
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-step preparation of novel 1-(N-indolyl)-1,3-butadienes by base-catalysed isomerization of alkynes as an access to 5-(N-indolyl)-naphthoquinones†
C. M. Pis Diez ab,
J. F. Fernandezab,
G. Di Venosac,
A. Casasc,
R. Pis Diez*d and
J. A. Palermo*ab
ab,
J. F. Fernandezab,
G. Di Venosac,
A. Casasc,
R. Pis Diez*d and
J. A. Palermo*ab
aUniversidad de Buenos Aires, Departamento de Química Orgánica, Facultad de Ciencias Exactas y Naturales, Ciudad Universitaria, Pabellón 2 – (1428), Buenos Aires, Argentina. E-mail: palermo@qo.fcen.uba.ar
bCONICET-Universidad de Buenos Aires, Unidad de Microanálisis y Métodos Físicos en Química Orgánica (UMYMFOR), Buenos Aires, Argentina
cCentro de Investigaciones sobre Porfirinas y Porfirias (CIPYP) CONICET and Hospital de Clínicas José de San Martin, Universidad de Buenos Aires, Córdoba 2351, 1er subsuelo, Ciudad de Buenos Aires, 1120AAF Buenos Aires, Argentina
dCEQUINOR (CONICET-CCT-La Plata, UNLP), Bvd 120 N1465, 1900 La Plata, Argentina
First published on 23rd October 2018
Abstract
A series of novel 1-(N-indolyl)-1,3-butadienes, as (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixtures of the (E) and (Z) dienes, was prepared in one step by base-catalysed isomerization of N-alkylindoles with a terminal butyne chain. The reaction conditions are mild, and in all cases the yields were very high (>90%). The (E) and (Z) dienes were separable by preparative TLC and could be fully characterized. This isomerization proceeded readily in the case of a butynyl chain, but didn't take place with a pentynyl chain. A mechanism was proposed for this reaction, based on previous studies on the isomerization of alkynes in basic media, and a key intermediate that supports the proposed mechanism could be isolated and fully characterized. A theoretical study of the proposed mechanism was performed by computational methods and the results validated the proposal. The reactivity of the synthesized dienes was studied in Diels–Alder reactions with p-benzoquinone, to obtain a small library of new 5-(N-indolyl)-1,4-naphthoquinones.The lack of reactivity in the case of the (Z) isomers was explained by calculation of the rotational curves of the central bond of the (Z) and (E) dienes. Finally, the cytotoxicity of the new 5-(N-indolyl)-1,4-naphthoquinones was tested against a panel of three cell lines.
:1) mixtures of the (E) and (Z) dienes, was prepared in one step by base-catalysed isomerization of N-alkylindoles with a terminal butyne chain. The reaction conditions are mild, and in all cases the yields were very high (>90%). The (E) and (Z) dienes were separable by preparative TLC and could be fully characterized. This isomerization proceeded readily in the case of a butynyl chain, but didn't take place with a pentynyl chain. A mechanism was proposed for this reaction, based on previous studies on the isomerization of alkynes in basic media, and a key intermediate that supports the proposed mechanism could be isolated and fully characterized. A theoretical study of the proposed mechanism was performed by computational methods and the results validated the proposal. The reactivity of the synthesized dienes was studied in Diels–Alder reactions with p-benzoquinone, to obtain a small library of new 5-(N-indolyl)-1,4-naphthoquinones.The lack of reactivity in the case of the (Z) isomers was explained by calculation of the rotational curves of the central bond of the (Z) and (E) dienes. Finally, the cytotoxicity of the new 5-(N-indolyl)-1,4-naphthoquinones was tested against a panel of three cell lines.
Introduction
Substituted 1,3-butadienes are versatile building blocks in organic synthesis, and the preparation of these compounds is still an active field in organic chemistry.1 However, to date there have been no reports on the preparation of 1-(N-indolyl)-1,3-butadienes. During the course of a project of structural diversification of indole alkaloids by means of click reactions, it was necessary to prepare N-substituted indoles with alkyl chains bearing terminal triple bonds. The typical N-substitution reaction in DMSO and base (with the use of NaOH instead of NaH) with 5-Cl-1-pentyne as alkylating agent proceeded smoothly. However, when the reaction was attempted using 4-Br-1-butyne, the outcome was different, and a (1:1) mixture of (Z) and (E) 1-N-indolyl-1,3-butadienes was obtained instead (Fig. 1). Taking into account that the indole moiety is a frequent motif in many natural products and bioactive drugs,2 the use of this reaction was explored for the preparation and characterization of a series of dienes with different substituted indoles or carbazole at C-1 (1–7a,b Fig. 2). Furthermore, the reactivity of these dienes in Diels Alder reactions was explored using benzoquinone as dienophile to obtain a family of new cytotoxic 5-N-indolyl-1,4-naphthoquinones.3
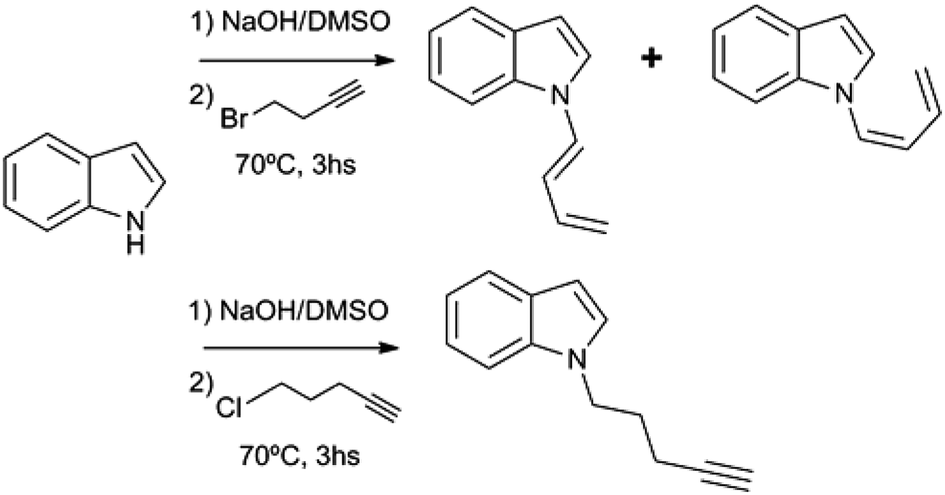 | ||
| Fig. 1 N-alkylation of indole with terminal C-4 and C-5 haloalkynes. | ||
 | ||
| Fig. 2 Synthesis of 1-(N-indolyl) and 1-(N-carbazolyl)-1,3-butadienes. | ||
Results and discussion
