
A turn-on AIE active fluorescent sensor for Hg2+ by combination of 1,1-bis(2-pyridyl)ethylene and thiophene/bithiophene fragments†
Moustafa T.
Gabr
and
F.
Christopher Pigge
 *
*
Department of Chemistry, University of Iowa, Iowa City, Iowa 52242, USA. E-mail: chris-pigge@uiowa.edu
First published on 27th April 2017
Abstract
Fluorescent sensors for Hg2+ that combine aggregation-induced emission (AIE) activity of tetraarylethylenes with metal chelating 1,1-bis(2-pyridylethylene) fragments and thiophene/bithiophene substituents have been prepared and characterized. The sensors exhibit red-shifted and enhanced emission in the presence of Hg2+ in aqueous solution while exhibiting little to no change in fluorescence in the presence of other metal ions. Job plot analyses indicate 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 sensor:Hg2+ binding stoichiometries in solution. 1H-NMR spectroscopy was also employed to investigate solution phase binding interactions between the sensors and Hg(ClO4)2, and a chelated HgI2–bis(pyridyl) complex has been characterized by X-ray crystallography. The limit of detection for Hg2+ was determined to be 48 nM. In contrast, the fluorescence of structurally analogous materials possessing quinoline rings in place of pyridine groups is completely quenched in the presence of Hg(ClO4)2. The high sensitivity and selectivity displayed by these sensors for Hg2+ over other metal ions may enable monitoring of mercury in aqueous environments.
:1 sensor:Hg2+ binding stoichiometries in solution. 1H-NMR spectroscopy was also employed to investigate solution phase binding interactions between the sensors and Hg(ClO4)2, and a chelated HgI2–bis(pyridyl) complex has been characterized by X-ray crystallography. The limit of detection for Hg2+ was determined to be 48 nM. In contrast, the fluorescence of structurally analogous materials possessing quinoline rings in place of pyridine groups is completely quenched in the presence of Hg(ClO4)2. The high sensitivity and selectivity displayed by these sensors for Hg2+ over other metal ions may enable monitoring of mercury in aqueous environments.
Introduction
Fluorescence-based probes capable of sensing and/or imaging metal ions are important analytical tools in diverse areas of research, including molecular biology, clinical diagnostics, biotechnology, and environmental science.1–4 Among the metal ions targeted for selective sensor development, Hg2+ continues to receive considerable attention because of its non-biodegradable nature and its extreme toxicity toward human health and harmful effects on the environment.5 Inorganic Hg2+ released into the environment can be converted to methyl mercury by multiple species of bacteria. Methyl mercury readily bio-accumulates in tissues of living animals and plants, and enters the human body through the food chain (e.g. edible fish). Elevated Hg2+ levels in humans produce acute neurological effects, kidney failure, and brain damage.6 Therefore, it is important to develop sensitive and selective methods for detection of Hg2+ in environmental and biological samples.7Traditional methods of Hg2+ detection, such as atomic absorption spectroscopy (AAS) and inductively coupled plasma mass spectrometry (ICP-MS) are limited by the need for expensive and complex instrumentation. In contrast, fluorescence-based methods hold promise as simple, sensitive, non-invasive, portable, and relatively inexpensive alternatives for detection and sensing of Hg2+ in biological and environmental samples.8 Since Hg2+ is a heavy metal, its interaction with organic fluorophores often results in fluorescence quenching as a consequence of mercury's large spin–orbit coupling constant.9 Consequently, many fluorescent sensors developed for Hg2+ exhibit negative or “turn-off” chelation-enhanced quenching (CHEQ) effects.10,11 Turn-off fluorescence sensors, however, can suffer from high rates of false negative results. Accordingly, fluorescence “turn-on” sensors are more desirable because of their high sensitivity and elevated signal-to-noise ratio (S/N).
There are, however, relatively few examples of “turn-on” fluorescent receptors suitable for detection of Hg2+, especially in aqueous systems. Lippard and Nolan have characterized several Hg2+ sensors based on fluorescein derivatives with attached Hg-chelating groups featuring coordinating thioether groups.12 Similarly, a fluorescein-based sensor functionalized with an azathia-crown ether group has been developed for specific sensing of Hg2+ in aqueous solution at ppm concentrations.13 Additionally, fluorescent Hg2+ sensors based on modulating the dye-quenching ability of gold nanoparticles (AuNP) have been reported with sensitivities as low as 2 ppb in aqueous solution.14
Increasingly, compounds that display aggregation-induced emission (AIE) are being used as components of turn-on fluorescent sensors. Unlike conventional fluorophores that exhibit aggregation-caused quenching (ACQ), compounds with aggregation-induced emission (AIE) characteristics typically have weak fluorescence in dilute solution but become highly emissive upon aggregation in the presence of poor solvents or in the solid state.15 This intrinsic behaviour of AIE active compounds is largely attributed to restriction of intramolecular motions that occur upon aggregation.15g Several turn-on fluorescence sensors featuring AIE characteristics have been developed for monitoring Hg2+ in aqueous solution.8a,16 For example, tetraphenylethylene (TPE) based compound 1 (Fig. 1) was designed as a turn-on AIE active sensor that utilizes thymine for selective coordination of Hg2+.16c Additionally, the intense red emission of aggregated TPE-functionalized quinolinium salt 2 that is quenched in the presence of iodide anion can be fully recovered upon addition of Hg2+, allowing fluorescence off–on detection of HgI2.8a More recently, rhodamine-TPE derivative 3 has been reported as a ratiometric Hg2+ sensor.16b
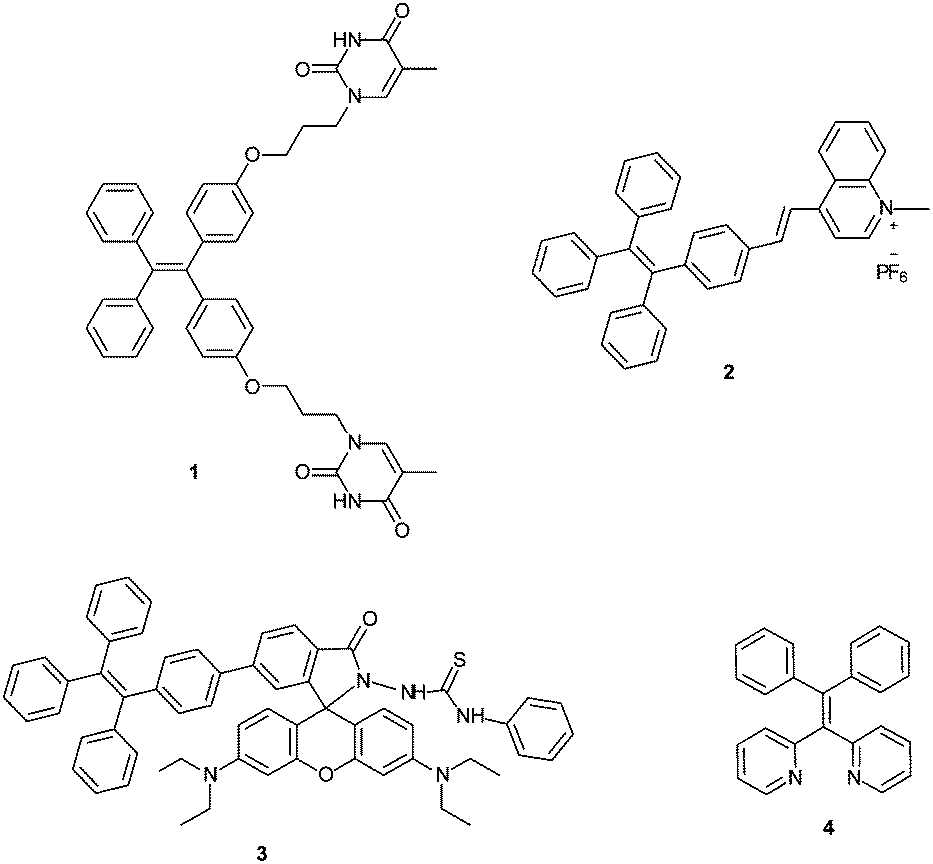 | ||
| Fig. 1 Examples of TPE derivatives as fluorescent metal sensors (1–4). | ||
In addition to AIE-based systems, chelation-induced fluorescence enhancement is also a useful strategy to develop emissive sensors for Hg2+.17 In this context, we recently reported 1,1-bis(2-pyridyl)-2,2-diphenylethylene (4) as a selective fluorescent sensor for Zn2+ ions based on the combination of AIE properties and metal chelating ability.18 In continuing research aimed at further modifying AIE active sensor 4 for sensing of other biologically and environmentally relevant metal ions, we sought to target Hg2+ using an AIE active tetraarylethylene that combines a metal-chelating 1,1-bis(2-pyridyl)ethylene fragment with thioarenes. Specifically, we envisioned that derivatives bearing thiophene or bithiophene moieties might display selectivity for Hg2+ over other metals because of the thiophilic nature of Hg2+.19,20 We report here the synthesis and characterization of two new 1,1-bis(2-pyridyl) tetraarylethylenes (6 and 7) and two new 1,1-bis(2-quinolinyl) tetraarylethylenes (10 and 11) that incorporate thiophene or bithiophene units as part of the tetraarylethylene framework. The AIE activity of these compounds and their ability to function as selective fluorescent sensors for detection of Hg2+ ions in aqueous solution has been evaluated.
