
Batch–flow hybrid synthesis of the antipsychotic clozapine†
N. C.
Neyt
and
D. L.
Riley
 *
*
Department of Natural and Agricultural Sciences, University of Pretoria, Pretoria, 0028, South Africa. E-mail: darren.riley@up.ac.za
First published on 22nd November 2017
Abstract
The development of batch–flow hybrid processes is becoming an attractive prospect through which chemists can make use of the best aspects of both technologies. We have reported the implementation of an on-the-fly purification by trituration which can also be utilised to perform solvent swaps. We have demonstrated this concept through the synthesis of the antipsychotic clozapine. In addition, we report a novel means of performing a reduction of an aryl nitro group under flow conditions and an overall improved process route for the total synthesis of clozapine.
Introduction
The development of new and efficient synthetic routes to complex molecules today requires the use of both the latest synthetic approaches and technologies.1–5 The modern process chemist needs to be able to select the best batch and flow approaches for a particular synthetic target and combine these to afford robust, safe and economical processes. As such, the distinction lines between traditional batch chemistry and flow chemistry are starting to blur with a move towards the development of hybrid batch–flow processes that can ideally be conducted on single platforms.Recently, the Ley group has been able to elegantly demonstrate this concept by developing a hybrid platform on which automated batch and flow reactions are performed including downstream processing steps such as solvent swaps and extractions.6,7 The de Oliveira group in a similar vein also showed the strength of combining batch and flow processes in their synthesis of naturally occurring curcuminoids.8
The integration of such downstream processing techniques, although reasonably simple under batch conditions at the bench scale, can be particularly challenging under flow conditions. Exploration of the integration of said downstream processes has been investigated but most traditional batch purification methods which include distillation, recrystallization, column chromatography, extraction and trituration are not always easily translated to flow.9,10 In contrast, flow chemistry typically relies on the use of inline scavengers and catch-and-release approaches which require distinct functional group differences between species that are to be separated.11–15 The use of such techniques has certain advantages, but is arguably more limited than batch purification which can be manipulated based upon both functional group differences and physical differences such as boiling point and solubility. That being said, examples of continuous crystallisation using mixed-suspension, mixed-product removal vessels, extractions using counter current extraction, in-line liquid–liquid extraction across hydrophobic membranes and micro-distillations have been reported, though in almost all cases the implementation of such systems is arguably not straightforward.16–18 The development of hybrid processes and platforms, however, affords the chemist the opportunity to make use of both batch and flow type purifications within the context of a continuous process.
In line with the thinking of Ley's group, we also strongly believe in the importance of being able to integrate batch and flow processes.3,6,7,9,10,19,20 An important aspect of this will require not only the simple linking of a batch or flow step to subsequent batch or flow steps, but also the incorporation of in-line and on-the-fly work-up and purification protocols (Fig. 1). Such protocols should ideally be incorporated in such a manner so as to allow i) continuous production and ii) safe production without the need to manually handle reaction intermediates or products, and should take advantage of simple small-scale batch work-up and purification techniques as a means of process intensification negating the need to perform challenging and time consuming batch based scale-ups. One should rather focus on simply performing these processes continuously on a smaller, more manageable scale.
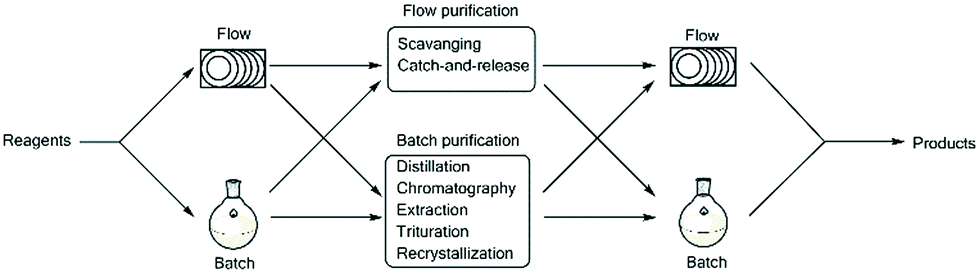 | ||
| Fig. 1 The chemist should be able to seamlessly choose and integrate between either a flow or batch type process within the context of continuous processing. | ||
One of the simplest purification processes involves the trituration of a pure material from a complex reaction matrix typically afforded by the addition of a reaction mixture to a solvent system in which the desired product is insoluble and the undesired impurities are soluble. Described below is our attempt to incorporate continuous on-the-fly trituration within the construct of a batch–flow hybrid process using both standard and custom batch techniques coupled to a Uniqsis FlowSyn™ flow reactor platform.
