
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVanadyl phthalocyanines on graphene/SiC(0001): toward a hybrid architecture for molecular spin qubits†
I.
Cimatti
 a,
L.
Bondì
a,
G.
Serrano
*b,
L.
Malavolti
cd,
B.
Cortigiani
a,
E.
Velez-Fort
e,
D.
Betto
e,
A.
Ouerghi
f,
N. B.
Brookes
e,
S.
Loth
cdg,
M.
Mannini
*a,
F.
Totti
*a and
R.
Sessoli
a
a,
L.
Bondì
a,
G.
Serrano
*b,
L.
Malavolti
cd,
B.
Cortigiani
a,
E.
Velez-Fort
e,
D.
Betto
e,
A.
Ouerghi
f,
N. B.
Brookes
e,
S.
Loth
cdg,
M.
Mannini
*a,
F.
Totti
*a and
R.
Sessoli
a
aDepartment of Chemistry “Ugo Schiff” and INSTM RU of Florence, University of Florence, Via della Lastruccia 3-13, 50019, Sesto Fiorentino, FI, Italy. E-mail: matteo.mannini@unifi.it; federico.totti@unifi.it
bDepartment of Industrial Engineering (DIEF) and INSTM RU of Florence, University of Florence, Via di S. Marta, 3, 50139, Firenze, FI, Italy. E-mail: giulia.serrano@unifi.it
cMax Planck Institute for the Structure and Dynamics of Matter, Luruper Chaussee 149, 22761 Hamburg, Germany
dMax-Planck Institute for Solid State Research, Heisenbergstrasse 1, 70569 Stuttgart, Germany
eEuropean Synchrotron Radiation Facility, 71, avenue des Martyrs CS 40220, 38043 Grenoble Cedex 9, France
fCentre de Nanosciences et de Nanotechnologies (C2N), CNRS, Univ Paris Sud, Université Paris-Saclay, 91120 Palaiseau, France
gInstitute for Functional Matter and Quantum Technologies, University of Stuttgart, Pfaffenwaldring 57, 70569 Stuttgart, Germany
First published on 7th June 2019
Abstract
Vanadyl phthalocyanine (VOPc) contains a highly coherent spin S = ½, which is of interest for applications in quantum information. Preservation of long coherence times upon deposition on conductive materials is crucial for use of single-spin in devices. Here, we report a detailed investigation of the structural, electronic and magnetic properties of a hybrid architecture constituted by a monolayer film of VOPc molecules deposited on graphene/SiC(0001). Graphene (Gr) is a two-dimensional conductor with exceptional chemical stability, a property which we exploited here to preserve the spin of VOPc. Low temperature-scanning tunneling microscopy supported by density functional theory (DFT) simulations revealed that VOPc molecules were adsorbed intact on the Gr/SiC(0001) surface in a planar geometry assuming a unique configuration in which the vanadyl group is projected out toward the vacuum, different to that found commonly on other conductive surfaces. Furthermore, X-ray photoelectron spectroscopy and UV-photoelectron spectroscopy (flanked theoretically by DFT) showed that VOPc interact weakly with the Gr/SiC(0001) substrate to preserve its electronic configuration with the unpaired electron located on the V ion. These findings were confirmed by X-ray magnetic circular dichroism, revealing that the S = ½ character of the VOPc assembly on Gr/SiC(0001) was preserved, in agreement with the theoretical prediction. Hence, molecules could be adsorbed and used as qubits on substrates of technological importance, such as graphene. This new hybrid architecture could be employed for local investigation of static and dynamic spin properties and as molecular qubits for spintronic applications.
New conceptsPreserving the functional properties of molecular building blocks in hybrid materials for molecular spintronics is a key issue and it is strongly related to their adsorption geometry and specific interactions with the surface. Among molecular systems, vanadyl phthalocyanines (VOPc) have garnered great interest as potential candidates for qubits. The asymmetric geometry of the vanadyl group is crucial because it induces non-equivalent adsorption configurations on surfaces, some of which may strongly alter molecular properties with detrimental effects for their use in real devices. Here, we explore VOPc deposition on graphene (Gr) grown on SiC(0001). The latter is a highly interesting substrate for technological applications thanks to its structural and electronic properties and because of its predominant nuclear spin-free isotopic composition. We show that Gr/SiC(0001) induces the optimal orientation of VOPc molecules required for technological purposes: a flat lying configuration with the vanadyl group exposed to the vacuum. We demonstrate unambiguously that VOPc molecules are not altered by the substrate and maintain their electric dipole and S = ½ properties. We show that VOPc deposited on Gr/SiC(0001) is a hybrid system that can be used to address single spin and for investigation of the coherent spin-transport phenomena in quantum-information applications. |
Introduction
Quantum technologies have attracted great interest in the last few decades for the development of new devices which, by exploiting quantum coherent processes, can reach record calculation efficiency or advanced sensing properties.1 In particular, quantum information technology is based on the use of “qubits” as building blocks for logic processes. Specific molecular systems, such as metal phthalocyanines (Pc), have shown suitable behavior as a S = ½ qubit, with a long spin coherence time.2,3 In its crystalline form, vanadyl phthalocyanine (VOPc) has shown record room-temperature quantum coherence among molecular spin systems, as well as coherent spin-photon coupling when inserted in a superconducting resonator.4 These features, together with its high thermal stability and processability, makes this compound one of the most attractive molecular systems for spintronic applications and quantum computation purposes.3 VOPc is a nonplanar metal-phthalocyanine in which the vanadyl ion (VO2+), coordinated by four nitrogen donor atoms of the dianionic phthalocyaninato ligand, points away from the plane of the ligand (see scheme in Fig. 1a). VOPc presents a distorted square-pyramidal coordination with the V atom slightly above the basal plane (0.575(1) Å) and double-bonded with the oxo-ligand, which results in a V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O distance of 1.580(3) Å.5 VOPc deposition has been performed by thermal sublimation of the pristine powder on different substrates.6–11 The molecular orientation of the monolayer deposit is dependent on the specific surface reactivity, and it shows two preferential orientations of the vanadyl group: pointing away (O-up) or toward (O-down) the substrate.6,7,10 The latter configuration has been found to be favored for specific adsorption sites on gold10 or in presence of strong chemical interactions with the substrate, as in the case of Si(111).6 Both adsorption geometries have been found for Pb(111), showing very different interactions of the molecular spin with the Cooper pairs of the superconducting substrate.12 A strong interaction of the vanadyl group with the surface can also alter the electronic molecular structure, which undergoes partial charge transfer, as observed for VOPc monolayer deposited on Ni(111).7 In the latter case, use of graphene as an intermediate layer between the molecules and metal surface prevents such alterations.7