Results and discussion
As illustrated in Scheme 1, the synthesis of the putative fluorescent sensors 6 and 7 was easily accomplished using procedures analogous to those we recently reported for preparation of 4.18 Double Suzuki coupling of 5 proceeded smoothly with either thiophene-2-boronic acid or 2,2′-bithiophene-5-boronic acid pinacol ester as cross-coupling partners to furnish 6 and 7 in reasonable yields.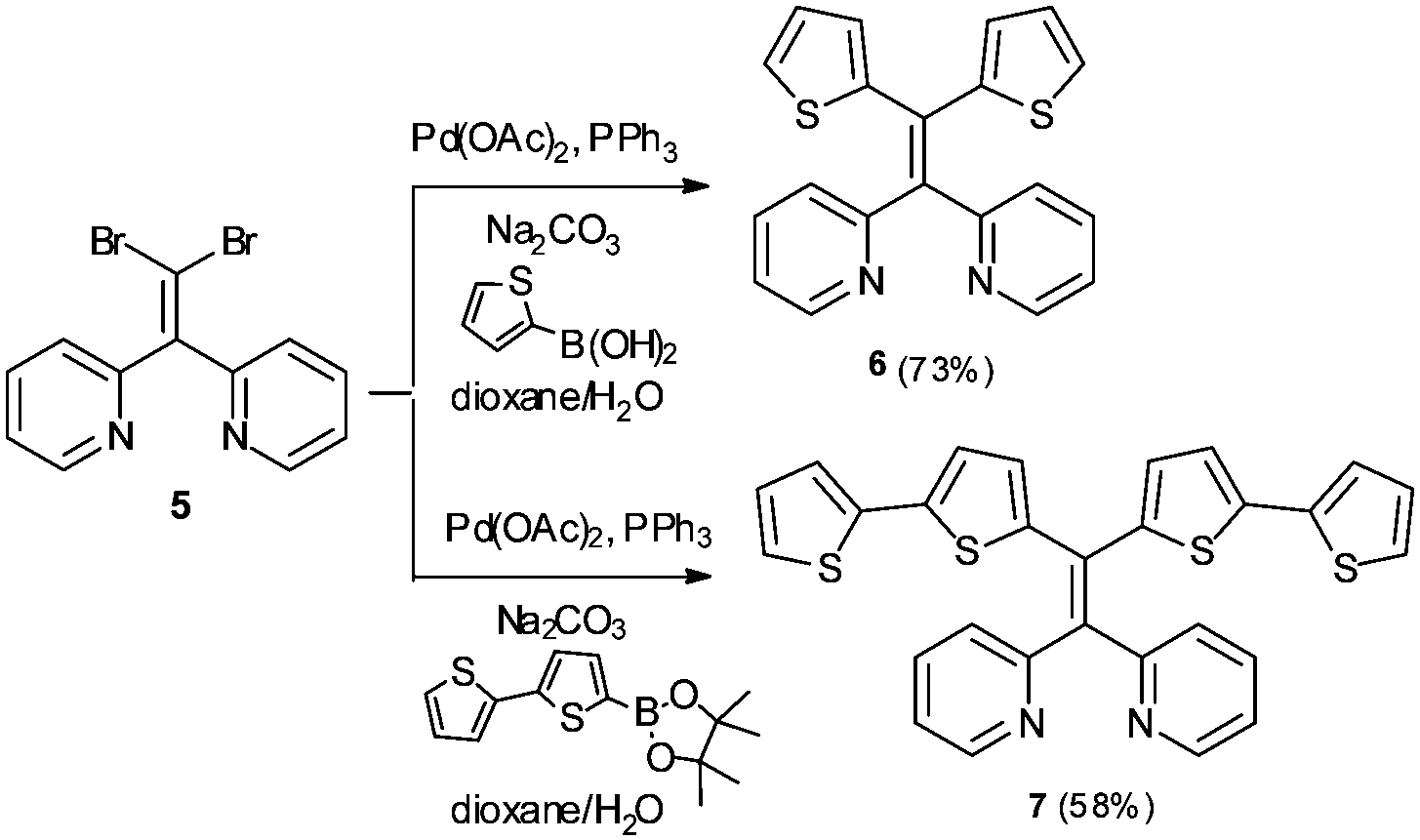 | ||
| Scheme 1 Synthesis of thiophene and bithiophene 1,1-bis(2-pyridyl)tetraarylethylenes 6 and 7. | ||
Both compounds 6 and 7 exhibited similar UV-visible absorption spectra in CH3CN solution, with longest wavelength absorption appearing at ∼340 nm (see ESI†). However, their fluorescence properties under conditions leading to aggregation induced emission displayed significant differences. The fluorescence spectrum of 6 in pure CH3CN revealed a weak emission at ∼512 nm (Fig. 2A). Incremental addition of H2O (a poor solvent for 6) initially produced a slight red-shift in the emission wavelength followed by a decrease in emission intensity at ∼520 nm and the appearance of a new blue-shifted emission peak at ∼404 nm that increases in intensity as a function of added H2O (Φ404 = 6.3%, 9:1 H2O:CH3CN). The emission profile of 6 as a function of solvent polarity and aggregation state may reflect transition from a locally excited state fluorophore to emission from a charge-transfer state.21 In contrast, 7 exhibits a more or less conventional AIE effect that features a marked increase in fluorescence emission (λem ∼ 503 nm) upon incremental addition of H2O (Fig. 2B, Φ503 = 7.5%, 9:1 H2O:CH3CN).