During the course of the reaction of indole and 4-Br-1-butyne, the formation of an initial product was detectable by TLC at early stages, before complete conversion of the starting compound. This product was isolated and identified as 1-N-(2-butynyl)-indole (12), an isomer of the expected product (8). However, when the reaction was taken to completion, compound 12 was replaced by a less polar product. Analysis of this product by 1H NMR showed that it was more complex than expected, with no observable signals of protons bound to sp3 carbons. Instead, two dienic systems were clearly observable, and a complete structural elucidation by 2D NMR proved that these dienes were bound to different indole nitrogens. Especially notable were the differences in the geometry of the 1,2 double bond of the dienes. In one case a J = 17 Hz clearly indicated an E geometry, while in the other case a coupling constant of 10 Hz was indicative of a Z configuration. In this way the product was identified as a (1:1) mixture of (Z) and (E) 1-(N-indolyl)-1,3-butadiene (1a,b). Surprisingly, these dienes had never been synthesized. There are previous syntheses of a 1-(N-carbazolyl)-1,3-butadiene and 1-(N-phtalimido)-1,3-butadiene, albeit by a completely different methodology, based on cross-coupling reactions of N-vinyl derivatives.4 Since the obtained yield in the present work was higher than 90%, a family of these compounds was prepared in this way from different substituted indoles, and also from carbazole. The reaction was not feasible using K2CO3 as base, either in acetone or DMSO, while the use of NaH or stronger bases would form alkyne salts. The presence of a strong electron-attracting substituent such as a nitro group (as in 3-nitrocarbazole) produces the failure of the N-alkylation step. In all the cases where the isomerization took place, the (Z) and (E) dienes were separable by preparative TLC, and completely characterized by NMR and MS.
The complete isomerization of the alkynes to form these butadienes were unexpected results, although it is known that the reaction between a terminal alkyne and a catalytic amount of base gives rise to a mixture of isomers. This process was described by Favorskii as early as 1887, and its mechanism has been revised several times.5 In the case of hydrocarbon-type alkynes, which require strong bases and high temperatures for isomerization, the main products are generally disubstituted acetylenes and allenes. Quite surprisingly, conjugated dienes, which would be the thermodynamically favored species, are only formed under forcing temperature conditions.6 In the case of acetylenic nitrogen-substituted compounds, the required conditions for the isomerization are generally milder, and the prevailing product depends on the electronic nature of the nitrogen atom. In a study of the behaviour of different N-2-propinylamines under basic catalysed isomerization conditions, it was observed that a basic nitrogen atom as in N,N-dialkylamino substituents, produced the central, isomerized alkyne almost exclusively. However, with several heterocyclic substituents (1-N-pyrrolyl, 1-N-imidazolyl, 1-N-pyrazolyl) the main product was the allenic isomer. In the case of a N-carbazolyl group, the terminal alkyne was 100% isomerized to the corresponding allene.7 However, no similar studies were carried out with N-substituted butynes, so there are no reports on the influence of the nitrogen atom on the isomerization of a C-4 alkyne to a conjugated diene.
In the present work, the isomerization proceeded under mild conditions (70 °C, NaOH in DMSO), with nearly quantitative yields of the dienes. In the case of the butyne chain, the isomerization is so favourable that the initial N-alkylation product without isomerization of the triple bond (8), was only detected as a trace component of the crude reaction mixture, and only when the reaction was stopped at an early stage. This behaviour was not observed when 5-Cl-1-pentyne was used as alkylating agent. In this case, the reaction proceeded as a normal N-alkylation without any isomerization of the triple bond under the same conditions and reaction times. It is well known that alkyne-diene isomerizations in basic media usually take place via a previous isomerization to a more substituted alkyne, which in turn takes place via allenic intermediates.8 Although no allenic intermediates could be isolated from the crude mixture in this work, the finding of the isomerized central alkyne supports this proposal. A possible mechanism for this reaction is shown in Fig. 3. This mechanism is inspired in the accepted route for alkyne isomerizations in basic media, which occur by a series of equilibria that involve resonance-stabilized carbanions.9 The initial N-substitution product 8, forms in basic medium a resonance-stabilized propargyl carbanion (9). Allenic carbanions are particularly stable, and subsequent protonation gives the allene 10. A similar allene-alkyne isomerization via carbanion 11 originates the disubstituted alkyne 12, which could be isolated and fully characterized. This isomerization is kinetically favoured, and so the disubstituted alkyne is the first product observed in this reaction. The loss of a proton “α” to the allene group in compound 10, would generate a carbanion (13), which in turn could lead to the diene formation. In previous studies such as in the case of unsubstituted C-5 alkynes, the formation of this anion was extremely slow and the conjugated diene was generally not observed. This was attributed to a lack of acidity of the “α” proton due to an inefficient resonance stabilization by the allene group. The presence of an indole moiety helps to stabilize the anion at the “α” position of the allene, and so the formation of the corresponding anion and subsequent isomerization gives rise to the dienes 1a,b as the final, thermodynamically stable products, albeit at a slower rate.
