a glass-ceramic named Fotoceram in 1957, later called Pyroceram,2 coining the term glass-ceramic and defining it as: “…made by first melting and forming special glasses containing nucleating agents and then causing controlled crystallization of the glass particles”.2 For many years, this definition only included materials containing more than 50% of crystals. However, GCs with lower crystal fraction have also been developed in the last 60 years. The definition was updated after the “12th International Symposium on Crystallization in Glasses and Liquids”, organized by TC07 of the International Commission on Glass (ICG), to include different technologies and processing methods.
“Glass-ceramics are inorganic, non-metallic materials prepared by controlled crystallization of glasses via different processing methods. They contain at least one type of functional crystalline phase and a residual glass. The volume fraction crystallized may vary from ppm to almost 100%”.3
Particular attention is currently devoted to the preparation of oxide glasses containing fluoride nanocrystals (NCs). Such materials, known as oxyfluoride glass-ceramics (OxGCs), were obtained for the first time in 1998 by Dejneka (1998). Fluoride nanocrystals add unique properties to oxide glasses, making them attractive for photonic applications. They combine the very low phonon energy (300–450 cm−1) of fluoride nanocrystals with the high chemical, mechanical and thermal stability of the glass matrices, thereby increasing the luminescence efficiency.4–6 The resulting oxyfluoride glass-ceramics have notable advantages compared with pure glass or ceramic materials. The LaF3–SiO2 system has been investigated due to the behaviour of the LaF3 crystals as rare-earth (RE) hosts. LaF3 has superior solid solubility compared with other fluoride crystal phases.7,8
Among the different techniques employed for the preparation of OxGCs, glass melting-quenching (MQ) is the most relevant. However, high melting temperatures (1400–1700 °C) cause significant fluorine loss, limiting the final fluorine content of the crystal phase and resulting in uncontrollable compositions with respect to fluorine and lanthanide (Ln3+) ions. Many researchers have proposed alternative processing methods to overcome these limitations9 such as Spark Plasma Sintering (SPS). The preparation of xZrO2–(100–x)SiO2 glass-ceramics by SPS with a high content of crystalline phase (higher than 45 mol%)10 has been reported, but LaF3–SiO2 materials have not yet been reported. Moreover, the SPS process is rather expensive and the size and shape of samples are quite limited.10
On the other hand, the sol–gel process is a promising route for production of glass-ceramic materials, which avoids the drawbacks of melting-quenching, with the versatility to allow differently shaped products such as thin films, powders, and bulk materials obtained at low temperatures. Further advantages of the method include homogeneity and high purity of the resultant materials. However, sol–gel was not widely applied for glass-ceramic processing until significant improvements in structural and optical properties appeared in the late nineties. Precise control of the synthesis and crystallisation process is required to obtain transparent oxyfluoride glass ceramics, which avoids both uncontrolled particle growth and the formation of undesirable phases. Careful process design is, therefore, critical for obtaining well-ordered, transparent, and efficient optical glass-ceramics.
The most studied oxyfluoride compositions prepared by sol–gel are LnF3 (Ln = La, Y, Gd) and RLnF4 (R = Na, K, Li; Ln = Gd, Y), doped with different rare-earth (RE3+) ions (Er3+, Eu3+, Nd3+, etc.).11,12
The first papers reporting the processing of pure LaF3 on silica glass substrates by sol–gel were published in 1998 by Fuhijara and Tada (1999), who showed that control of the synthesis parameters and heat-treatment were essential to obtain transparent LaF3 materials avoiding the precipitation of other phases such as LaOF.13,14
Up to 2018, the literature typically reported the preparation of oxyfluoride glass-ceramics with nominal contents of 5–10 mol% of active crystal phases, with little innovation related to synthesis and processing conditions.13,15 Nevertheless, in the past decade, the GlaSS Group of the Instituto de Cerámica y Vidrio (CSIC) has optimised the sol–gel process to obtain oxyfluoride glass-ceramics with high active-phase contents (up to 20 mol%) by sol–gel. Quantitative Rietveld refinement of 80SiO2–20LaF3 bulk samples doped with 0.5Er3+ and treated at 550 °C for 1 min indicated a crystal fraction ∼18 wt%,13 the highest reported in the literature for transparent oxyfluoride glass-ceramics prepared by sol–gel and the highest compared to any glass-ceramics prepared by melting. However, photoluminescence emission and excitation spectra of SiO2:LaF3 samples exhibited only a few, broad structured bands due to the small size of the nanocrystals (∼2 nm for thin films and 8 nm for bulk samples). Gorni et al.15 proposed that the weak luminescence properties are due to uncontrolled nucleation and growth of the LaF3 crystals in the sol–gel matrix, because, at high temperatures, the crystals tends dissolve and reach chemical-potential equilibrium. It is essential, therefore, to improve the optical properties by obtaining larger nanocrystal sizes, which is difficult using current synthesis methods based on a two-step chemical process.
An alternative, recently proposed procedure to obtain glass-ceramics is the production of nano-crystalline powders followed by their dispersion in a sol–gel matrix. Although this method is promising, the luminescence studies are not conclusive.16
The aim of the current work is the preparation of Nd3+ doped-LaF3 nanoparticle suspensions and their incorporation into silica (SiO2) sols prepared by acid catalysis. The dispersion of the nanoparticles and stability of the LaF3 and LaF3–SiO2 suspensions were studied, confirming that LaF3 appears as the only crystalline phase. Xerogels and glass-ceramic powders were produced by thermal treatment and characterized by different techniques with promising luminescence results.
Experimental
Synthesis of LaF3 and Nd3+:LaF3 nanoparticle suspensions
Lanthanum chloride (LaCl3, Alfa Aesar) and ammonium fluoride (NH4F, Merck) were used as reagents without further purification and mixed with deionized water to a La3+ concentration of 0.04 M.17 The solution was maintained at 75 °C for 2 h with continuous stirring to obtain the LaF3–0.04 M nanoparticle suspension with a final pH = 7. Following the same process, neodymium acetate (Nd(CH3CO2)3, Alfa Aesar) was added to NH4F/LaCl3/H2O solution in a molar ratio Nd3+/LaCl3 = 0.9. The solution was maintained at 75 °C for 2 h under continuous stirring to obtain a 0.9Nd3+–LaF3–0.04 M nanoparticle suspension.Polivinylpirrolidone (PVP) dispersant was added to LaF3–0.04 M and 0.9Nd3+–LaF3–0.04 M nanoparticle suspensions with concentrations of LaF3 nanoparticles in the range 1–10 wt%. Subsequently, the LaF3–0.04 M and 0.9Nd3+–LaF3–0.04 M suspensions were sonicated using an ultrasound probe (Ultrasonication Probe, GM 2200, Bandelin electronic, Germany) for 2 min. The resulting suspensions were labelled as LaF3–0.04 M–PVP(X) or 0.9Nd3+–LaF3–0.04 M–PVP(X), where X indicates the wt% of added PVP.