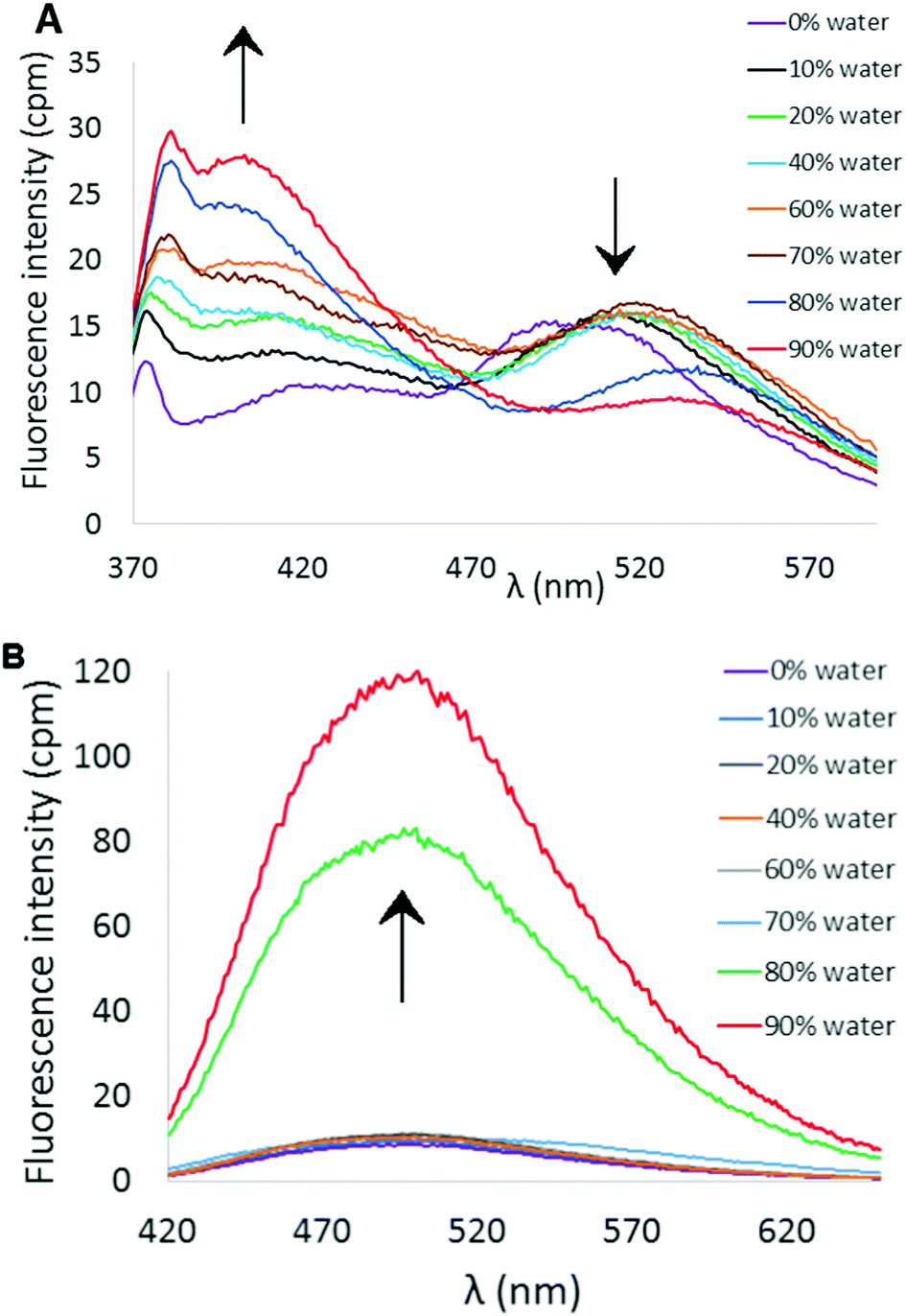 | ||
| Fig. 2 (A) AIE profile of 6 in CH3CN/H2O mixtures (λex = 338 nm, [6] = 10 μM). (B) AIE profile of 7 in CH3CN/H2O mixtures (λex = 351 nm, [7] = 10 μM). | ||
The fluorescence response of 6 in 9:1 H2O:CH3CN was then measured in the presence of 2 equivalents of different metal ions (Hg2+, Mg2+, Ca2+, Ba2+, Co2+, Cd2+, Pb2+, Cu2+, Ni2+, Zn2+, Fe3+, Fe2+, Mn2+, Na+, and K+), and the results are shown in Fig. 3A (perchlorate counterion(s) in each case). Most metal ions produced little to no change in the fluorescence of the solution. Addition of either Cu2+ or Fe3+ resulted in fluorescence quenching, whereas Cd2+ and Zn2+ elicited a slight increase in fluorescence in the 400–450 nm range. The effect of added Hg2+ ions, however, was much more dramatic as an intense red-shifted emission centered at 600 nm was observed. The fluorescence response of 6 at 600 nm as a function of increasing amounts of Hg(ClO4)2 was measured in 9:1 H2O:CH3CN (Fig. 3B). Emission intensity was observed to increase rapidly up to 2 equivalents Hg2+ (Φ = 15.7%), and then gradually plateau at ∼4 equivalents.
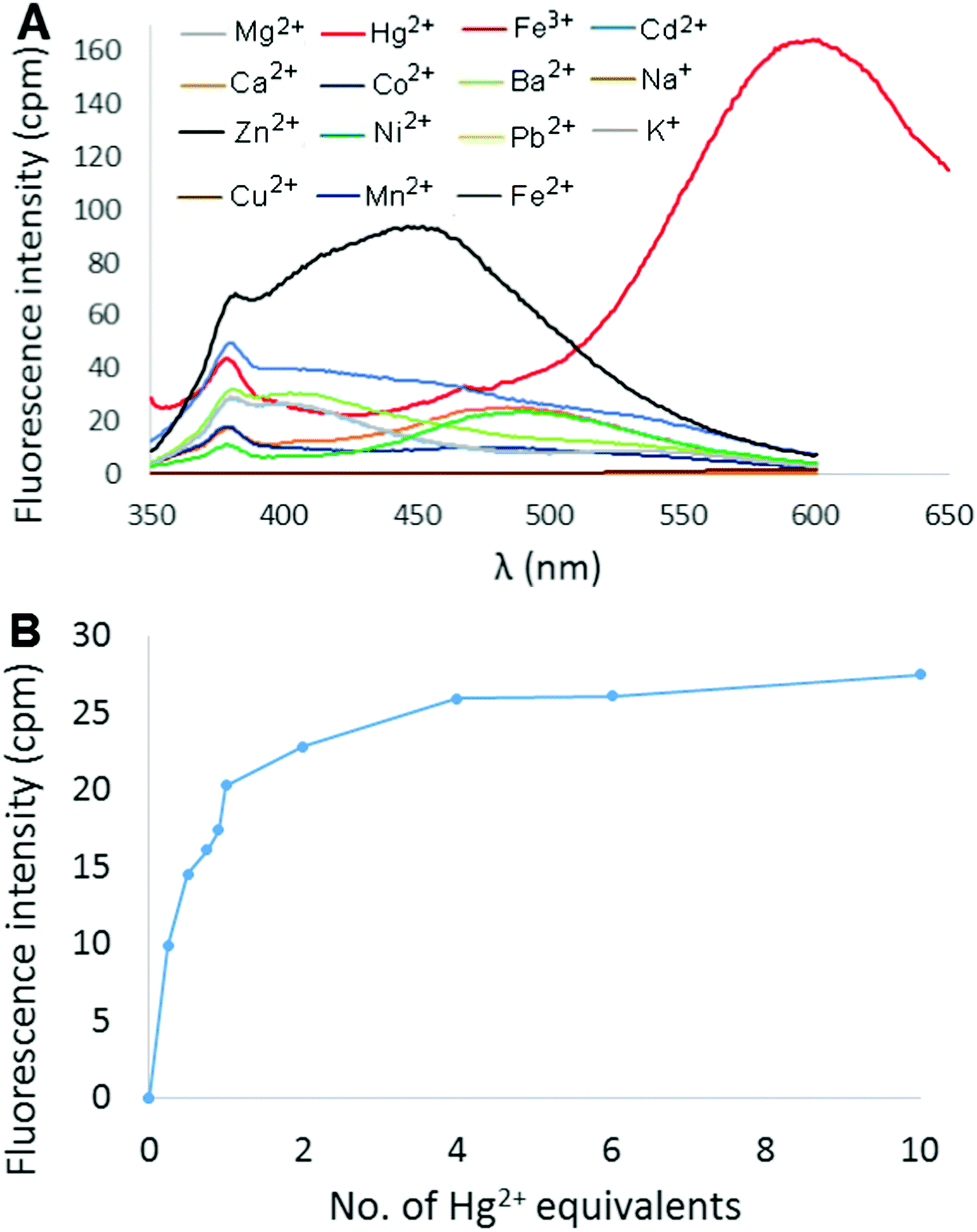 | ||
| Fig. 3 (A) Fluorescence spectra of 6 in the presence of different metal perchlorate salts (2 equivalents). (B) Relative change in fluorescence intensity at 600 nm of 6 as a function of added Hg2+ (λex = 338 nm, [6] = 10 μM). | ||
The selective red-shifted and enhanced fluorescence response of 6 in the presence of Hg2+ is attributed to the presence of both the chelating 2-pyridyl groups as well as the peripheral thiophene rings. Indeed, we have previously shown that addition of Hg(ClO4)2 to 4 (possessing the 1,1-bis(2-pyridyl)ethylene fragment but lacking thiophene substituents) quenches the fluorescence under conditions similar to those outlined above.18 Likewise, exposure of 1,1-bis(2-thiophenyl)-2,2-diphenylethylene (an analogue of 6 in which the pyridine rings have been replaced with phenyl groups)22 also fails to produce a fluorescence response (see ESI†). In order to gain structural insight into possible adducts between 6 and Hg2+, we unsuccessfully attempted to grow crystals from solutions containing 6 and Hg(ClO4)2. Efforts to obtain crystals from other Hg(II) salts (Hg(OAc)2, Hg(NO3)2, HgCl2) were also unsuccessful. X-Ray quality crystals were obtained, however, from slow evaporation of a THF/MeOH solution of 6 and HgI2. The molecular structure of the resulting complex features chelation of HgI2 by the bis(2-pyridyl)ethylene moiety (see ESI†). Presumably the strong affinity of Hg2+ for iodide anions8a,16d precludes halide dissociation to allow for potential thiophene ring interactions or formation of 2:1 ligand:Hg2+ complexes.