Process description
To demonstrate an integrated batch–flow process in which a continuous purification by trituration is implemented in a stop-go fashion, we selected clozapine [1], an antipsychotic, as a challenging flow target.21 The current synthetic approaches to clozapine are somewhat limited with most being simply variations of each other.22 The most commonly encountered route22 (Scheme 1) was validated in-house under traditional batch conditions affording clozapine in an overall yield of 27% vs. 25.8% as previously reported with a slightly longer reaction residence time of 132.25 h vs. 112.25 h (see the ESI† for stage by stage comparison). The process suffers from severe solubility issues (stages 1 and 4), solvent incompatibilities between stages 1, 2 and 3 and poor molecular differentiation between stages to allow for easy purification by scavenging or catch-and-release techniques. Under batch conditions, however, the differences in solubility of the reagents and products in different solvent systems can be manipulated to allow efficient purification by trituration (stages 1 and 2).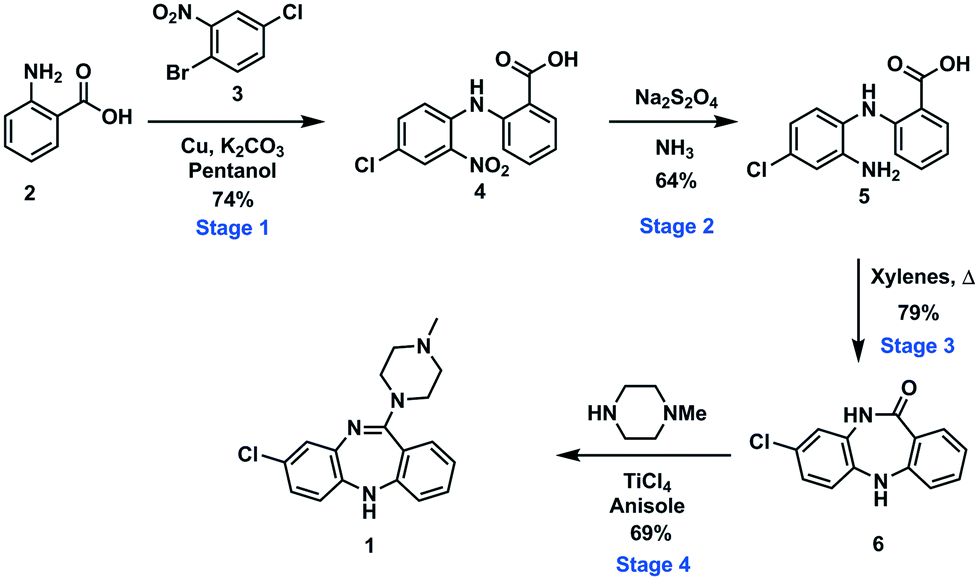 | ||
| Scheme 1 Synthetic route to the antipsychotic clozapine. | ||
The designed process is composed of four distinct stages that can be performed separately or as part of a linked hybrid process (Scheme 2). The design allows modification and optimisation of individual steps without affecting the overall process. Stages 1 and 2 consist of a batch and flow process respectively; in both cases the downstream processing and purification is facilitated by the use of an on-the-fly batch–flow trituration and solvent swap. Stage 3 is a purely flow process and stage 4 a purely batch process.
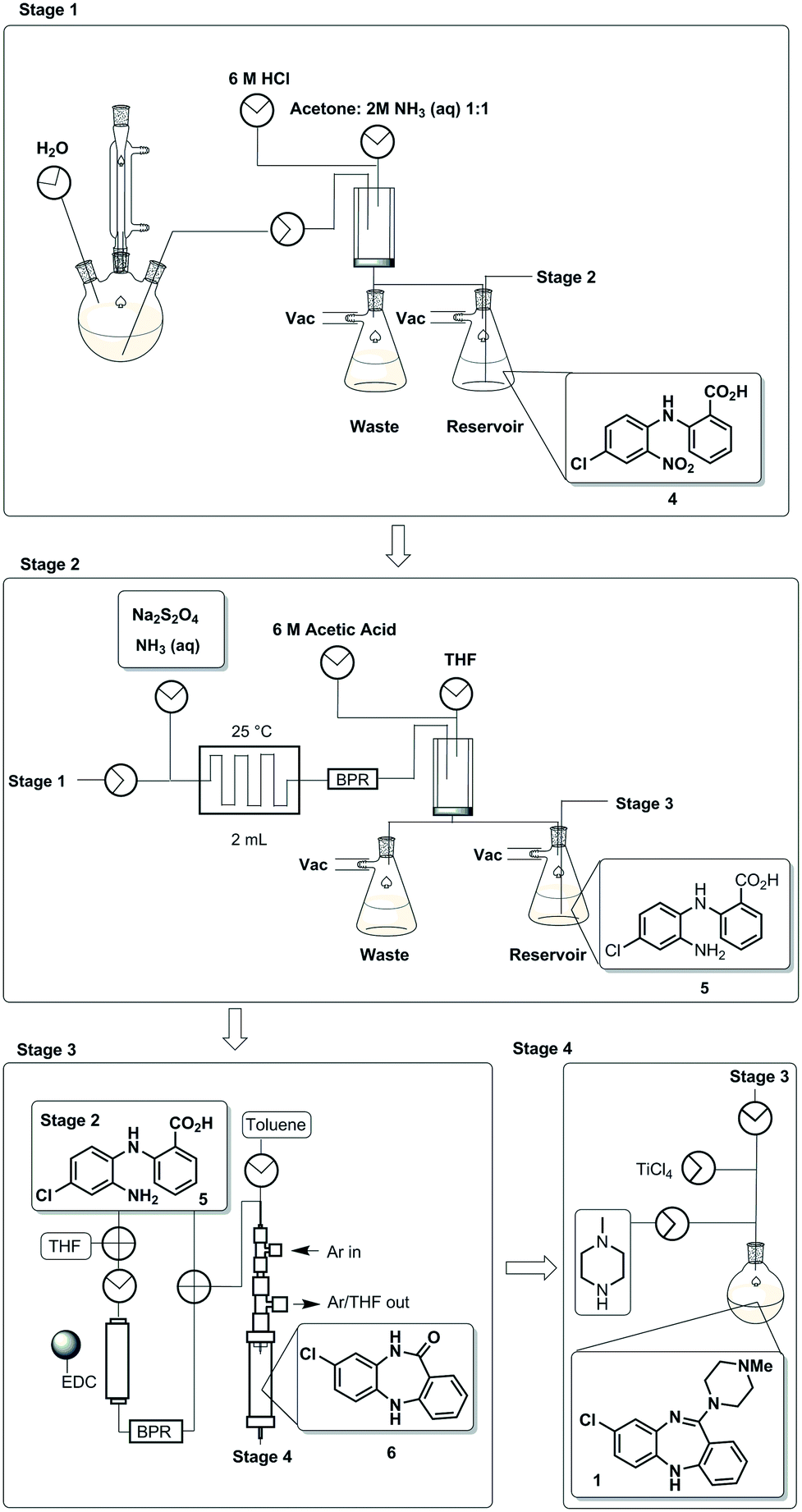 | ||
| Scheme 2 A schematic representation of the telescoped four stage batch–flow hybrid process, incorporating on-the-fly purification by trituration and in-line solvent swapping. | ||
Integration of the trituration process