O distance of 1.580(3) Å.5 VOPc deposition has been performed by thermal sublimation of the pristine powder on different substrates.6–11 The molecular orientation of the monolayer deposit is dependent on the specific surface reactivity, and it shows two preferential orientations of the vanadyl group: pointing away (O-up) or toward (O-down) the substrate.6,7,10 The latter configuration has been found to be favored for specific adsorption sites on gold10 or in presence of strong chemical interactions with the substrate, as in the case of Si(111).6 Both adsorption geometries have been found for Pb(111), showing very different interactions of the molecular spin with the Cooper pairs of the superconducting substrate.12 A strong interaction of the vanadyl group with the surface can also alter the electronic molecular structure, which undergoes partial charge transfer, as observed for VOPc monolayer deposited on Ni(111).7 In the latter case, use of graphene as an intermediate layer between the molecules and metal surface prevents such alterations.7
 | ||
Fig. 1 (a) Molecular structure of VOPc. Color code: white, hydrogen; brown, carbon; blue, nitrogen; light blue, vanadium; red, oxygen. The isodensity of the surface (ψ2 = 0.06 e Bhor−3), of the SOMO for VOPc@Gr is represented in yellow and light blue. (b) STM image of the Gr/SiC(0001) surface (10 × 10 nm2, −2 V, filled states, 70 pA, 1.8 K); the hexagonal atomic periodicity of the graphene layer is highlighted in the inset (2.5 × 2.5 nm2); (c and d) STM images of the VOPc submonolayer deposited on Gr/SiC(0001) at two magnifications. (c) STM image 3.2 × 3.2 nm2 (2 V, filled states, 11 pA, 1.7 K). The structure of a VOPc molecule is superimposed on the bottom-right corner as a visual guide. (d) STM image 10 × 10 nm2 (2 V, filled states, 11 pA, 1.7 K). White arrows represent the unit-cell vectors of the molecular lattice (![[a with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0061_20d1.gif) and and ![[b with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0062_20d1.gif) ). ). | ||
Here, we report detailed characterization of a VOPc monolayer deposited on graphene by combining theory with experimental results. Graphene is a two-dimensional conductor made of a single sheet of C atoms. It has garnered considerable attention because of its remarkable structural and electronic properties13,14 (which have been exploited recently for spintronic applications15) and has been used as a templating substrate for deposition of magnetic molecules.7,16 Here, we used the Gr/SiC(0001) substrate, which features large graphene areas with high structural and electronic quality,17,18 and the capability to preserve the magnetic properties of single-molecule magnetic systems directly deposited upon it.16 The inert and templating properties featured by Gr/SiC(0001)16 were exploited to deposit VOPc molecules and assess the potentialities of such a hybrid system for molecular qubit applications. We show that VOPc molecules, unlike on most conductive surfaces, retain their structural and magnetic properties after adsorption and self-assembly on the Gr/SiC(0001) substrate, maintaining a unique configuration with the VO group pointing away from the surface and featuring weak interactions with the substrate. The structural and chemical properties were studied by X-ray photoelectron spectroscopy (XPS) and low temperature-scanning tunneling microscopy (LT-STM). Density functional theory (DFT) calculations were used to investigate adsorption of VOPc molecules on the graphene framework. The electronic properties of the molecular assembly were studied at local and non-local scales by scanning tunneling spectroscopy (STS) and ultraviolet photoelectron spectroscopy (UPS), respectively. By comparing the calculated density of states (DOS) with the UPS results, we derived the electronic structure of the VOPc layer on graphene, showing that the electronic structure of the VOPc was preserved and that the V ion retained one unpaired electron. Study of the magnetic properties of the VOPc assembly on Gr/SiC(0001) by X-ray magnetic circular dichroism (XMCD) measurements confirmed retention of the S = ½ character on this substrate.
Results and discussion
The Gr/SiC(0001) substrate was studied by STM at low temperature before and after VOPc deposition. Fig. 1b shows the clean surface at atomic-scale resolution, with two hexagonal periodicities superimposed over the whole area. The larger periodicity correspond to the (6 × 6) cell of the 6√3 reconstruction of the SiC(0001) surface (highlighted in the image by a dashed line). The smaller periodicity is attributed to the continuous graphene layer lying on top having a periodicity of 2.4 ± 0.5 Å, in agreement with the (1 × 1) graphene lattice grown on SiC(0001).19–21 The inset in Fig. 1b shows a close-up view of the area where the graphene lattice is better resolved (the red hexagon highlights one cell). The image was filtered using a fast Fourier transform method. The presence of a different number of graphene layers can be excluded because of the strong intensity of the (6 × 6) with respect to the graphene pattern.19–22Fig. 1c and d show the surface after deposition of a submonolayer of VOPc molecules. In the left of Fig. 1c, the clean Gr/SiC(0001) substrate remains visible. VOPc molecules are visible on the right side. Each VOPc molecule appears as a small central bright spot surrounded by an inner set of eight lobes and an outer set of four lobes that have a four-fold rotation symmetry. In the bottom-right corner of Fig. 1c, the stick structure of the VOPc molecule