All the suspensions were concentrated using a rotary evaporator (R-210 with vacuum pump V-700, Buchi) to reach a concentration of 0.25 M, and labelled as LaF3–0.25 M–PVP(X) and 0.9Nd3+–LaF3–0.25 M–PVP(X).
Characterisation of Nd3+–LaF3 nanoparticle suspensions
The dispersibility and stability of a LaF3–0.04 M nanoparticle suspension was studied through the variation of zeta potential as a function of pH using a Zetasizer Nano ZS instrument (Malvern S, UK). Suspensions of different pH in the range 3–12 were prepared on addition of nitric acid (HNO3, Sigma Aldrich) and tetramethylammonium hydroxide (TMHA, Merck) then stabilised for 12 h.Particle size was also measured as a function of wt% PVP for LaF3–0.04 M–PVP(X) nanoparticle suspensions (X = 1, 3, 5, 7, 9 and 10) using the Zetasizer Nano ZS instrument.
LaF3–0.04 M and 0.9Nd3+–LaF3–0.25 M–PVP(X) suspensions were centrifuged at 6000 rpm for 5 min, and the resulting powders rinsed with deionized water; the process was repeated three times to remove all the organic components. Powders were further dried at 80 °C for 12 h and characterized by X-ray diffraction (XRD). Diffractograms were acquired in the range 20–70° 2θ with a step size of 0.02° and 1 second of integration time using a D8 Advance diffractometer (Bruker, MA). The crystal size was calculated by applying Scherrer's equation,
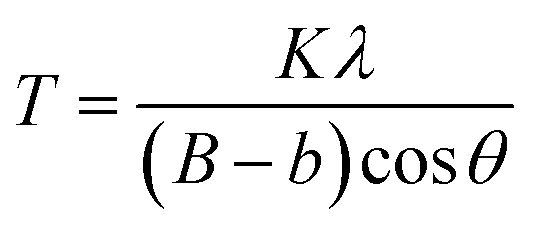
High Resolution Transmission Electron Microscopy (HRTEM) and Energy-dispersive X-ray spectroscopy (EDX) were used to characterize 0.9Nd3+–LaF3–0.04 M and 0.9Nd3+–LaF3–0.25 M–PVP(X) nanoparticles dried at 80 °C for 12 h and redispersed in ethanol employing a JEOL 2100F microscope. The samples were prepared by dropping the suspensions onto a carbon-coated copper grid (Lacey Carbon, LC-200-Cu 25/pk). HRTEM images were processed using ImageJ® software. The lattice parameters of LaF3 nanoparticles in 0.9Nd3+–LaF3–0.25 M–PVP(10) were determined from the corresponding electron-diffraction pattern.
The crystal fraction of 0.9Nd3+–LaF3–0.25 M–PVP(10) nanoparticles in a glass ceramic treated at 450 °C for 6 h was estimated by the Rietveld method as described previously4 using the FULLPROF program,18 with NaF as internal weight standard in an appropriate quantity (5 wt%). The XRD data were collected in the range 20° ≤ 2θ ≤ 120° in a step width of 0.0167° employing a PHILIPS X-PERT PRO θ/2θ diffractometer operating at 45 kV and 40 mA.
Synthesis of 0.9Nd3+–20LaF3:80SiO2 particulate sols
20LaF3–80SiO2 particulate sols (weight relation 80/20 between SiO2 and LaF3) with and without Nd3+ doping were prepared from 14.5 ml of tetraethyl orthosilicate (TEOS, Sigma Aldrich) and 13 ml of methyl-triethoxysilane (MTES, ABCR) mixed with continuous stirring under ambient conditions. Subsequently, 28 g of a concentrated suspension of 0.9Nd3+–LaF3–0.25 M–PVP(X) nanoparticles was incorporated and concentrated; hydrochloric acid (HCl, Sigma Aldrich) was then added under vigorous stirring to produce the hydrolysis and condensation reactions,17 and achieve 0.9Nd3+–80SiO2:20LaF3–PVP(X) particulate sols on rapid cooling in ice baths for one minute; a final concentration of 171 g l−1 SiO2 was attained on diluting with 11 ml of absolute ethanol.Two further sols of composition 0.9Nd3+–5LaF3:95SiO2 and 0.9Nd3+–10LaF3–90SiO2 were also synthesised following the same process, incorporating the corresponding molar ratios of 0.9Nd3+–LaF3–0.25 M–PVP(10) nanoparticle suspensions to TEOS and MTES solutions.
Characterisation of 0.9Nd3+–20LaF3:80SiO2 materials
The stability of the 0.9Nd3+–80SiO2:20LaF3 particulate sols was determined using an AND Vibro Viscometer on measuring the viscosity once a day for five days.Differential thermal analysis (DTA) and thermogravimetric analysis (TGA) were performed with a SDT Q600 instrument (TA Instruments, New Castle, DE, USA). Approximately 15 g of 0.9Nd3+–20LaF3:80SiO2 xerogel, dried at 80 °C for 12 h, was measured in air from room temperature to 800 °C with a heating rate of 10 °C min−1.
The compositional and morphological characterization of the xerogels and glass-ceramics obtained from particulate sols was performed by HRTEM and EDX spectroscopy (JEOL 2100F). The 0.9Nd3+–20LaF3:80SiO2 particulate sol was dried and treated at 450 °C for 6 h. The powder was re-dispersed in ethanol and a drop of the suspension deposited onto carbon-coated copper grids (Lacey Carbon, LC-200-Cu 25/pk); the size distribution of the particles was determined using ImageJ® software.
The quantitative crystalline fraction of 0.9Nd3+–20LaF3:80SiO2 samples treated at 450 °C for 6 h was determined by the Rietveld method using the parameters and internal weight standard indicated earlier.
Optical characterisation of LaF3 nanoparticles and Nd3+–LaF3–SiO2 glass-ceramics
0.9Nd3+–LaF3–0.25 M–PVP(10) nanoparticle suspensions without any additive were dried at 80 °C and treated at 450 °C before pressing in a uniaxial press for 3 min at 1000 MP for luminescence analysis. The final compact samples were excited with a tuneable Ti-sapphire ring laser (0.4 cm−1 linewidth) in the 770–920 nm spectral range. The emitted light was analysed with a single grating monochromator (focal length 0.25 m), detected by an extended IR Hamamatsu H10330A-75 photomultiplier, and amplified by a standard lock-in technique. Luminescence decay curves were obtained by exciting the samples with a Ti-sapphire laser pumped by a pulsed frequency-doubled Nd:YAG laser (9 ns pulse width), and detecting the emission with a Hamamatsu H10330A-75 photomultiplier. All measurements were performed at room temperature.Optical characterisation of the glass-ceramics was performed by luminescence measurements on samples of 0.9Nd3+–5LaF3:95SiO2, 0.9Nd3+–10LaF3:90SiO2 and 0.9Nd3+–20LaF3–80SiO2 powders treated for 6 h at 450 °C and then pressed at 1000 MP for 3 min. The steady-state emission spectra and luminescence decay curves were collected as described above.