An integral part of the synthetic design involved the development of a process which limited chemical and solvent exposure to the chemist, reduced the number of manual operations and eliminated down-time between operations; as such the need to perform on-the-fly trituration, filtration, drying and solvent swaps as a single integrated operation was viewed as being critical. Several approaches utilising tubular continuous-flow crystallizers,23–29 stirred tank crystallizers30 or rotating sintered glass disks onto which a solvent stream containing the precipitate is fed have been reported.20,31 In the latter case, a solid material accumulates on the disc and is continuously removed by scraping the disc with a fixed PTFE arm. Although elegant, these solutions all required the pumping of slurries and did not afford us the degree of process intensification which would allow purification by trituration linked to filtration, drying and solvent swapping in a single affordable piece of hardware. In order to facilitate this need, a single new piece of glassware based upon a stirrer tank crystallizer was designed which allowed us to efficiently perform all four processes in a continuous stop-go fashion.The glassware developed consisted of a cylindrical two-layer jacketed vessel fitted with a sintered glass funnel at the bottom connected to a single B19 ground glass joint via a Rotoflo tap (Fig. 2). The double-layered vessel was fitted with inlet and outlet points to allow the heating or cooling of the vessel by connection to a suitable water circulator and heater/chiller system. The top end of the vessel was fitted with a B24 ground glass joint which could be used to facilitate the insertion of an overhead stirrer and if required could be connected to a reaction manifold for processing under inert conditions using a standard stirrer bearing adaptor. The glassware is connected to a reservoir via the bottom B19 joint which itself is connected to a vacuum pump or aspirator. Alternatively, a Y-piece joint can be connected to the B19 joint allowing two reservoirs to be attached simultaneously facilitating the separation of waste and product streams.
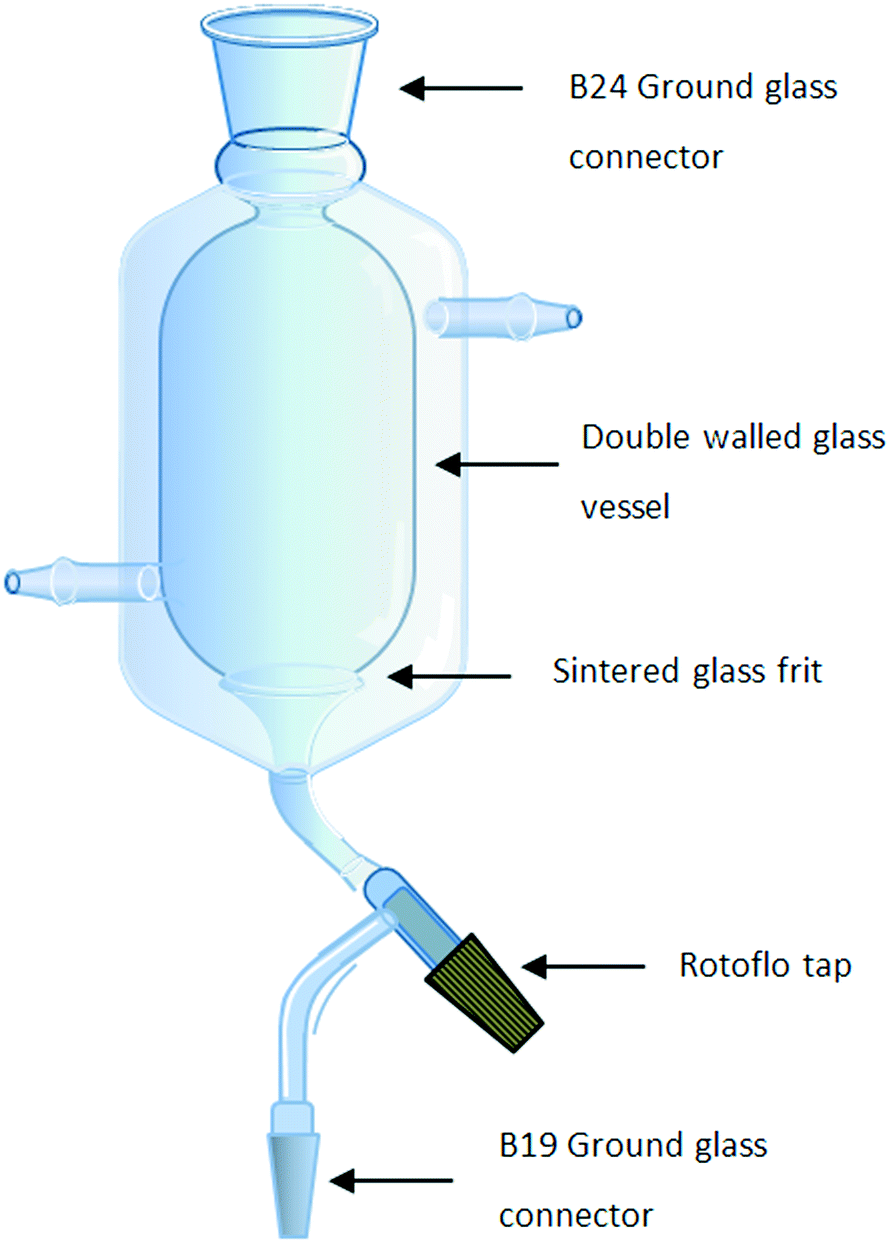 | ||
| Fig. 2 Schematic of the glassware developed for on-the-fly triturations and filtrations. | ||
Operation was afforded by priming the vessel with an anti-solvent in which the product of interest is insoluble; thereafter run-off from a reaction is pumped into the vessel under rapid overhead stirring resulting in the trituration of the compound of interest. Typically, the outer jacket would be connected to a water chiller to cool the vessel and promote efficient trituration at this stage.