is superimposed over the STM image as a visual guide. The observed structure corresponds to that reported for a VOPc monolayer deposited on highly oriented pyrolytic graphite (HOPG).11,23 The molecules lie flat on the surface (i.e., with the phthalocyanine plane parallel to the substrate) and the oxygen of the vanadyl group points away from the surface into a vacuum. The STM image in Fig. 1d shows that the molecules self-assemble laterally in a closely packed layer on top of graphene to form large “molecular islands”. Molecules are packed in a square-like lattice with unit cell vectors of equal magnitude, || = || (Fig. 1d), of ∼14 Å, in good agreement with reports11,23,24 and compatible with the lateral dimensions of VOPc (14.85 Å).5 This result provides evidence that the VOPc layer was incommensurate with the graphene lattice underneath. This is a strong experimental indication of the weak interaction between VOPc and the surface. Hence, intermolecular forces dominate the ordering process instead of molecule–substrate forces. The height of the VOPc layer, evaluated by STM characterization, is 3.5 ± 0.2 Å, i.e., about half of the height reported for a bilayer of VOPc molecules on HOPG24 and, therefore, compatible with the presence of a single VOPc layer. The dI/dV (V) spectra (see ESI,† Fig. S1.2) recorded on a single molecule within the molecular layer are consistent with the spectra recorded for VOPc with an O-up configuration on HOPG, which differed from those recorded on the O-down configuration on HOPG.23
Deposition of VOPc molecules on the Gr/SiC(0001) surface was studied by XPS to evaluate the effect of adsorption on the electronic structure of the molecular film. The XPS spectra of a submonolayer of VOPc are shown in Fig. 2 (bottom panels) and compared with those obtained on a thick film (∼3 monolayers) grown on Au(111) (upper panels). The XPS region between 510 eV and 535 eV show the core levels of V2p and O1s (Fig. 2a, bottom). The V2p3/2 contribution (red) is found at 516.4 eV, which is 7.3 eV away from the V2p1/2 component (blue), in agreement with the expected spin–orbit splitting.7 A broad peak centered at 520.0 eV (green), attributed to a satellite contribution of the V2p, has been included according to earlier reports.7 The O1s peak (orange) is found at 531.0 eV, 14.6 eV away from the V2p3/2 component. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio between the areas of the V2p peak and O1s peak expected from molecular stoichiometry is respected within the error limit of the technique. The N1s region (Fig. 2b, bottom) presents a main peak at 398.9 eV (red) and a small shake-up satellite. The C1s component of the phthalocyanine is expected to be hidden by a graphene peak (Fig. S1.1 in ESI†) and, therefore, has not been taken into account for the analysis.6 This dataset is in good agreement with previous reports of XPS measurements on VOPc thin films6,7 and with the data analysis of the thick film shown in the upper panels of Fig. 2. In particular, absence of additional contribution to the vanadium and nitrogen peaks indicates no significant alterations of the molecular electronic structure after deposition on graphene, thereby indicating weak interaction with the substrate.6,7 In earlier reports, the appearance of additional components in V1s and N1s regions was attributed to charge transfer or stronger surface–molecule interaction due to the coexistence of O-up and O-down configurations.6,7,12
:1 ratio between the areas of the V2p peak and O1s peak expected from molecular stoichiometry is respected within the error limit of the technique. The N1s region (Fig. 2b, bottom) presents a main peak at 398.9 eV (red) and a small shake-up satellite. The C1s component of the phthalocyanine is expected to be hidden by a graphene peak (Fig. S1.1 in ESI†) and, therefore, has not been taken into account for the analysis.6 This dataset is in good agreement with previous reports of XPS measurements on VOPc thin films6,7 and with the data analysis of the thick film shown in the upper panels of Fig. 2. In particular, absence of additional contribution to the vanadium and nitrogen peaks indicates no significant alterations of the molecular electronic structure after deposition on graphene, thereby indicating weak interaction with the substrate.6,7 In earlier reports, the appearance of additional components in V1s and N1s regions was attributed to charge transfer or stronger surface–molecule interaction due to the coexistence of O-up and O-down configurations.6,7,12
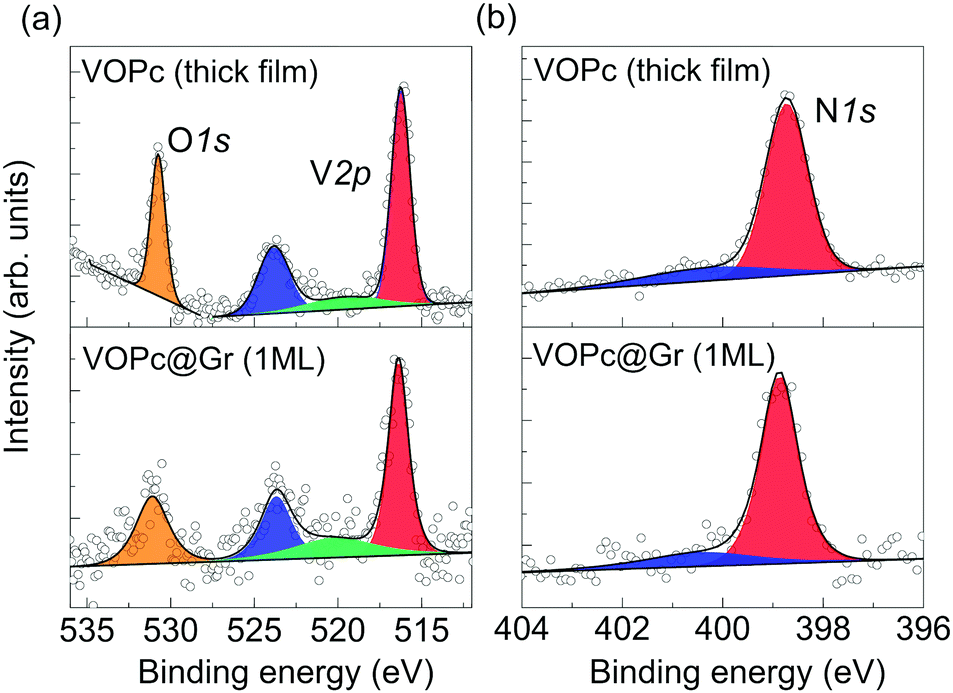 | ||
| Fig. 2 XPS spectra of a VOPc thick film (top panels) and in the first monolayer on Gr/SiC(0001) (bottom panels): core-level spectra of O1s and V2p (a) and N1s (b). The region has been fitted using a linear background. (a) Red and blue components correspond to the V2p3/2 and V2p1/2 signals, a satellite peak is shown in green; the O1s region is fitted with a single component (orange). (b) N1s results in a main component (red) and a satellite peak (blue) at higher binding energy. | ||
Our experiments, therefore, indicates that VOPc molecules adsorb intact on Gr/SiC(0001) lying flat with an O-up configuration, as suggested by STM characterization. This configuration was observed predominately on HOPG,11,23 even if both O-up and O-down configurations were found to be stable and reversibly switchable upon STM manipulation.23 Reports of VOPc films on Gr/Ni(111) have not shown topographical evidence of the orientation of the vanadyl group, and an O-up configuration was inferred only from the comparison with the HOPG case.7