Results and discussion
Characterisation of LaF3 and Nd3+:LaF3 nanoparticle suspensions
Fig. 1a shows the variation of zeta potential as a function of pH for the LaF3–0.04 M nanoparticle suspension from an initial pH of 7, corresponding with a zeta potential of −22 mV. The addition of HNO3 modified the zeta potential to more positive values, from −22 mV (pH 7) to 10 mV (pH 3), reaching the isoelectric point (ξ-potential = 0) at a pH of 4.8. On the other hand, the zeta potential remains practically constant, ∼−25 mV, with the addition of TMAH. The variation of the zeta potential provides information on the stability of the nanoparticle suspension, which greatly depends on the pH. A high zeta potential, in absolute values, often indicates more stable suspensions, hence a pH of 7 was selected to prepare the LaF3–0.04 M nanoparticle suspension, corresponding with a zeta potential of −22 mV.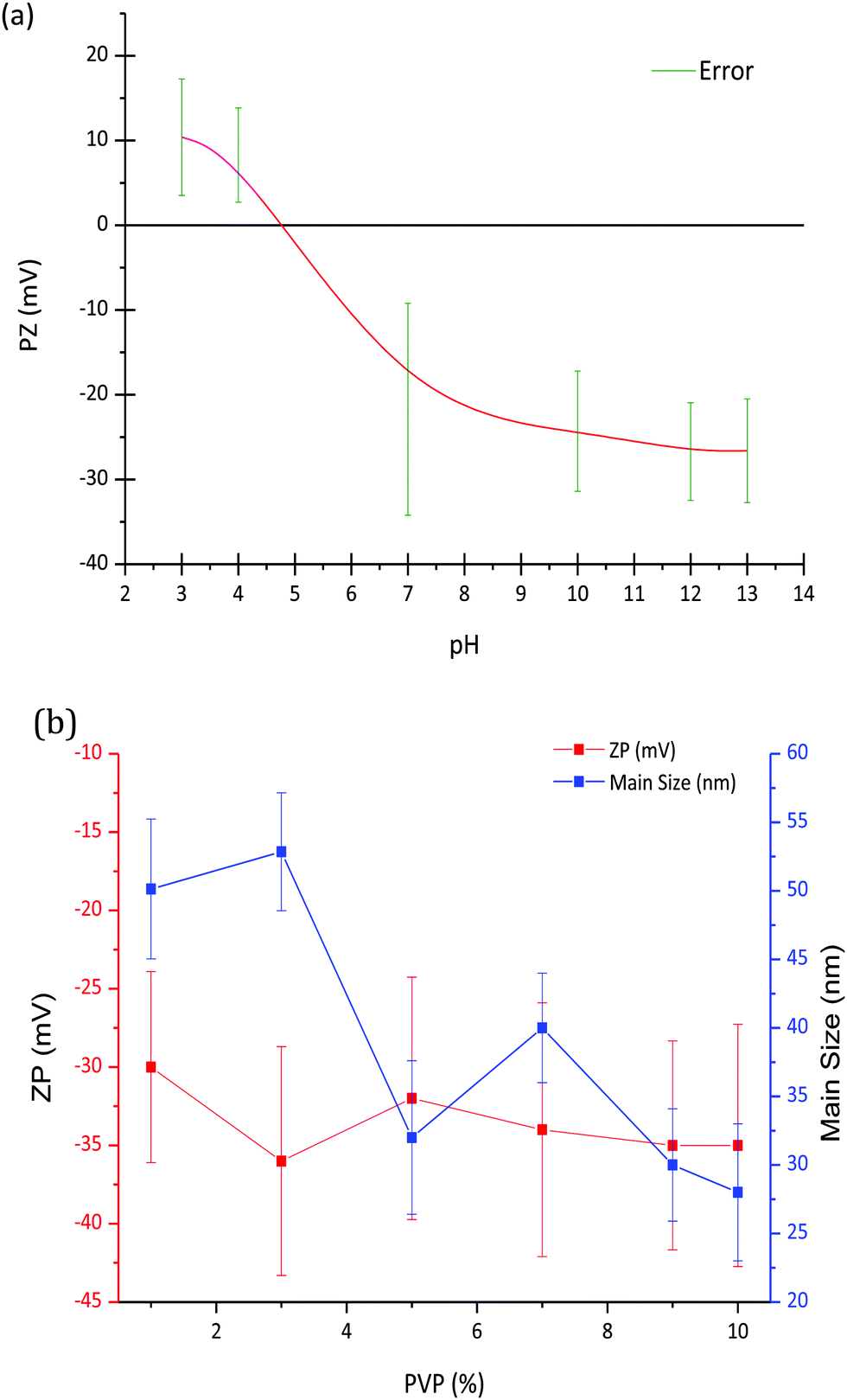 | ||
| Fig. 1 (a) Variation of the ZP (Mv) of LaF3–0.04 M nanoparticle suspensions as a function of pH and (b) variation of the zeta potential and particle size of LaF3–0.04 M nanoparticle suspensions at pH = 7 as a function of PVP wt%. | ||
Zeta potential and particle size of LaF3–0.04 M nanoparticle suspensions at pH 7 were measured as a function of PVP wt%, Fig. 1b. Although a slight decrease of the zeta potential is observed up to 3 wt% PVP, it remains constant, around −35 mV, with the increment of PVP. However, as observed in Fig. 1b, the hydrodynamic particle size of the nanoparticles decreases with the addition of PVP. In general, the addition of PVP to LaF3–0.04 M–PVP(X) nanoparticle suspensions produces a significant increase in its stability, related to the repulsive forces from the hydrophobic carbon chains of the PVP molecules.19 In comparison with previous works of G. Gorni et al.,16 starting from a two-step process with further precipitation of NP, larger particle sizes have been obtained using this new method. No limitation to crystal growth occurs because crystallization occurs in the liquid phase and the ion mobility facilitates the diffusion and growth of NPs. A maximum hydrodynamic size of 27 nm was obtained, which is an order of magnitude greater than that reported by G. Gorni.22 Concentrations of 0 and 10 wt% PVP were selected to prepare the corresponding suspensions that were later added to the SiO2 sol.
Fig. 2 shows the XRD patterns of LaF3 nano powders obtained after drying the LaF3–0.04 M and 0.9Nd3+–LaF3–0.25 M–PVP(10) suspensions at 80 °C. The XRD analysis revealed crystallisation of LaF3 with hexagonal symmetry (JCPDS 03-1013); no other crystalline phases were detected. The desired crystallisation was achieved for the LaF3–0.04 M suspension as well as for the doped and concentrated suspensions. This confirms that nanocrystal growth is affected neither by the incorporation of the rare-earth acetates nor by the dispersant. Moreover, the nanocrystals remain stable in size after the concentration of the suspensions from 0.04 to 0.25 M in the rotavapor.