Once the vessel is full, the Rotoflo tap is opened and the waste filtrate evacuated under vacuum. The compound remaining in the vessel is then re-solubilised in the vessel by priming with a solubilising solvent (this can be performed with heating of the vessel jacket if required). Once solubilised, the solution can be evacuated into a reagent reservoir under vacuum prior to further processing. The process can be run continuously in a stop-go fashion with the vessel being re-primed with the anti-solvent after evacuation of the solution of the solubilised product. The approach would also be applicable to systems in which it was possible to triturate out unwanted by-products from a reaction stream.
Process description
Stage 1
The first synthetic stage involved a copper mediated Ullmann/Goldberg coupling between anthranilic acid [2] and 2-bromo-4-chloronitrobenzene [3]. Previously, such transformations have been reported using copper coils to catalyse the reaction under flow conditions,32 however, in the case in hand solubility issues precluded the efficient translation to flow. The use of polar protic solvents (pentanol, isopropanol or ethanol) allowed solubilisation under dilute conditions (0.17 M), unfortunately when using a packed-bed reactor charged with various copper catalysts and bases, unwanted precipitation of the diaryl product [4] inevitably led to reactor fouling with yields in the range of 30–38% (see the ESI†).As a result, we opted to perform the reaction in batch with an integrated in-line work-up and purification (Scheme 3). The batch reaction was performed by charging a standard 100 mL three-necked round bottom flask with isopropanol in which anthranilic acid [2] was reacted with 1-bromo-4-chloro-2-nitrobenzene [3] in the presence of copper metal and potassium carbonate under reflux. A batch optimisation indicated that the reaction conversion slowed down after 8 h, and as such the process was typically halted after 8 h to 12 h of reaction.
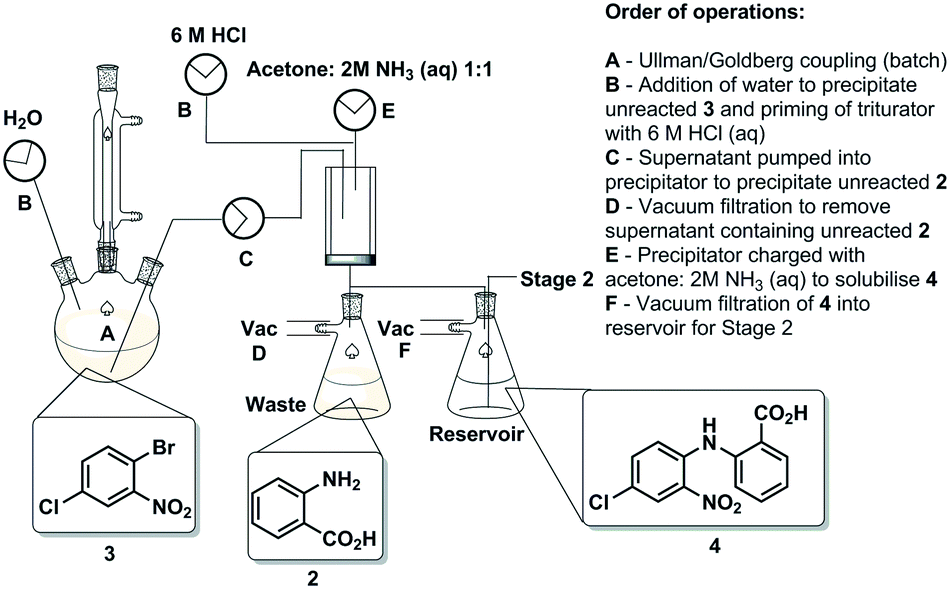 | ||
| Scheme 3 Stage 1 schematic with the order of operations. | ||
After the reaction heating was stopped, the vessel was primed with water to facilitate the precipitation of any unreacted 1-bromo-4-chloro-2-nitrobenzene [3]. Simultaneously, the triturating vessel was primed with 6 M HCl(aq), and once the precipitation of [3] in the reaction vessel was deemed to be complete the contents were pumped into the cooled triturator under rapid overhead stirring resulting in the precipitation of the desired diarylamine [4]. The subsequent evacuation of the filtrate which contains unreacted anthranilic acid [2] left behind pure coupled [4] that was then allowed to dry under vacuum before being re-solubilised in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetone:NH3(aq) solution, and evacuated into a reagent reservoir. As a standalone step the process allows the isolation of [4] in 80% yield (Fig. 3).
