
Titania supported on silica as an efficient catalyst for deep oxidative desulfurization of a model fuel with exceptionally diluted H2O2†
C. G.
Piscopo
 *a,
J.
Tochtermann
a,
M.
Schwarzer
a,
D.
Boskovic
a,
R.
Maggi
b,
G.
Maestri
b and
S.
Loebbecke
a
*a,
J.
Tochtermann
a,
M.
Schwarzer
a,
D.
Boskovic
a,
R.
Maggi
b,
G.
Maestri
b and
S.
Loebbecke
a
aFraunhofer Institute for Chemical Technology, Energetic Materials, Joseph-von-Fraunhoferstr. 7, 76327 Pfinztal, Germany. E-mail: calogero.piscopo@ict.fraunhofer.de
bClean Synthetic Methodology Group, Department of Chemical and Life Sciences and Environmental Sustainability, University of Parma, Parco Area delle Scienze 17A, 43124 Parma, Italy
First published on 15th January 2018
Abstract
Deep oxidative desulfurization has been carried out, achieving the production of a model fuel with minimal sulphur content (0–15 ppm). This desulfurization process has been developed in continuous flow, using a titanium supported heterogeneous catalyst and exceptionally low hydrogen peroxide concentration (0.77% w/w).
Due to the tight sulphur limits in transportation fuels, deep desulfurization of liquid fuels is becoming a critical topic in environmental sciences. The society at large is stepping on the road to zero sulphur fuel, therefore the implementation of effective desulfurization methods, able to replace the classical hydrodesulfurization (HDS), which is associated with harsh reaction conditions, is a crucial goal in technical chemistry.1 An alternative to HDS is provided by adsorptive desulfurization, and the scope of this methodology is focused mainly on jet fuels.2
The most promising desulfurization methodology is undoubtedly the oxidative desulfurization (ODS), which consists of two steps: (i) the oxidation of sulphur compounds (sulphides to sulfones) and (ii) the extraction of oxidation products.3 Both reactions can be performed under mild conditions, using hydrogen peroxide as the oxidant;4 in particular, biphasic systems5 together with suitable catalysts6 have been investigated, providing satisfactory results. The selection and the preparation of the catalyst have obviously a prominent role in the development of an efficient ODS route; indeed a huge scientific effort has been made in testing different catalytic materials, ranging from classical mixed metal oxides7 to polyoxometalates,8 metal–organic frameworks9 and biomimetic materials.10 Excellent results have been achieved using titanium based catalysts, which is so far the most commonly employed metal in ODS.11,12
Hydrogen peroxide is the oxidant of choice for ODS since it is a cheap, mild and environmentally benign reagent.13 Nevertheless, the manufacture, transport and handling of concentrated H2O2 is associated with safety issues and considerable logistical efforts, limiting the implementation of hydrogen peroxide routes in industry. A promising solution to this problem is the in situ generation of hydrogen peroxide14 and its direct combination with selective oxidation reactions, increasing the environmental and economic attractiveness of the use of H2O2 as a green oxidant.15
Since the concentration of in situ generated H2O2, e.g. by electrochemical synthesis, is typically rather low, the development of a catalytic material which is active in the presence of diluted H2O2 represents a new challenge that has been rarely addressed. The use of diluted hydrogen peroxide for the selective oxidation of thioanisole to its sulfoxide has been already reported;16 however, in order to perform the oxidation of refractory sulphides such as dibenzothiophene (DBT), an even more efficient catalyst has to be designed and tested. ODS has been recently used as a suitable example for the coupling of hydrogen peroxide synthesis and utilization in oxidation reactions.17 Indeed, some interesting results using tandem Pd/Ti (ref. 18) and Au/Ti (ref. 19) catalysts have been already reported.
Herein, the development of an efficient solid catalyst for the ODS of a model fuel with diluted hydrogen peroxide is described.