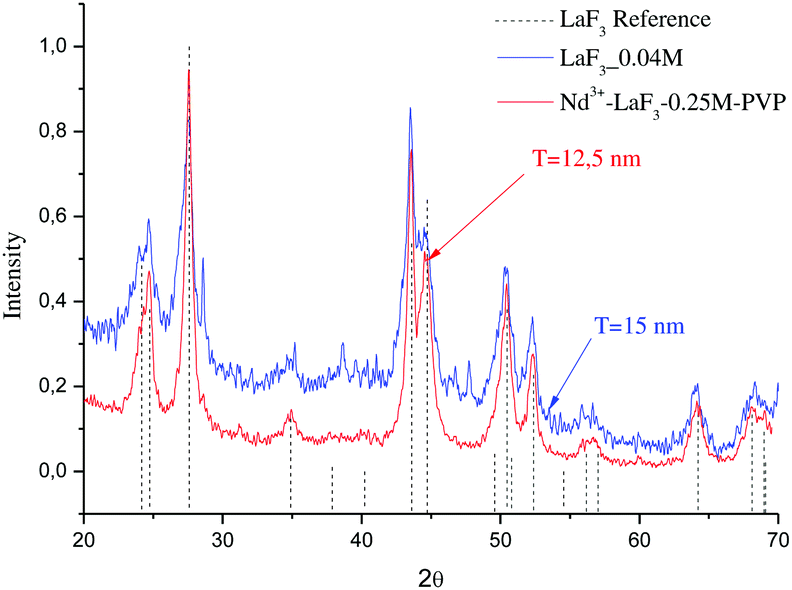 | ||
| Fig. 2 XRD patterns of LaF3–0.04 and Nd3+–LaF3–0.25 M–PVP(10) nanoparticle suspensions dried at 80 °C for 12 h. | ||
The particle size (T) of the powders determined using the Debye–Scherrer equation, was 12 and 15 nm respectively, much higher than particle sizes reported in the literature.20–22
0.9Nd3+–LaF3–0.04 M and 0.9Nd3+–LaF3–0.25 M–PVP(10) NP suspensions were dried at 80 °C and analysed by HRTEM to complete characterisation of the morphology. Fig. 3a shows the presence of aggregated LaF3 nanoparticles with a tubular form and average size, ∼18 nm. In the case of 0.9Nd3+–LaF3–0.25 M–PVP(10), the increase in concentration from 0.04 to 0.25 M produces greater aggregation of the LaF3 nanoparticles (Fig. 3b), although the morphology and the particle size remained constant. The crystal phase was analysed with the ImageJ® software (Fig. 3c), giving a planar distance (d spacing) of 0.325nm, which corresponds to the lattice distance between the (101) planes of the LaF3 hexagonal phase. A Fast Fourier Transform (FFT) also performed with ImageJ®, Fig. 3(d), shows the corresponding diffraction rings and white spots in the electron-diffraction pattern where (101) is identified at 2θ = 27.42°.
 | ||
| Fig. 3 HRTEM images of (a) Nd3+–LaF3–0.04 M powder and (b) Nd3+–LaF3–PVP(10)–0.25 M powder (c) single nanocrystal showing a lattice spacing of 0.325 nm for sample 0.9Nd3+–LaF3–0.25 M–PVP(10) (d) FFT pattern corresponding to (c) and (e) EDX analysis of the region. | ||
Fig. 3e shows the EDX analysis of the 0.9Nd3+–LaF3–0.25 M–PVP(10) sample corresponding to the area shown in Fig. 3c. The chemical analysis reveals the presence of F, La and Nd, providing clear evidence of Nd3+ incorporation in the nanoparticles.
Quantitative Rietveld refinement of LaF3 (not shown) was performed for 0.9Nd3+–LaF3–0.25 M–PVP(10) nanoparticles treated at 450 °C for 6 h. A crystallised fraction of 13 wt% was determined, below that expected and indicating that the LaF3 nanoparticles are highly amorphous. It should also be considered that a lower limit of detection occurs when the peaks become so broad due to the small particle size that they disappear into the background radiation. Although there is no exact limit to define when this occurs, particles of low size are likely to appear amorphous and special conditions such as a slow stem scan are required.
Characterisation of 0.9Nd3+–20LaF3:80SiO2 particulate sol
The stability of the sol with time was studied through the evolution of viscosity. Fig. 4 shows the variation of the viscosity with aging time of the 0.9Nd3+–20LaF3:80SiO2 particulate sol up to its gelification. The initial viscosity, measured two hours after the sol synthesis, was 4.1 mPa s. increasing to 4.2 mPa s. after 42 hours. During the following days, the viscosity was measured once per day achieving a value of 8.5 mPa s on the fifth day, before gelation occurred the following day. Although these values are high for depositing thin films, they are suitable for producing xerogels in a shorter time from stable sols.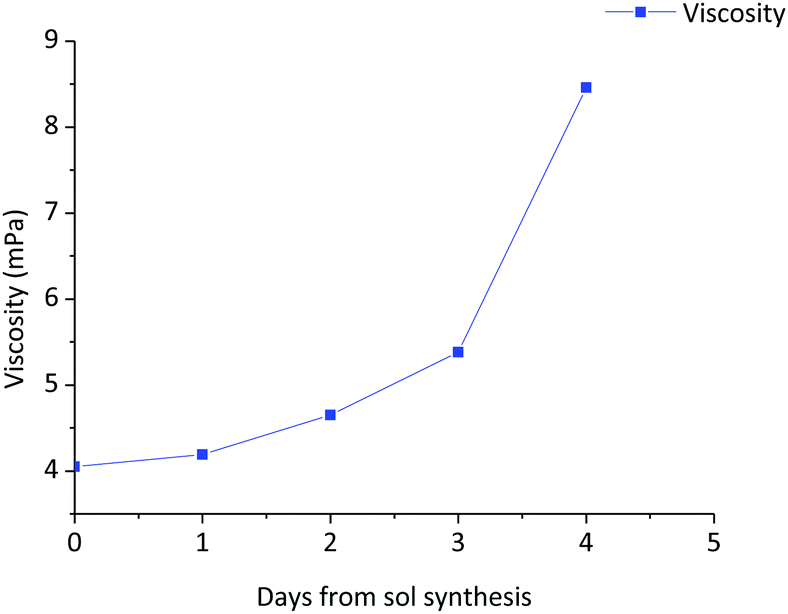 | ||
| Fig. 4 Variation of the viscosity of the 0.9Nd3+–80SiO2:20LaF3 particulate sol as a function of aging time up to the gelification point. | ||
Fig. 5 shows the DTA and TGA analysis for the 0.9Nd3+–20LaF3:80SiO2 xerogel measured in air. Weight loss occurs in four different steps. The first step, around 25–100 °C, is associated with an endothermic process at 76.7 °C, corresponding to the elimination of water and adsorbed ethanol, confined in large pores. The OH groups of the silica network generate hydrogen bridges, increasing the energy required to desorb the ethanol and water.23 The second step, between 100–200 °C, is usually assigned to the evaporation of solvents absorbed in small pores. The combustion of remaining organic material is responsible for the third step, occurring in the range 250–500 °C, along with final elimination of trapped solvents and water molecules in nanopores up to 500 °C.24 A sharp exothermic peak at 607 °C appears associated with the final weight loss event, corresponding to the crystallisation of LaOF, as confirmed by XRD (not shown).