To shed light on the adsorption preferences of VOPc@Gr, we carried out DFT ab initio calculations. The potential energy surface described by different possible adsorption sites was evaluated by carrying out several geometrical optimizations. Four sites were chosen for the O-up configuration (Fig. 3a) and were named after the position of the V atom and the angle (α) defined as the angle between the N2VN4 axis of the molecule and the C–C bond of the graphene framework (dashed lines). They were V positioned: (a) at the center of a graphene aromatic ring with the N2VN4 axis aligned with the C–C bond of graphene (α = 0°) (ring-centered, RC); (ii) on top of a C atom of graphene with α = 0° (atom-centered, AC); (iii) at the center of a C–C bond of graphene with α = 0° (CC0); (iv) in the middle of a C–C bond of graphene with α = 45° (CC45). In addition, we considered two O-down geometries, RC and AC, because they are the two electron-poor sites and, therefore, interact efficiently with the electron-rich oxygen atom of the vanadyl group. The computed energy values for the different configurations are reported in Table 1 and compared with the isolated VOPc molecule. The adsorption process is very exothermic, with ∼43 kcal mol−1 recorded for the most stable adsorption site for O-up (CC0) and ∼30 kcal mol−1 for the corresponding O-down configuration (RC). These results support the STM observations that suggested the O-up to be the preferred adsorption configuration. The potential energy surface for adsorption is very flat, showing variation in adsorption energy of <2 kcal mol−1 (Table 1), which is in agreement with the observed high molecular mobility observed (see below). This result is not surprising considering that, for both VOPc geometries, the energy landscape is dominated by extended π–π interactions between the phthalocyanine and graphene, and that no covalent bond is formed during adsorption. As such, the geometrical structure of the VOPc do not show significant differences among the various adsorption sites (see the root mean square deviation (RMSD) values in Table 1). Conversely, in the adsorption process, a sizable geometrical rearrangement (RMSD = 2.316 Å), corresponding to flattening of the phthalocyanine structure, is observed to maximize the π–π interactions with graphene. Such distortion, however, do not alter the electronic structure of the vanadyl group, as indicated by the identical computed value of the V(IV) spin density for the VOPc in different geometries. Aware also of the important role of interface dipoles in “tailoring” the alignment of interfacial energy levels in organic molecules,25,26 we computed the electrical properties of VOPc after adsorption in the most energetically stable configuration (O-up). We show that the electric dipole of the molecule remain aligned to the vanadyl group with negligible variations in magnitude or direction (∼4.5% and ∼2.0%, respectively, Table 2). The computed values are in good agreement with the experimental data of μ0 = 2.7(4) D.27
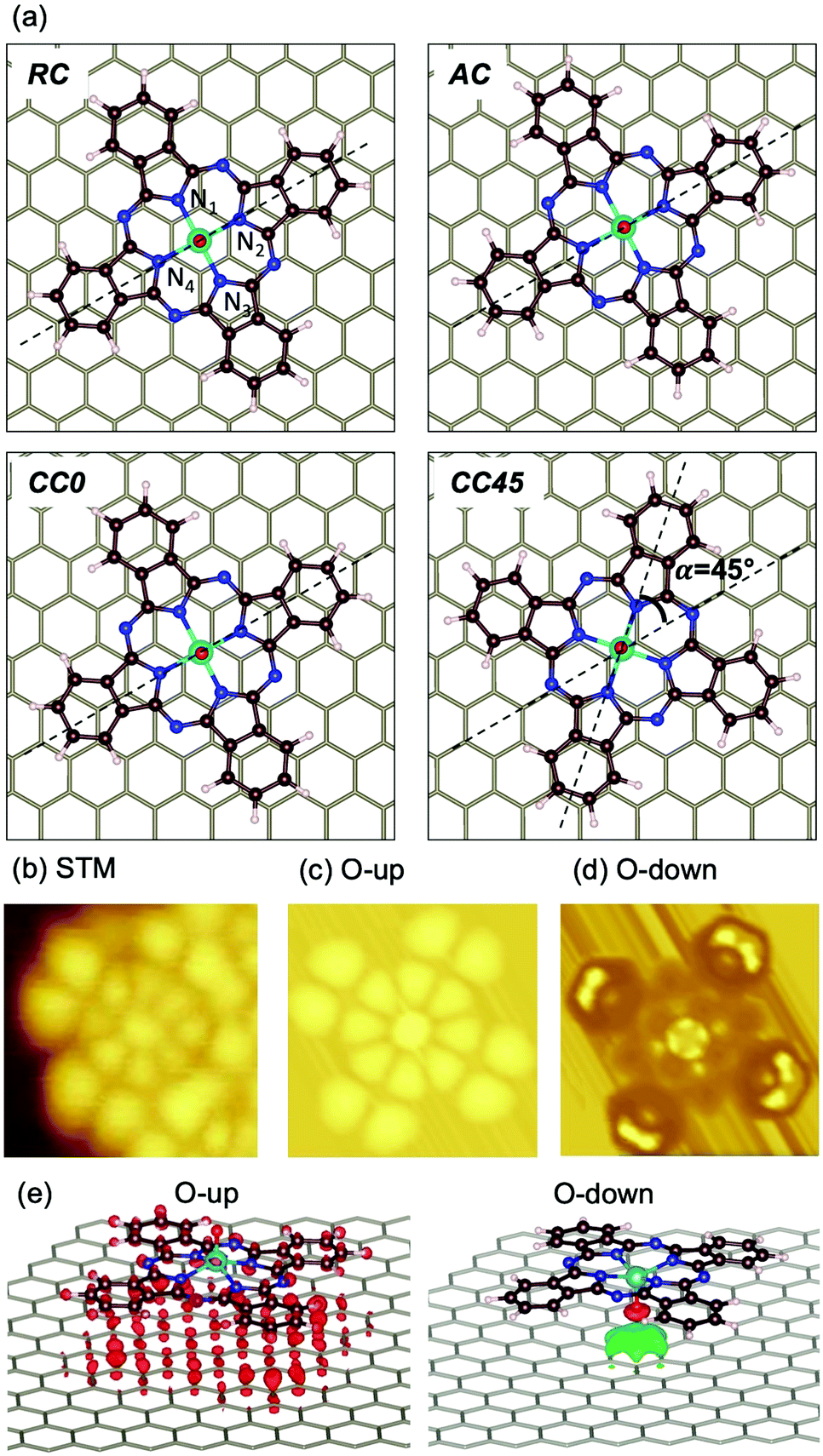 | ||
| Fig. 3 (a) Configuration of VOPc on graphene considered for theoretical calculations. From top-left to bottom-right: ring-centered (RC), atom-centered (AC), C–C α = 0° (CC0) and CC/α = 45° (CC45). Nitrogen atoms coordinated with vanadium atoms are numbered; the N2VN4 axis and the direction of graphene C–C bonds are indicated by dashed lines. (b) Experimental STM image of VOPc on Gr/SiC(0001) measured at Vb = 2 V (filled states); (c) simulated STM image of VOPc@Gr at a RC site having an O-up configuration and (d) O-down configuration (V = −1.7 V, filled states). (e) Difference electronic density plots for O-up (left) and O-down (right) configurations. | ||
| Energya (kcal mol−1) | RMSDc (Å) | Spind (e−) | |
|---|---|---|---|
| a Adsorption energies of each configuration with respect to the most stable site, CC0, for the O-up geometry. b In brackets is the energy loss as a result of the flattening of the phthalocyanine ring in order to maximize interaction with the surface with respect to its isolated structure (VOPc) (the Pc has been constrained in a flattened configuration while all the other degrees of freedom are left free to relax). c Structural changes are parametrized by the RMSD with respect to the most stable configuration (CC0). d Value computed from the Mulliken spin population analysis. | |||
| VOPc | +42.88 (−0.63)b | 2.316 | 1.19 |
| O-Up | |||
| CC0 | 0 | 0 | 1.19 |
| RC | +1.41 | 0.010 | 1.19 |
| AC | +1.41 | 0.007 | 1.19 |
| CC45 | +0.44 | 0.006 | 1.19 |
| O-Down | |||
| RC | +13.34 | ||
| AC | +13.96 | ||
| μ x | μ y | μ z | |μ| | |
|---|---|---|---|---|
| VOPc | 0.002 | −0.02 | −2.47 | 2.47 |
| O-Up | ||||
| CC0 | 0.090 | −0.10 | −2.46 | 2.46 |
| RC | 0.010 | −0.02 | −2.36 | 2.36 |
| AC | 0.09 | −0.04 | −2.39 | 2.40 |
| CC45 | 0.008 | −0.11 | −2.47 | 2.47 |
To clarify the results of high-resolution STM on VOPc deposited on graphene (Fig. 3b), we reported the simulated images for the RC O-up and O-down configurations (Fig. 3c and d, respectively). No significant changes are observed among the computed STM images for the different geometries (RC, AC, CC0 and CC45) in the O-up configuration. Conversely, the computed STM images for O-up and O-down configurations present different characteristic features, which are in agreement with previous theoretical studies.23 The O-up configuration shows strong matching with the main features of experimental STM images. Filled states correspond to negative biases for the theoretical calculations and to positive biases for the experimental image of Fig. 3b. A deviation of ∼300(±1) mV between experimental and simulated image bias voltages is observed. Such an energy mismatch is dependent upon the non-local exchange (i.e., inclusion of exact Hartree–Fock exchange or Hubbard corrections) and correlations present in the function used.23,28
To gain a more detailed picture of the electronic properties of the molecular assembly, we compared the calculated DOS and projected density of states (pDOS) with experimental UPS measurements carried out on the Gr/SiC(0001) substrate before and after adsorption of a VOPc submonolayer (Fig. 4). The He(II) UPS spectrum of the clean graphene (black curve in Fig. 4a) is characterized by a main broad peak with a maximum centered at −8.4 eV and less intense features around −6.2, −4.5 and −2.5 eV (black asterisks), in agreement with previous reports for graphene/Ni(111).7 The red curve in Fig. 4a represents the UPS spectrum of the surface after adsorption of the VOPc submonolayer. The persistence of the peak around −8.4 eV is observed, along with the appearance of pronounced features at higher energies (marked with red asterisks), in particular at −6.2 eV, −3.3 eV and close to the Fermi level. Such peaks are ascribed to the molecular orbitals of VOPc, in agreement with the literature,7,29 as confirmed further by the UPS spectrum of a thick film of VOPc (∼3.5 monolayers) deposited on Au(111) reported in the ESI† (Fig. S1.3).