To gain insight into the solution phase behaviour of 6 and Hg(ClO4)2, a Job plot was constructed by monitoring the change in fluorescence intensity at 600 nm as a function of mole fraction 6 (Fig. 4). The maximum emission response was observed at 0.67 mole fraction of 6, which indicates a 2:1 (6:Hg2+) binding stoichiometry. Furthermore, NMR titration experiments between 6 and Hg(ClO4)2 in D2O/CD3CN revealed a downfield shift of the 2-pyridyl ortho hydrogens, indicating the coordination of Hg2+ to the pyridine nitrogen atoms (see ESI†).
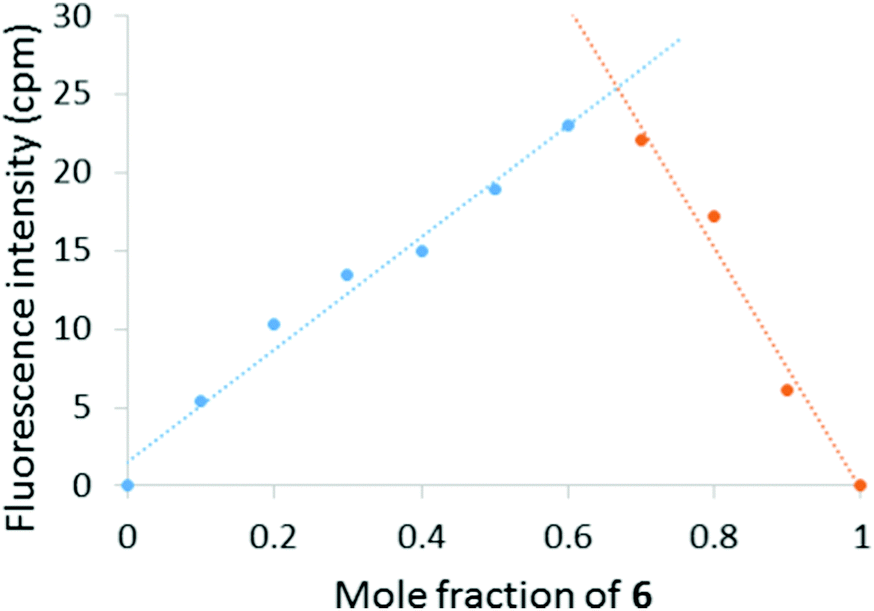 | ||
| Fig. 4 Job plot of 6 and Hg(ClO4)2 binding in 9:1 H2O:CH3CN indicating 2:1 (6:Hg2+) binding stoichiometry. The relative concentrations of 6 and Hg(ClO4)2 were varied while the total concentration [6 + Hg(ClO4)2] remained constant (20 μM). | ||
The effect of counterion on the turn-on fluorescence response of 6 in the presence of Hg2+ was also probed (Fig. 5). Addition of Hg(OAc)2 and HgCl2 to solutions of 6 produced a fluorescence response comparable to that observed upon addition of Hg(ClO4)2. Inclusion of nitrate counterions resulted in diminished fluorescence, and addition of HgI2 resulted in fluorescence quenching.
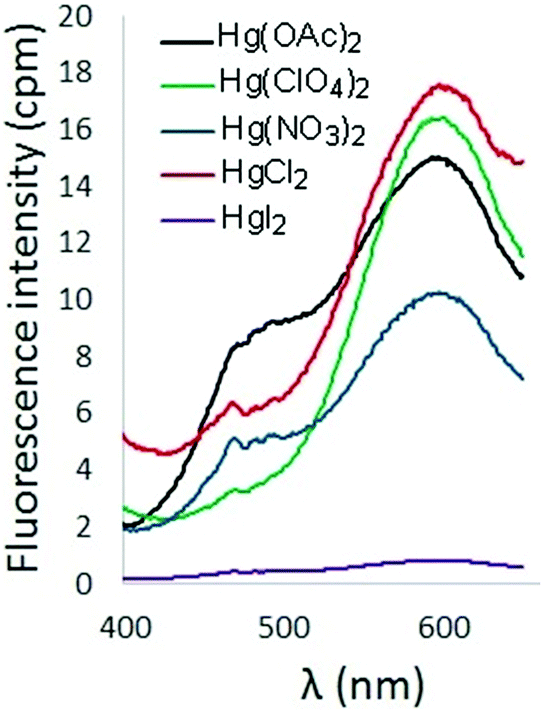 | ||
| Fig. 5 Fluorescence spectra of 6 in the presence of Hg2+ with different counterions (λex = 338 nm, [6] = 10 μM). | ||
The fluorescence response of bithiophene derivative 7 in 9:1 H2O:CH3CN was also examined in the presence of 2 equivalents of various metal ions. Once again, most metal ions elicited little to no change in fluorescence, including Zn2+ and Cd2+ (which did produce a fluorescent response with 6). Addition of Hg(ClO4)2, however, resulted in a significantly red-shifted and enhanced fluorescence signal centered at 633 nm (Φ = 20.8%, Fig. 6A). As illustrated in Fig. 6B, the selective turn-on response of 7 to Hg2+ is not affected by the presence of Group I and Group II metal ions (Na+, K+, Ca2+, Mg2+ and Ba2+). Moreover, the Hg2+-selective fluorescence response of 7 is also unaffected by the presence of other transition metal or heavy metal ions, with the exception of Cu2+ and, to a lesser extent, Fe3+.