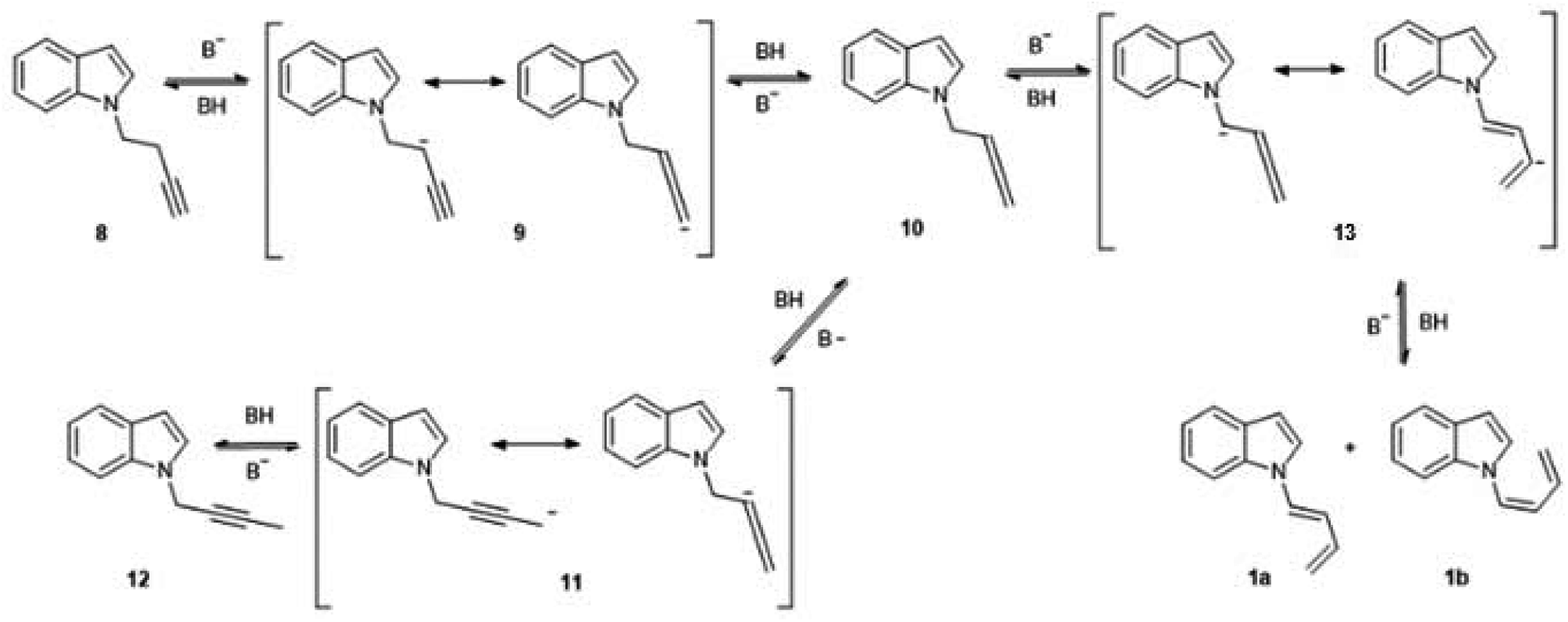 | ||
| Fig. 3 Mechanism for the alkyne-diene isomerization of compound 8. | ||
In order to validate the proposed mechanism, computational calculations were performed, starting from the initial non-isomerized alkyne 8, with the use of the ORCA package.10 These calculations were done for both the C-4 and the C-5 alkyne chain. First, the conformational space of the different compounds involved in the proposed mechanism was explored. Several starting structures were generated by selected modifications of dihedral angles of the side chain. The resulting starting geometries were optimized using the hybrid GGA exchange-correlation functional M06-2X,11 with the Def2-TZVP basis set for all atoms.12 To alleviate the computational cost, the resolution of the identity in its RIJCOSX version was used,13 together with the corresponding basis functions for the calculation of the Coulomb and the exchange-correlation terms.14 After optimization, the Hessian matrix of the energy with respect to the nuclear coordinates was constructed and diagonalized, and its eigenvalues were used to verify whether the geometries are local minima or saddle points on the potential energy surface of the molecules. Only the lowest-energy conformer of every species involved in the mechanism was used for the calculation of reaction or barrier energies. Solvent effects (DMSO) were considered implicitly through the Conductor-like Screening Model for geometry optimizations.15
Geometric parameters (bond lengths and dihedral angles) were calculated for the lowest-energy conformations of all the molecules involved in the proposed mechanism for the reaction of indole with the C-4 alkyne. It is interesting to note that in the final diene, the bond lengths of the N–C1′, C7a–N and C2–N bonds are almost identical (1.383, 1.382 and 1.385 angstroms respectively). This means that the N–C1′ bond has a marked double bond character.
Fig. 4 shows the ΔGs of the different species for the C-4 chain, and it is evident the large energy drop to the final dienes, which makes this last step almost irreversible under thermodynamic conditions. In this scheme, the final intermediate is a dienic carbanion (13), which adds a proton to give the final dienes. The small energy differences (0.4 kcal mol−1) between these final anions 13, as well as of the corresponding transition states (0.7 kcal mol−1) which lead to the final (E) or (Z) butadienes, determine the observed (1:1) ratio of these compounds.