:1 acetone:NH3(aq) solution, and evacuated into a reagent reservoir. As a standalone step the process allows the isolation of [4] in 80% yield (Fig. 3).
 | ||
| Fig. 3 Stage 1 stand-alone set-up. The triturator can be seen on the left with precipitation of the bright orange couple product [4] clearly seen; the batch reaction vessel is out of shot on the right. | ||
An analysis of [4] was undertaken using scanning electron microscopy (SEM) to gain insight into the morphology of the material prepared and to determine if the process was a true trituration or a crystallisation. Microscopy images of samples prepared by standard recrystallization techniques (Fig. 4A) and using our trituration approach (Fig. 4B) show clear differences. Sample B was characteristic of a trituration with significantly smaller particles with good uniformity ranging in size from 1–3 μM; furthermore, energy-dispersive X-ray spectroscopy (EDX) performed concurrently was used to determine the amounts of residual copper left over from stage 1. In the case of the triturated sample B no copper was observed; in contrast traces (∼0.1 wt%) were observed in the recrystallised sample A.
 | ||
| Fig. 4 SEM scans (1000× magnification) of diarylamine [4] prepared under standard batch recrystallization (A) and under the hybrid trituration process (B). Additional images are available in the ESI.† | ||
Stage 2
The pure stock solution of [4] from stage 1 was subjected to a flow based reduction of the aryl nitro group to an aryl amine under a novel flow reduction process involving mixing with sodium dithionite/aqueous ammonia in a 2 mL mixing chip at 25 °C. The process allowed high conversion with a residence time of only 92 s affording an isolated yield of 79% as a standalone step (Scheme 4). Again, the process purification was afforded by an on-the-fly trituration utilising 6 M acetic acid to affect the trituration of pure [5] (Fig. 5). The precipitated [5] was dried under vacuum and re-solubilised in anhydrous THF prior to passage into stage 3. In comparison, the validated batch procedure utilising sodium dithionite is also relatively rapid with a 15 min residence time, however, a best isolated yield of only 65% was achieved under these conditions. SEM assessment of the morphology of [5] revealed irregular sized rod-shaped particles ranging from 2 to 10 μM in length (Fig. 6).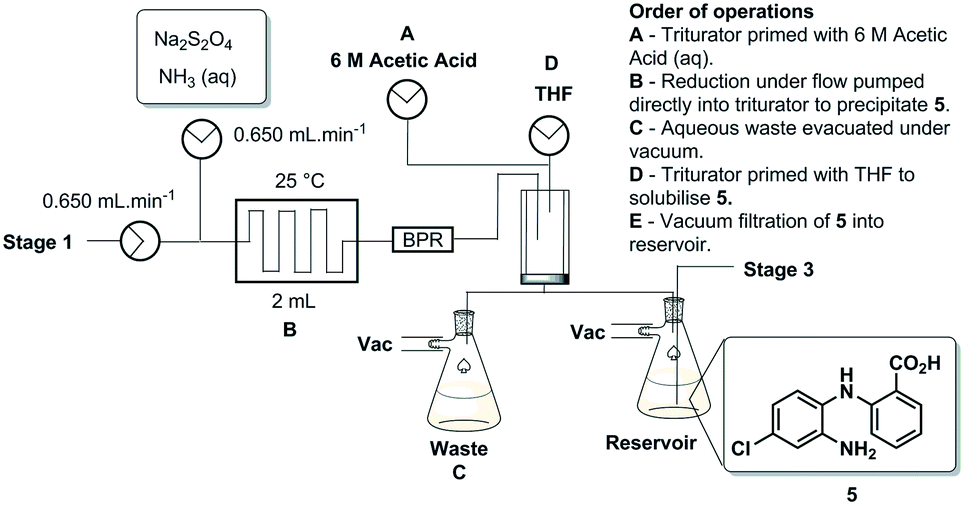 | ||
| Scheme 4 Stage 2 schematic. | ||
 | ||
| Fig. 5 Stage 2 stand-alone set-up, the mixing chip in which the reduction occurs can be seen on the left and the triturator with the reduced [4] can be seen on the right. | ||
 | ||
| Fig. 6 SEM scans (1000× magnification) of [5]. Additional images available in the ESI.† | ||
Stage 3
Stage 3 consisted of a simple intramolecular amide coupling. Initial attempts to utilise ground-up activated molecular sieves (4 Å) housed in a packed bed reactor to drive the process through the removal of water proved unsuccessful.Passage across polymer supported EDC, however, afforded good clean conversions (Scheme 5). The process was initially run with a packed bed reactor containing 1.5 eq. of polymer supported EDC at 0.15 M concentration relative to [5]. A single pass through the reactor with a residence time of ∼30 min afforded pure [6] in 70% isolated yield. The introduction of a recycle loop allowed us to increase the residence time without having to either increase the amount of polymer supported EDC used or reduce the flow rate (Fig. 7). Under these conditions, conversion by TLC appeared to be complete (3.5 h residence time, 0.25 mL min−1) affording an isolated yield of 76% when run as a standalone reaction. NMR analysis of the crude reaction stream at this stage, however, suggested close to quantitative conversion and as such the reaction stream was deemed pure enough for passage directly into stage 4 without chromatographic purification.