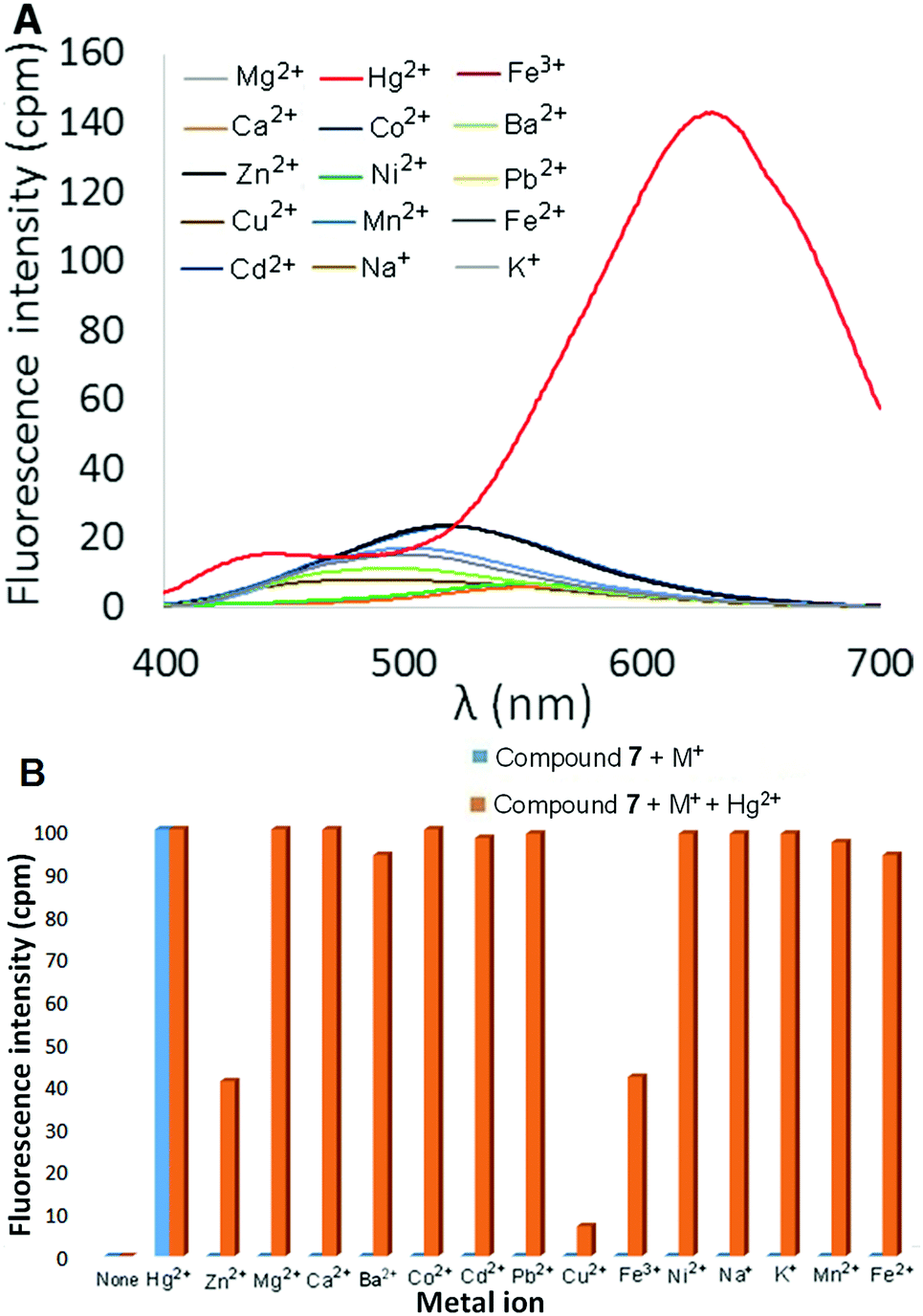 | ||
| Fig. 6 (A) Fluorescence spectra of 7 in the presence of various metal ions (2 equivalents). (B) Emission of 7 at 633 nm in the presence of various metal ions and Hg2+–metal ion combinations (normalized to emission in the presence of Hg2+). λex = 351 nm, [7] = 10 μM. | ||
A Job plot based on the emission response of 7 in presence of different mole fractions of Hg2+ in 9:1 H2O:CH3CN was almost identical to the plot constructed for 6 (Fig. 4) and indicated a similar 2:1 (7:Hg2+) binding stoichiometry (see ESI†). The effect of pH on the turn-on response of 7 to Hg2+ was additionally probed over a wide range of pH (Fig. 7). At acidic pH (pH < 5), protonation of the (2-pyridyl) groups will affect the ability of 7 to chelate Hg2+ and consequently no emission was observed. At basic pH, the intensity of emission at 633 nm is significantly diminished, which is likely due to formation of Hg(OH)2. The maximum emission response was observed between pH ∼ 6–9, which indicates that 7 is suitable for sensing Hg2+ at physiological pH. Finally, the limit of detection of 7 for Hg2+ was determined to be 48 nM (ESI†).
 | ||
| Fig. 7 Fluorescence intensity at 633 nm of 7 in the absence and presence of Hg2+ (2 equivalents) as a function of pH (λex = 351 nm, [7] = 10 μM). | ||
Solution phase binding interactions between 7 and Hg(ClO4)2 were further investigated by NMR. Fig. 8 shows a series of 1H-NMR spectra of 7 obtained in the absence and presence of increasing amounts of Hg(ClO4)2. These experiments were performed in 1:1 D2O:CD3CN in order to maintain solubility of 7 at the concentration needed for NMR spectroscopy. The signal corresponding to the ortho-H of the 2-pyridyl group (doublet at 8.42 ppm) is incrementally shifted downfield upon addition of up to 0.5 equivalent of Hg(ClO4)2. This shift is attributed to coordination of Hg2+ to the pyridine nitrogen atoms and is in agreement with our previous results observed upon coordination of Zn2+ to 4.18,23 Further addition of Hg(ClO4)2 (beyond 0.5 equivalents) results in significant shifting and broadening of all peaks in the aromatic region, including signals for the bithiophene hydrogens. The downfield shifts of all aryl hydrogen resonances and peak broadening might reflect formation of ternary ligand:metal complexes, consistent with the results of Job plot analysis which indicates a 2:1 ligand:Hg2+ stoichiometry. Qualitatively similar peak shifting/broadening as a function of [Hg2+] was also observed in NMR titrations of 6 (see Fig. S13, ESI†).
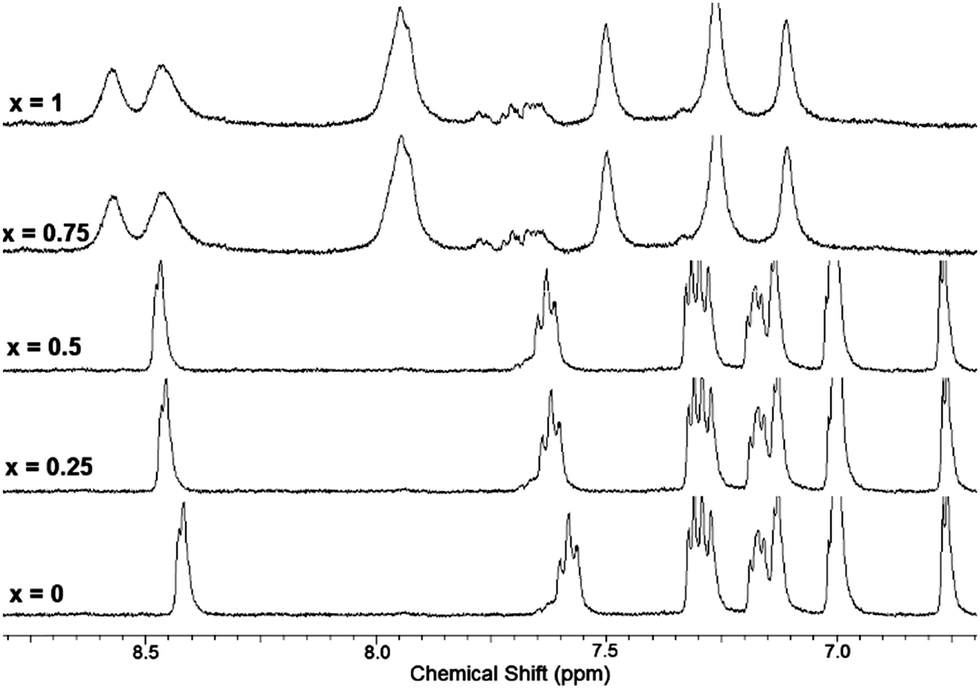 | ||
| Fig. 8
1H NMR spectra of 7 + Hg(ClO4)2 at different equivalents of Hg(ClO4)2 (x = number of Hg(ClO4)2 equivalents) in D2O:CD3CN (1:1), [7] = 3 mM. | ||
Variation in N-heterocycle was also briefly examined through preparation of tetraarylethylenes possessing 2-quinolinyl substituents in place of 2-pyridyl groups. Dibromoalkene 9 was obtained upon treatment of di(quinolin-2-yl)methanone (8)24 with CBr4 and PPh3. Subsequent double Suzuki reaction of 9 with either thiophene-2-boronic acid or 2,2′-bithiophene-5-boronic acid pinacol ester afforded 10 and 11 in serviceable yields (Scheme 2). The AIE properties of 10 and 11 were examined in CH3CN/H2O mixtures. The AIE profile of 10 mirrors that of 6 in that incremental addition of H2O results in enhanced and blue-shifted emission at ∼462 nm compared to emission at ∼515 nm in 100% CH3CN (Fig. 9A). Compound 11 exhibited a much reduced AIE effect in CH3CN/water mixture compared to the other compounds examined in this study. A weak emission at ∼413 nm in pure CH3CN was found to slightly shift to 435 nm upon incremental addition of H2O, but fluorescence intensity was only modestly enhanced (Fig. 9B).