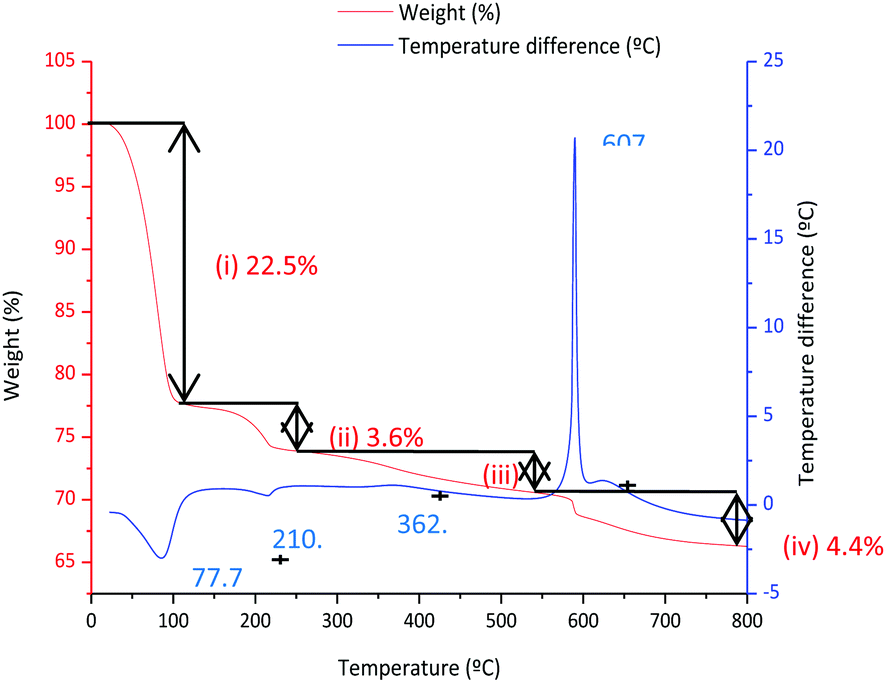 | ||
| Fig. 5 TGA and DTA analysis of the 80 °C–12 h dried 80SiO2:20LaF3 particulate sol. | ||
The structural characterisation of LaF3 nanoparticles incorporated in the 0.9Nd3+–20LaF3:80SiO2 particulate sol was analysed by HRTEM. Fig. 6a shows the HRTEM image of the 0.9Nd3+–20LaF3:80SiO2 sample treated at 450 °C for 6 h, showing well-dispersed nanoparticles. The average particle size is ∼16 nm according to the size distribution given in Fig. 6b. This particle size is consistent with that determined by XRD and HRTEM (Fig. 2 and 3). Furthermore, the LaF3 nanoparticles maintained their oval shape after incorporation into the silica sol. Fig. 6c shows a planar distance of 0.32 nm corresponding to the (101) plane for the nanocrystalline particles, similar to that of the 0.9Nd3+–LaF3 nanoparticles before their incorporation into the silica sol. Fig. 6d confirms the presence of Nd accompanying La and F, measured by STEM analysis.
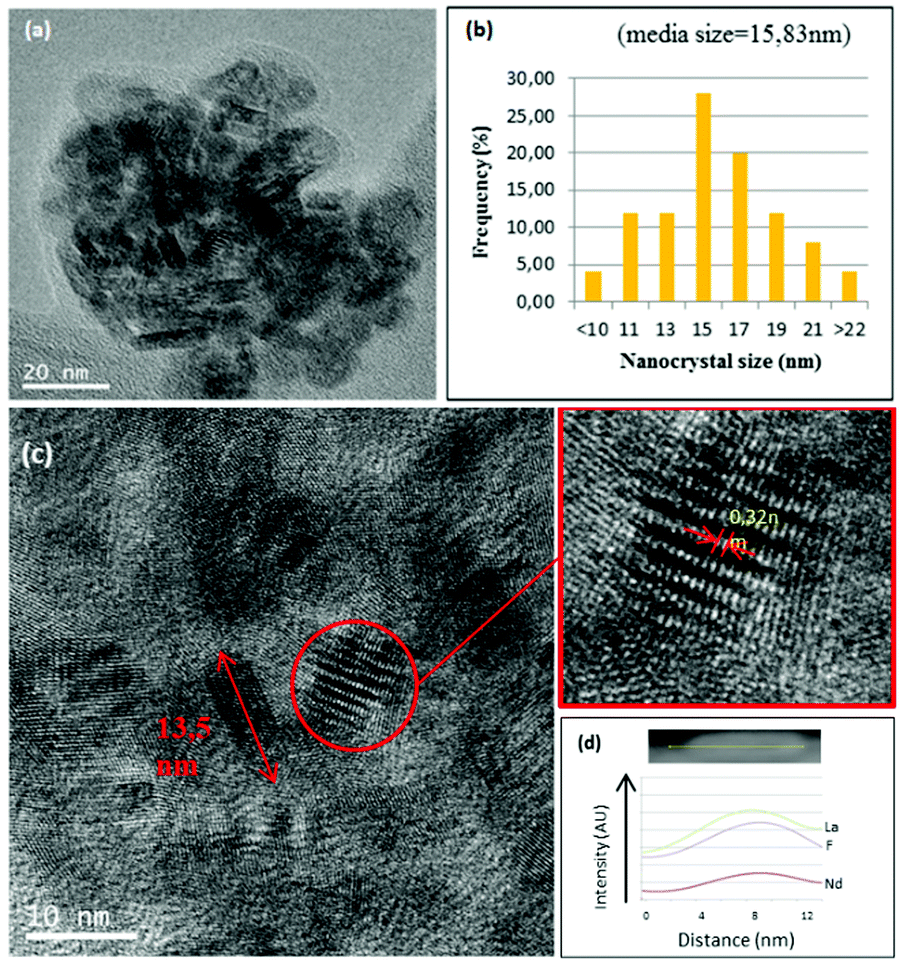 | ||
| Fig. 6 (a) HRTEM image of 0.9Nd3+–LaF3 nanoparticles in the 0.9Nd3+–80SiO2:20LaF3 sol. (b) Distribution of nanocrystal size of the nanoparticles in the sol. (e) EDX of the corresponding 0.9Nd3+–LaF3 nanoparticle and its scanning lines. | ||
It is confirmed that no new crystallisation process occurs, and that the shape and size of the LaF3 nanoparticles are not affected either by incorporation into the silica sol or by the heat treatment at 450 °C for 6 h. Incorporation in the silica matrix does not produce agglomerations or growth of the nanoparticles.