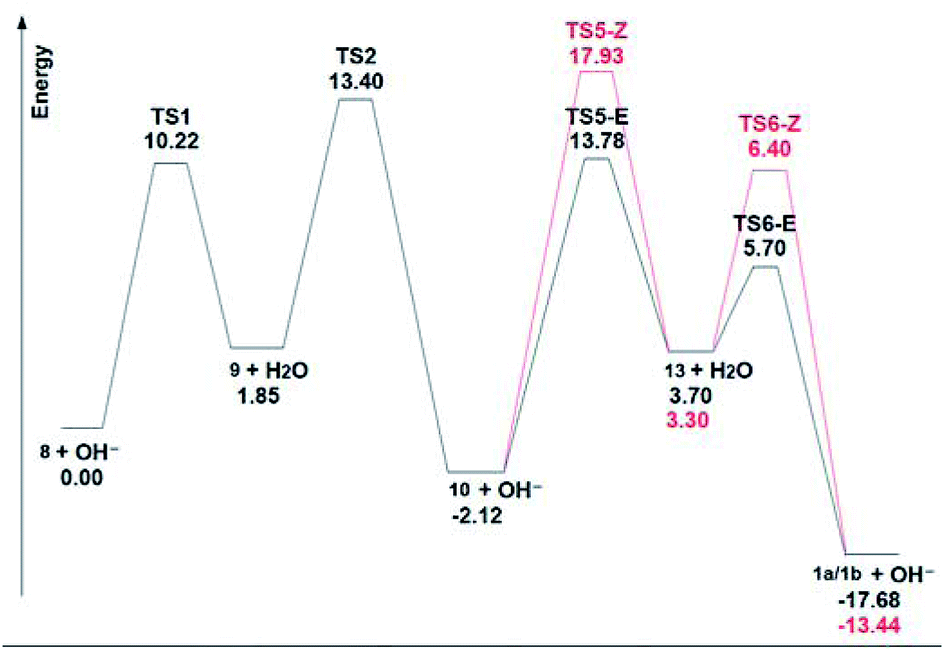 | ||
| Fig. 4 Calculated ΔG for the route to the C-4 dienes. | ||
When the energy values are compared for the C-4 and C-5 chains (see ESI†) of the central disubstituted alkynes, which are the kinetically favoured products, it is evident that they are approximately equally stable. As for the final dienes, the final energy drop is also considerable. However, there are marked energy differences when carbanions 9 and 13 are compared. In the case of the butyne chain, carbanion 9 lies 1.85 kcal mol−1 higher than the terminal alkyne 8, with an energy barrier of 10.22 kcal mol−1. On the other hand, in the case of the pentyne chain, the energy difference between the terminal alkyne 8 and carbanion 9 is 6.86 kcal mol−1, while the energy barrier is in this case of 14.47 kcal mol−1. These values indicate that the anion 9 from the pentyne chain is less stabilized than the corresponding anion from the butyne chain, thus preventing the initial isomerization under the mild conditions of this study. This stabilization of the C-4 anion 9 is probably caused by an inductive effect of the polarizable indole moiety.
This is in accordance with previous results obtained with an N-substituted indole bearing a C-3 alkyne. In this case, the isomerization occurred readily under very similar conditions to those of the present study, and gave rise to a mixture of both alkynes and the corresponding allene.6 However, with this substrate, the reaction ended up in a mixture of isomers since it was not possible to reach the final diene, which is the irreversible step in the case of the butyne chain.
The possible use of the new dienes in Diels–Alder reactions was explored, using p-benzoquinone as dienophile (Fig. 5). Since the final product of this reaction under oxidizing conditions would be a substituted 1,4-naphthoquinone, namely a 5-(N-indolyl)-1,4-naphthoquinone, there was no need of a previous separation of the diene mixture. However, under all the different assayed conditions, the yields of substituted naphthoquinones were lower than 50%, and the conversion of the dienes was not complete. The recovered unreacted diene was in all cases the (Z) isomer, which accounts for the low yields of the reaction. This reaction was also attempted with the purified (Z) diene with no success, and, under forcing conditions, the compound decomposed readily.
 | ||
| Fig. 5 Diels Alder reaction between the E diene and p-benzoquinone. | ||
It is known that (Z) isomers of 1-substituted 1,3-butadienes are much less reactive, or even totally unreactive in Diels Alder reactions compared to the (E) isomers. In (Z) dienes, the steric interaction between the substituent at C-1 and H-4 of the diene while adopting the s-cis configuration, produces a deviation from coplanarity of the diene system, a key requirement for the DA reaction, by rotation of the C2–C3 bond of the diene.16 In the case of a methyl group at C-1, this deviation is small, but as the volume of the substituent increases, this interaction prevents coplanarity almost completely and makes the DA reaction extremely sluggish under thermal conditions. However, there are some recent examples of successful DA reactions of (Z) dienes using Lewis acid catalysis.17 Unfortunately, in the present case, the use of BF3·O(Et)2 and other Lewis acids was unsuccessful.