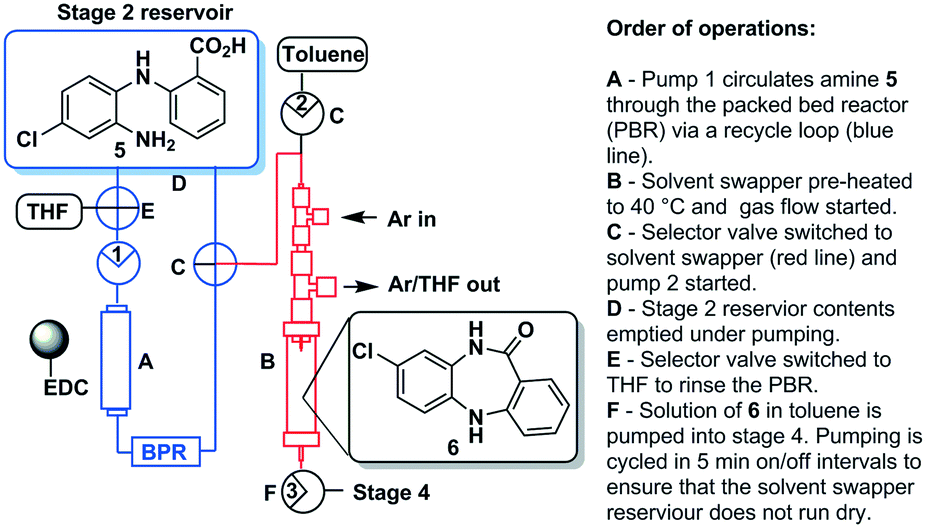 | ||
| Scheme 5 Stage 3 schematic. | ||
 | ||
| Fig. 7 Stage 3 with the EDC packed column as well as the in-line solvent swapper connected to an argon bottle. | ||
The final batch-stage, however, required heating in excess of 100 °C and as such a solvent swap from THF to toluene was required. In preparation, the reaction mixture was recycled through the packed bed reactor for 6 hours at a flow rate of 0.25 mL min−1 after which time the solution of [6] was fed directly into a solvent swapper based on the design reported by the Ley group.33,34 The solution was concentrated in the evaporator (1 mL min−1) at 60 °C and enriched with toluene (4:1 toluene/THF) allowing the complete removal of THF. The remaining toluene solution containing product [6] was collected in a 50 mL round bottom flask for use in stage 4 (Fig. 7).
In contrast, under batch conditions heating with mechanical removal of water utilizing a Dean and Stark apparatus afforded a comparable isolated yield of 74% but required a reaction time of up to 96 hours. The use of EDC as a coupling reagent resulted in poorer isolated yields in the range of 55–62% and required careful chromatographic purification.
Stage 4
Stage 4 involved a titanium tetrachloride mediated chlorination of [6] followed by immediate displacement of the chloride with methyl piperazine (Scheme 6). The extensive precipitation of titanium salts and limited alternative options with regard to the chlorination of [6] prompted us to run the stage as a batch based process as described by Capuano et al. Titanium tetrachloride was added to the reagent stock from stage 3 at 0 °C, followed by N-methylpiperazine. The reaction mixture was refluxed at 110 °C for 24 hours. A manual work-up and purification by column chromatography afforded clozapine [1] in 72% yield across the final two steps (stage 3 and stage 4).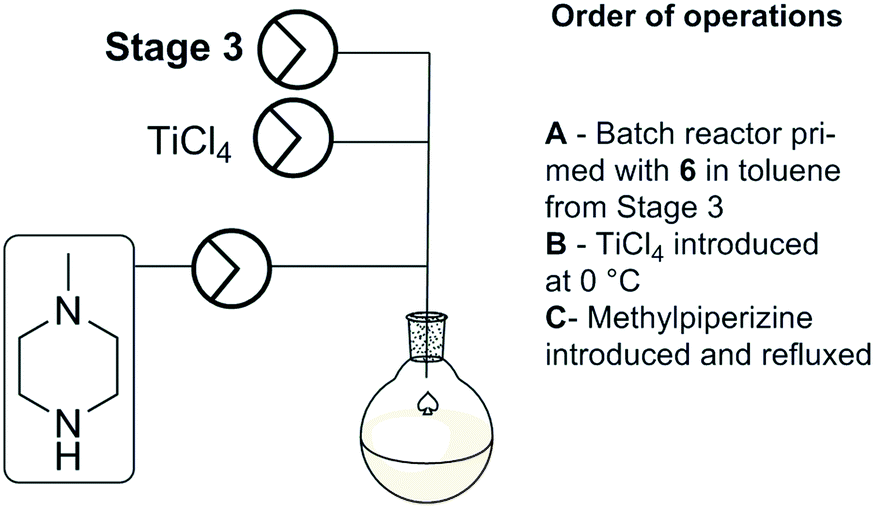 | ||
| Scheme 6 Stage 4 schematic. | ||
Telescoped process
A key aspect of the research was to ascertain if a fully continuous start to end synthesis of the target molecule could be achieved with limited manual processing and purifications.In stages 1 and 2, the use of the in-line triturator effectively allowed us the freedom to both purify the products and perform solvent swaps. In the case of the telescoped process, the precipitated products [4] and [5] had to be dried in the triturator under vacuum prior to passage into the subsequent stages. On the demonstration scale, only a single cycle of operation was required for both triturations, however, it is anticipated that on a larger scale simple numbering up of the triturators would allow continuous supply of feedstocks between stages one, two and three.
In the case of stage 3, we chose to employ the recycle loop approach as opposed to a single pass through the packed bed reactor. This approach allowed us to maximise the conversion without having to resort to the use of excessive amounts of polymer supported EDC. Utilisation of this approach followed by in-line solvent swapping from THF to toluene allowed direct telescoping into stage 4.
The final stage was run under standard batch conditions and in our case we elected to purify the final product [1] utilising column chromatography.
The telescoped process was successfully demonstrated on a 1 g scale based on anthranilic acid [2] affording 1.084 g of clozapine representing an overall yield of 45.5% in approximately 44 h for the hybrid process.