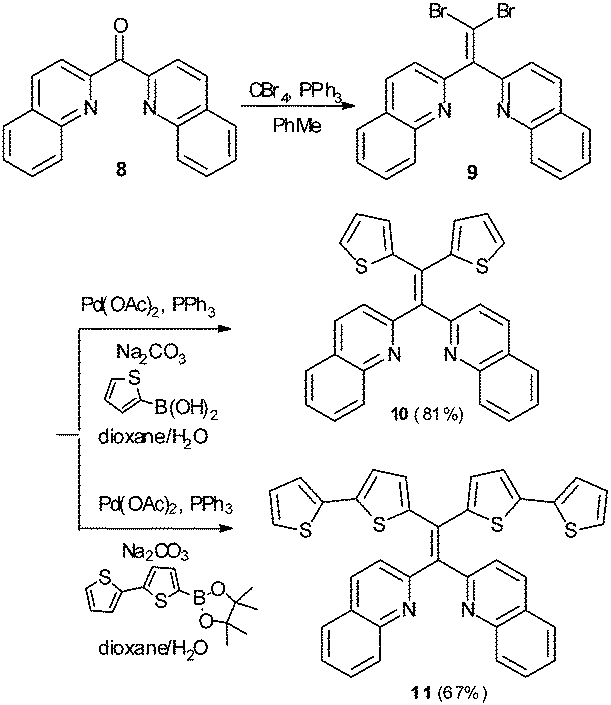 | ||
| Scheme 2 Synthesis of 2,2′-bis(quinolinyl) tetraarylethylenes 10 and 11. | ||
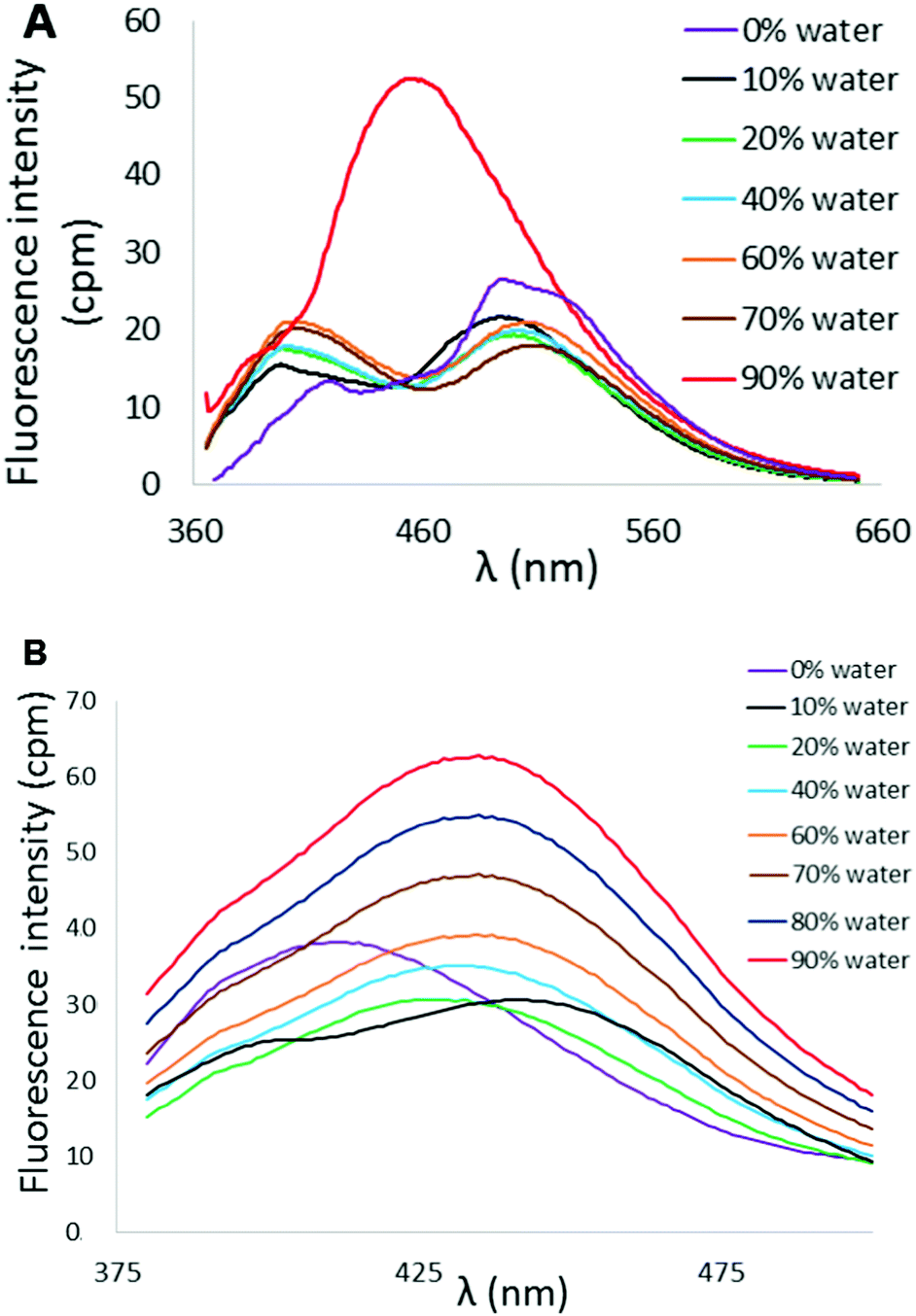 | ||
| Fig. 9 (A) AIE profile of 10 in CH3CN/H2O mixtures (λex = 353 nm, [10] = 10 μM). (B) AIE profile of 11 in CH3CN/H2O mixtures (λex = 364 nm, [11] = 10 μM). | ||
In contrast to the selective turn-on fluorescence response observed for 6 and 7 upon exposure to Hg2+, addition of Hg(ClO4)2 (2 equivalents) to 10 in 90% aqueous CH3CN resulted in complete fluorescence quenching (Fig. 10). A similar result was obtained with compound 11 as well (ESI†). The behaviour of 10 and 11 is attributed to chelation-enhanced quenching (CHEQ) effects, which are often observed upon coordination of Hg2+ to fluorophores.9,11 Interestingly, only the most downfield hydrogens in the 1H-NMR spectra of 10 and 11 (corresponding to the H4 and H5 signals of the quinoline rings) are shifted upon addition of up to 1 equivalent Hg(ClO4)2, and the remaining quinoline and thiophene/bithiophene signals are unaffected by the presence of mercuric ion (Fig. S14 and S15, ESI†). This behaviour contrasts with the spectra of 6 or 7 + Hg2+ in which all the 1H-NMR signals exhibited appreciable downfield shifting and broadening (see Fig. 8 and Fig. S13, ESI†). While uncovering the exact mechanisms underlying the divergent fluorescence and NMR responses exhibited by 6/7 and 10/11 toward Hg2+ ions requires additional research, steric hindrance and/or extended π systems introduced through incorporation of quinoline rings into the tetraarylethylene framework has clearly altered the photophysical response of 10 and 11 toward Hg2+, and the bis(2-pyridyl)ethylene fragment in 6 and 7 appears crucial for selective turn-on fluorescence detection of Hg2+.