The rotational energy curves of the central bond of the diene to go from the s-trans to the s-cis configuration, were calculated for the (Z) and (E) isomers of 1-(N-indolyl)-1,3 butadiene (Fig. 6). In the case of (E)-butadiene, the calculated skew angle was 25.4° at the lowest energy conformation for the s-cis conformation. The energy barrier between this minimum and the coplanar conformation is just 0.56 kcal mol−1. On the other hand, in the case of the (Z)-butadiene, the calculated skew angle was −56.5°, and the access to the coplanar s-cis diene from the lowest energy conformation had an energy penalization of 5.59 kcal mol−1. This gives a total energy difference for the planar s-trans – planar s-cis configurations of 10.17 kcal mol−1 for the (Z) isomer and of 4.39 kcal mol−1 for the (E) isomer, which explains the difference in reactivity of both compounds in thermal Diels–Alder reactions. A series of new 5-(N-indolyl)-1,4-naphthoquinones was synthesized from benzophenone and some of the new dienes (Fig. 7). Several natural and synthetic naphthoquinones have shown interesting pharmacological activities,18 and, for this reason, the cytotoxicity of the synthesized compounds was evaluated against a small panel of cell lines (Table 1).
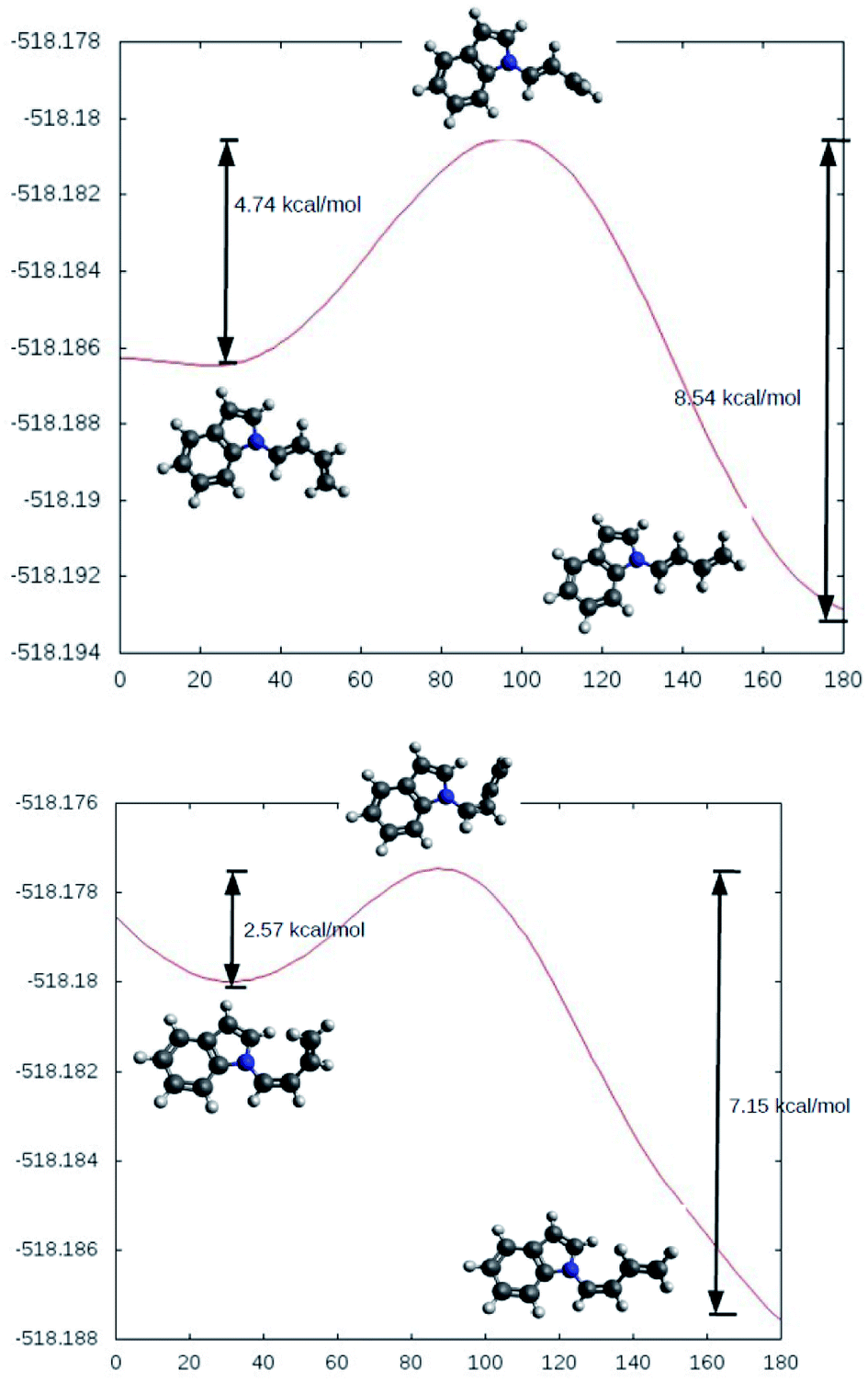 | ||
| Fig. 6 Calculated rotational energy curves for the central bond of the E (top) and Z (bottom) dienes. | ||
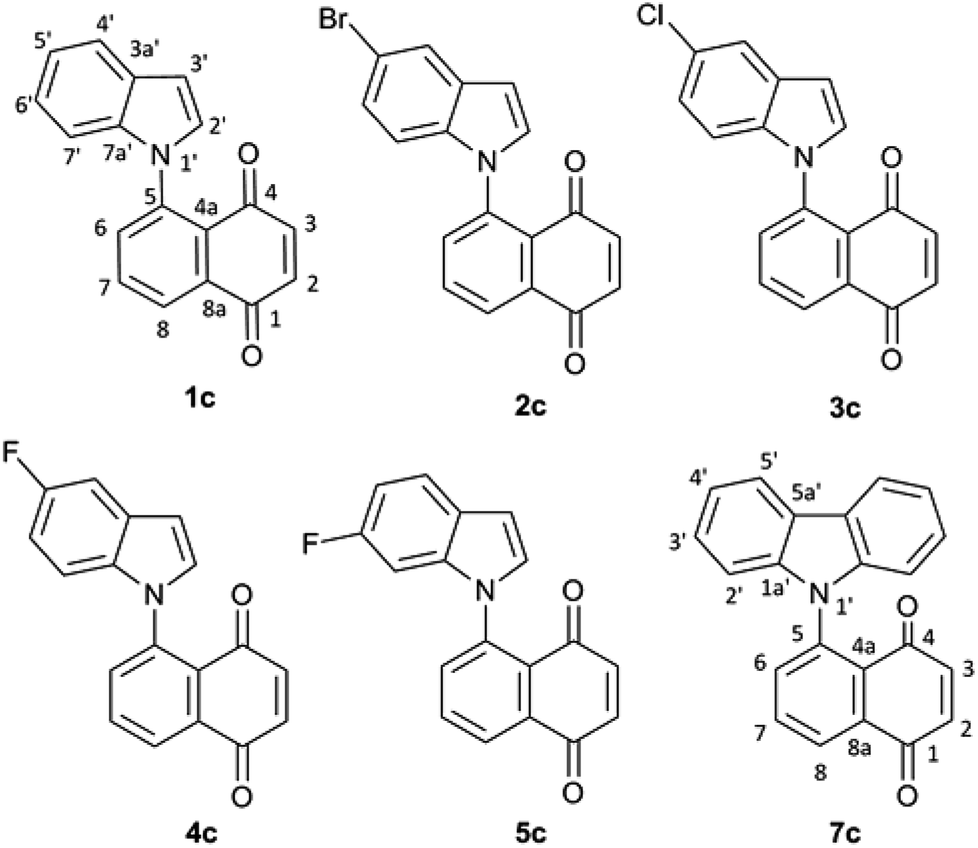 | ||
| Fig. 7 Diels–Alder reaction products of compounds 1a–5a and 7a with p-benzoquinone. | ||
| IC50 (μM) | |||
|---|---|---|---|
| Comp. | LM2 | HaCat | K562 |
| 1c | 7.15 | 4.02 | 5.91 |
| 2c | 18.33 | 4.13 | 6.80 |
| 3c | 7.54 | 4.24 | 3.96 |
| 4c | 4.78 | 7.12 | 8.23 |
| 5c | 3.31 | 3.22 | 2.34 |
| 7c | 3.51 | 3.72 | 6.23 |
These results show that the new naphthoquinones are strongly cytotoxic, and, although additional tests should be performed against a wider panel of cell lines, these preliminary results may indicate a tendency to be generally toxic. However, naphthoquinones are interesting scaffolds for further derivatization, and it is possible that additional synthetic modifications may modulate the cytotoxicity of these new derivatives.
Conclusions
In this work, a base-catalysed isomerization of N-alkylated indoles with a terminal butyne chain produced a series of 1-(N-indolyl)-1,3-butadienes, as (1:1) mixtures of chromatographically separable (E) and (Z) dienes. The same methodology was applicable to other heterocycles having an acidic N-bound proton, such as carbazole.