Conclusions
We have been able to successfully demonstrate an integrated batch–flow process which utilises a process intensified on-the-fly purification by trituration and affords the opportunity to perform solvent swaps.We performed and optimised all processes as stand-alone stages and then telescoped each step into the next. The approach, although manually controlled, greatly reduces the chemist's exposure to chemicals and solvents with no physical contact required between the preparation of the initial stock solutions and the final stage 4 work-up and purification. We believe that the process would also lend itself well to a full automated approach in a similar vein to those described by the Ley group.6,7,19
The second stage of the process also represents a novel approach for performing reductions under flow conditions, and to the best of our knowledge is the first demonstration of a flow reduction not making use of metals and/or molecular hydrogen.
The telescoped process affords a complete synthesis of the antipsychotic clozapine in an overall yield of 45.5% in comparison with the 27% yield under purely batch conditions. Improvements in yield are primarily due to the flow conversion of stages 2 and 3 affording a yield increase from 64% to 79% for stage 2 and 54.5% to 72% for stage 3/4.
Furthermore, the hybrid process afforded a significantly reduced reaction time equating to a production time of ∼44 h vs. 132 h for the purely batch route when tested on a 1 g scale.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of South Africa (grant number 87893), the University of Pretoria (University, Science Faculty Research Councils and Research and Development Program), and Pelchem Pty Ltd. Opinions expressed in this publication and the conclusions arrived at are those of the authors, and are not necessarily attributed to the NRF. The authors would like to gratefully acknowledge Eric Palmer and Mamoalosi Selepe for NMR spectroscopy services, Madelien Wooding for mass spectroscopy services and Uniqsis Ltd for discounted flow equipment.Notes and references
- M. Baumann and I. R. Baxendale, The synthesis of active pharmaceutical ingredients (APIs) using continuous flow chemistry, Beilstein J. Org. Chem., 2015, 11, 1194–1219 CrossRef CAS PubMed.
- G. M. Whitesides, Reinventing Chemistry, Angew. Chem., Int. Ed., 2015, 54(11), 3196–3209 CrossRef CAS PubMed.
- D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, Enabling Technologies for the Future of Chemical Synthesis, ACS Cent. Sci., 2016, 2(3), 131–138 CrossRef CAS PubMed.
- V. Sans and L. Cronin, Towards dial-a-molecule by integrating continuous flow, analytics and self-optimisation, Chem. Soc. Rev., 2016, 45(8), 2032–2043 RSC.
- R. Porta, M. Benaglia and A. Puglisi, Flow Chemistry: Recent Developments in the Synthesis of Pharmaceutical Products, Org. Process Res. Dev., 2016, 20(1), 2–25 CrossRef CAS.
- D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, A Novel Internet-Based Reaction Monitoring, Control and Autonomous Self-Optimization Platform for Chemical Synthesis, Org. Process Res. Dev., 2016, 20(2), 386–394 CrossRef CAS.
- D. E. Fitzpatrick and S. V. Ley, Engineering chemistry: integrating batch and flow reactions on a single, automated reactor platform, React. Chem. Eng., 2016, 1(6), 629–635 CAS.
- C. C. Carmona-Vargas, L. D. C. Alves, T. J. Brocksom and K. T. de Oliveira, Combining batch and continuous flow setups in the end-to-end synthesis of naturally occurring curcuminoids, React. Chem. Eng., 2017, 2(3), 366–374 CAS.
- S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Organic Synthesis: March of the Machines, Angew. Chem., Int. Ed., 2015, 54(11), 3449–3464 CrossRef CAS PubMed.
- S. V. Ley, D. E. Fitzpatrick, R. M. Myers, C. Battilocchio and R. J. Ingham, Machine-Assisted Organic Synthesis, Angew. Chem., Int. Ed., 2015, 54(35), 10122–10136 CrossRef CAS PubMed.
- I. R. Baxendale, S. V. Ley, C. D. Smith and G. K. Tranmer, A flow reactor process for the synthesis of peptides utilizing immobilized reagents, scavengers and catch and release protocols, Chem. Commun., 2006,(46), 4835–4837 RSC.
- M. Caldarelli, I. R. Baxendale and S. V. Ley, Clean and efficient synthesis of azo dyes using polymer-supported reagents, Green Chem., 2000, 2(2), 43–45 RSC.
- S. V. Ley, I. R. Baxendale, R. N. Bream, P. S. Jackson, A. G. Leach, D. A. Longbottom, M. Nesi, J. S. Scott, R. I. Storer and S. J. Taylor, Multi-step organic synthesis using solid-supported reagents and scavengers: a new paradigm in chemical library generation, J. Chem. Soc., Perkin Trans. 1, 2000,(23), 3815–4195 RSC.
- A. Palmieri, S. V. Ley, A. Polyzos, M. Ladlow and I. R. Baxendale, Continuous flow based catch and release protocol for the synthesis of alpha ketoesters, Beilstein J. Org. Chem., 2009, 5, 23 Search PubMed.
- C. Wiles, P. Watts and S. J. Haswell, The use of solid-supported reagents for the multi-step synthesis of analytically pure alpha,beta-unsaturated compounds in miniaturized flow reactors, Lab Chip, 2007, 7(3), 322–330 RSC.
- S. Mascia, P. L. Heider, H. T. Zhang, R. Lakerveld, B. Benyahia, P. I. Barton, R. D. Braatz, C. L. Cooney, J. M. B. Evans, T. F. Jamison, K. F. Jensen, A. S. Myerson and B. L. Trout, End-to-End Continuous Manufacturing of Pharmaceuticals: Integrated Synthesis, Purification, and Final Dosage Formation, Angew. Chem., Int. Ed., 2013, 52(47), 12359–12363 CrossRef CAS PubMed.