 | ||
| Fig. 10 Fluorescence spectra of 10 in 9:1 H2O:CH3CN in the absence and presence of Hg(ClO4)2 (2 equivalents). λex = 353 nm, [10] = 10 μM. | ||
Conclusions
In summary, two new fluorescent turn-on probes (6 and 7) have been developed for sensing of Hg2+ in aqueous solution. These probes were designed by combining metal chelating 1,1-bis(2-pyridylethylene) fragments and thiophene/bithiophene substituents in a single AIE-active platform. Job plots and 1H-NMR titrations were used to investigate solution phase binding of the new sensors and Hg2+. An X-ray crystal structure of the complex between 6 and HgI2 confirmed the Hg2+-chelating ability of the 1,1-bis(2-pyridyl)ethylene moiety. Replacement of the 2-pyridyl groups with 2-quinoline rings (as in 10 and 11), however, completely abolished the selective fluorescence turn-on response toward Hg2+. Examples of turn-on fluorescent sensors for Hg2+ are rare, and the utilization of an AIE-active scaffold in 6 and 7 eliminates the need for incorporation of additional fluorophores.8a,12–16 Further modification of the relatively simple tetraarylethylene framework in 6 and 7 to provide even greater selectivity and sensitivity toward Hg2+ is being examined, as is the design and construction of analogues that are expected to exhibit altered metal ion selectivities. Indeed, combining the AIE activity of tetraarylethylenes with metal ligating/chelating groups offers a wealth of opportunity for development of selective metal ion sensors with numerous potential biomedical and environmental applications.Experimental
General
All commercially available starting materials, reagents, and solvents were used as supplied unless otherwise stated. Reported yields are isolated yields. Proton (1H) and carbon (13C) NMR were collected on Bruker NMR spectrometers at 300 MHz or 400 MHz for 1H and 100 MHz or 75 MHz for 13C. Chemical shifts (δ) are reported in parts-per million (ppm) relative to residual undeuterated solvent. Melting points were recorded using a capillary melting point apparatus and are uncorrected. High resolution mass spectra were obtained in positive ion mode using electrospray ionization (ESI) on a double-focusing magnetic sector mass spectrometer. UV-visible spectra were obtained using quartz cuvettes on a Varian Cary 100-Scan dual-beam spectrophotometer. Each measurement was done in duplicate and compared to solvent blank. Blank samples were prepared using HPLC grade acetonitrile. Fluorescence spectra were obtained in air at room temperature using a Horiba Jobin Yvon Fluoromax-4 spectrofluorimeter using 3 mL quartz cuvettes. Quantum yields (Φ) were determined relative to quinine sulfate. Samples were prepared using HPLC grade solvents. X-Ray diffraction data were collected on a Nonius Kappa CCD diffractometer equipped with Mo Kα radiation with λ = 0.71073 Å. Structures were solved by direct methods and data was refined by full-matrix least squares refinement on F2 against all reflections.Synthetic procedures
:water (4:1). The flask was charged with Na2CO3 (1.38 g, 10.00 mmol), Pd(OAc)2 (100 mg, 0.43 mmol), PPh3 (470 mg, 1.80 mmol) and thiophene-2-boronic acid (1.28 g, 10.00 mmol). The reaction was heated to reflux under argon overnight. After cooling, the reaction mixture was diluted with water and extracted with ethyl acetate (2 × 50 mL) and the combined organic fractions were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography using ethyl acetate/hexane 1:2 and 2:1 as eluent to yield 6 (506 mg, 73%) as a yellow solid. Mp 191–193 °C. 1H NMR (400 MHz, CDCl3) δ 6.81–6.84 (m, 4H), 7.06–7.10 (m, 2H), 7.22–7.24 (m, 2H), 7.26–7.29 (m, 2H), 7.65–7.70 (m, 2H), 8.54 (d, J = 5.4 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 122.7, 127.2, 127.5, 129.5, 129.6, 131.3, 133.1, 137.1, 145.3, 150.5, 161.6. HRMS (ESI): calcd for C20H15N2S2 [M + H]+, 347.0677; found, 347.0672.
:4, 10 ml), HgI2 (26 mg, 0.058 mmol) was added. The solution was allowed to slowly evaporate over 2 days to give 6·HgI2 (25.1 mg, 54%) as yellowish brown crystals.25 Mp >200 °C.
:1 as eluent to yield 9 (1.47 g, 56%) as dark yellow solid. Mp 183–185 °C. 1H NMR (300 MHz, CDCl3) δ 7.52–7.58 (m, 2H), 7.72–7.86 (m, 6H), 8.14–8.26 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 99.4, 124.7, 129.7, 129.9, 130.3, 132.3, 132.5, 139.3, 149.5, 150.5, 160.4. HRMS (ESI): calcd for C20H13N2Br2 [M + H]+, 438.9445; found, 438.9452.
Job plots
Job plots were constructed to determine the binding stoichiometry between sensors and Hg(ClO4)2 by monitoring the change in fluorescence emission intensity as a function of sensor/Hg2+ mole fraction. Stock solutions (20 μM each) of sensor and Hg(ClO4)2 were prepared in H2O:CH3CN (9:1). Samples were prepared with different mole fractions of sensor and Hg(ClO4)2 while maintaining the total concentration of ([sensor] + [Hg(ClO4)2]) for each sample at 20 μM.
Competition experiments
Hg(ClO4)2 (2 equivalents) was added to solutions of sensor (10 μM) and the tested metal ion (2 equivalents) in H2O:CH3CN (9:1). Test solutions were stirred for 5 min and then allowed to stand at room temperature for 30 min. Fluorescence measurements were done at λex = 338 nm for 6, and λex = 351 nm for 7.
Acknowledgements
We thank the Department of Chemistry and the Graduate College of the University of Iowa for support. We thank Dr Dale Swenson for assistance with X-ray crystallography.Notes and references
- B. L. Sui, S. Tang, T. H. Liu, B. Kim and K. D. Belfield, ACS Appl. Mater. Interfaces, 2014, 6, 18408–18412 CAS.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- R. Miao, L. X. Mu, H. Y. Zhang, G. W. She, B. J. Zhou, H. T. Xu, P. F. Wang and W. S. Shi, Nano Lett., 2014, 14, 3124–3129 CrossRef CAS PubMed.
- G. Sener, L. Uzun and A. Denizli, ACS Appl. Mater. Interfaces, 2014, 6, 18395–18400 CAS.
- (a) H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC; (b) E. M. Nolan and S. Lippard, Chem. Rev., 2008, 108, 3443–3480 CrossRef CAS PubMed; (c) H. H. Harris, I. J. Pickering and G. N. George, Science, 2003, 301, 1203 CrossRef CAS PubMed; (d) D. W. Boening, Chemosphere, 2000, 40, 1335–1351 CrossRef CAS PubMed; (e) Z. Yan, M. Yeun, L. Hu, P. Sun and C. Lee, RSC Adv., 2014, 4, 48373–48388 RSC.
- (a) K. Bera, A. K. Das, M. Nag and S. Basak, Anal. Chem., 2014, 86, 2740–2746 CrossRef CAS PubMed; (b) F. Wang, S. W. Nam, Z. Guo, S. Park and J. Yoon, Sens. Actuators, B, 2012, 161, 948–953 CrossRef CAS; (c) J. Jiang, W. Liu, J. Cheng, L. Yang, H. Jiang, D. Bai and W. Liu, Chem. Commun., 2012, 48, 8371–8373 RSC; (d) B. Weiss, Toxicol. Sci., 2007, 97, 223–225 CrossRef CAS PubMed.