Quantitative Rietveld refinement of the 0.9Nd3+–20LaF3:80SiO2 glass-ceramic treated at 450 °C for 6 h provided a weight fraction of 5.2 wt% of crystalline phase (Fig. 7). Considering the low crystalline fraction obtained for the nanopowders of LaF3 (13%), it is likely that incorporation in the silica sol and further thermal treatment increases the average particle size and improves crystallisation of the NPs, leading to a higher relative crystalline fraction.
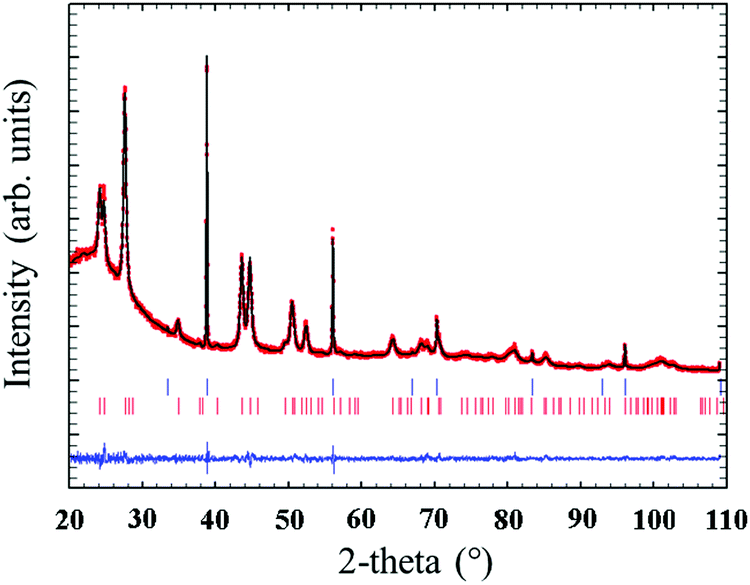 | ||
| Fig. 7 Observed (red circles), calculated (continuous black line) and difference (continuous blue line) X-ray diffraction profiles of 0.9Nd3+–80SiO2:20LaF3 particulate sol powders treated at 450 °C for 6 h with 5% NaF as internal standard. The Bragg peaks of NaF and LaF3 are indicated by top and bottom vertical bars, respectively. | ||
Optical characterisation of Nd3+:LaF3 nanoparticles before and after their incorporation in a silica matrix
The near-infrared emission spectra corresponding to the 4F3/2 → 4I11/2 laser transition were obtained for all samples at room temperature by exciting at 786 nm in the 4I9/2 → 4F5/2 absorption band. Fig. 8a shows the emission spectrum of the 0.9Nd3+–LaF3 nanoparticle suspension treated at 80 °C, together with the experimental decay of the 4F3/2 level. The emission spectrum shows similar spectral features to the Nd3+ emission of the LaF3 crystal. However, the experimental decay of the 4F3/2 level obtained under excitation at 786 nm with collection of luminescence at 1064 nm shows a different behaviour. The decay deviates from a single exponential function and the lifetime is unexpectedly short. The average lifetime.where I(t) represents the luminescence intensity at time t corrected for the background, is reduced to 61 μs.
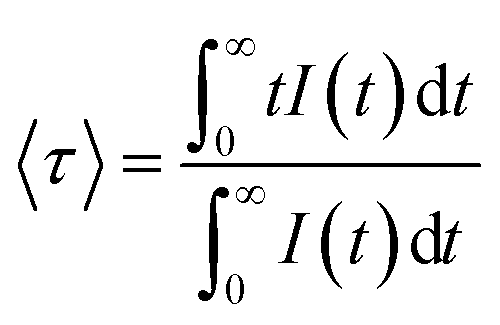
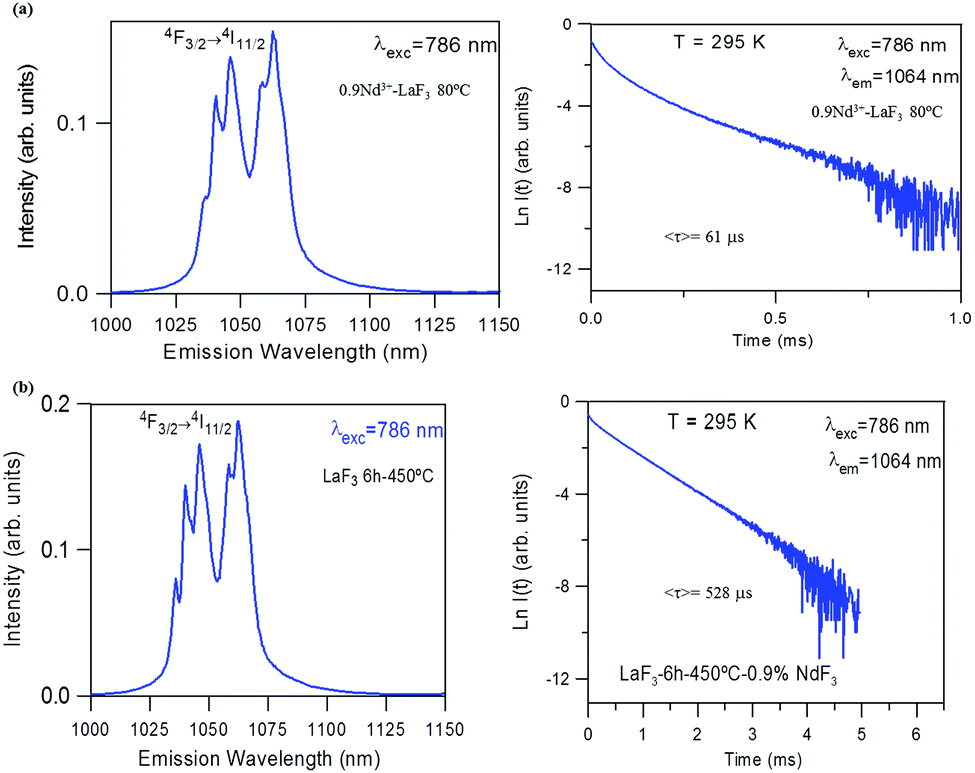 | ||
| Fig. 8 (a) Emission spectrum of 0.9Nd3+–LaF3 nanoparticles treated at 80 °C and corresponding semi-logarithmic plot of the experimental decay under excitation at 786 nm collecting the luminescence at 1064 nm. (b) Emission spectrum of 0.9Nd3+–LaF3 nanoparticles treated at 450 °C for 6 h obtained under excitation at 786 nm and corresponding semi-logarithmic plot of the experimental decay obtained after excitation at 786 nm collecting the luminescence at 1064 nm. | ||
On the other hand, the near-infrared emission spectrum of 0.9Nd3+–LaF3 nanoparticles treated at 450 °C for 6 h (Fig. 8b) shows a series of peaks with emissions of comparable intensity, around 1047 and 1064 nm. The spectral features correspond to the emission of Nd3+ ions in LaF3,25 which confirms the incorporation of the rare-earth ion in the LaF3 NPs. Moreover, the fluorescence decay curve of the 4F3/2 level obtained under excitation at 786 nm on collecting the luminescence at 1064 nm can be described to a good approximation by a single exponential function with a lifetime of 528 μs, which is close to the lifetime reported for Nd3+ in a LaF3 crystal (522 μs). These results unambiguously confirm the incorporation of Nd3+ ions in the LaF3 NPs.