Considering the reported results on the use of sulfonic resins as heterogeneous catalysts,16 the first batch experiments were performed using Amberlite IR 120H as the catalyst for the oxidation of dibenzothiophene (DBT) with 3% H2O2. The poor solubility of the DBT oxidation products (dibenzothiophene sulfoxide and sulfone) in water necessitates the use of acetonitrile as a co-solvent in the polar phase in order to extract the sulphur from the fuel. A biphasic reaction mixture was prepared combining 3% w/w aqueous hydrogen peroxide, acetonitrile (final H2O2 concentration: 0.77% w/w) and the selected catalyst with a model fuel (DBT solution in dodecane, with a sulphur content of 1000 ppm). By varying the reaction parameters, with the use of tenfold excess hydrogen peroxide at 60 °C for 24 hours, Amberlite still gave 345 ppm residual sulphur in the model fuel solution in the form of DBT (see the ESI†). The water phase contains the oxidation products dibenzothiophene sulfoxide and sulfone, allowing a coherent mass balance.
On the basis of these results, a variety of heterogeneous catalysts, commercially available or prepared in our labs, were tested under the same reaction conditions; the results are presented in Table 1.
| Entry | Catalysta | Residual sulphurb (ppm) |
|---|---|---|
| a Reaction conditions: 2 ml of DBT solution in dodecane (0.024 mol L−1), 2 ml of acetonitrile, 250 mg of the catalyst, 0.533 ml of H2O2 solution at 3%, 60 °C, 24 h. b Determined by HPLC analysis. | ||
| 1 | — | 770 |
| 2 | Amberlite IR 120H | 345 |
| 3 | Nafion | 464 |
| 4 | Aquivion | 432 |
| 5 | TiO2 (rutile) | 445 |
| 6 | TiO2 (anatase) | 242 |
| 7 | Ti-SBA-15 | 335 |
| 8 | TS-1 | 205 |
| 9 | TS-1 B-doped | 231 |
| 10 | TiO2@SiO2 | 6 |
The HPLC results show that in the absence of any catalyst, hydrogen peroxide is not able to reduce the sulphur content, likely owing to the high dilution of the mixture. In a blank experiment without any catalyst, 770 ppm residual sulphur was detected, which indicates that partial desulfurization occurs solely due to the extraction of DBT by acetonitrile. Perfluorinated sulfonic resins such as Nafion and Aquivion were slightly less active than Amberlite. Switching to inorganic catalysts, interesting results were obtained using titanium-based materials: anatase performed significantly better than the polymorphous rutile, showing a residual sulphur content of 242 and 445 ppm, respectively. On the basis of these considerations, a series of catalysts with 3.5 wt% titanium content were tested.12,20 Ti-SBA-15, a mesoporous silica incorporating titanium atoms, gave an unsatisfactory sulphur oxidation (335 ppm residual sulphur), whereas with TS-1 and its boron-doped counterpart,21 residual sulphur values similar to that of anatase were achieved (205 and 231 ppm, respectively). In order to obtain a more efficient catalyst, TiO2 was anchored on the surface of silica by grafting of TiCl4 and subsequent calcination at 450 °C; the prepared catalyst TiO2@SiO2 allowed almost complete desulfurization of the model fuel (6 ppm residual sulphur). These results suggest the need of well dispersed and accessible catalytic sites on the surface of the silica to promote the oxidation process. To further examine this hypothesis, a TiO2@SiO2 material was prepared, avoiding the final calcination step; the solid material, heated in an oven at 100 °C merely to remove the organic solvents, did not oxidize DBT at all. XRD analyses of both uncalcined and calcined TiO2@SiO2 materials provide clarifying results (Fig. 1). A completely amorphous material has been obtained by the grafting of Ti onto silica; nevertheless, the calcined catalyst showed the presence of the typical anatase peaks. These results show that a phase transition, which occurs during the calcination step, is responsible for the formation of titania nanocrystals dispersed on amorphous silica, representing the catalytic active sites.
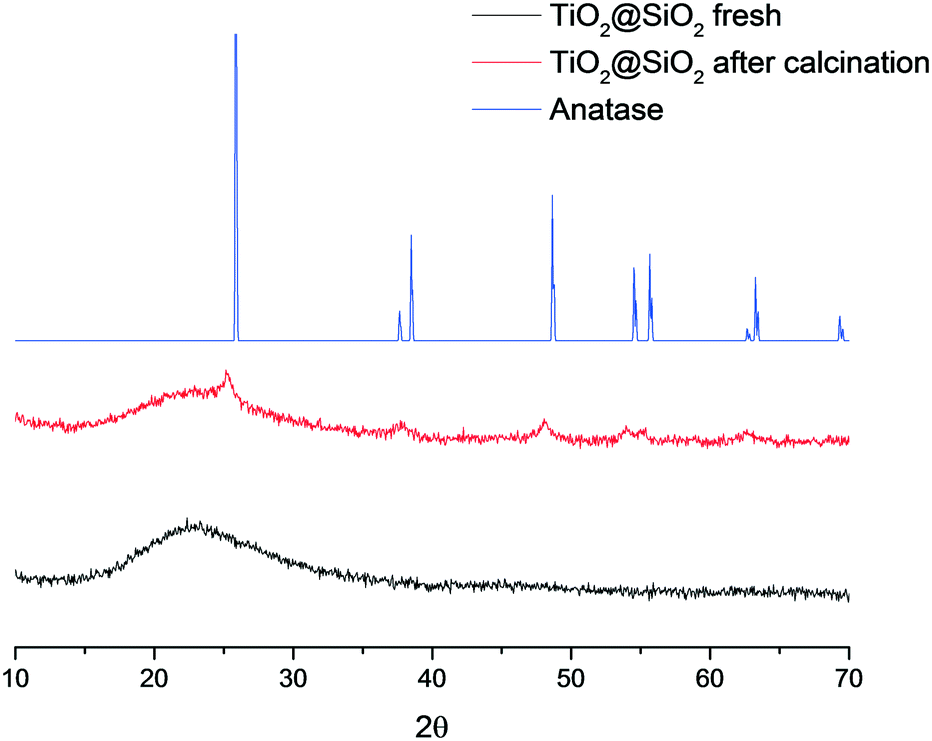 | ||
| Fig. 1 XRD spectra of anatase (blue), fresh (black) and calcined (red) TiO2@SiO2. | ||
The desulfurization progress with time has been monitored to optimize the reaction parameters (Fig. 2). Surprisingly, the ODS process is almost complete in just 20 minutes. Considering that all the other materials tested were unable to attain complete oxidation even after 24 hours, these results clearly show the great potential of this catalyst. However, the quality of a heterogeneous catalyst is related to its reusability. To test its reusability, the catalyst was filtered off after the reaction, washed with acetonitrile, dried at 80 °C and reused in successive runs. The results for the first 6 runs are reported in Table 2.
 | ||
| Fig. 2 Reaction trend for the batch oxidation of DBT with 3% aqueous hydrogen peroxide catalysed by TiO2@SiO2 under optimized conditions. | ||
A partial catalyst deactivation started already from the second run (234 ppm residual sulphur); later from the 4th to 6th runs, the residual sulphur content attained a plateau at around 420 ppm, revealing a marked decrease of the catalyst activity but not a complete inactivity. The hot filtration test22 and the ICP analysis of the solution obtained after catalyst filtration confirmed the absence of any leaching. Hence, the decrease in the activity of TiO2@SiO2 could be ascribable to the poisoning of titanium catalytic sites due to water coordination or to the presence of reaction products within the catalyst's pores.23
To test whether it would be possible to avoid deactivation, the process was operated under continuous flow conditions;24 the setup was assembled as schematized in Fig. 3.
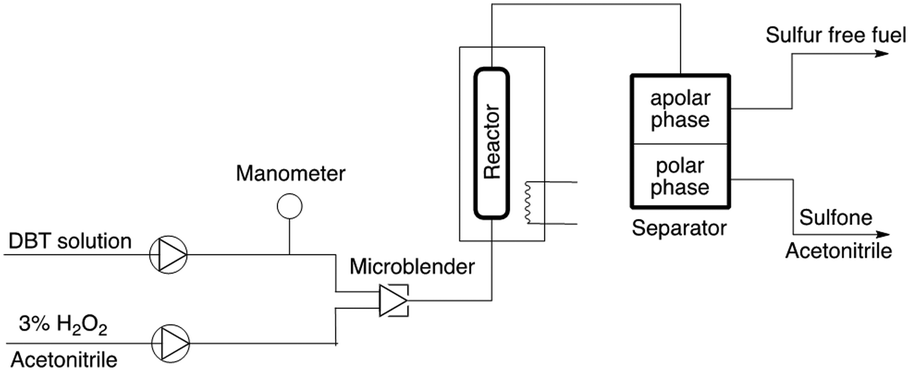 | ||
| Fig. 3 Schematic diagram of the setup for the continuous flow oxidation of DBT with hydrogen peroxide. | ||
Preliminary test showed the need to homogenize the DBT and hydrogen peroxide solutions prior to entering the reactor in order to obtain a better oxidative extraction.25 The reactor consists of a tubular glass column filled with TiO2@SiO2 dispersed with glass beads (≈1/2 w/w) heated at 60 °C. In a typical experiment, the reactor was filled with 1 gram of the catalyst, and the continuous reaction was run for 1 hour; the results are listed in Table 3.
| Entry | Model fuel flow (mL min−1) | AcCN/H2O2 flow (mL min−1) | Residence time (min) | Residual sulphurb (ppm) |
|---|---|---|---|---|
a Reaction conditions: DBT solution in dodecane (0.024 mol L−1), acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3% aqueous H2O2 3.75/1 v/v, 1 g of TiO2@SiO2, temperature: 60 °C, reaction time: 1 h.
b Determined by HPLC analysis.
c Reactor was washed with AcCN every 10 min for 10 min.
d Reactor was washed with AcCN/H2O2 every 10 min for 10 min.
e Reaction was carried out with silanized TiO2@SiO2 and the reactor was washed with AcCN/H2O2 every 20 min for 10 min.
f Reaction was carried with 5 g of silanized TiO2@SiO2 and the reactor was washed with the polar phase every 20 min for 10 min; the result is the average of 2 experiments. :3% aqueous H2O2 3.75/1 v/v, 1 g of TiO2@SiO2, temperature: 60 °C, reaction time: 1 h.
b Determined by HPLC analysis.
c Reactor was washed with AcCN every 10 min for 10 min.
d Reactor was washed with AcCN/H2O2 every 10 min for 10 min.
e Reaction was carried out with silanized TiO2@SiO2 and the reactor was washed with AcCN/H2O2 every 20 min for 10 min.
f Reaction was carried with 5 g of silanized TiO2@SiO2 and the reactor was washed with the polar phase every 20 min for 10 min; the result is the average of 2 experiments.
|
||||
| 1 | 0.04 | 0.14 | 5 | 225 |
| 2 | 0.04 | 0.14 | 10 | 160 |
| 3 | 0.08 | 0.28 | 5 | 138 |
| 4c | 0.08 | 0.28 | 5 | 63 |
| 5d | 0.08 | 0.28 | 5 | 86 |
| 6e | 0.06 | 0.22 | 10 | <LOD |
| 7f | 0.16 | 0.58 | 10 | 15 |
By carrying out the process with a residence time of 5 minutes, modest results were achieved (a final sulphur content of 225 ppm); moreover, when either the residence time was increased up to 10 minutes or the polar flow was doubled, only modest improvements were detected (a final sulphur content of 160 and 138 ppm). Additionally, by prolonging the reaction time from 1 to 5 hours, a substantial worsening was observed (544 ppm residual sulphur), confirming that also under continuous flow conditions, poisoning of the active sites is occurring.
With the aim of restoring the initial activity of the catalyst, the reactor was purged with polar solutions, namely acetonitrile, or with the polar solution of the feed (3% aqueous hydrogen peroxide in acetonitrile – Table 3, entries 4 and 5). A remarkable decrease in the final sulphur content was observed: 63 and 86 ppm, respectively.
These data support the hypothesis of polar species (DBT oxidation products) as catalyst poisoning agents, and suggest that a less polar material could prove to be more robust. Therefore, the silanization of TiO2@SiO2 by treatment with ethyltrimethoxysilane was considered. This procedure allows the conversion of the free silanols on the catalyst's surface to silanes, drastically reducing the polarity and the hydrophilicity of the material. According to the described hypothesis, the tailored catalyst became more robust without losing its outstanding activity, delivering a sulphur-free model fuel (Table 3, entry 6) and requiring fewer washing steps with the polar phase (every 20 min instead of 10 min). The reduced final sulphur content (15 ppm) has been accomplished also in an up-scaled experiment (Table 3, entry 7).
Conclusions
A TiO2@SiO2 catalyst has been developed and successfully applied in the deep desulfurization of a model fuel. The presence of anatase nanocrystals appear to be decisive in producing an active catalyst, and additional silanization of the material substantially increases its lifetime. Achieving the ODS with 0.77% hydrogen peroxide provides a useful application for in situ generated H2O2, which, to date, can be produced only in such a range of concentrations. The realization of an ODS demonstrator for the direct utilization of H2O2, produced through an electrochemical process, is currently ongoing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Fraunhofer Internal Program: Leitprojekt “Strom als Rohstoff”. The authors are grateful to Prof. D. Cauzzi and A. Polyzoidis for measuring and discussing XRD and B.E.T. data.Notes and references
- A. Stanislaus, A. Marafi and M. S. Rana, Catal. Today, 2010, 153, 1 CrossRef CAS.
- (a) J. M. Palomino, D. T. Tran, J. L. Hauser, H. Dong and S. R. J. Oliver, J. Mater. Chem. A, 2014, 2, 14890–14895 RSC; (b) J. M. Palomino, D. T. Tran, A. R. Kareh, C. Miller, J. M. V. Gardner, H. Dong and S. R. J. Oliver, J. Power Sources, 2015, 278, 141–148 CrossRef CAS.
- J. M. Campos-Martin, M. C. Capel-Sanchez, P. Perez-Presas and J. Fierro, J. Chem. Technol. Biotechnol., 2010, 85, 879 CrossRef CAS.
- F. M. Collins, A. R. Lucy and C. Sharp, J. Mol. Catal. A: Chem., 1997, 117, 397 CrossRef CAS.
- (a) D. Zhao, J. Wang and E. Zhou, Green Chem., 2007, 9, 1219 RSC; (b) L. Xia, H. Zhang, Z. Wei, Y. Jiang, L. Zhang, J. Zhao, J. Zhang, L. Dong, E. Li, L. Ruhlmann and Q. Zhang, Chem. – Eur. J., 2017, 23, 1920 CrossRef CAS PubMed; (c) H. Lu, J. Gao, Z. Jiang, Y. Yang, B. Song and C. Li, Chem. Commun., 2007, 150 RSC.
- (a) K.-G. Haw, W. A. W. A. Bakar, R. Ali, J.-F. Chong and A. A. A. Kadir, Fuel Process. Technol., 2010, 91, 1105 CrossRef CAS; (b) Y. Hu, Q. He, Z. Zhang, N. Ding and B. Hu, Chem. Commun., 2011, 47, 12194 RSC; (c) P. Yang, S. Zhou, Y. Du, J. Li and J. Lei, RSC Adv., 2016, 6, 53860 RSC; (d) A. E. S. Choi, S. Roces, N. Dugos and M.-W. Wan, Sustainable Environ. Res., 2016, 26, 184 CrossRef CAS.
- J. L. García-Gutiérrez, G. A. Fuentes, M. E. Hernández-Terán, F. Murrieta, J. Navarrete and F. Jiménez-Cruz, Appl. Catal., A, 2006, 305, 15 CrossRef.
- J. L. García-Gutiérrez, G. C. Laredo, P. García-Gutiérrez and F. Jiménez-Cruz, Fuel, 2014, 138, 118 CrossRef.
- (a) S. Ribeiro, C. M. Granadeiro, P. Silva, F. A. Almeida Paz, F. F. de Biani, L. Cunha-Silva and S. S. Balula, Catal. Sci. Technol., 2013, 3, 2404 RSC; (b) X.-S. Wang, L. Li, J. Liang, Y.-B. Huang and R. Cao, ChemCatChem, 2017, 9, 971 CrossRef CAS.
- H. Liu, S. Bao, Z. Cai, T. Xu, N. Li, L. Wang, H. Chen, W. Lu and W. Chen, Chem. Eng. J., 2017, 317, 1092 CrossRef CAS.
- (a) S. Du, F. Li, Q. Sun, N. Wang, M. Jia and J. Yu, Chem. Commun., 2016, 52, 3368 RSC; (b) A. Corma, P. Esteve, A. Martinez and S. Valencia, J. Catal., 1995, 152, 18 CrossRef CAS; (c) L. Kong, G. Li and X. Wang, Catal. Today, 2004, 93–95, 341 CrossRef CAS; (d) K. Leng, X. Li, G. Ye, Y. Du, Y. Sun and W. Xu, Catal. Sci. Technol., 2016, 6, 7615 RSC; (e) Y. Qin, S. Xun, L. Zhan, Q. Lu, M. He, W. Jiang, H. Li, M. Zhang, W. Zhu and H. Li, New J. Chem., 2017, 41, 569 RSC; (f) C. Shen, Y. J. Wang, J. H. Xu and G. S. Luo, Green Chem., 2016, 18, 771 RSC; (g) S. Du, X. Chen, Q. Sun, N. Wang, M. Jia, V. Valtchev and J. Yu, Chem. Commun., 2016, 52, 3580 RSC.
- R. Castillo, B. Koch, P. Ruiz and B. Delmon, J. Mater. Chem., 1994, 4, 903 RSC.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977 RSC.
- R. Dittmeyer, J.-D. Grunwaldt and A. Pashkova, Catal. Today, 2015, 248, 149 CrossRef CAS.
- B. Puértolas, A. K. Hill, T. García, B. Solsona and L. Torrente-Murciano, Catal. Today, 2015, 248, 115 CrossRef.
- R. Maggi, S. Chitsaz, S. Loebbecke, C. G. Piscopo, G. Sartori and M. Schwarzer, Green Chem., 2011, 13, 1121 RSC.
- Positionspapier Katalytische Oxidationsreaktionen als Schlüsseltechnologie, ed. Dechema, 2015 Search PubMed.
- S. K. Karmee, L. Greiner, A. Kraynov, T. E. Muller, B. Niemeijer and W. Leitner, Chem. Commun., 2010, 46, 6705 RSC.
- (a) S. Ma, G. Li and X. Wang, Chem. Eng. J., 2010, 156, 532 CrossRef CAS; (b) H. Song, G. Li, X. Wang and Y. Chen, Microporous Mesoporous Mater., 2011, 139, 104 CrossRef CAS.
- A. Y. Shan, T. I. M. Ghazi and S. A. Rashid, Appl. Catal., A, 2010, 389, 1 CrossRef CAS.
- D. T. On, S. Kaliaguine and L. Bonneviot, J. Catal., 1995, 157, 235 CrossRef CAS.
- H. Lempers and R. A. Sheldon, J. Catal., 1998, 175, 62 CrossRef CAS.
- (a) A. M. Cojocariu, P. H. Mutin, E. Dumitriu, F. Fajula, A. Vioux and V. Hulea, Appl. Catal., B, 2010, 97, 407 CrossRef CAS; (b) X. Kong, C. Zeng, X. Wang, J. Huang, C. Li, J. Fei, J. Li and Q. Feng, Sci. Rep., 2016, 6, 29049 CrossRef CAS PubMed , EP -.
- H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noel, Chem. Soc. Rev., 2016, 45, 83 RSC.
- S. Doherty, J. G. Knight, M. A. Carroll, J. R. Ellison, S. J. Hobson, S. Stevens, C. Hardacre and P. Goodrich, Green Chem., 2015, 17, 1559 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00192d |
| This journal is © The Royal Society of Chemistry 2018 |