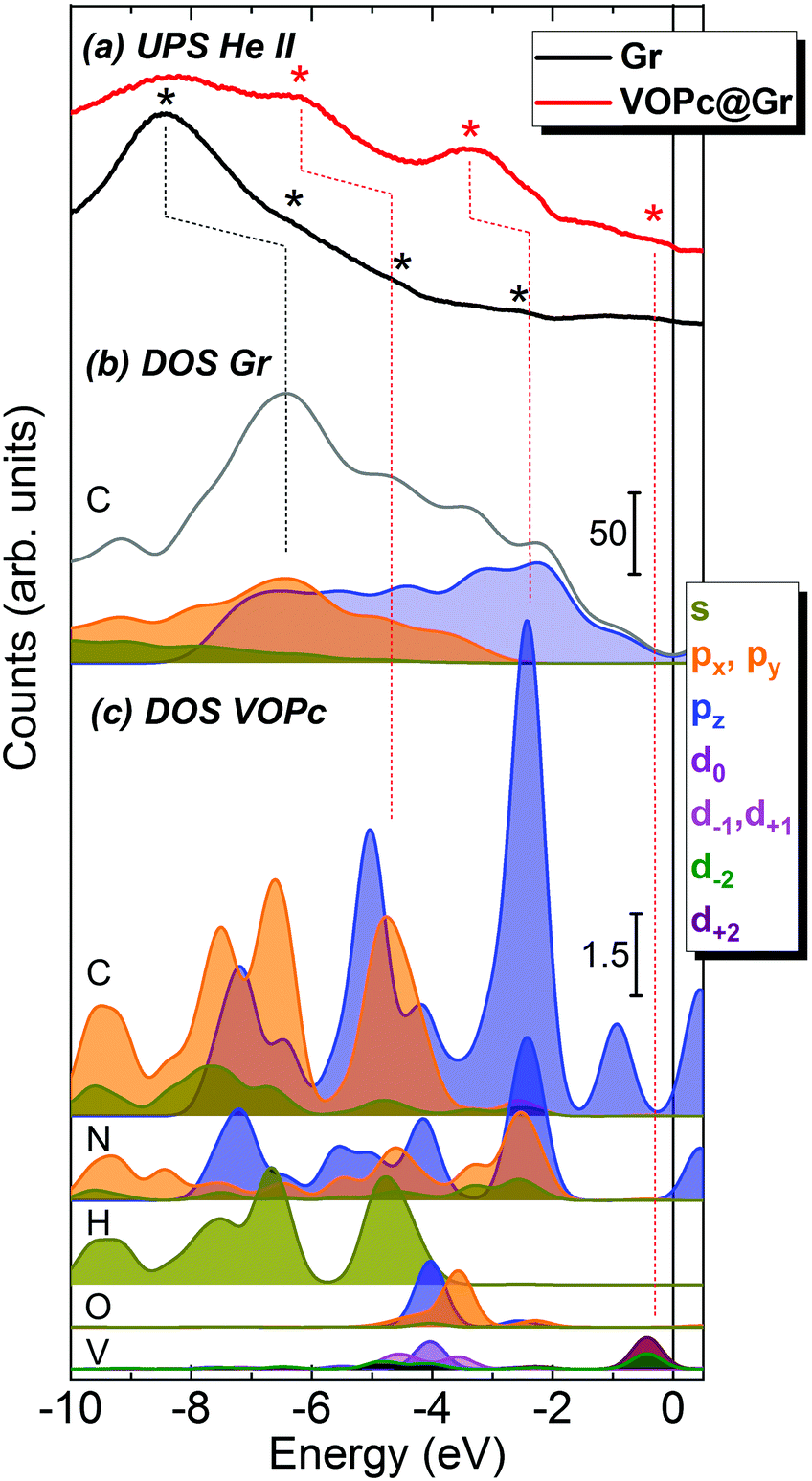 | ||
| Fig. 4 (a) UPS spectra of VOPc@Gr (red) and clean Gr/SiC(0001) (black). Because of the reduced signal intensity after molecular deposition, the intensity scale of the VOPc@Gr spectrum has been multiplied by a factor of 3 with respect to the clean graphene for clarity. Asterisks indicate the main peaks in each UPS spectrum. (b) DOS and pDOS of graphene: the gray line represents the total DOS of graphene whereas colored filled lines show the pDOS contribution of the carbon atoms in graphene. (c) pDOS for each atom of the VOPc molecule in the O-up configuration. The contribution of graphene is not taken into account so that all the pDOS intensity of the VOPc atoms (panel c) are significantly lower in intensity than the one for graphene (panel b), as shown by the scale bars. Dotted lines are superimposed on the figure as a guide for the peak attribution. The color codes of the pDOS are indicated in the legend and are the same for all panels. Equivalent orbital contribution (e.g., px and py) resulted in doubled intensities in the final contribution. | ||
The calculated total DOS of clean graphene (Fig. 4b, gray curve) correctly reproduces the shape of the experimental UPS spectrum for graphene (Fig. 4a, black curve). The pDOS of the C atoms of graphene are presented in Fig. 4b as filled curves. In the calculated spectra, a progressive blue-shift is observed moving from the Fermi level to the core regions. For the most prominent peak at −8.4 eV, the energy shift is ∼2.0 eV (a black dotted line is shown as a visual guide). However, this discrepancy could be accounted for by an expansion of the DOS energy scale by a factor of 1.3 eV, in agreement with previous reports.30
To disentangle the features of VOPc molecules to the UPS spectrum of VOPc@Gr (red curve, Fig. 4a), the pDOS for each atom constituting the VOPc molecule in the O-up configuration was calculated and reported in Fig. 4c excluding the contributions of graphene orbitals. Indeed, a direct comparison between the computed total DOS of VOPc@Gr and the bare graphene do not provide significant indications due to the low molecular coverage (i.e., a single VOPc molecule) necessary to keep the computational cost tolerable. Therefore, the calculated contribution of the molecule would be overwhelmed by the graphene contribution. The UPS signals of graphene and VOPc@Gr (Fig. 4a) are dominated by the p orbitals of the different elements in the valence region, whereas the s orbitals (green curves) are located at lower energies. The in-plane components (degenerate px,y orbitals) of C atoms (orange curves) of graphene and VOPc are the main contributions to the peaks at −8.4 eV and at −6.2 eV in the UPS spectra. The pz orbitals (blue curve), which are responsible for the π interactions, enrich both of the above-mentioned peaks, and their contribution extend to the spectral region closer to the Fermi level. In addition, the UPS feature at −6.2 eV and the peak around −3.3 eV for VOPc@Gr (red curve, Fig. 4a) originate by the p orbitals of N atoms (dotted red lines are superimposed as a visual guide). Importantly, a significant contribution in the region close to the Fermi level for VOPc@Gr UPS is provided by the d orbitals (d−2, d+2 components) of the V atom. As such, the semi-occupied molecular orbital (SOMO) possesses a strong d orbital character and the unpaired electron is located in the dxy orbital of the V ion, as expected by ligand-field considerations (Fig. 1a). Further insights on the nature of the interaction between the VOPc molecule and graphene are given by the difference densities plots (see Methods) reported in Fig. 3e. A net increase of electronic density in Pc and graphene π systems is evidenced for the O-up configuration (as expected for a direct interaction between two π systems). For the O-down configuration, we observed a decrease in the electronic density of charge at the O atom, whereas an increase was observed for the π system of the six graphene carbon atoms directly interacting with the VO group and for the vanadium dxy orbital, too. A through overlap direct communication channel was, therefore, evidenced for both configurations.