There are previous examples of N-alkylations with 4-bromo-1-butyne or the corresponding triflate, of 2-bromoindoles, cytosines, and imidazole and pyridine moieties in more complex molecules.19–21 Interestingly, all these N-alkylations were performed using K2CO3 as base either in acetone or in DMF, and in all cases the alkyne isomerization was not observed. The lack of isomerization was probably due to the weaker basicity of K2CO3, which was unable to form anion 9 and start the chain of isomerization equilibria. With the heterocyclic substrates used in the present work, N-alkylation did not take place with the use of K2CO3 as base. The lack of reactivity of 3-nitrocarbazole towards N-alkylation with 4-bromo-1-butyne in NaOH/DMSO is also remarkable, since the formation of the anion was evidenced by a color change. This reaction was effective (although at a slow rate) with saturated alkyl halides such as n-butylbromide. The presence of a strong electron-attracting substituent probably reduces the nucleophilicity of the anion of 3-nitrocarbazole, slowing the rate of the N-alkylation up to a point where an elimination reaction on the haloalkyne (to give a conjugated product) competes successfully.
Compared to the previous synthesis of 1-(N-indolyl)-carbazole in ref. 4, the present methodology is simpler, and does not require the use of expensive organometallic catalysts or inert atmosphere. Besides, this is a one-step process, a shorter route than the sequence of 2 or 3 steps in ref. 4. However, with the use of organometallic chemistry, the configuration of the diene can be controlled as almost exclusively (E), while in this work a (1:1) mixture of (Z) and (E) isomers are obtained, which requires a chromatographic separation to recover the pure dienes. Anyway, the present work has the advantage of simplicity, very high yields, and the possibility to prepare simultaneously both stereoisomers in one step. A possible mechanism was proposed for the alkyne-diene isomerization, and computational studies were performed to validate this proposal. A key intermediate for this mechanism, the central alkyne, could be isolated and fully characterized.
The obtained butadienes were used in Diels–Alder reactions to obtain a family of new cytotoxic 5-(N-indolyl)-1,4-naphthoquinones. These structures are as well an interesting scaffold for further synthetic transformations. The lack of reactivity in the DA reaction of the (Z) dienes, was explained by the energy values obtained from the rotational energy curves. Besides their use in DA reactions, the preparation of these new dienes opens new possibilities for further synthetic transformations, such as the synthesis of polymers, and also as substrates for additional transformations by organometallic chemistry.