The lifetime of NPs treated at 80 °C is nearly one order of magnitude shorter than that observed in the NPs heat treated for 6 h at 450 °C. This strong quenching of the lifetime could be related to the presence of aggregated LaF3 nanoparticles; however, this is not appreciated in the HRTM micrographs. The most probable explanation is that NPs treated at 80 °C still contain a high percentage of PVP, which is eliminated on treatment at 450 °C.
For the 0.9Nd3+–20LaF3:80SiO2 sample treated at 450 °C for 6 h, both the emission spectrum and the lifetime (Fig. 9) correspond to those obtained for LaF3 NPs with the same treatment. This result agrees with the structural characterisation, which indicates that LaF3 nanoparticles are not affected by their incorporation in the silica sol, maintaining their high efficiency as active phase.
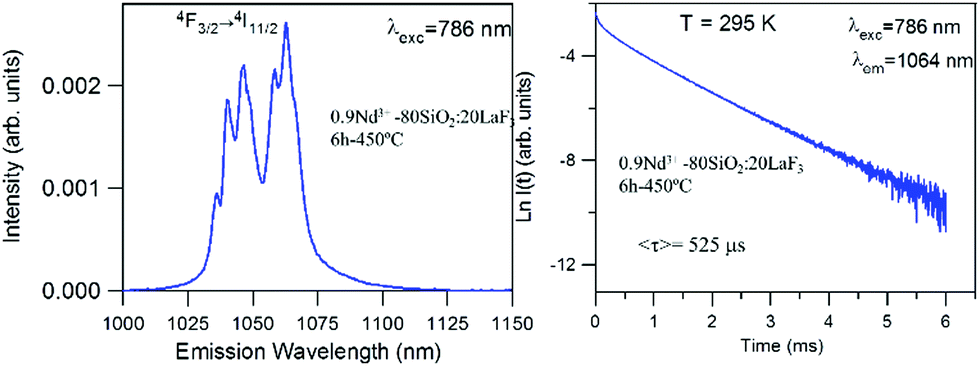 | ||
| Fig. 9 Emission spectrum of 450 °C–6 h 0.9Nd3+–80SiO2:20LaF3 sol and its corresponding semi-logarithmic plot of the experimental decay under excitation at 786 nm collecting the luminescence at 1064 nm. | ||
On the other hand, emission measurements performed in 0.9Nd3+–LaF3:SiO2 glass-ceramics with three different LaF3 concentrations (5, 10 and 20%), treated at 450 °C for 6 h, confirmed that the increase in the crystalline fraction results in a nearly linear increase of the emission intensity (Fig. 10).
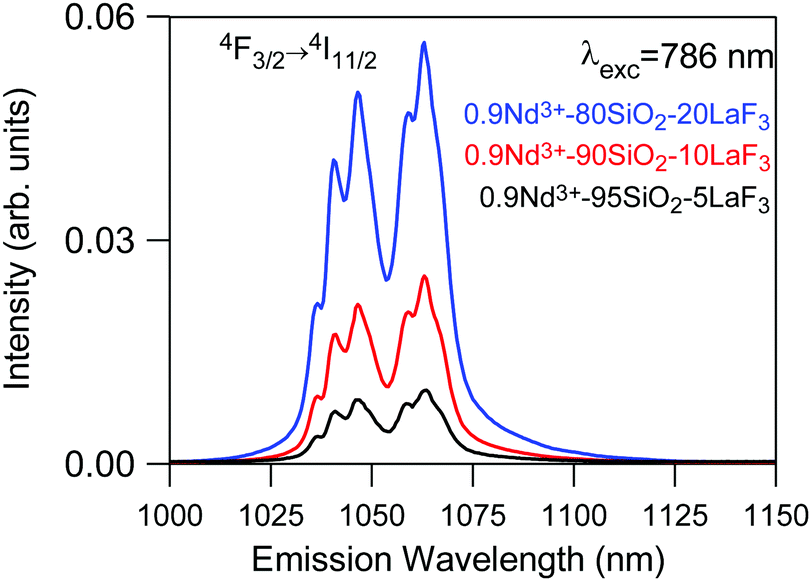 | ||
| Fig. 10 Emission spectrum of 450 °C–6 h 0.9Nd3+–SiO2:LaF3 GCs with three different crystalline fractions 5% (black), 10% (red) and 20% (blue) obtained under 786 nm excitation. | ||
This promising innovative process which produces optically active oxyfluoride glass-ceramics is under further investigation to produce optically efficient transparent glass-ceramics coatings with wide and diverse applications.
Conclusions
Stable Nd3+-doped LaF3 nanoparticle suspensions with particle size ∼ 16 nm, were obtained using a rather simple precipitation process. The incorporation of PVP as dispersant increases the stability of the LaF3 NP suspensions.The incorporation of Nd3+ ions into the LaF3 nanoparticles, as well as in SiO2–LaF3 powders, was confirmed by HRTEM, STEM and optical characterisation. The luminescence spectra of Nd3+-doped LaF3 nanoparticles treated at 450 °C for 6 h confirms the incorporation of Nd3+ ions in the nanoparticles and the complete elimination of PVP after suitable heat treatment. A lifetime of 528 μs was determined, similar to that of Nd3+ in pure LaF3 crystals.
Moreover, stable SiO2–LaF3 sols with and without Nd3+ were prepared by mixing the LaF3 NP suspension with a silica sol, up to a maximum molar ratio of 20% LaF3 NPs. To the best of our knowledge, this is the first time that Nd3+ NPs incorporated into a glass-ceramic has been produced by this route.
Rietveld refinement indicated that LaF3 NPs present a crystalline fraction of 13%, likely related to a high percentage of amorphous fraction. However, the relative crystallised fraction in the silica matrix with 20% of NP determined by the same method increases to ∼5%.