- K. P. Cole, J. M. Groh, M. D. Johnson, C. L. Burcham, B. M. Campbell, W. D. Diseroad, M. R. Heller, J. R. Howell, N. J. Kallman, T. M. Koenig, S. A. May, R. D. Miller, D. Mitchell, D. P. Myers, S. S. Myers, J. L. Phillips, C. S. Polster, T. D. White, J. Cashman, D. Hurley, R. Moylan, P. Sheehan, R. D. Spencer, K. Desmond, P. Desmond and O. Gowran, Kilogram-scale prexasertib monolactate monohydrate synthesis under continuous flow CGMP conditions, Science, 2017, 356(6343), 1144–1150 CrossRef CAS PubMed.
- E. Y. Kenig, Y. H. Su, A. Lautenschleger, P. Chasanis and M. Grunewald, Micro-separation of fluid systems: A state-of-the-art review, Sep. Purif. Technol., 2013, 120, 245–264 CrossRef CAS.
- R. J. Ingham, C. Battilocchio, D. E. Fitzpatrick, E. Sliwinski, J. M. Hawkins and S. V. Ley, A Systems Approach towards an Intelligent and Self-Controlling Platform for Integrated Continuous Reaction Sequences, Angew. Chem., Int. Ed., 2015, 54(1), 144–148 CrossRef CAS PubMed.
- S. V. Ley, R. J. Ingham, M. O'Brien and D. L. Browne, Camera-enabled techniques for organic synthesis, Beilstein J. Org. Chem., 2013, 9, 1051–1072 CrossRef CAS PubMed.
- J. Crilly, The history of clozapine and its emergence in the US market: a review and analysis, Hist. Psychiatry, 2007, 18(1), 39–60 CrossRef PubMed.
- F. Hunziker, E. Fischer and J. Schmutz, 11-Amino-5h-Dibenzo[B,E]-1,4-Diazepine .10. Mitteilung Uber Siebengliedrige Heterocyclen, Helv. Chim. Acta, 1967, 50(6), 1588–1599 CrossRef CAS.
- M. Jiang, Z. L. Zhu, E. Jimenez, C. D. Papageorgiou, J. Waetzig, A. Hardy, M. Langston and R. D. Braatz, Continuous-flow tubular crystallization in slugs spontaneously induced by hydrodynamics, Cryst. Growth Des., 2014, 14(2), 851–860 CAS.
- A. J. Alvarez and A. S. Myerson, Continuous Plug Flow Crystallization of Pharmaceutical Compounds, Cryst. Growth Des., 2010, 10(5), 2219–2228 CAS.
- S. Lawton, G. Steele, P. Shering, L. H. Zhao, I. Laird and X. W. Ni, Continuous Crystallization of Pharmaceuticals Using a Continuous Oscillatory Baffled Crystallizer, Org. Process Res. Dev., 2009, 13(6), 1357–1363 CrossRef CAS.
- R. Vacassy, J. Lemaitre, H. Hofmann and J. H. Gerlings, Calcium carbonate precipitation using new segmented flow tubular reactor, AIChE J., 2000, 46(6), 1241–1252 CrossRef CAS.
- R. M. del Rio and R. W. Rousseau, Batch and tubular-batch crystallization of paracetamol: Crystal size distribution and polymorph formation, Cryst. Growth Des., 2006, 6(6), 1407–1414 Search PubMed.
- R. J. P. Eder, S. Radl, E. Schmitt, S. Innerhofer, M. Maier, H. Gruber-Woelfler and J. G. Khinast, Continuously Seeded, Continuously Operated Tubular Crystallizer for the Production of Active Pharmaceutical Ingredients, Cryst. Growth Des., 2010, 10(5), 2247–2257 CAS.
- S. Ferguson, G. Morris, H. X. Hao, M. Barrett and B. Glennon, In-situ monitoring and characterization of plug flow crystallizers, Chem. Eng. Sci., 2012, 77, 105–111 CrossRef CAS.
- S. Y. Wong, J. Chen, L. E. Forte and A. S. Myerson, Compact Crystallization, Filtration, and Drying for the Production of Active Pharmaceutical Ingredients, Org. Process Res. Dev., 2013, 17(4), 684–692 CrossRef CAS.
- C. S. Elmore, S. Landvatter, P. N. Dorff, M. E. Powell, D. Killick, T. Blake, J. Hall, J. R. Heys, J. Harding, R. Urbanek and G. Ernst, Synthesis of three alpha 7 agonists in labeled form, J. Labelled Compd. Radiopharm., 2014, 57(5), 342–349 CrossRef CAS PubMed.
- Y. Zhang, T. F. Jamison, S. Patel and N. Mainolfi, Continuous Flow Coupling and Decarboxylation Reactions Promoted by Copper Tubing, Org. Lett., 2011, 13(2), 280–283 CrossRef CAS PubMed.
- B. J. Deadman, C. Battilocchio, E. Sliwinski and S. V. Ley, A prototype device for evaporation in batch and flow chemical processes, Green Chem., 2013, 15(8), 2050–2055 RSC.
- C. Battilocchio, B. J. Deadman, N. Nikbin, M. O. Kitching, I. R. Baxendale and S. V. Ley, A Machine-Assisted Flow Synthesis of SR48692: A Probe for the Investigation of Neurotensin Receptor-1, Chem. - Eur. J., 2013, 19(24), 7917–7930 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00146k |
| This journal is © The Royal Society of Chemistry 2018 |