- (a) H. Ding, C. Zheng, B. Li, G. Liu, S. Pu, D. Jia and Y. Zhou, RSC Adv., 2016, 6, 80723–80728 RSC; (b) L. Lindenburg and M. Merkx, ChemBioChem, 2012, 13, 349–351 CrossRef CAS PubMed; (c) Z. Guo, G. Chen, G. Zeng, Z. Li, A. Chen, M. Yan, L. Liu and D. Huang, RSC Adv., 2014, 4, 59275–59283 RSC; (d) M. I. J. Stich, L. H. Fischer and O. S. Wolfbeis, Chem. Soc. Rev., 2010, 39, 3102–3114 RSC; (e) X. Cheng, Q. Li, C. Li, J. Qin and Z. Li, Chem. – Eur. J., 2011, 17, 7276–7281 CrossRef CAS PubMed; (f) M. H. Lee, S. W. Lee, S. H. Kim, C. Kang and J. S. Kim, Org. Lett., 2009, 11, 2101–2104 CrossRef CAS PubMed; (g) S. K. Kim, K. M. Swamy, S.-Y. Chung, H. N. Kim, M. J. Kim, Y. Jeong and J. Yoon, Tetrahedron Lett., 2010, 51, 3286–3289 CrossRef CAS; (h) M. G. Choi, Y. H. Kim, J. E. Namgoong and S.-K. Chang, Chem. Commun., 2009, 3560–3562 RSC.
- (a) R. X. Zhang, P. F. Li, W. J. Zhang, N. Li and N. Zhao, J. Mater. Chem. C, 2016, 4, 10479–10485 RSC; (b) D. Dinda, B. K. Shaw and S. K. Saha, ACS Appl. Mater. Interfaces, 2015, 7, 14743–14749 CrossRef CAS PubMed; (c) G. Chen, Z. Guo, G. Zeng and L. Tang, Analyst, 2015, 140, 5400–5443 RSC; (d) J. Ding, H. Li, C. Wang, J. Yang, Y. Xie, Q. Peng, Q. Li and Z. Li, ACS Appl. Mater. Interfaces, 2015, 7, 11369–11376 CrossRef CAS PubMed; (e) P. Mahato, S. Saha, P. Das, H. Agarwalla and A. Das, RSC Adv., 2014, 4, 36140–36174 RSC.
- (a) A. T. Afaneh and G. Schreckenbach, J. Phys. Chem. A, 2015, 119, 8106–8116 CrossRef CAS PubMed; (b) G. Aragay, J. Pons and A. Merkoci, Chem. Rev., 2011, 111, 3433–3458 CrossRef CAS PubMed.
- (a) J. Hu, J. Li, J. Qi and J. Chen, New J. Chem., 2015, 39, 843–848 RSC; (b) H. Wang and S. Wu, Tetrahedron, 2013, 69, 1965–1969 CrossRef CAS; (c) P. Srivastava, R. Ali, S. S. Razi, M. Shahid and A. Misra, Sens. Actuators, B, 2013, 181, 584–595 CrossRef CAS; (d) P. Srivastava, R. Ali, S. S. Razi and M. Shahid, Tetrahedron Lett., 2013, 54, 3688–3693 CrossRef CAS; (e) K. Kanagaraj, K. Bavanidevi, T. J. Chow and K. Pitchumani, RSC Adv., 2014, 4, 11714–11722 RSC; (f) C. Zhou, Y. Song and Y. Li, RSC Adv., 2014, 4, 33614–33618 RSC.
- For a discussion of factors contributing to chelation-enhanced quenching effects in Hg2+ sensors, see H. Lee, H.-S. Lee, J. H. Reibenspies and R. D. Hancock, Inorg. Chem., 2012, 51, 10904–10915 CrossRef CAS PubMed.
- (a) E. M. Nolan and S. J. Lippard, J. Am. Chem. Soc., 2003, 125, 14270–14271 CrossRef CAS PubMed; (b) E. M. Nolan and S. J. Lippard, J. Am. Chem. Soc., 2007, 129, 5910–5918 CrossRef CAS PubMed.
- S. Yoon, E. W. Miller, Q. He, P. H. Do and C. J. Chang, Angew. Chem., Int. Ed., 2007, 46, 6658–6661 CrossRef CAS PubMed.
- (a) C.-C. Huang and H.-T. Chang, Anal. Chem., 2006, 78, 8332–8338 CrossRef CAS PubMed; (b) H. Wang, Y. Wang, J. Jin and R. Yang, Anal. Chem., 2008, 80, 9021–9028 CrossRef CAS PubMed.
- (a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (b) Z. Zhao, B. He and B. Z. Tang, Chem. Sci., 2015, 6, 5347–5365 RSC; (c) D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed; (d) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; (e) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC; (f) M. T. Gabr and F. C. Pigge, RSC Adv., 2015, 5, 90226–90234 RSC; (g) N. L. C. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Y. Lam and B. Z. Tang, Chem. – Eur. J., 2014, 20, 15349–15353 CrossRef CAS PubMed.
- (a) L. N. Neupane, E.-T. Oh, H. J. Park and K.-H. Lee, Anal. Chem., 2016, 88, 3333–3340 CrossRef CAS PubMed; (b) Y. Chen, W. Zhang, Y. Cai, R. T. Kwok, Y. Hu, J. W. Lam, X. Gu, Z. He, Z. Zhao, X. Zheng, B. Chen, C. Gui and B. Z. Tang, Chem. Sci., 2017, 8, 2047–2055 RSC; (c) L. Liu, G. Zhang, J. Xiang, D. Zhang and D. Zhu, Org. Lett., 2008, 10, 4581–4584 CrossRef CAS PubMed; (d) N. Zhao, J. W. Y. Lam, H. H. Y. Sung, H. M. Su, I. D. Williams, K. S. Wong and B. Z. Tang, Chem. – Eur. J., 2014, 20, 133–138 CrossRef CAS PubMed.
- (a) B. N. Ahamed and P. Ghosh, Dalton Trans., 2011, 40, 12540–12547 RSC; (b) B. N. Ahamed, I. Ravikumar and P. Ghosh, New J. Chem., 2009, 33, 1825–1828 RSC.
- M. T. Gabr and F. C. Pigge, Dalton Trans., 2016, 45, 14039–14043 RSC.
- (a) S. Park, W. Kim, K. M. Swamy, H. Y. Lee, J. Y. Jung, G. Kim, Y. Kim, S.-J. Kim and J. Yoon, Dyes Pigm., 2013, 99, 323–328 CrossRef CAS; (b) J. H. Huang, Y. F. Xu and X. H. Qian, J. Org. Chem., 2009, 74, 2167–2170 CrossRef CAS PubMed.
- (a) D. Singhal, N. Gupta and A. K. Singh, RSC Adv., 2015, 5, 65731–65738 RSC; (b) W. Huang, X. Zhu, D. Wua, C. He, X. Hu and C. Duan, Dalton Trans., 2009, 10457–10465 RSC; (c) A. K. Shigemoto, C. N. Virca, S. J. Underwood, L. R. Shetterly and T. M. McCormick, J. Coord. Chem., 2016, 69, 2081–2089 CrossRef CAS; (d) A. Lannes, A. Manceau, M. Rovezzi, P. Glatzel, Y. Joly and I. Gautier-Luneau, Dalton Trans., 2016, 45, 14035–14038 RSC.
- R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Lam, H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Pena-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CAS.
- G.-F. Zhang, H. Wang, M. P. Aldred, T. Chen, Z.-Q. Chen, X. Meng and M.-Q. Zhu, Chem. Mater., 2014, 26, 4433–4446 CrossRef CAS.
- G. Orellana, C. A. Ibarra and J. Santoro, Inorg. Chem., 1988, 27, 1025–1030 CrossRef CAS.
- C. T. Burns and R. F. Jordan, Organometallics, 2007, 26, 6737–6749 CrossRef CAS.
- CCDC 1533224.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data, crystallographic data. CCDC 1533224. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00085e |
| This journal is © the Partner Organisations 2017 |