The Nd3+–LaF3 nanoparticles are not affected by their incorporation into the silica sol, maintaining their composition, shape and size. The luminescence response of 0.9Nd3+–20LaF3:80SiO2 glass-ceramic treated at 450 °C for 6 h confirms the efficiency of the active phase.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from MINECO under projects MAT2017-87035-C2-1-P/-2-P (AEI/FEDER, UE), Basque Country University GIU17/014 and Basque Government PIBA2018-24. This article is a part of the dissemination activities of the project FunGlass, which has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement no. 739566. The authors also acknowledge the support of the Transmission Electron Microscope (TEM) service of the Institute of Catalysis and Petrochemistry (CSIC).Notes and references
- G. Gorni, M. J. Pascual and A. Caballero, Effect of processing on the structure and properties of glass and glass-ceramics for photonic applications Gorni, Madrid, Instituto de Cerámica y Vidrio-Universidad Autonoma de Madrid, 2019 Search PubMed.
- S. D. Stookey, History of the Development of Pyroceram, Res. Management, 1958, 1(3), 155–163 Search PubMed.
- J. Deubener, M. Allix, M. J. Davis, A. Durán, T. Höche and T. Honma, et al., Update definition of glass-ceramics, J. Non-Cryst. Solids, 2018, 501, 3–10 CrossRef CAS.
- D. de Pablos-Martín and M. J. Pascual, Nanocrystallisation in oxyfluoride systems: mechanisms of crystallisation and photonic properties, Int. Mater. Rev., 2012, 57(3), 165–186 CrossRef.
- M. J. Dejneka, The luminescence ans structure of novel transparent oxyfluoride glass-ceramics, J. Non-Cryst. Solids, 1998, 239(1–3), 149–155 CrossRef CAS.
- Y. Wei and C. Xian Nian, Enhaced green upconversion in Tb3+–Yb3+-Co-doped oxyfluoride glass-ceramics containing LaF33 nanocrystals, J. Lumin., 2013, 137, 70–72 CrossRef CAS.
- V. D. Rodríguez, J. Del Castillo, A. C. Yanes, J. Méndez-Ramos, M. Torres and J. Peraza., Luminescence of Er3+ -doped nanostructured SiO2-LaF3 glass ceramics prepared by the sol–gel method, Opt. Mater., 2007, 29(11), 1557–1561 CrossRef.
- B. Singarapu, D. Galusek, A. Durán and M. J. Pascual, Glass-Ceramics processed by Spark Plasma Sintering (SPS) for optical applications, Appl. Sci., 2020, 10, 8 Search PubMed.
- Z. Shan, D. Chen, Y. Yu, P. Huang, H. Lin and Y. Wang, Luminescence in rare earth-doped transparent glass ceramics containing GdF3 nanocrystals for lighting applications, J. Mater. Sci., 2010, 45, 2775–2779 CrossRef CAS.
- L. Fu, H. Engqvist and W. Xia, Highly translucent and strong ZrO2–SiO2 nanocrystalline glass ceramic prepared by sol–gel method and spark plasma sintering with fine 3D microstructure for dental restoration, J. Eur. Ceram. Soc., 2017, 37(13), 4067–4081 CrossRef CAS.
- S. Tanabe, H. Hideaki, T. Hanada and N. Onodera, Fluorescence properties of Er3+ ions in glass ceramics containing LaF3 nanocrystals, Opt. Mater., 2002, 19, 3 CrossRef.
- A. Biswas, G. S. Maciel, C. S. Friend and P. N. Prasad, Upconversion properties of a transparent Er3+–Yb3+ co-doped LaF3–SiO2 glass ceramics prepared by sol-gel method, J. Non-Cryst. Solids, 2003, 316(2-3), 393–397 CrossRef CAS.
- S. Fujihara, C. Mochizuki and T. Kimura, Formation of LaF3 microcrystals in sol–gel silica, J. Non-Cryst. Solids, 1999, 244(2–3), 267–274 CrossRef CAS.
- M. Tada, S. Fujihara and T. Kimura, Sol–gel processing and characterization of alkaline earth and rare earth fluoride thin films, J. Mater. Res., 1999, 14(4), 1610–1616 CrossRef CAS.
- B. Szipikowska-Sroka, L. Zur, R. Czoik, T. Goryczka, A. S. Swinarew and M. Zadlo, et al., Long-lived emission from Eu3+:PbF2 nanocrystals distributed into sol–gel silica glass, J. Sol-Gel Sci. Technol., 2013, 68, 278–283 CrossRef.
- G. Gorni, J. J. Velázquez, G. C. Mather, A. Durán, G. Chen and M. Sundararajan, et al., Selective excitation in transparent oxyfluoride glass-ceramics doped with Nd3+, J. Eur. Ceram. Soc., 2017, 37(4), 1695–1706 CrossRef CAS.
- G. Gorni, M. J. Pascual, A. Caballero, J. J. Velázquez, J. Mosa and Y. Castro, et al., Crystallization mechanism in sol–gel oxyfluoride glass-ceramics, J. Non-Cryst. Solids, 2018, 201, 145–152 CrossRef.
- G. Gorni, J. J. Velázquez, J. Mosa, R. Balda, J. Fernandez and A. Durán, et al., Transparent Glass-cercamics produced by sol–gel: a suitable alternative for photonic materials, Materials, 2018, 11(212), 1–30 Search PubMed.
- F. Wang, Y. Zhang, X. Fan and M. Wang, Facile synthesis of water-soluble LaF3:Ln3+ nanocrystals, J. Mater. Chem., 2006, 16(11), 1031–1034 RSC.
- J. Rodríguez-Caravajal, Recent advances in magnetic structure determination by neutron powder diffraction, Phys. B, 1993, 192(1–2), 55–69 CrossRef.
- K. Koczkur, S. Mourdikoudis, L. Polavarapu and S. E. Skrabalak, Polyvinylpyrrolidone (PVP) in nanoparticle synthesis, R. Soc. Chem., 2015, 44, 17883–17905 CAS.
- G. Gorni, J. J. Velázquez, J. Mosa, G. Mather, A. Serrano and M. Vila, et al., Transparent Sol–Gel oxyfluoride glass-ceramics with gigh crystalline fraction and study of RE incorporation, Nanomaterials, 2019, 9, 530 CrossRef CAS.
- J. J. Velázquez, J. Mosa, G. Gorni, R. Balda, J. Fernández and L. Pascual, et al., Transparent SiO2–GdF3 sol–gel nano-glass ceramics for optical applications, J. Sol-Gel Sci. Technol., 2018, 89, 322–332 CrossRef.
- B. Szpikowska-Sroka, N. Pawlik, L. Zur, R. Czoik, T. Goryczka and W. Pisarski, Effect of fluoride ions on the optical properties of Eu3+:PbF2 nanocrystals embedded into sol–gel host materials, Mater. Chem. Phys., 2016, 174, 138–142 CrossRef CAS.
- D. Aguilar, L. C. Torres-Gonzalez, L. Torres-Martinez, T. Lopez and A. Quintana, Study of the Crystallization of ZrO2 in the Sol–Gel System: ZrO2–SiO2, J. Solid State Chem., 2001, 158(2), 349–357 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |