
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and photocatalytic CO2 reduction performance of aminated coal-based carbon nanoparticles
Halidan Maimaiti *,
Abuduheiremu Awati,
Dedong Zhang,
Gunisakezi Yisilamu and
Bo Xu
*,
Abuduheiremu Awati,
Dedong Zhang,
Gunisakezi Yisilamu and
Bo Xu
Institute of Chemistry and Chemical Industry, Xinjiang University, Urumqi 830046, Xinjiang, China. E-mail: m15899160730@163.com
First published on 23rd October 2018
Abstract
To obtain high-efficiency, low-cost, environmentally friendly carbon-based photocatalytic material, we synthesized coal-based carbon dots with sp2 carbon structure and multilayer graphene lattice structure by the hydrogen peroxide (H2O2) oxidation method to strip nano-scale crystalline carbon in the coal structure and link with oxygen-containing groups such as the hydroxyl group. N, S co-doped aminated coal-based carbon nanoparticles (NH2-CNPs) were then obtained by thionyl chloride chlorination and ethylenediamine passivation. The physical properties and chemical structure of the synthesized NH2-CNPs were studied and the photocatalytic CO2 reduction performance was tested. The results show that NH2-CNPs are vesicle-type spherical particles with particle size of 42.16 ± 7.5 nm and have a mesoporous structure that is capable of adsorbing CO2. A defect structure was formed on the surface of the NH2-CNPs due to the doping of N and S elements, thereby significantly improving the ability to photogenerate electrons under visible light along with the ability to efficiently separate the photo-generated carriers. The photocatalytic reduction products of CO2 over NH2-CNPs were CH3OH, CO, C2H5OH, H2 and CH4. After 10 hours of reaction, the total amount of products was 807.56 μmol g−1 cat, the amount of CH3OH was 618.7 μmol g−1 cat, and the calculated selectivity for conversion of CO2 to CH3OH was up to 76.6%.
1. Introduction
The photocatalytic reduction of CO2 into hydrocarbon fuels has motivated the research and development of various potentially high-efficiency photocatalysts. The joint efforts of scholars around the world have resulted in new photocatalytic materials for photocatalytic reduction of CO2.1 However, most of the developed catalysts are noble metal- and metal oxide semiconductor-based photocatalysts, such as Ag, TiO2 and ZnO, etc., which have wide bandgaps, are expensive, show low efficiency for visible light utilization, and pollute the environment. As environmentally friendly substances, carbon-based nanomaterials such as graphene, carbon nanoparticles and activated carbon are abundant and easy to obtain, with low cost and low toxicity.2 Carbon nanoparticles (CNPs) are a class of zero-dimensional fluorescent nanomaterials that were accidentally discovered by Scrivens et al. during the separation and purification of single-wall carbon nanotubes.3 CNPs have fluorescence properties like traditional quantum dots and are also excellent electron acceptors and donors. After being excited by light, CNPs can generate electrons and holes and, at the same time, can suppress the combination of the photogenerated electrons and holes by their own surface defects.4–6 Therefore, they can be used as carriers for photocatalytic materials such as noble metals or semiconductors or can be used as a photocatalyst after being modified by hetero elements.7–11Coal is a natural and inexpensive carbon source, whose microstructure contains amorphous carbon regions consisting of aliphatic groups and nanoscale crystalline carbon formed by polyaromatic groups.12 The crystalline carbon regions are abundant and each crystalline carbon has the size of a quantum dot, suitable for obtaining CNPs by various methods.13,14 In our previous study,15–17 we prepared coal-based CNPs used HNO3 pretreated coal samples from the Wucaiwan coalmine in Xinjiang, China. We stripped crystalline carbon from the coal by acid oxidation and hydrogen peroxide oxidation methods and linked oxygen-containing groups such as hydroxyl groups on the CNPs. We found that the prepared coal-based CNPs had a multi-layered graphene structure with sp2 carbon and also contained some metal and non-metallic elements such as Fe, Ca, Al, Si, K, S, etc., thereby having the unique photoelectric properties of metal oxide semiconductor materials.
Modification of carbon nanomaterials and metal oxide semiconductors by doping (especially doping with non-metallic elements such as N, S) is a method for controlling the photoelectric properties of the products. For carbon nanomaterials, the sp2 hybrid carbon skeleton can be doped to obtain unpaired electrons, thereby generating a delocalized Π negative charge system and promoting the electron transport properties of the materials.18 For metal oxide semiconductors, creating states in the band gap that absorb visible light by doping can improve the visible light efficiency of the materials.19 Since coal-based CNPs have the characteristics of both general carbon nanomaterials and metal oxide semiconductors, they are expected to simultaneously increase their electron transport properties and visible light efficiency if they are doped with N and S elements. Thus, in this paper, sulfonyl chloride chlorination and ethylene diamine passivation were adopted to dope the synthesized CNPS with nitrogen and sulfur, and aminated coal-based carbon nanoparticles (NH2-CNPs) were obtained. The structure and photocatalytic CO2 reduction performance were investigated.
2. Experimental section
2.1 Raw materials and reagents
The coal samples were acquired from Wucaiwan coalmine in Zhundong coalfield, Xinjiang, China. The samples were ground and filtered by a 200 mesh, followed by drying at 105 °C for 4 h. The analytical grade sulfonyl chloride (SOCl2) was purchased from Aladdin Reagent Co. Ltd. The analytical grade diamine (EDA) was purchased from Tianjin Zhiyuan Chemical Reagent Co. Ltd. Analytical grade DMF, THF, H2O2, and HNO3 were purchased from common chemical stores.Ready-to-use dialysate bags (intercepted molecular weight of 500–1000, Spectrum Labs, U.S.) were purchased from Shanghai Tong Shan Biological Science and Technology Co., Ltd.
2.2 Preparation of coal-based CNPs
Reflux treatment was conducted on coal samples from Wucaiwan, Xinjiang with 2.5 M HNO3 for 24 h. The pre-treated samples were placed in a ball mill with distilled water, and ground for 3 h at room temperature and 600 rpm. After drying, 0.5 g of ultra-fine coal powder was dispersed in 10 mL of deionized water by sonication, then 30 mL of 30% H2O2 was added to the solution, followed by constant magnetic stirring and reaction at 85 °C for 3 h (5 mL of 30% H2O2 was added to the solution every 10 min for a total of four times). After the reaction stopped, the reaction liquid was transferred to a centrifuge tube for centrifuge separation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min). The supernatant was blown dry by N2, and the black-brown coal-based CNPs were obtained.
000 rpm for 10 min). The supernatant was blown dry by N2, and the black-brown coal-based CNPs were obtained.
2.3 Fabrication of NH2-CNPs
CNPs (1 g) were placed in a reaction bottle with 20 mL DMF and dispersed by sonication for 2 h at room temperature. After the CNPs were fully dispersed to form a suspension, 15 mL of SOCl2 was added to the solution, followed by reflux-stirring and reaction for 12 h at 65 °C. DMF and unreacted SOCl2 were removed by centrifugation. The reaction liquid was washed twice with THF to obtain the chloridized intermediate CNPs-Cl and mixed with 30 mL EDA for reflux reaction for 12 h at 130 °C. The temperature of the reaction liquid was decreased to 60 °C, and 30 mL of distilled water was added to the mixture with constant stirring for 3–4 hours. Next, the reaction liquid was centrifuged at 10000 rpm for 10 min. The supernatant was placed in a dialysate bag and dialyzed for 72 h (with constant water changing) and the aminated coal-based CNPs (NH2-CNPs) water dispersion was obtained. Finally, the NH2-CNPs were separated by blowing dry the solution with N2.
2.4 Structural characterization of the product
An H-600 transmission electron microscope (TEM) was employed to measure the morphology of the product. The elemental analysis was performed by an EDX energy spectrometer (HITACHI-SU8010). The thermogravimetric (TG-DTG) analysis was performed by a thermal analyzer (PE-DTA/1700) with temperature scanning range of 25–800 °C. The XRD patterns of the product were acquired by an X-ray diffractometer (M18XHF22-SRA). The experimental conditions were as below: Cu Kα radiation (λ = 0.154056 nm), and scan range 2θ = 10–80°. The confocal Raman spectra of the product were acquired by a Raman spectrometer (BRUKER VERTEX 70). The N2 adsorption–desorption performance was tested by a specific surface and aperture distribution meter (Autosorb-IQ2), and the specific surface and aperture distribution were calculated by the BET and BJH method. The FTIR spectrum of the product was obtained by a Fourier transform infrared spectrometer (FTIR, Bruker-EQUINOX 55). The surface states of the product were analyzed by an X-ray photoelectron spectrometer (XPS, ESCALAB 250). The CO2 temperature programmed desorption performance (CO2-TPD) was measured by a TP-5080 automatic adsorption–desorption dynamic analyzer. The ultraviolet-visible diffuse reflection spectrum (UV-vis DRS) of the product was measured by a UV-vis DRS spectrometer (Shimadzu UV-3100). The electrochemical impedance (EIS) and photoionization density were measured by an electrochemical workstation (CHI 660C).2.5 Photocatalytic CO2 reduction by visible light
NH2-CNPs (0.2 g) were added to 100 mL of 0.1 M NaOH solution and dispersed by sonication for 15 min in the dark. The suspension was transferred to a quartz bottle in a photocatalytic reduction reactor, as shown in Fig. 1.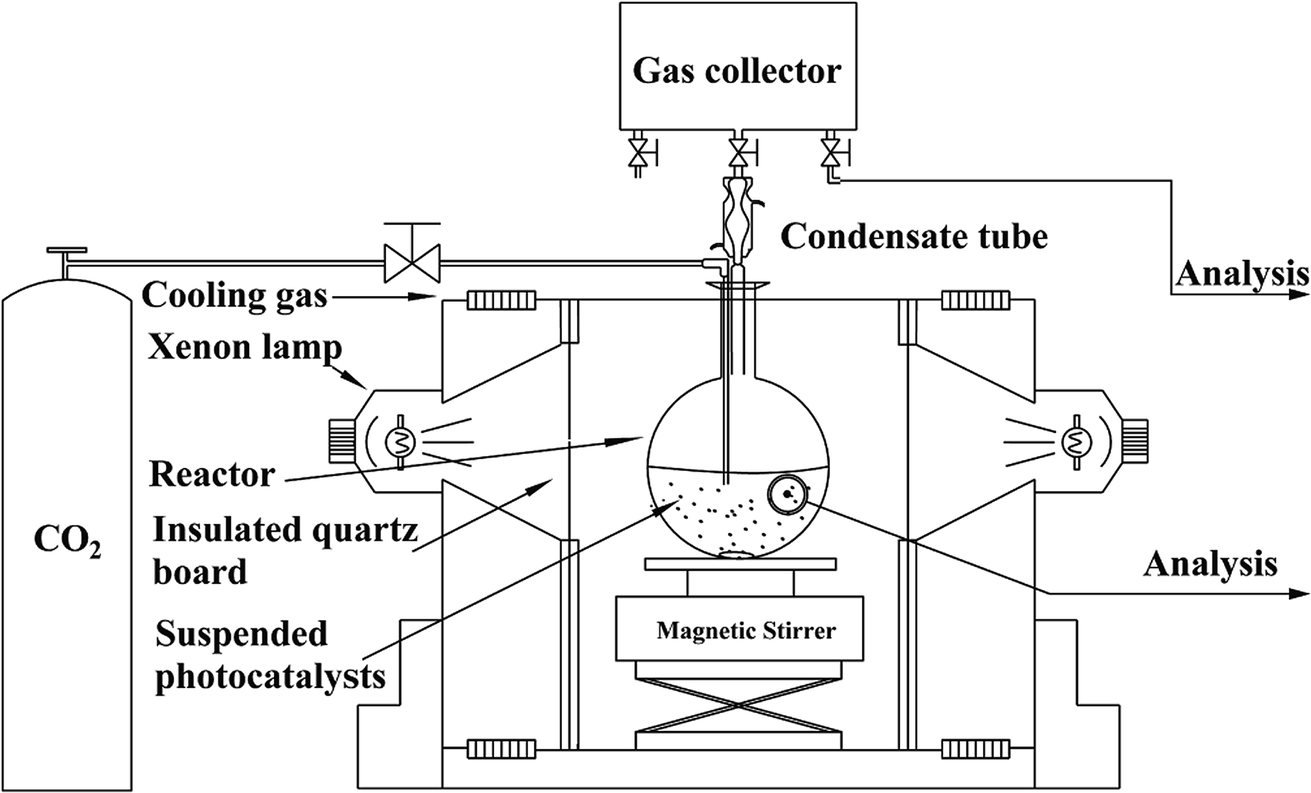 | ||
| Fig. 1 Schematic of the experimental setup for the photoreduction of CO2 with H2O. | ||
The system was placed under vacuum several times to remove air, and high-purity CO2 gas was flowed into the suspension at 1 atm with stirring (flow rate of CO2 was 20 mL min−1 for 30 min). Next, the whole reaction system was sealed, and illuminated with a 300 W xenon lamp (with a UV cut-off filter to obtain visible light with wavelength over 420 nm) to start the photocatalytic CO2/H2O reduction reaction. After reaction for 6 h, the liquid and gas products were acquired separately and subjected to 1H NMR and GC-MS to identify the species generated in the photocatalytic CO2 reduction by NH2-CNPs. The above CO2/H2O photocatalytic reduction was then repeated, and the samples were acquired every 2 h and injected into a gas chromatograph; the liquid and gas products were analyzed by the standard curve method (liquid phase: FID detector, PEG-21 filling column, gas phase: TCD detector, TDX-01 filling column).
3. Results and discussions
3.1 Structure of NH2-CNPs
According to Fig. 2a, coal powder has an irregular size distribution and shape, and it cannot be dispersed in water. However, the CNPs synthesized by H2O2 have good water dispersion and spherical shape with a diameter of 4.51 ± 0.82 nm (Fig. 2b). The high magnification TEM image (inset) shows that the surface of the CNPs has clear lattice strips. The spacing between the strips is 0.21 nm, close to the (100) plane of graphite,20 indicating that the coal-based CNPs have a graphite-like crystalline carbon structure. It can be seen from Fig. 2c that the NH2-CNPs are spherical particles with vesicle structure, formed by EDA linking and entangling with CNPs treated by SOCl2; the particle size of 42.16 ± 7.5 nm is larger compared to the CNPs. The NH2-CNPs can also be uniformly dispersed in water.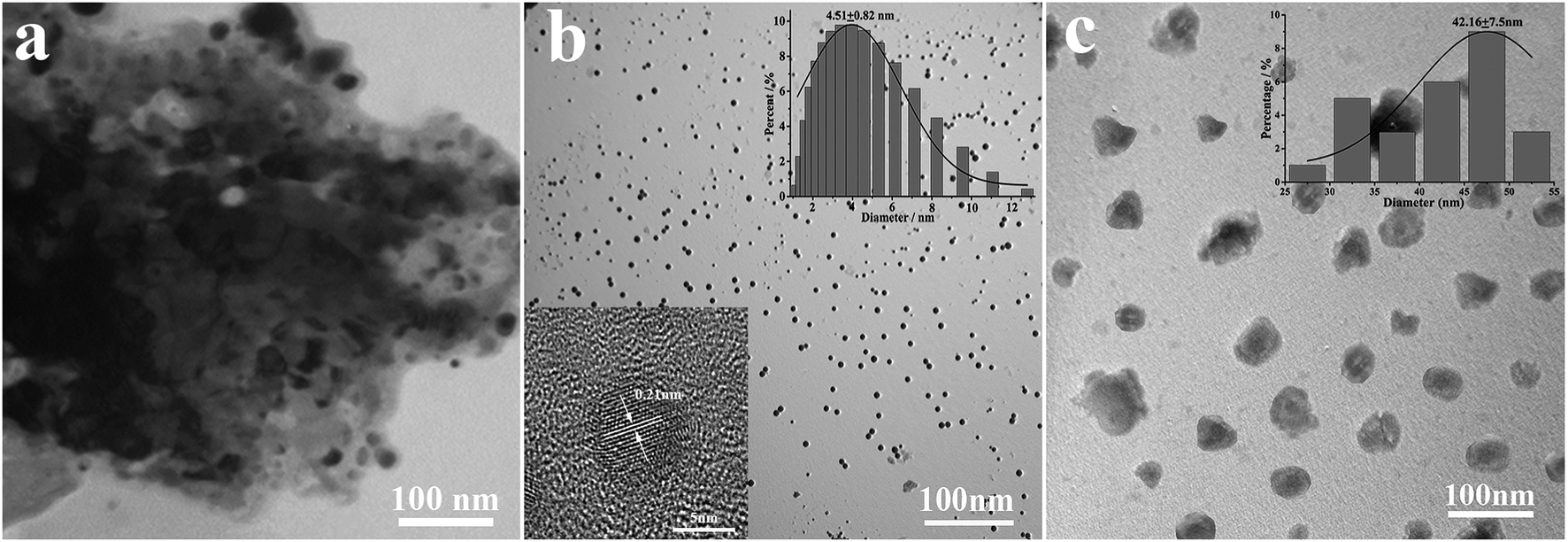 | ||
| Fig. 2 TEM images of (a) coal powder, (b) as-grown CNPs and (c) NH2-CNPs. | ||
From the EDX results for the coal powder, product CNPs and NH2-CNPs shown in Table 1, it can be seen that the O and N content in the coal-based CNPs are significantly higher compared to the coal powder, while the C content is decreased. This could be the result of the crystalline carbon being stripped out from the pulverized coal during the HNO3 pretreatment, ball milling, tank grinding, and H2O2 oxidation processes, and linked to N and O containing groups such as –NO3, –COOH, –OH. Compared to CNPs, NH2-CNPs have lower inorganic particle content and apparently higher N, S, and Cl content, which is due to the aminated reaction.
| Sample | Processing method | Elemental analysis,a w% | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| C | N | O | Al | Si | S | Ca | Fe | Cl | ||
| a By EDX. | ||||||||||
| Coal powder | Coal samples were ground and filtered by a 200 mesh | 75.04 | 0.58 | 17.79 | 0.89 | 3.35 | 0.52 | 0.71 | 1.12 | 0 |
| CNPs | As Section 2.2 | 47.34 | 6.65 | 43.40 | 0.24 | 1.36 | 0.14 | 0.28 | 0.59 | 0 |
| NH2-CNPs | As Section 2.3 | 40.64 | 14.43 | 36.45 | 0.18 | 0.93 | 6.31 | 0.17 | 0.21 | 0.68 |
Fig. 3A shows the TG-DTG results for coal powder, CNPs, and NH2-CNPs. We can see from the published literature21 that the first weight loss peak of coal powder under 100 °C is caused by water volatilization, and the second peak at 445 °C corresponds to the weight loss peak of devolatilization during the pyrolysis process. As the temperature increases, especially exceeding 600 °C, the weight loss is relatively flat, mainly caused by the polycondensation stage.
The coal-based CNPs also showed a peak for the loss of water under 100 °C but there was also a large weight loss peak at 288 °C, which was obviously different from the weight loss behavior of raw coal, corresponding to the volatilization of small molecular compounds and the decomposition or dehydration of oxygen-containing groups such as hydroxyl or carboxyl groups.22 This indicates that a large amount of active oxygen-containing functional groups appeared in the coal structure as a result of oxidative stripping by H2O2. In addition, the coal-based CNPs showed a weight loss peak between 500–600 °C, corresponding to the condensation and dehydrogenation of the aromatic structures in the coal; the peak position is lower than the pyrolysis temperature of the coal powder (600–800 °C). The possible reason is that the CNPs contain more oxygen-containing groups, which can induce pyrolysis of the organic compounds, thereby lowering the pyrolysis temperature.23 The results indicate that the coal-based CNPs obtained by the H2O2 oxidation method mainly contain aromatic structures linked by oxygen-containing groups.
The primary weight loss of NH2-CNPs has four stages. The first stage occurs below 100 °C with a loss amount of 2.84%, which is attributed to the water in the sample. The second stage happens at around 192 °C with a loss of 9.63%, corresponding to the heat cracking of EDA groups linked to the CNPs surface.24 In the third stage, the TG loss is 8.22%, which occurs at around 267 °C and can be attributed to the decomposition of oxygen-containing groups such as carboxyl and hydroxyl into CO2 or CO at high temperatures.25 The oxygen-containing groups were introduced in the synthesis process (such as coal pre-treatment by HNO3 or H2O2 oxidation). The fourth stage happened at near 551 °C with a loss amount of 28.28%, corresponding to the condensation dehydrogenation of the aromatic structure into semicoke.16 Above 600 °C, there was 57.30% undecomposed remnant, which was made up of the graphitized carbon and a small amount of inorganic particles in NH2-CNPs.26 The TG-DTG results further prove that the CNPs were successfully aminated. Fig. 3B shows the XRD patterns of coal powder, CNPs and NH2-CNPs. According to the figure, CNPs demonstrate a broad diffraction peak at 15–40°, corresponding to the (002) crystal plane of graphite;27 this characteristic peak does not appear in the XRD pattern of the coal sample. After amination, the corresponding graphite (002) peak in the NH2-CNPs shifted toward lower angle. This is because the surface of CNPs is linked to NH2– and other groups containing N and S, leading to the larger particle size of NH2-CNPs compared to CNPs.
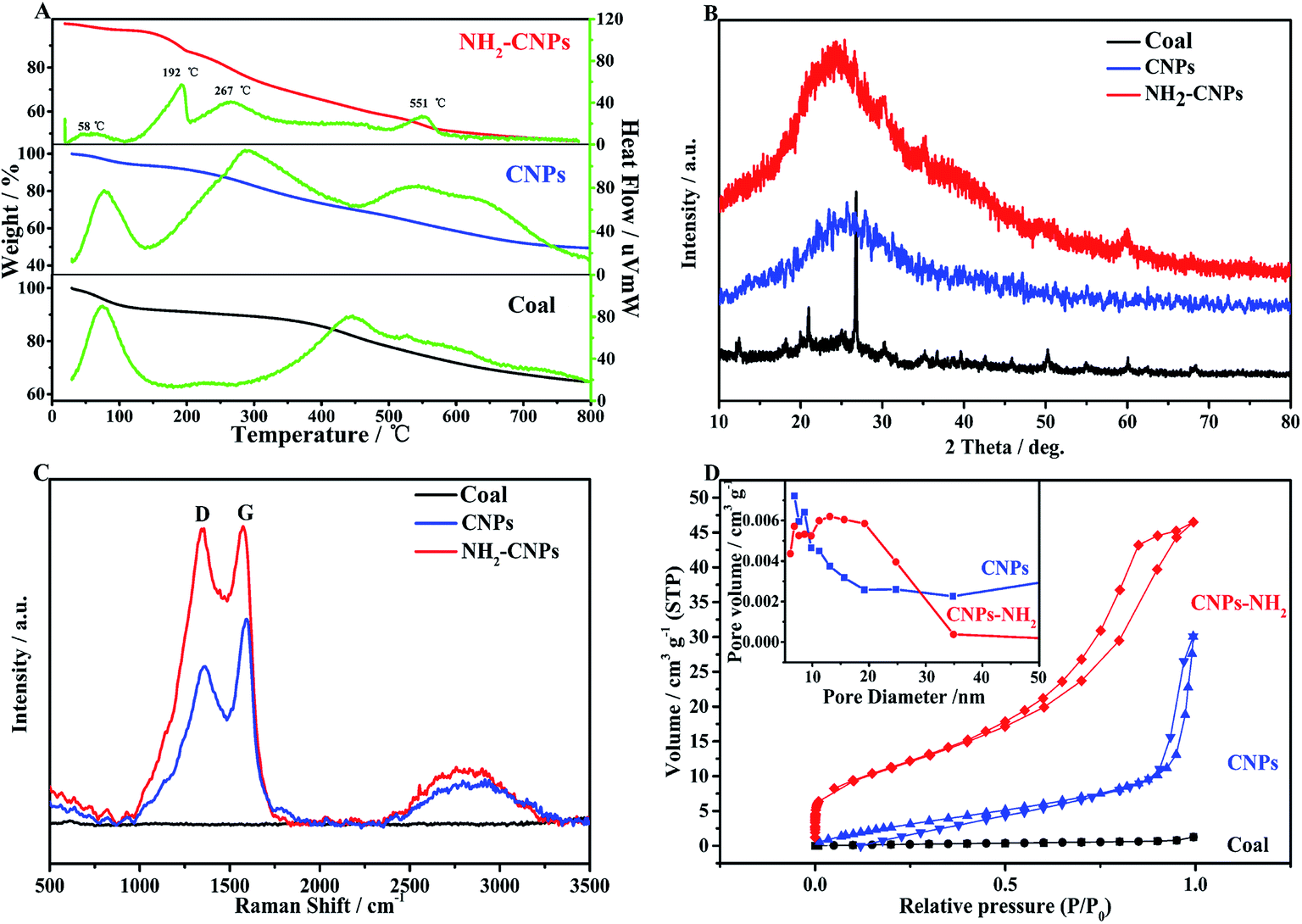 | ||
| Fig. 3 Physical characterizations of coal powder, CNPs, and NH2-CNPs. (A) TG-DTG; (B) XRD; (C) Raman; (D) BET. | ||
Raman spectrometry is an effective method for evaluating the structure and performance of carbon nanoparticles. As shown in the Raman spectrum in Fig. 3C, both CNPs and NH2-CNPs demonstrate two obvious peaks at 1350 and 1580 cm−1, corresponding to the D band and G band, respectively; however, the coal powder does not display the characteristic peaks. The D band is related to the defects in carbon materials, and the G band reflects the sp2 hybrid carbon atom content in carbon material.28 The ratio of the peak intensity (ID/IG) between the D band and G band can be used to evaluate the graphitization and defects of the carbon material. Using the raw data from Fig. 3C, the calculated ID/IG for CNPs and NH2-CNPs is 0.76 and 0.98, respectively. The ID/IG was apparently increased after the CNPs were aminated, suggesting their high microstructural integrity. The aminated material also has defect edges, which favor the effective separation of photogenerated electrons and holes,29 leading to enhanced photocatalytic CO2 reduction activity.
N2 adsorption–desorption tests were performed on coal powder, CNPs and NH2-CNPs. The specific surface area of the sample was calculated using the BET equation, and the pore size and distribution were calculated by the BJH equivalent cylinder model; the results are shown in Fig. 3D. The hysteresis loop of coal powder is not obvious, indicating that coal has a complex pore structure that cannot be described by the BET theory. The adsorption isotherms of the coal-based CNPs belong to I-type isotherms, indicating that CNPs are microporous structural materials. The N2 adsorption–desorption isotherms for NH2-CNPs are type-IV isotherms, indicating the mesoporous structure. From the pore distribution graph (inset), the CNPs have a pore size of 3–12 nm, and NH2-CNPs have small pores of 2–9 nm and large pores of 10–60 nm. The corresponding BET specific surface areas are 29.36 m2 g−1 for CNPs, and 172.65 m2 g−1 for NH2-CNPs. The total pore volume for CNPs and NH2-CNPs is 0.0114 cm3 g−1 and 0.320 cm3 g−1, respectively. Generally, a larger specific surface area and pore volume are favorable for reactant adsorption, which can significantly enhance the photocatalytic activity.30
Fig. 4A shows the infrared spectrum of CNPs and NH2-CNPs. The CNPs synthesized by H2O2 oxidation and separation show the stretching–vibration peak of the hydroxyl groups at 3400 cm−1 (curve a). At 1635 cm−1, the peak corresponds to the stretching–vibration absorption peak of –C–O–. The peak at 1350 cm−1 corresponds to the stretching–vibration peak of COO–. The peak at 1093 cm−1 is the stretching–vibration peak of C–O. The peak at 797 cm−1 is the stretching–vibration peak of C–NO2. The absorption peak at 605 cm−1 corresponds to inorganic particles. For NH2-CNPs, besides the above characteristic peaks, there are apparent amido (CONH–) peaks at 3245 and 2922 cm−1. At 1520, 1443 and 1219 cm−1, the absorption peaks correspond to the amide I band, amide II band and amide III band, respectively. The peak at 583 cm−1 corresponds to the O–S absorption peak. The results suggest that CNPs have successfully reacted with EDA to give the aminated product NH2-CNPs. There is no apparent absorption peak for inorganic particles or free ethylenediamine, suggesting the inorganic content was lowered and the EDA was removed by dialysis.
XPS can be used to detect the valence states of elements in nanomaterials. To further determine the product chemical structure, we used XPS to analyze the surface states of CNPs and NH2-CNPs, as shown in Fig. 4(B–F). According to the full XPS spectra of CNPs and NH2-CNPs (Fig. 4B), the two products show similar XPS patterns, with four characteristic peaks at 160.0, 286.0, 401.0 and 530.9 eV, corresponding to S2p, C1s, N1s and O1s. The results show that CNPs and NH2-CNPs are mainly composed of C, N, O, and S, and the N and S contents in NH2-CNPs are apparently higher as compared to those in CNPs. The O content of NH2-CNPs is lower than CNPs, possibly due to the replacement of oxygen-containing groups by NH2– groups. Fig. 4C shows the high-resolution XPS spectrum of C1s for CNPs and NH2-CNPs. The NH2-CNPs have five splitting peaks at 284.8 eV, 286.1 eV, 287.3 eV, 288.0 eV and 292.4 eV, corresponding to C–C, C–O, C–O–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and OC–O bonds, respectively. The C–C, C–O and CO bond have high content and are consistent with the high-resolution XPS spectrum of C1s for CNPs (inset). This shows that the skeleton of the NH2-CNPs comes from CNPs. Fig. 4D shows the high-resolution XPS spectrum of O1s for CNPs and NH2-CNPs. The CNPs have peaks at 530.0 eV and 532.8 eV, which correspond to the C–O and CO groups of the graphene structure (inset).31 NH2-CNPs have a single peak between 530.0 eV and 532.8 eV, which is attributed to C–O–H. This suggests that the oxygen content is lowered during amination. Fig. 4E shows the high-resolution XPS spectrum of N1s for NH2-CNPs. There are three spitting peaks at 398.28 eV, 399.98 eV and 401.68 eV. The peaks at 398.28 eV and 399.98 eV have low binding energies, corresponding to the amide bond (CONH) and an amine with a positive charge (–NH3+).32 The peak at 401.68 eV corresponds to graphite nitrogen, which is formed by replacing the carbon atoms with nitrogen atoms.33 Fig. 4F is the high-resolution XPS spectrum of S2p in NH2-CNPs. It can be seen that the products have four types of S structure. The peaks at 163.96 eV, 164.9 eV, 168.4 eV, and 169.7 eV correspond to C–S–C, CS, C–SO2–C, and C–SO3–C groups,34–37 respectively.
O and OC–O bonds, respectively. The C–C, C–O and CO bond have high content and are consistent with the high-resolution XPS spectrum of C1s for CNPs (inset). This shows that the skeleton of the NH2-CNPs comes from CNPs. Fig. 4D shows the high-resolution XPS spectrum of O1s for CNPs and NH2-CNPs. The CNPs have peaks at 530.0 eV and 532.8 eV, which correspond to the C–O and CO groups of the graphene structure (inset).31 NH2-CNPs have a single peak between 530.0 eV and 532.8 eV, which is attributed to C–O–H. This suggests that the oxygen content is lowered during amination. Fig. 4E shows the high-resolution XPS spectrum of N1s for NH2-CNPs. There are three spitting peaks at 398.28 eV, 399.98 eV and 401.68 eV. The peaks at 398.28 eV and 399.98 eV have low binding energies, corresponding to the amide bond (CONH) and an amine with a positive charge (–NH3+).32 The peak at 401.68 eV corresponds to graphite nitrogen, which is formed by replacing the carbon atoms with nitrogen atoms.33 Fig. 4F is the high-resolution XPS spectrum of S2p in NH2-CNPs. It can be seen that the products have four types of S structure. The peaks at 163.96 eV, 164.9 eV, 168.4 eV, and 169.7 eV correspond to C–S–C, CS, C–SO2–C, and C–SO3–C groups,34–37 respectively.
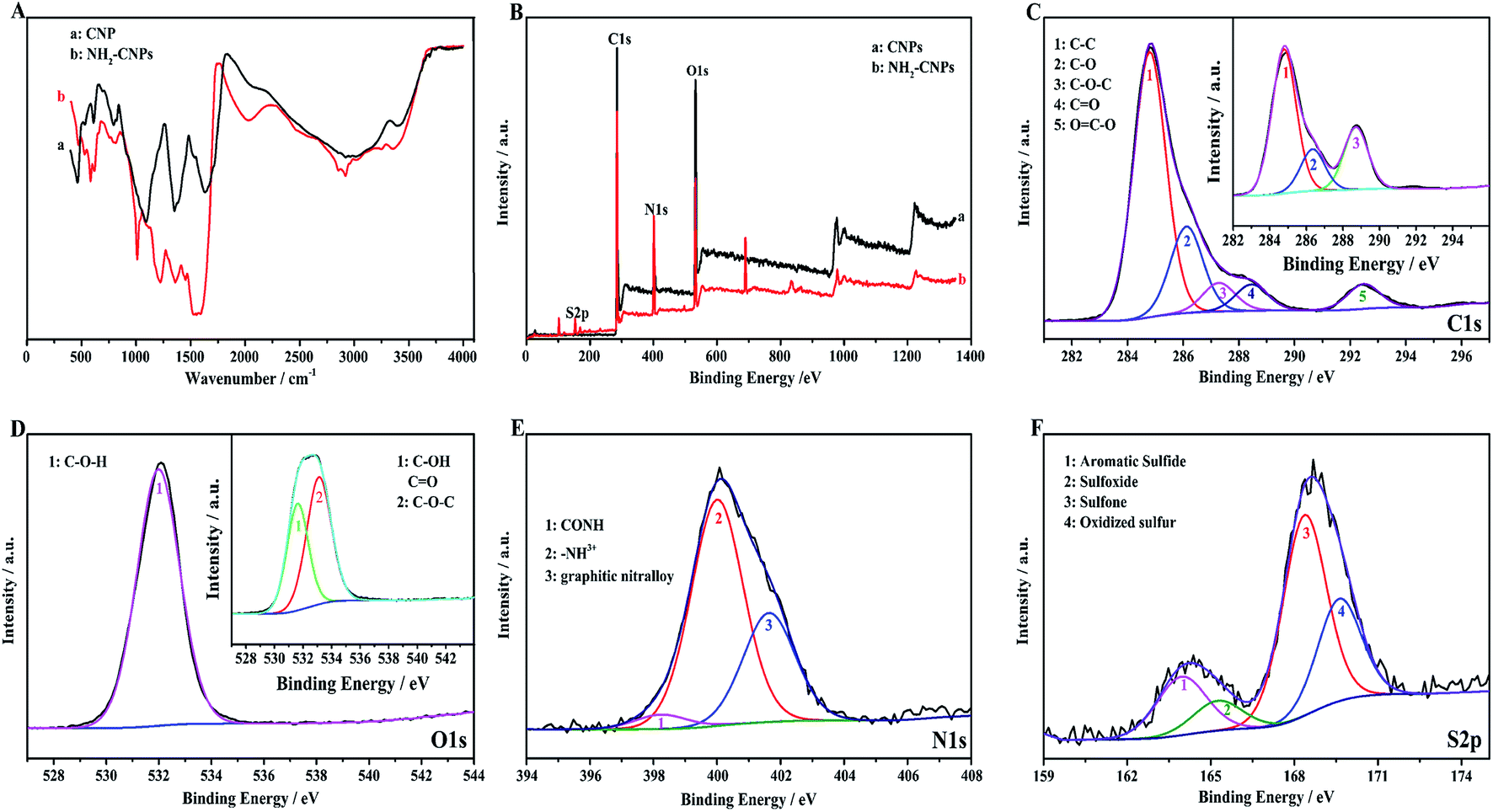 | ||
| Fig. 4 Structural characterizations of CNPs and NH2-CNPs: (A) FT-IR spectra; (B–F) XPS spectra. | ||
3.2 Photocatalytic CO2 reduction performance by NH2-CNPs
To confirm the photocatalytic products and selectivity of the NH2-CNPs, we used 1H NMR and GC-MS to analyze the liquid and gas products obtained after reaction for 6 hours.Fig. 5A shows the 1H NMR results for the liquid products from the photocatalytic CO2 reduction by NH2-CNPs. The single peak at δ = 3.34 ppm is the chemical shift of hydrogen protons at –CH3 of CH3OH. The triple peak at δ = 1.17 ppm and the quadruple peak at δ = 3.67 ppm represent the hydrogen protons at –CH2 of CH3CH2OH. The single peak at δ = 4.79 ppm is the D2O solvent. This result shows that the liquid products of photocatalytic CO2 reduction are CH3OH and C2H5OH.
We also identified the gas products by GC-MS. The results show that the gas products of photocatalytic CO2 reduction by NH2-CNPs contain H2, CO, and CH4. Fig. 5B shows the GC-MS results of the gas products when we used the isotopic marker 13CO2 as the only carbon source in the system for photocatalytic CO2 reduction. According to the results, m/z = 17 and 29 represent 13CH4 and 13CO, respectively, which indicate that the CO and CH4 come from photocatalytic CO2 reduction by NH2-CNPs.
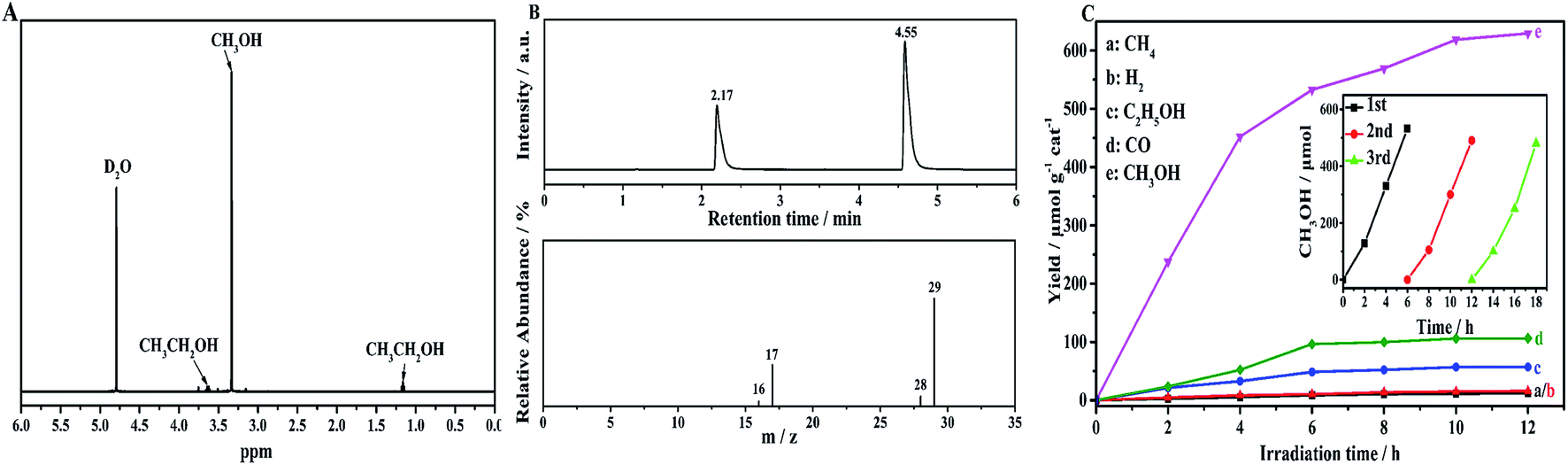 | ||
| Fig. 5 (A) 1H NMR spectra of the aqueous samples after 6 h of irradiation. (B) GC-MS spectral (m/z = 16, 17, 28, 29) analyses of the carbon source for the evolved CH4 and CO in the photocatalytic conversion of 13CO2 on NH2-CNPs. (C) Time-dependent photocatalytic conversion of CO2 into hydrocarbons over NH2-CNPs (the inset shows the long-term time curve for the recovered NH2-CNPs from the photocatalytic CO2 synthesis of CH3OH, with replacement of the solution and photocatalyst (recovered) every 6 h). | ||
Fig. 5C shows the time-dependent photocatalytic conversion of CO2 into hydrocarbons over NH2-CNPs. It can be seen that the yield of CH3OH is significantly higher than that of CH4, H2, C2H5OH and CO. In addition, the amount of the photocatalytic product was substantially unchanged after 10 hours of reaction, indicating that NH2-CNPs have a faster photocatalytic reaction rate. After the 10 hours of reaction, the total amount of products was 807.56 μmol g−1 cat, and the amount of CH3OH was 618.7 μmol g−1 cat. Therefore, the calculated selectivity for the conversion of CO2 to CH3OH was 76.6%. The inset of Fig. 5C shows the change in CH3OH yield produced by photocatalytic CO2 reduction using NH2-CNPs after three cycles. In each cycle, the reaction liquid was changed, and the illumination time was 6 h. The results show that after reusing 3 times, NH2-CNPs gave a stable performance with repeatable CH3OH production. The CH3OH yield in the third cycle was 89.4% of the yield in the first cycle. This suggests that the NH2-CNPs are suitable for photocatalytic CO2 reduction. We also examined the non-aminated CNPs photocatalytic CO2 reduction products. It was found that the CH3OH yield in the liquid products was only 91.70 μmol g−1 cat, about 1/6th of the CH3OH yield when using the NH2-CNPs photocatalyst.