Experimental procedure
General procedures
All solvents used for chromatography were distilled from glass. All NMR spectra were recorded on Bruker AC-200 (200.13 MHz), Fourier 300 (300 MHz) and Bruker Avance III (500.13 MHz) spectrometers, using the signal of residual non deuterated solvent as internal reference; J values are given in Hz. All 2D NMR experiments (COSY, DEPT-HSQC, HMBC) were performed using standard pulse sequences. ESI-HRMS spectra were recorded on a Bruker micrOTOF-Q II spectrometer. UV spectra were obtained on a Hewlett Packard 8453 spectrophotometer. TLC separations and analyses were carried out on Merck Sílicagel 60 F254 plates. TLC plates were observed under UV light (254 nm) or sprayed with 2% vanillin in concentrated H2SO4. Merck Silica gel (230–400 mesh) was used for column chromatography. Sephadex LH-20 was obtained from GE Healthcare. All the reagents for synthesis were purchased from Sigma-Aldrich.Synthesis of compounds 1a/b–7a/b
The indole or carbazole (1–7) was added to a solution of excess NaOH in DMSO (0.5 mL per 20 mg indole) in a reaction vial at room temperature. The reaction mixture was stirred for 10 minutes at 70 °C, after which 2.0 equivalents of 1-Br-4-butyne were added. The temperature was kept at 70 °C until total disappearance (by TLC) of the starting material, and a darkening of the reaction medium. The crude reaction mixture was then partitioned between EtOAc and brine, and the organic phase was washed twice again with brine, and finally with distilled water. The organic layer was then taken to dryness, and the 1:1 mixture of (E) and (Z) dienes (by NMR inspection) was obtained without further purification. In all cases, the yields were higher than 85–90%. The individual (E) and (Z) dienes were purified by preparative TLC on silica, using typically cyclohexane as mobile phase and characterized by 1D and 2D NMR and HRMS.
Synthesis of compounds 1c–5c and 7c
Compounds 1a/b–5a/b or 7a/b were added to a solution of 2 eq. of p-benzoquinone in ethanol. The reaction mixture was kept at room temperature for 4 hs, after which the ethanol was evaporated. The solid residue was dissolved in MeOH, permeated through a Sephadex LH-20 column using MeOH as mobile phase, and finally purified by flash column chromatography using cyclohexane/EtOAc 9:1 as eluant.
Compounds 1a–1b
45.6 mg as mixture, yield 95.0%. Oil.Compounds 2a–2b
29.8 mg as mixture, yield 96.9%. Oil.Compounds 3a–3b
27.4 mg as mixture, yield 96.8%. Oil.Compounds 4a–4b
27.2 mg as mixture, yield 97.0%. Oil.Compounds 5a–5b
26.8 mg as mixture, yield 96.5%. Oil.Compounds 6a–6b
26.8 mg as mixture, yield 96.5%. Oil.Compounds 7a–7b
22.1 mg as mixture, yield 86.2%. Oil.Isolation of compound 12a
When the basic catalyzed isomerization of indole (1) was quenched after 45 minutes, compound 12a could be isolated by preparative TLC using cyclohexane/ethyl acetate (9:1) as mobile phase and characterized by 1D and 2D NMR and HRMS.
Cytotoxicity assays
Human leukemic K562 cells were obtained from ATCC, the murine mammary adenocarcinoma LM2 cells were provided by Dr A. Eijan (InstitutoRoffo, Argentina), while the human normal keratinocytes HaCaT were obtained from Dr M. Quintanilla (Instituto de Investigaciones Biomedicas, Madrid, Spain). The cells were cultured in RPMI 1640 medium, supplemented with 5% fetal bovine serum (LM2) and 10% (K562 and HaCat), 2 mM L-glutamine, 100 IU mL−1 penicillin, 100 μg mL−1 streptomycin and maintained at 37 °C in a 5% CO2 atmosphere. Cells were detached with 0.1% trypsin–EDTA, and viable cells were counted using a hematocytometer. Cells (70000 to 10000 cells per mL) were seeded in 48-well microtiter plates and incubated at 37 °C. After 24 h, the cells were treated with different concentrations (1–100 μM) of the test compounds initially dissolved in DMSO (5 mM), and further diluted in medium to produce the desired concentrations. Final DMSO concentrations were lower than 2%. The plates were incubated for another 48 h at 37 °C. Immediately after treatment, cell viability was measured employing the MTT method.22 Briefly, 25 μL of MTT (2.5 mg ml−1) was added to the wells and incubated for 1 h. After DMSO dissolution of the resulting formazan crystals, absorbance was recorded at 570 nm in a microplate reader. The concentrations required to inhibit cell growth by 50% (IC50) were calculated from the abscissa intercept from logistic curves constructed by plotting cell survival (%) versus drug concentration (μM).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research at the University of Buenos Aires was supported by grants from CONICET (PIP 2014–2016 No. 11220130100523CO), UBA (UBACyT 2014–2017 No. 20020130100457BA) and ANPCyT (PICT 2014–2063). We thank Dr Gabriela Cabrera (UMYMFOR–CONICET) for recording the mass spectra and Eng. José Gallardo and Lic. Gernot Eskuche (UMYMFOR–CONICET) for the NMR spectra. G. C., G. D. V., R. P. D. and J. A. P are researchers of CONICET. C. P. D. thanks CONICET for a doctoral fellowship, and J. F. thanks the University of Buenos Aires for an undergraduate research fellowship.Notes and references
- For recent examples of syntheses of substituted 1,3-butadienes, see: (a) V. Bandi, V. Kavala, C. H. Hsu, A. Konala, B. K. Villuri, T. Kotipalli, C. W. Kuo and C. F. Yao, RSC Adv., 2017, 7, 46704–46712 RSC; (b) S. Zhou, E. Sánchez-Larios and M. Gravel, J. Org. Chem., 2012, 77, 3576–3582 CrossRef CAS PubMed; (c) L. Wang and M. E. Welker, J. Org. Chem., 2012, 77, 8280–8286 CrossRef CAS PubMed; (d) Q. Zhao, V. Tognetti, L. Joubert, T. Besset, X. Pannecouke, J. P. Bouillon and T. Poisson, Org. Lett., 2017, 19, 2106–2109 CrossRef CAS PubMed; (e) J. Sudkowska-Frątczak, B. Marciniec, G. Hreczycho, M. Kubicki and P. Pawluć, Org. Lett., 2015, 17, 2366–2369 CrossRef PubMed; (f) J. Sudkowska-Frątczak, M. Taczała and P. Pawluć, Materials, 2015, 8, 7250–7256 CrossRef PubMed; (g) T. Liu, J. Dong, S. J. Cao, L. C. Guo and L. Wu, RSC Adv., 2014, 4, 61722–617261 RSC; (h) X. M. Zhang, G. Q. Wu and W. Z. Chen, Chin. J. Chem., 2007, 25, 1722–1727 CrossRef CAS.
- N. Chadha and O. Silakari, Eur. J. Med. Chem., 2017, 134, 159–184 CrossRef CAS PubMed.
- (a) C. W. Kuo, A. Konala, L. Ling, T. T. Chiang, C. Y. Huang, T. H. Yang, V. Kavala and C. F. Yao, Chem. Commun., 2016, 52, 7870–7873 RSC; (b) C. C. Nawrat and C. J. Moody, Angew. Chem., Int. Ed., 2014, 53, 2056–2077 CrossRef CAS PubMed.
- (a) P. Pawluć, A. Franczyk, J. Walkowiak, G. Hreczycho, M. Kubicki and B. Marciniec, Org. Lett., 2011, 13, 1976–1979 CrossRef PubMed; (b) P. Pawluć, A. Franczyk, J. Walkowiak, G. Hreczycho, M. Kubicki and B. Marciniec, Tetrahedron, 2012, 68, 3545–3551 CrossRef; (c) J. Sudkowska-Frątczak, A. Ryba, A. Franczyk, J. Walkowiak, M. Kubicki and P. Pawluć, Appl. Organomet. Chem., 2014, 28, 137–139 CrossRef.
- H. Pines and W. Stalick, Base catalyzed reaction of hydrocarbons and related compounds, Chapter 3: Isomerization of Acetylenes and Allenes, Academic Press, New York, 1977, pp. 124–204 Search PubMed.
- W. Smadja, C. R. Acad. Sci. Paris, 1963, 257, 3950–3952 CAS.
- (a) A. J. Hubert and H. G. Viehe, J. Chem. Soc. C, 1968, 228–230 RSC; (b) A. J. Hubert and H. Reimlinger, J. Chem. Soc. C, 1968, 606–608 RSC.
- (a) T. L. Jacobs, R. Akawie and R. G. Cooper, J. Am. Chem. Soc., 1951, 73, 1273–1276 CrossRef CAS; (b) S. W. Benson, F. R. Cruickshank, D. M. Golden, G. R. Haugen, H. E. O'Neal, A. S. Rogers, R. Shaw and R. Walsh, Chem. Rev., 1969, 69, 279–324 CrossRef CAS.
- J. Dale, Properties of acetylenic compounds, in Chemistry of Acetylenes, ed. H. G.Viehe, Dekker, New York, 1969, p. 78 CrossRef CAS PubMed; C. Bernasconi and P. Wenzel, J. Am. Chem. Soc., 2001, 123, 7146–7153 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. Sinnecker, A. Rajendran, A. Klamt, M. Diedenhofen and F. Neese, J. Phys. Chem. A, 2006, 110, 2235–2245 CrossRef CAS PubMed.
- R. Huisgen, R. Grashey and J. Sauer, Cycloaddition Reactions of Alkenes, in The chemistry of alkenes, ed. S.Patai, ch. 11, Interscience Publishers, 1964, pp. 739–954 Search PubMed.
- (a) W. Roush and D. Barda, J. Am. Chem. Soc., 1997, 119, 7402–7403 CrossRef CAS; (b) H. Adams, J. C. Anderson, R. Bell, D. N. Jones, M. R. Peel and N. C. O. Tomkinson, J. Chem. Soc., Perkin Trans. 1, 1998, 3967–3973 RSC; (c) N. Yakelis and W. Roush, Org. Lett., 2001, 3, 957–960 CrossRef CAS PubMed.
- H. Y. Qiu, P. F. Wang, H. Y. Lin, C. Y. Tang, H. L. Zhu and Y. H. Yang, Chem. Biol. Drug Des., 2018, 91, 681–690 CrossRef CAS PubMed.
- A. P. Dobbs, K. Jones and K. T. Veal, Tetrahedron, 1998, 54, 2149–2160 CrossRef CAS.
- P. Kielkowski, H. Cahová, R. Pohl and M. Hocek, Bioorg. Med. Chem., 2016, 24, 1268–1276 CrossRef CAS PubMed.
- M. Čížková, D. Šaman, D. Koval, V. Kašička, B. Klepetářová, I. Císařová and F. Teplý, Eur. J. Org. Chem., 2014, 5681–5685 CrossRef.
- F. Denizot and R. Lang, J. Immunol. Methods, 1986, 89, 271–277 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, energy curves for C-5 alkyne. See DOI: 10.1039/c8ra05208e |
| This journal is © The Royal Society of Chemistry 2018 |