An experimental confirmation of the order of molecular deposition and persistence of localization of the unpaired electron on the molecule was observed using synchrotron light. A VOPc@Gr sample was prepared in situ at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France (as reported in the Method section). X-ray absorption spectroscopy (XAS) signal was measured between 505 eV and 535 eV to observe the L2,3 edges (L3 at 513–520 eV and L2 at 522–527 eV) of V and K edge (at 530 eV) of O. Initially, absorption spectra were recorded with linear vertical (σV) and horizontal (σH) polarization of the incoming X-rays to obtain the X-ray natural linear dichroism (XNLD) signal (σV–σH), which provides information about molecular ordering on surfaces.31,32 The XNLD spectrum (ESI,† Fig. S1.4) showed a strong dichroic signal at the L2,3 edges of V, thereby confirming the high ordering of the molecular layer in agreement with STM results. A similar shape of the XNLD line was found for a VOPc layer on Pb(111).12 Subsequently, XAS was performed using circular left (σ+) and right (σ−) polarizations (Fig. 5) for different X-ray incidence angles (θ) with respect to the normal sample surface. The XMCD signal was obtained as the difference between right and left circular polarizations (XMCD = (σ− − σ+))33 and is reported as percent dichroism (% XMCD), calculated by normalization with respect to the L3 edge-jump of the isotropic spectrum, (σ+ + σ−)/2. Measurements were taken at 7 K using a magnetic field of 9 T aligned parallel to the beam. The XAS spectra reported in Fig. 5 show the typical line shape of VOPc thin films.6,7,9,34 At 517 eV, the XMCD has a strong dichroic signal at normal incidence (θ = 0°, green line in Fig. 5) and at grazing incidence (θ = 45°, ESI,† Fig. S1.5), fully consistent with data reported in the literature.9,12 The XMCD of the VOPc monolayer was compared with that recorded on a dropcast of VOPc on Au/mica (black line in Fig. 5). The dropcast contains a large number of molecules (see Methods) and could, therefore, be used as a “fingerprint” of the VOPc molecules at the L2,3 edges of V. The shape of the XMCD line of the VOPc monolayer on graphene is in good agreement with the dropcast one, except for small differences attributed to the ordered configuration of the molecules deposited on graphene.31,32 At θ = 0°, the %XMCD is −44% and −40% for the monolayer deposited on graphene and the dropcast, respectively. The XMCD data confirm the persistence of localized spin on the V atom after molecular adsorption on the Gr/SiC(0001) surface. Therefore, VOPc molecules maintain their magnetic properties, in agreement with the theoretical calculation reported above.
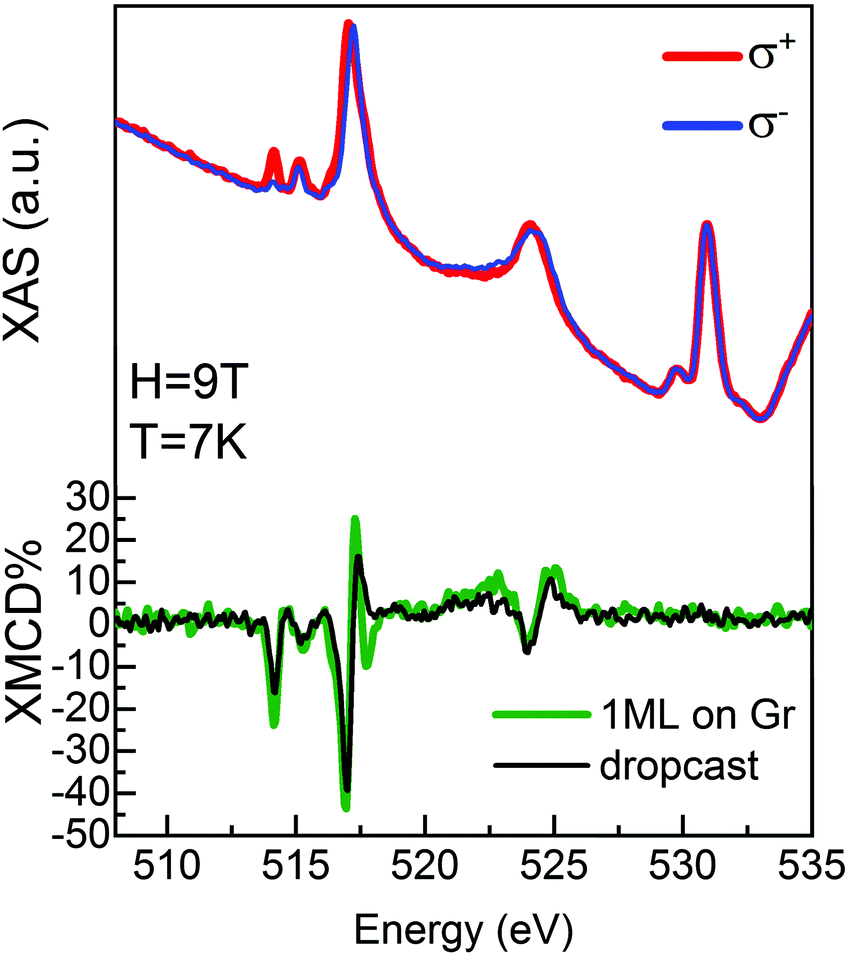 | ||
| Fig. 5 XAS and XMCD spectra for a VOPc monolayer deposited on Gr/SiC(0001) recorded at 9 T and 7 K for normal incidence of the X-ray beam with respect to the surface plane. Blue and red lines show the XAS spectra measured with left and right circular polarized X-rays, respectively, on the VOPc monolayer on Gr/SiC(0001), whereas the XMCD signal is shown in green. The XMCD spectrum of a thick dropcast film of VOPc on Au/mica is superimposed for comparison (black line). | ||
Conclusions
VOPc molecules were adsorbed intact on a Gr/SiC(0001) surface in a flat-lying orientation with the vanadyl group pointing out from the surface. Combined UPS and computed DOS analyses showed that the electronic structure of the VOPc monolayer was dominated by the p orbitals of C and N atoms at low energies, whereas the d orbitals of V contributed mainly to the Fermi-level region. Experimental magnetic characterization indicated that the S = ½ character of the molecule was preserved upon adsorption on graphene, thereby opening up the prospects of using VOPc deposited on graphene for spintronic applications. This concept was corroborated by DFT calculations, which also confirmed the preservation of the electric dipole of the molecule upon deposition. Our results demonstrated that a submonolayer of VOPc molecules maintained unaltered structural, electronic and magnetic properties and selectively assumed an O-up configuration on the Gr/SiC(0001) surface, unlike that seen with metal substrates. The Gr/SiC substrate employed was constituted exclusively by elements shown to have superior capabilities for retention of a long coherence time of spin impurities, thanks also to their predominant nuclear spin-free isotopic composition. Because the vanadyl group that carries the spin is projected toward the vacuum, the VOPc@Gr/SiC system could be used for local investigation of the spin properties and dynamics of a molecular qubit with S = ½ by scanning-probe methods.35,36Methods
Experimental methods
Graphene was grown on silicon carbide to obtain large-area and high-quality epitaxial graphene (Gr/SiC(0001)). Preparation and characterization of the substrate has been reported elsewhere.16–18 After insertion in ultra-high vacuum (UHV), the substrate was annealed at 770 K for 30 min and substrate cleanliness was checked by XPS and STM. The XPS spectrum of the C1s core level of the clean Gr/SiC(0001) substrate, measured before deposition, confirmed the expected chemical structure (Fig. S1.1 in ESI†). This showed three contributions: SiC (at 283.4 eV, red line), graphene (at 284.3 eV, blue line) and the buffer layer (at 285.0 eV, green line), in agreement with the literature.16–18,22VOPc powders (dye content >90%) used for deposition were purchased from Sigma-Aldrich and purified before deposition by prolonged thermal treatments using a resistively heated quartz crucible in UHV. Molecular sublimation was achieved at 615 K (P ∼ 5 × 10−7 mbar). The temperature was monitored using a K-thermocouple buried into the molecular powder. A quartz crystal microbalance was used to monitor the growth of molecular films. The same quartz crucible and deposition parameters were used for all experiments.
Optimization of the deposition conditions and spectroscopic characterization (UPS and XPS) were carried out at the University of Florence. Deposition and coverage of molecules was estimated using a variable-temperature STM system (Omicron) operated at 30 K. UPS spectra were recorded using the He(II) line (40.8 eV) from a non-monochromatized gas discharge lamp (22-101; VG Scientific) and a multichannel detector electron analyser (model SPECS Phoibos 150 1DLD) using a pass energy of 10 eV and entrance and exit slits of 3 × 20 mm. A fixed bias of −30 V was applied to the sample to ensure detection of all photoelectrons. Spectra were measured in normal emission and calibrated at the Fermi energy. XPS data were acquired using a micro-focused monochromatic Al K-alpha radiation source (model SPECS XR-MS Focus 600) and the same electron analyser used for UPS measurements. A pass energy of 40 eV was used to ensure appropriate resolution. The spectra were measured in normal emission with the X-ray source mounted at 54.44° with respect to the analyzer. The binding energy scale was calibrated using the C1s peak of the SiC contribution: 283.4 eV.22 Spectra were deconvoluted using CasaXPS software introducing mixed Gaussian and Lorentzian contributions for each component. The background was fitted using the Shirley or linear method.
LT-STM measurements were carried out at the Max Planck Institute (Hamburg, Germany) using a Unisoku USM 1300 3He STM setup connected with a UHV platform for sample preparation. The STM system was equipped with a 3He pumped cryostat, and measurements were carried out between 0.5 K and 1.8 K. The bias voltage could be applied to the tip or to the sample side. Both configurations were used for experiments, so we specified whether filled or empty states were imaged.
XAS and XMCD measurements were made at the ID32 beamline of ESRF. Samples were prepared in UHV exploiting the preparation chamber of the beamline connected to the cryostat used for measurements. Absorption spectra were acquired using the total-electron-yield method. As a reference sample, a bulk deposit of VOPc on gold on mica was prepared by drop-casting a 1 mM CH2Cl2 solution of VOPc and measuring using the same setup.
Theoretical methods
We employed CP2K37 and ORCA38 packages for the DFT studies. For CP2K, the electronic structure and nuclear forces were calculated at the GGA DFT level of theory (revPBE)39 with periodic boundary conditions (PBC) applying Gaussian and plane wave (GPW) methods. The electronic density obtained by the Kohn–Sham orbitals on a GTO basis set was mapped to an auxiliary plane wave basis set.40–42 The molecular optimized DZVP-MOLOPT-GTH basis sets were used for all atoms (except for V, where the DZVP-MOLOPT-SR-GTH basis set was applied), with an energy cutoff of 300 Ry for plane wave expansion.43 Tests on corrections for dispersive forces were performed using the Grimme D3 model44 or rVV10 model.45,46 The computational setup was then validated on geometrical and electronic properties reproducing X-ray and UPS data for crystalline VOPc (see Paragraph 2.1 in ESI,† for details) following the computational protocol used elsewhere.43–45 In our tests, the revPBE + rVV10 functional performed better and was used throughout the calculations for isolated VOPc and VOPc@Gr.To study the adsorption process of VOPc on graphene, we selected a model constituted by a single VOPc molecule adsorbed on a single sheet of graphene characterized by a hexagonal cell of 12-condensed rings edge (32.11 Å × 32.11 Å × 40.00 Å) instead of considering the whole substrate bulk structure of Gr/SiC(0001). Such an approximation is supported by the presence of the buffer layer on the SiC(0001) surface, which makes the graphene layer virtually insulated (no direct bonds) from the inner substrate (detailed description reported in Paragraph 2.2 of ESI†). The cell parameters used for the VOPc@Gr system were also used for the isolated single molecule (VOPc) and graphene. Each geometry was fully optimized with convergence criteria of 10−6 Hartree for the wavefunction (SCF) and at 10−3 Hartree Bohr−1 for atomic forces. The cutoff energy for the plain-wave auxiliary basis set was set to 300 Ry.
STM images were simulated on the optimized VOPc@Gr geometries according to the Tersoff–Hamann approximation47 as implemented in CP2K. The computed bias ranged from −4.0 eV to −0.5 eV for O-up and O-down structures. The approximation of using a single molecule to obtain STM images as representative of the monolayer was justified by the rigidity of the geometry and, therefore, by considering side interactions to be negligible. Moreover, such an approach has been validated in recent studies for the same molecule on different surfaces47,48 or more complex molecules.12,49,50 Differential density plots were evaluated by subtracting the electron densities computed singularly on the geometries of the extrapolated VOPc molecule and the related graphene from the electron density of whole VOPc@Gr.
To estimate the adsorption energy, the total DFT energies of optimized graphene and VOPc refer to the energy minima found for VOPc@Gr in different scenarios. CP2K-optimized structures where the VO group was aligned along the z direction and the V ion was at the origin, were used for single point energy calculations performed with the ORCA package to calculate the electric dipole (with respect to the origin). The graphene sheet had to be resized in a five-condensed rings edge hexagonal shape (detailed description reported in Paragraph 2.3 of ESI†) to reduce the computational cost. For the calculations performed using the ORCA code, the def2-TZVPP basis set was used for all elements,51 along with the def2/J auxiliary basis52 set to take advantage of the RI-J approximation;38 the revPBE39 functional and VV1045,46 correction on dispersive forces were employed.
DOS and pDOS were computed with the CP2K package. To model the broadening of the experimental UPS spectrum, DOS curves were calculated using Gaussian functions with a full-width half-maximum of 0.6 eV. The d orbitals were expressed in the spherical harmonics as d−2, d−1, d0, d1 and d2, where the subscript number is the m quantum number. In a Cartesian reference system, the dxy, dyz, dz2, dxz and dx2−y2 orbitals were expressed as d−2 − d2, d−1 + d1, d0, d−1 − d1, and d−2 + d2, respectively.
Author contributions
I. C. and L. B. contributed equally to this work. G. S., L. B. and F. T. initially wrote the manuscript and all the authors contributed. G. S. coordinated the manuscript drafting. I. C. and L. M. carried out the LT-STM and STS experiments. I. C. and B. C. undertook the XPS and UPS measurements. G. S., I. C., E. V. F., L. M., M. M. and D. B. carried out the synchrotron experiments. E. V. F. and A. O. fabricated the graphene substrate. L. B. and F. T. did the theoretical calculations. M. M., F. T., S. L., N. B. B. and R. S. supervised the work. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Italian MIUR through PRIN 2015 HYFSRT project, from European QuantERA through the SUMO project, and from Fondazione CR Firenze is acknowledged. The research leading to these results received funding from the European Research Council (ERC-2014-StG-633818-dasQ). The experiments were carried out at the ID32 beamline of the European Synchrotron Radiation Facility synchrotron, Grenoble, France. We acknowledge the COST action for funding the Short-Term Scientific Mission (COST-STSM-CA15128-34550).References
- A. Acín, I. Bloch, H. Buhrman, T. Calarco, C. Eichler, J. Eisert, D. Esteve, N. Gisin, S. J. Glaser and F. Jelezko, et al. , New J. Phys., 2018, 20, 080201 CrossRef.
- M. Warner, S. Din, I. S. Tupitsyn, G. W. Morley, A. M. Stoneham, J. A. Gardener, Z. Wu, A. J. Fisher, S. Heutz and C. W. M. Kay, et al. , Nature, 2013, 503, 504 CrossRef CAS PubMed.
- M. Atzori, L. Tesi, E. Morra, M. Chiesa, L. Sorace and R. Sessoli, J. Am. Chem. Soc., 2016, 138, 2154–2157 CrossRef CAS PubMed.
- C. Bonizzoni, A. Ghirri, M. Atzori, L. Sorace, R. Sessoli and M. Affronte, Sci. Rep., 2017, 7, 13096 CrossRef CAS PubMed.
- R. F. Ziolo, C. H. Griffiths and J. M. Troup, J. Chem. Soc., Dalton Trans., 1980, 2300 RSC.
- K. Eguchi, Y. Takagi, T. Nakagawa and T. Yokoyama, J. Phys. Chem. C, 2013, 117, 22843–22851 CrossRef CAS.
- H. Adler, M. Paszkiewicz, J. Uihlein, M. Polek, R. Ovsyannikov, T. V. Basova, T. Chassé and H. Peisert, J. Phys. Chem. C, 2015, 119, 8755–8762 CrossRef CAS.
- D. A. Duncan, W. Unterberger, K. A. Hogan, T. J. Lerotholi, C. L. A. Lamont and D. P. Woodruff, Surf. Sci., 2010, 604, 47–53 CrossRef CAS.
- K. Eguchi, Y. Takagi, T. Nakagawa and T. Yokoyama, J. Phys. Chem. C, 2014, 118, 17633–17637 CrossRef CAS.
- T. Niu, C. Zhou, J. Zhang, S. Zhong, H. Cheng and W. Chen, J. Phys. Chem. C, 2012, 116, 11565–11569 CrossRef CAS.
- T. Niu, J. Zhang and W. Chen, J. Phys. Chem. C, 2014, 118, 4151–4159 CrossRef CAS.
- L. Malavolti, M. Briganti, M. Hänze, G. Serrano, I. Cimatti, G. McMurtrie, E. Otero, P. Ohresser, F. Totti and M. Mannini, et al. , Nano Lett., 2018, 18, 7955–7961 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- K. S. Novoselov, Z. Jiang, Y. Zhang, S. V. Morozov, H. L. Stormer, U. Zeitler, J. C. Maan, G. S. Boebinger, P. Kim and A. K. Geim, Science, 2007, 315, 1379 CrossRef CAS PubMed.
- A. Candini, S. Klyatskaya, M. Ruben, W. Wernsdorfer and M. Affronte, Nano Lett., 2011, 11, 2634–2639 CrossRef CAS PubMed.
- G. Serrano, E. Velez-Fort, I. Cimatti, B. Cortigiani, L. Malavolti, D. Betto, A. Ouerghi, N. B. Brookes, M. Mannini and R. Sessoli, Nanoscale, 2018, 10, 2715–2720 RSC.
- D. Pierucci, H. Sediri, M. Hajlaoui, E. Velez-Fort, Y. J. Dappe, M. G. Silly, R. Belkhou, A. Shukla, F. Sirotti and N. Gogneau, et al. , Nano Res., 2015, 8, 1026–1037 CrossRef CAS.
- E. Pallecchi, F. Lafont, V. Cavaliere, F. Schopfer, D. Mailly, W. Poirier and A. Ouerghi, Sci. Rep., 2014, 4, 4558 CrossRef CAS PubMed.
- A. Ouerghi, M. G. Silly, M. Marangolo, C. Mathieu, M. Eddrief, M. Picher, F. Sirotti, S. El Moussaoui and R. Belkhou, ACS Nano, 2012, 6, 6075–6082 CrossRef CAS PubMed.
- P. Mallet, F. Varchon, C. Naud, L. Magaud, C. Berger and J.-Y. Veuillen, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 041403 CrossRef.
- P. Lauffer, K. V. Emtsev, R. Graupner, T. Seyller, L. Ley, S. A. Reshanov and H. B. Weber, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 155426 CrossRef.
- B. Lalmi, J. C. Girard, E. Pallecchi, M. Silly, C. David, S. Latil, F. Sirotti and A. Ouerghi, Sci. Rep., 2014, 4, 4066 CrossRef CAS PubMed.
- J. Zhang, Z. Wang, T. Niu, Z. Li and W. Chen, Appl. Phys. Lett., 2014, 104, 113506 CrossRef.
- W. Xie, J. Xu, J. An and K. Xue, J. Phys. Chem. C, 2010, 114, 19044–19047 CrossRef CAS.
- I. H. Campbell, S. Rubin, T. A. Zawodzinski, J. D. Kress, R. L. Martin, D. L. Smith, N. N. Barashkov and J. P. Ferraris, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, R14321–R14324 CrossRef CAS.
- H. Fukagawa, H. Yamane, S. Kera, K. K. Okudaira and N. Ueno, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 041302 CrossRef.
- M. L. Blumenfeld, M. P. Steele and O. L. A. Monti, J. Phys. Chem. Lett., 2010, 1, 145–148 CrossRef CAS.
- U. Becker and M. F. Hochella, Geochim. Cosmochim. Acta, 1996, 60, 2413–2426 CrossRef CAS.
- H. Fukagawa, H. Yamane, S. Kera, K. K. Okudaira and N. Ueno, J. Electron Spectrosc. Relat. Phenom., 2005, 144–147, 475–477 CrossRef CAS.
- N. Papageorgiou, Y. Ferro, E. Salomon, A. Allouche, J. M. Layet, L. Giovanelli and G. Le Lay, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 235105 CrossRef.
- C. Brouder, J. Phys.: Condens. Matter, 1990, 2, 701–738 CrossRef CAS.
- M. Perfetti, M. Serri, L. Poggini, M. Mannini, D. Rovai, P. Sainctavit, S. Heutz and R. Sessoli, Adv. Mater., 2016, 28, 6946–6951 CrossRef CAS PubMed.
- P. Totaro, L. Poggini, A. Favre, M. Mannini, P. Sainctavit, A. Cornia, A. Magnani and R. Sessoli, Langmuir, 2014, 30, 8645–8649 CrossRef CAS PubMed.
- Y. Zhang, T. Learmonth, S. Wang, A. Y. Matsuura, J. Downes, L. Plucinski, S. Bernardis, C. O’Donnell and K. E. Smith, J. Mater. Chem., 2007, 17, 1276 RSC.
- P. Willke, Y. Bae, K. Yang, J. L. Lado, A. Ferrón, T. Choi, A. Ardavan, J. Fernández-Rossier, A. J. Heinrich and C. P. Lutz, Science, 2018, 362, 336–339 CrossRef CAS PubMed.
- K. Yang, Y. Bae, W. Paul, F. D. Natterer, P. Willke, J. L. Lado, A. Ferrón, T. Choi, J. Fernández-Rossier and A. J. Heinrich, et al. , Phys. Rev. Lett., 2017, 119, 227206 CrossRef PubMed.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3641–3662 CrossRef CAS.
- M. Krack, Theor. Chem. Acc., 2005, 114, 145–152 Search PubMed.
- M. Guidon, J. Hutter and J. VandeVondele, J. Chem. Theory Comput., 2010, 6, 2348–2364 CrossRef CAS PubMed.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- E. Caldeweyher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2017, 147, 034112 CrossRef PubMed.
- R. Sabatini, T. Gorni and S. de Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 041108 CrossRef.
- O. A. Vydrov and T. Van Voorhis, J. Chem. Phys., 2010, 133, 244103 CrossRef PubMed.
- J. Tersoff and D. R. Hamann, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 805–813 CrossRef CAS.
- P. J. Blowey, R. J. Maurer, L. A. Rochford, D. A. Duncan, J.-H. Kang, D. A. Warr, A. J. Ramadan, T.-L. Lee, P. K. Thakur and G. Costantini, et al. , J. Phys. Chem. C, 2019, 123(13), 8101–8111 CrossRef CAS PubMed.
- L. Malavolti, V. Lanzilotto, S. Ninova, L. Poggini, I. Cimatti, B. Cortigiani, L. Margheriti, D. Chiappe, E. Otero and P. Sainctavit, et al. , Nano Lett., 2015, 15, 535–541 CrossRef CAS PubMed.
- G. Rajaraman, A. Caneschi, D. Gatteschi and F. Totti, J. Mater. Chem., 2010, 20, 10747 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional spectroscopic characterization; theoretical calculations’ details. See DOI: 10.1039/c9nh00023b |
| This journal is © The Royal Society of Chemistry 2019 |