To prove that the catalyst, illumination, and CO2 are the necessary requirements for photocatalytic CO2 reduction to CH3OH, CH4, H2, C2H5OH and CO, we conducted three control experiments under non-catalyst, non-illumination, and non-CO2 conditions. The experiments were performed according to Section 2.5, with illumination time (if any) of 6 h. No products were detected.
3.3 CO2 photocatalytic reduction mechanism for NH2-CNPs
To reveal that the NH2-CNPs have greater photocatalytic reduction of CO2 activity than CNPs, we first analyzed the type of basic sites and their distribution on the catalyst surface by CO2-TPD. The results are shown in Fig. 6A. Generally, the area of the desorption peak reflects the number of active sites on the catalyst surface, while the temperature of the desorption peak reflects the adsorption intensity of the corresponding species at active sites.38 NH2-CNPs exhibited a weak α peak in the low-temperature zone (162 °C), which is generally considered as the CO2 physical adsorption peak of the catalyst. At around 350 °C, the CO2-TPD spectra of both CNPs and NH2-CNPs showed a strong β peak, corresponding to a medium-strong adsorption of catalyst, indicating that both CNPs and NH2-CNPs have CO2 adsorption. NH2-CNPs demonstrated a broad γ peak at 460 °C, suggesting strong CO2 adsorption sites in NH2-CNPs. In addition, the adsorption peak area of NH2-CNPs is apparently larger than CNPs. Based on the above results, it can be concluded that NH2-CNPs have improved basic site types and distribution compared to CNPs. This is an important reason why NH2-CNPs have higher activity than CNPs in CO2 photocatalytic reduction.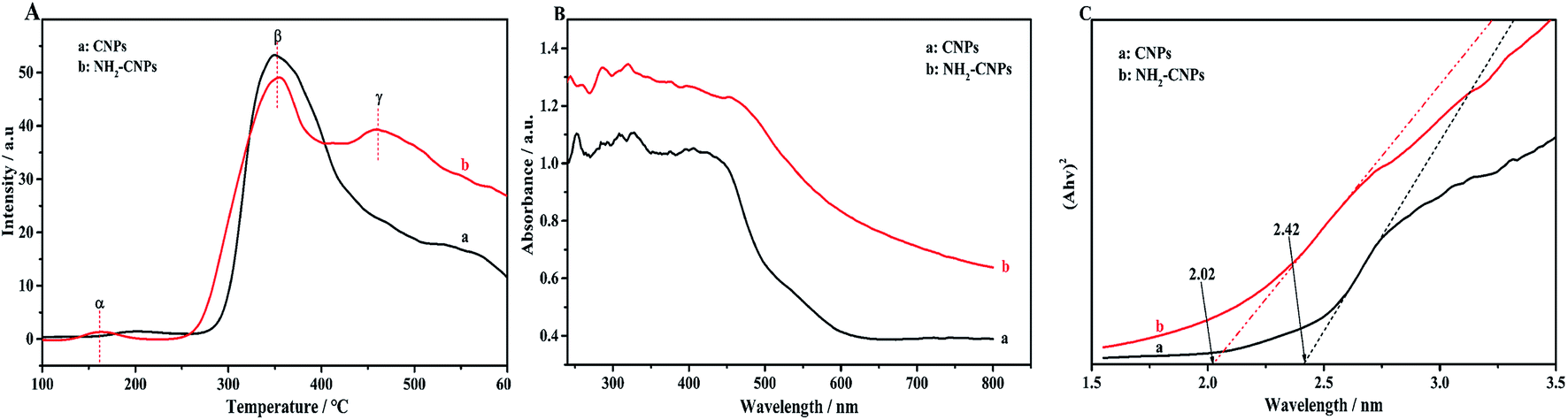 | ||
| Fig. 6 (A) The CO2-TPD profiles of the CNPs (a) and NH2-CNPs (b), (B) DR UV-vis spectra of the CNPs (a) and NH2-CNPs (b). (C) The calculation of the band gap of the CNPs (a) and NH2-CNPs (b) based on the (Ahv)2 versus hv curve. | ||
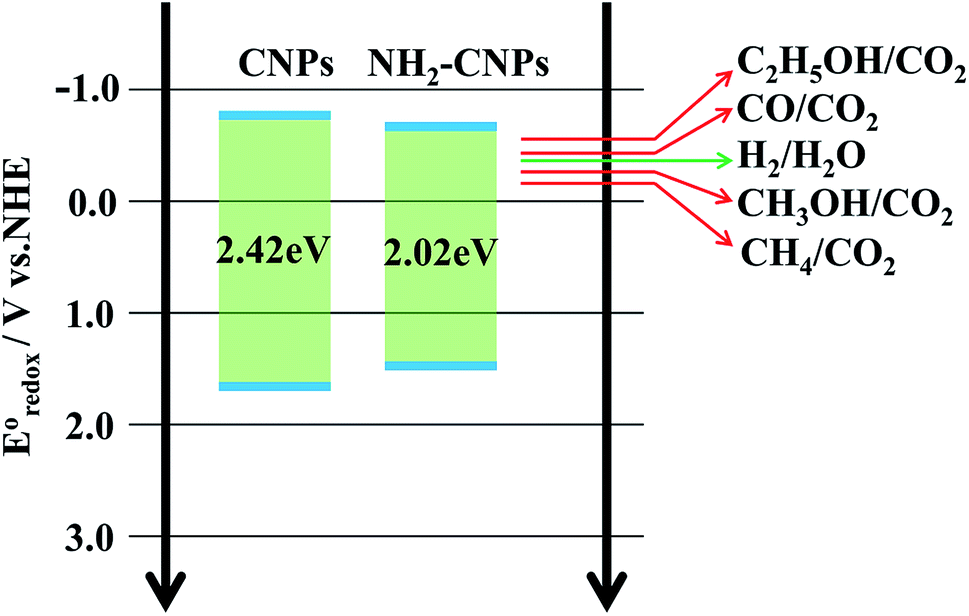
The photocatalytic process often involves the light excitation of the catalyst, photocarrier separation, migration, and recombination, as well as the generation of active species and the subsequent oxidation and reduction reactions. Among them, the absorption range and intensity are two important factors affecting the CO2 photocatalytic reduction performance. Fig. 6B shows the measured UV-vis spectra of CNPs and NH2-CNPs. According to the figure, both materials can absorb visible light, while NH2-CNPs have a broader absorption range and intensity than CNPs. Thus, after being aminated, the CNPs' response to visible light is enhanced. In this work, we employed excitation light with a wavelength greater than 420 nm for the photocatalytic reaction. The highest photon energy was 2.95 eV. The incident photon energy must be equal to or greater than the material band gap to excite electrons from the VB to CB and generate holes in the VB for the subsequent redox reaction.39 Thus, to prove that the CNPs and NH2-CNPs can be excited by visible light, we measured their DR UV-vis curves, and used eqn (1) to calculate and plot the relationship between (Ahv)2 and hv.40 By extending the straight-line portion of the curve to the x-axis, the energy band gaps (Eg) of CNPs and NH2-CNPs were obtained, which were 2.42 and 2.02 eV, respectively (Fig. 6C). This further suggests that NH2-CNPs have stronger visible light absorption than CNPs to generate electrons and holes. This is another important reason why NH2-CNPs have higher activity than CNPs in CO2 photocatalytic reduction.
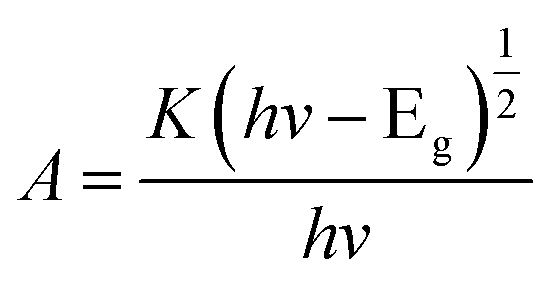 | (1) |
After the catalyst is excited by light, the photogenerated electrons and holes have two pathways: (i) migrating to the surface of the catalyst and triggering the subsequent photocatalytic reduction reaction; (ii) recombining with each other. Apparently, to enhance the photogenerated electron–hole pair separation efficiency, the catalyst surface must have clear pathways for electrons. It is generally considered that the semi-circle in the EIS curve represents the transfer resistance for electrons in the pores of the composite material. Semi-circles with larger diameters correspond to poorer electron transfer capability. Thus, to investigate the separation efficiency of photogenerated electron–hole pairs, we measured the EIS curves for CNPs and NH2-CNPs under the open circuit potential; the results are shown in Fig. 7A. According to the Nyquist curve, after the CNPs are aminated, the impedance of NH2-CNPs is significantly lower compared to the CNPs. This is likely due to the defects in NH2-CNPs, which can trap more photogenerated electrons via the quantum confinement effect, enhancing the separation efficiency of electron–hole pairs in the composite catalyst, leading to lower sample impedance.
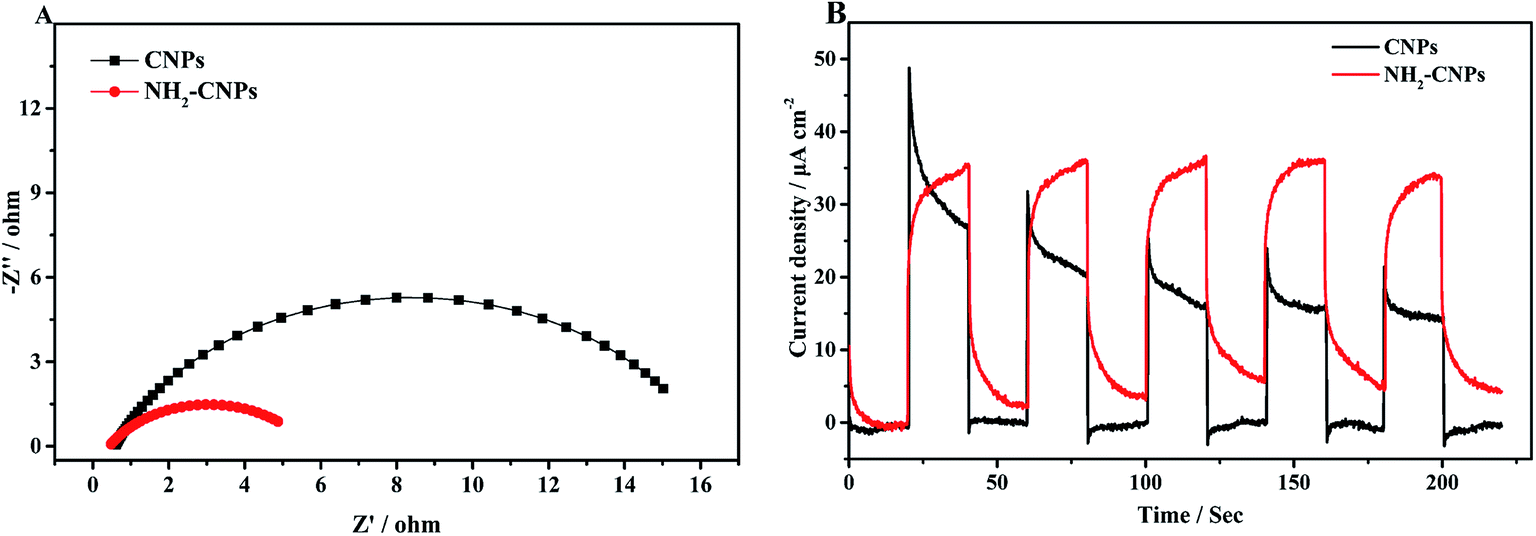 | ||
| Fig. 7 (A) Nyquist plots of the CNPs (a) and NH2-CNPs (b). (B) The transient photocurrent responses of the CNPs (a) and NH2-CNPs (b) in 0.5 M Na2SO4 aqueous solution under visible-light (λ > 420 nm) irradiation. | ||
The transient photocurrent–time curve is an effective way to study the transfer of photogenerated carriers on the surface of the catalyst. Fig. 7B shows the photocurrent–time curve of CNPs and NH2-CNPs electrodes in a 0.5 M Na2SO4 solution with illumination by a xenon lamp (λ > 420 nm) under open circuit potential and at room temperature. There are three differences between the transient photocurrent–time curves of CNPs and NH2-CNPs: (i) NH2-CNPs have larger steady-state photocurrent density than CNPs. This suggests that NH2-CNPs can excite more photogenerated electrons for photocurrent compared to CNPs. (ii) Upon turning on the light, the photocurrent of CNPs increases quickly, while the photocurrent of NH2-CNPs increases slowly. This is because upon illumination, CNPs can be quickly excited and generate electrons, while part of the carriers in NH2-CNPs are trapped and cannot transfer in time, leading to a slow increase to the maximum.41 (iii) Upon cutting-off the light, the photocurrent of CNPs quickly drops to zero, while the photocurrent of NH2-CNPs decreases exponentially to a “tail” rather than zero. This is because the previously trapped charges are gradually released, generating low current and slowing down the decrease of the photocurrent to a non-zero value.42 Based on the transient photocurrent–time curve, it can be concluded that NH2-CNPs can generate more photocarriers, and due to surface defects, the photo-generated carriers can be effectively separated. Even after the light is turned off, NH2-CNPs can still release a small amount of charges for photocatalytic CO2 reduction. This is another reason that NH2-CNPs have better photocatalytic performance.
Based on the above analysis, we concluded that the mechanism of photocatalytic CO2/H2O reduction by NH2-CNPs is as shown in Fig. 8. The analysis of UV-vis DRS and band gap curves revealed that the band gap of NH2-CNPs decreased from 2.42 eV for CNPs to 2.02 eV, indicating that the NH2-CNPs have enhanced response to visible light and can generate more electrons and holes (eqn (2)). The PL, EIS, and I–T results show that the photogenerated electron–hole pairs in NH2-CNPs can be easily separated, and the electrons can be transferred to the surface of the catalyst for the CO2 reduction reaction (eqn(3)–(8)). The reduction potential of NH2-CNPs and the generated electron quantities are likely to be the same as the reduction potential for CH3OH production and the required electron amount, respectively. Therefore, the primary product of photocatalytic CO2 reduction by NH2-CNPs is CH3OH.
| NH2-CNPs + hv → e− + h+ | (2) |
| H2O + e− → H+ + OH− | (3) |
| 2H+ + e−→ H2, E0redox = −0.41 V | (4) |
| CO2 + 2H+ + 2e− → CO + H2O, E0redox = −0.53 V | (5) |
| CO2 + 6H+ + 6e− → CH3OH + H2O, E0redox = −0.38 V | (6) |
| CO2 + 8H+ + 8e− → CH4+ 2H2O, E0redox = −0.24 V | (7) |
| 2CO2 + 12H+ + 12 e− → C2H5OH + 3H2O, E0redox = −0.74 V | (8) |
 | ||
| Fig. 8 Schematic of the mechanism of the photocatalytic CO2 reduction with water on NH2-CNPs. | ||
4. Conclusions
Using HNO3 pre-treated coal samples from Wucaiwan, Xinjiang, China, nano-scale crystalline carbon was fabricated from coal by the hydrogen peroxide oxidation method. After being linked with oxygen-containing groups such as hydroxyl, the coal-based CNPs with sp2 carbon architecture and multilayer graphene fragmentation structure were obtained. Next, sulfonyl chloride chlorination and ethylenediamine passivation were adopted to dope the CNPs with nitrogen and sulfur and produce aminated coal-based carbon nanoparticles (NH2-CNPs) for photocatalytic CO2 reduction. The results show that NH2-CNPs possess enhanced visible light response, leading to more photogenerated electrons that contribute to the photocurrent. Moreover, since the surface of NH2-CNPs has a defect structure, the photogenerated carriers can be effectively separated. Even upon turning off the illumination, the NH2-CNPs can still release a small amount of charge for photocatalytic CO2 reduction, improving the overall catalytic performance. This work contributes to better controlling the optoelectronic properties of carbon nanomaterials, and provides a new approach to further utilize coal resources for high-performance materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support of the National Science Foundation of China (21663028) and the Key Laboratory of Coal Clean Conversion & Chemical Engineering Process of Xinjiang Uygur Autonomous Region.References
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- H. Sun and S. Wang, Energy Fuels, 2014, 28, 22–36 CrossRef CAS.
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS PubMed.
- X. Wang, L. Cao, F. Lu, M. J. Meziani, H. Li, G. Qi, B. Zhou, B. A. Harruff, F. Kermarrec and Y.-P. Sun, Chem. Commun., 2009, 3774–3776, 10.1039/b906252a.
- L. Cao, X. Wang, M. J. Meziani, F. Lu, H. Wang, P. G. Luo, Y. Lin, B. A. Harruff, L. M. Veca, D. Murray, S.-Y. Xie and Y.-P. Sun, J. Am. Chem. Soc., 2007, 129, 11318–11319 CrossRef CAS PubMed.
- K. A. S. Fernando, S. Sahu, Y. Liu, W. K. Lewis, E. A. Guliants, A. Jafariyan, P. Wang, C. E. Bunker and Y.-P. Sun, ACS Appl. Mater. Interfaces, 2015, 7, 8363–8376 CrossRef CAS PubMed.
- S. Hu, R. Tian, L. Wu, Q. Zhao, J. Yang, J. Liu and S. Cao, Chem.–Asian J., 2013, 8, 1035–1041 CrossRef CAS PubMed.
- R. Wang, K.-Q. Lu, Z.-R. Tang and Y.-J. Xu, J. Mater. Chem. A, 2017, 5, 3717–3734 RSC.
- S. Hu, Q. Chang, K. Lin and J. Yang, Carbon, 2016, 105, 484–489 CrossRef CAS.
- J. Di, J. Xia, X. Chen, M. Ji, S. Yin, Q. Zhang and H. Li, Carbon, 2017, 114, 601–607 CrossRef CAS.
- H. Yu, R. Shi, Y. Zhao, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2016, 28, 9454–9477 CrossRef CAS PubMed.
- R. Ye, C. Xiang, J. Lin, Z. Peng, K. Huang, Z. Yan, N. P. Cook, E. L. Samuel, C. C. Hwang, G. Ruan, G. Ceriotti, A. R. Raji, A. A. Marti and J. M. Tour, Nat. Commun., 2013, 4, 2943 CrossRef PubMed.
- C. Hu, C. Yu, M. Li, X. Wang, J. Yang, Z. Zhao, A. Eychmuller, Y. P. Sun and J. Qiu, Small, 2014, 10, 4926–4933 CrossRef CAS PubMed.
- S. Hu, Z. Wei, Q. Chang, A. Trinchi and J. Yang, Appl. Surf. Sci., 2016, 378, 402–407 CrossRef CAS.
- B. Zhang, H. Maimaiti, Y. Zhang and M. Wei, Int. J. Coal Sci. Technol., 2017, 4, 342–353 CrossRef.
- Z. Dedong, H. Maimaiti, A. Awati, G. Yisilamu, S. Fengchang and W. Ming, Chem. Phys. Lett., 2018, 700, 27–35 CrossRef.
- F. Sun, H. Maimaiti, Y.-e. Liu and A. Awati, Int. J. Energy Res., 2018, 1–12 Search PubMed.
- K. A. Kurak and A. B. Anderson, J. Phys. Chem. C, 2009, 113, 6730–6734 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS PubMed.
- H. Krishna Sadhanala, A. Maddegalla and K. K. Nanda, New J. Chem., 2017, 41, 13742–13746 RSC.
- L. Shi, Q. Liu, X. Guo, W. Wu and Z. Liu, Fuel Process. Technol., 2013, 108, 125–132 CrossRef CAS.
- A. J. Fletcher, Y. Uygur and K. M. Thomas, J. Phys. Chem. C, 2007, 111, 8349–8359 CrossRef CAS.
- B. Ruiz, J. B. Parra, J. A. Pajares and J. J. Pis, J. Anal. Appl. Pyrolysis, 2006, 75, 27–32 CrossRef CAS.
- Z. Yun-fei, H. Maimaiti and Z. Bo, RSC Adv., 2017, 7, 2842–2850 RSC.
- Z. Li, L. Zhu, W. Wu, S. Wang and L. Qiang, Appl. Catal., B, 2016, 192, 277–285 CrossRef CAS.
- A. Sachdev and P. Gopinath, Analyst, 2015, 140, 4260–4269 RSC.
- Y. Badhe, K. Balasubramanian and R. Gupta, RSC Adv., 2015, 5, 23622–23634 RSC.
- S. Ms and S. Sankararaman, J. Mater. Sci. Nanotechnol., 2017, 5 DOI:10.15744/2348-9812.5.103.
- X. Wang, X. Zhao, D. Zhang, G. Li and H. Li, Appl. Catal., B, 2018, 228, 47–53 CrossRef CAS.
- J. Jin, J. Yu, D. Guo, C. Cui and W. Ho, Small, 2015, 11, 5262–5271 CrossRef CAS PubMed.
- T. Ishizaki, Y. Wada, S. Chiba, S. Kumagai, H. Lee, A. Serizawa, O. L. Li and G. Panomsuwan, Phys. Chem. Chem. Phys., 2016, 18, 21843–21851 RSC.
- M.-X. Wang, Z. Guo, Z.-H. Huang and F. Kang, Catal. Commun., 2015, 62, 83–88 CrossRef CAS.
- G. P. Mane, S. N. Talapaneni, K. S. Lakhi, H. Ilbeygi, U. Ravon, K. Al-Bahily, T. Mori, D. H. Park and A. Vinu, Angew. Chem., Int. Ed. Engl., 2017, 56, 8481–8485 CrossRef CAS PubMed.
- T. Zhang, C. Li, Y. Gu, X. Yan, B. Zheng, Y. Li, H. Liu, N. Lu, Z. Zhang and G. Feng, Talanta, 2017, 165, 143–151 CrossRef CAS PubMed.
- S. Bian, C. Shen, Y. Qian, J. Liu, F. Xi and X. Dong, Sens. Actuators, B, 2017, 242, 231–237 CrossRef CAS.
- S. C. Wuang, K. G. Neoh, E. T. Kang, D. W. Pack and D. E. Leckband, Adv. Funct. Mater., 2006, 16, 1723–1730 CrossRef CAS.
- Y.-T. Yang, X.-X. Yang, Y.-T. Wang, J. Luo, F. Zhang, W.-J. Yang and J.-H. Chen, Fuel, 2018, 219, 166–175 CrossRef CAS.
- T. Witoon, N. Kachaban, W. Donphai, P. Kidkhunthod, K. Faungnawakij, M. Chareonpanich and J. Limtrakul, Energy Convers. Manage., 2016, 118, 21–31 CrossRef CAS.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- F. Wang, P. Chen, Y. Feng, Z. Xie, Y. Liu, Y. Su, Q. Zhang, Y. Wang, K. Yao, W. Lv and G. Liu, Appl. Catal., B, 2017, 207, 103–113 CrossRef CAS.
- H. Zhang, L. Zhao, F. Geng, L.-H. Guo, B. Wan and Y. Yang, Appl. Catal., B, 2016, 180, 656–662 CrossRef CAS.
- Z. Li, J. Zhang, Y. Li, S. Zhao, P. Zhang, Y. Zhang, J. Bi, G. Liu and Z. Yue, Biosens. Bioelectron., 2018, 99, 251–258 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |