
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium catalyzed diastereoselective synthesis of 2,2,3,3-tetrasubstituted indolines from N-sulfonyl-1,2,3-triazoles and ortho-vinylanilines†
Dongari
Yadagiri
,
Angula Chandra Shekar
Reddy
and
Pazhamalai
Anbarasan
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: anbarasansp@iitm.ac.in; Web: http://chem.iitm.ac.in/faculty/anbarasan/profanbu/
First published on 24th May 2016
Abstract
An efficient diastereoselective rhodium catalyzed synthesis of indolines possessing two contiguous tetrasubstituted carbon centers has been achieved with good to excellent yields using ortho-vinylanilines and iminocarbenes derived from N-sulfonyl-1,2,3-triazoles. The reaction affords excellent cis-diastereoselectivity through the initial formation of a N-ylide followed by intramolecular trapping with unactivated alkenes via an ene-type reaction with a well-organized transition state, namely intramolecular carbenylative amination of alkenes. The developed transformation was further extended to the successful synthesis of tricyclic compounds, imidazoindolines, through reduction and hypervalent iodine mediated oxidative cyclization.
Introduction
2,3-Multisubstituted indolines are widely present in various alkaloid natural products and therapeutically important molecules possessing diverse biological activities.1 Representative examples include minfiensine, vindoline and strychnine, which exhibit significant anticancer activities (Fig. 1).2 Hence, over several decades 2,3-multisubstituted indolines have attracted great synthetic interest.3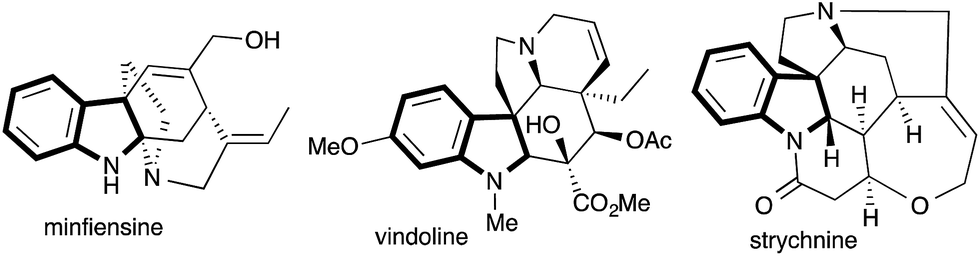 | ||
| Fig. 1 Representative examples of indoline alkaloids. | ||
Most of the known traditional methods involve different functionalizations of (ox)indole derivatives.4 Recently, these methods have been replaced with transition metal catalyzed5 or free radical6 construction of indoline either through C–C or C–N bond forming reactions.7 Hu and co-workers8 utilized the intramolecular trapping of ammonium ylides, generated from diazo carbonyl compounds and an amine, with either a polar double bond-like ketone or an activated alkene for the synthesis of 2,2,3-trisubstituted indolines. Although the intramolecular trapping of ammonium ylides with activated double bonds has been studied, trapping with an unactivated alkene, such as styrene which would afford 2,3-multisubstituted indolines, is very rare and highly desirable.
Over the past few years, N-sulfonyl-1,2,3-triazoles have emerged as a valuable resource for the generation of imino carbenes, a new reactive intermediate in organic synthesis.9 Unlike the traditional α-oxocarbenes, imino carbenes possess unique reactivity due to the presence of both an electrophilic carbene and nucleophilic nitrogen. This has been utilized in the construction of pharmaceutically important nitrogen based building blocks and heterocycles, through various carbene induced transformations.10 For instance, Fokin et al.11 and Yoo et al.12 demonstrated a formal stereoselective 1,3-insertion of rhodium imino carbenes into the N–H bond of amine derivatives through the generation of a N-ylide (Scheme 1).
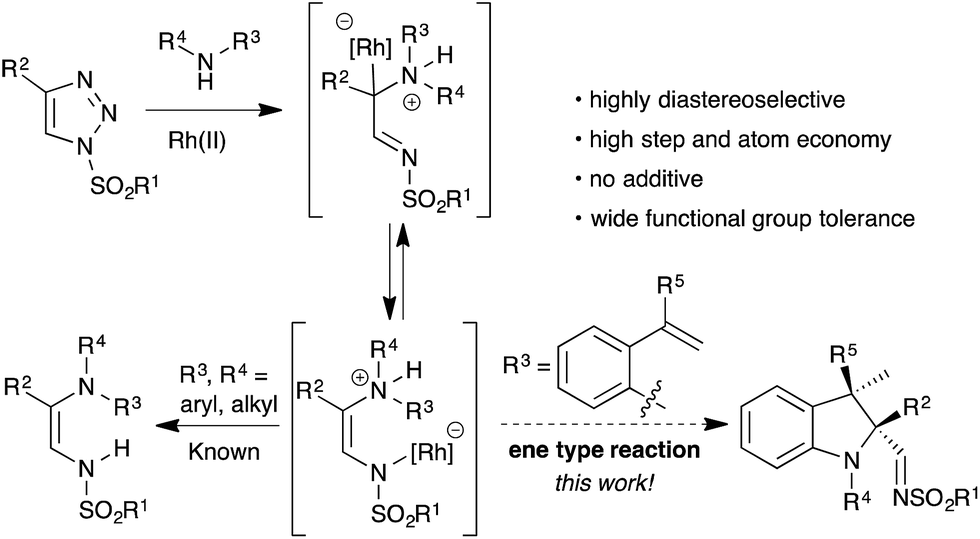 | ||
| Scheme 1 Rhodium-catalyzed reactions of N-sulfonyl-1,2,3-triazoles. | ||
Inspired by these studies and our continuous efforts for the functionalization of imino carbenes,13 we reasoned that ammonium ylides generated from suitably substituted ortho-vinylanilines and a rhodium imino carbene could undergo intramolecular trapping in a ene type fashion with an unactivated alkene (Scheme 1). This transformation would enable access to the stereoselective synthesis of 2,2,3,3-tetrasubstituted indoline motifs containing contiguous tetrasubstituted carbon centers via the formation of C–N and C–C bonds in a single operation, namely carbenylative amination of alkenes.
Results and discussion
Intramolecular carbenylative amination of ortho-vinylaniline 2a with N-sulfonyl-1,2,3-triazole 1a to provide 2,2,3,3-tetrasubstituted indoline 3aa was chosen at the start of our investigation as a model reaction. The reaction of 1 equivalent of 1a and 1.5 equivalents of 2a in the presence of 2 mol% of Rh2(OAc)4 in toluene at room temperature (∼35 °C) did not afford the expected product 3aa. To our delight, increasing the temperature to 90 or 100 °C furnished the tetrasubstituted indoline 3aa, wherein formation of 3aa was observed with a 15% yield at 100 °C as a single diastereoisomer, based on 1H NMR and HPLC analysis (Table 1, entries 1–2). NOE results suggested that the relative stereochemistry of the isolated diastereomer is cis, i.e. the methyl and imine substituents are cis in relation (see ESI†).|
|
||||
|---|---|---|---|---|
| Entry | Rh(II) | Solvent | Temp (°C) | Yielda (%) |
| a Isolated yields. b 1 mol% of Rh2(Piv)4. R = p-methylbenzyl. | ||||
| 1 | Rh2(OAc)4 | Toluene | 90 | <5 |
| 2 | Rh2(OAc)4 | Toluene | 100 | 15 |
| 3 | Rh2(Oct)4 | Toluene | 100 | 30 |
| 4 | Rh2(Piv)4 | Toluene | 100 | 45 |
| 5 | Rh2(TBSP)4 | Toluene | 100 | 18 |
| 6 | Rh2(S-NTTL)4 | Toluene | 100 | 8 |
| 7 | Rh2(Piv)4 | Benzene | 100 | 35 |
| 8 | Rh2(Piv)4 | 1,2-DCE | 100 | 29 |
| 9 | Rh2(Piv)4 | Toluene | 120 | 85 |
| 10 | Rh2(Piv)4 | Toluene | 120 | 75b |
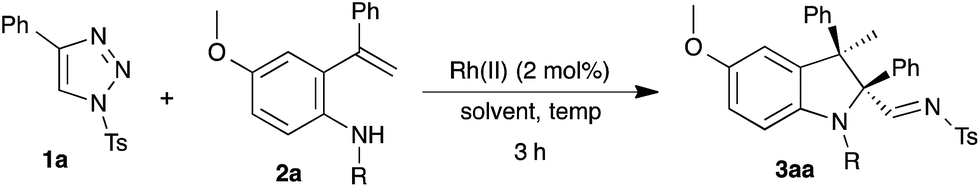
Encouraged by this result, next, the influence of the catalyst was examined. Changing Rh2(OAc)4 to a bulkier catalyst such as Rh2(Oct)4 or Rh2(Piv)4 showed a significant improvement in the yield (Table 1, entries 3–4). Next, screening of Rh2(TBSP)4 and Rh2(S-NTTL)4, chiral rhodium catalysts, under similar conditions afforded 3aa in diminished yield (Table 1, entries 5–6). Unfortunately, no appreciable enantioselectivity was observed. Similarly, a low yield of 3aa was observed with other solvents such as benzene and 1,2-DCE (Table 1, entries 7–8). Gratifyingly, further increasing the temperature to 120 °C with Rh2(Piv)4 provided the product 3aa in 85% yield, and the conditions used for this reaction were chosen as the optimized conditions for studying the reaction scope and limitations (Table 1, entry 9).
Having optimized the conditions, the generality of the present method was investigated with functionally different 1,2,3-triazoles 1. As can be seen in Scheme 2, various substituted triazoles 1 underwent efficient rhodium catalyzed intramolecular carbenylative amination with 2a to afford multisubstituted indolines 3. Changing the sulfonyl moiety of the triazole afforded the corresponding indolines 3aa–3ea in excellent yield. Interestingly, both electron donating and withdrawing groups as well as reactive functional groups like a nitro group and enolizable ketone were well tolerated under the optimized conditions to furnish the indolines (3fa–3ha, 3ja–3ma) with good to excellent yields. Medicinally important fluorinated and trifluoromethylated indolines (3ia, 3na, 3oa) were successfully obtained from the corresponding triazoles in 92%, 77% and 80% yield, respectively. Furthermore, thiophene substituted and 4,5-disubstituted triazoles also underwent the reaction smoothly to provide indoline 3pa and 3qa, respectively. It is important to note that in all cases the cis-diastereomer was the only detectable isomer.
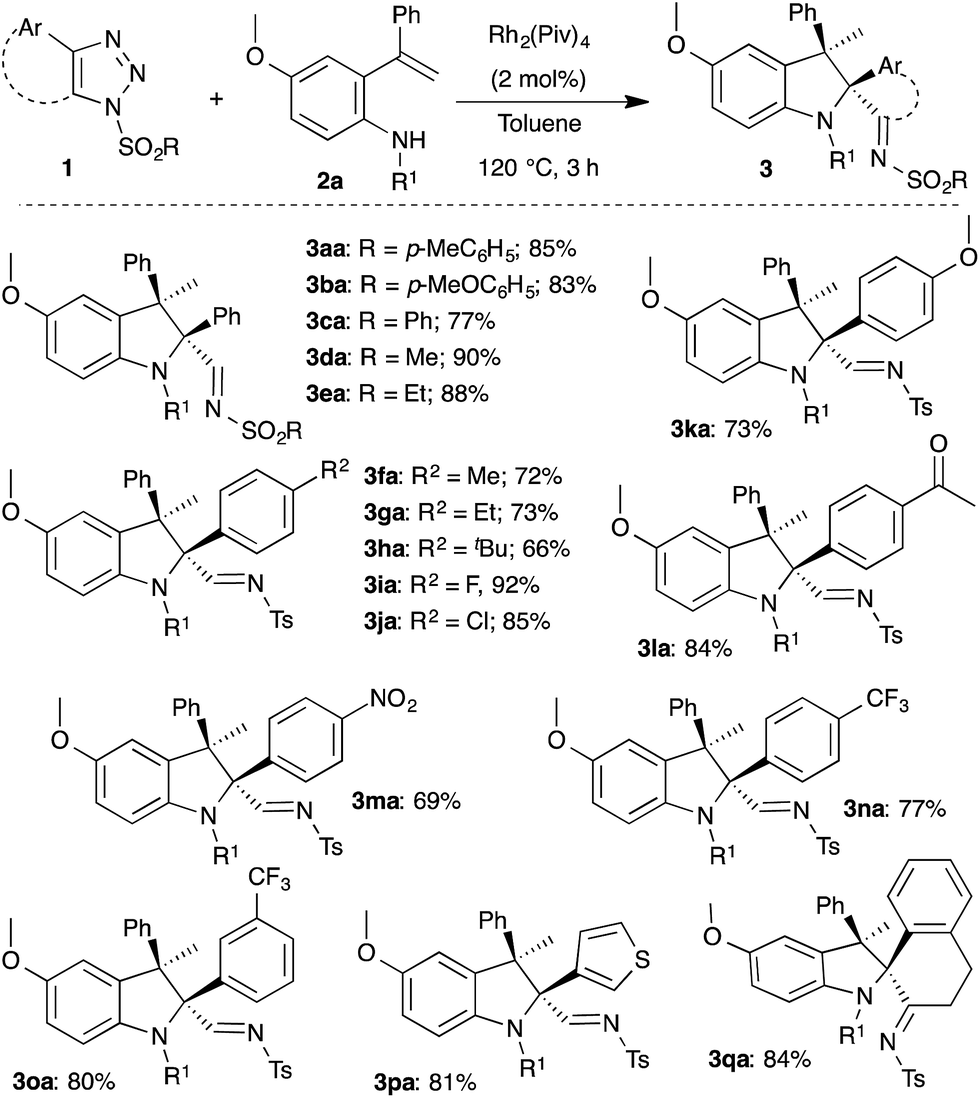 | ||
| Scheme 2 Rhodium catalyzed carbenylative amination: scope of the triazoles 1. R1 = p-methylbenzyl. | ||
Next, the scope in relation to ortho-vinylanilines 2 was investigated using the optimized conditions. Initially, the effect of the substituent on the nitrogen was examined. p-Methoxybenzyl and methyl substituted aniline derivatives provided indolines 3ab and 3ad in good yield (Scheme 3). The structure and relative stereochemistry of indole 3ad was unambiguously confirmed using single crystal X-ray analysis.14 A sterically hindered cyclohexyl substituted aniline provided 3ac in relatively low yield. Unfortunately, having an electron-withdrawing substituent on the nitrogen did not afford the expected product 3ae. Subsequently, screening of different substituents on the aromatic ring of the aniline was carried out. Electronically and sterically different substituents on the aromatic ring were well tolerated, especially, reactive functional groups such as acetal, sulfonate and silyl ether groups were untouched under the reaction conditions, and led to the formation of the corresponding indolines (3af–3ak) with good to excellent yields. Different C3-substituted indolines 3al and 3am were also obtained in 81% and 60% yield, respectively, from the corresponding alkene derivative. Furthermore, 2,2,3-trisubstituted indoline 3an was also synthesized, employing the present method, in 49% yield with excellent diastereoselectivity from a simple vinyl substituted aniline.
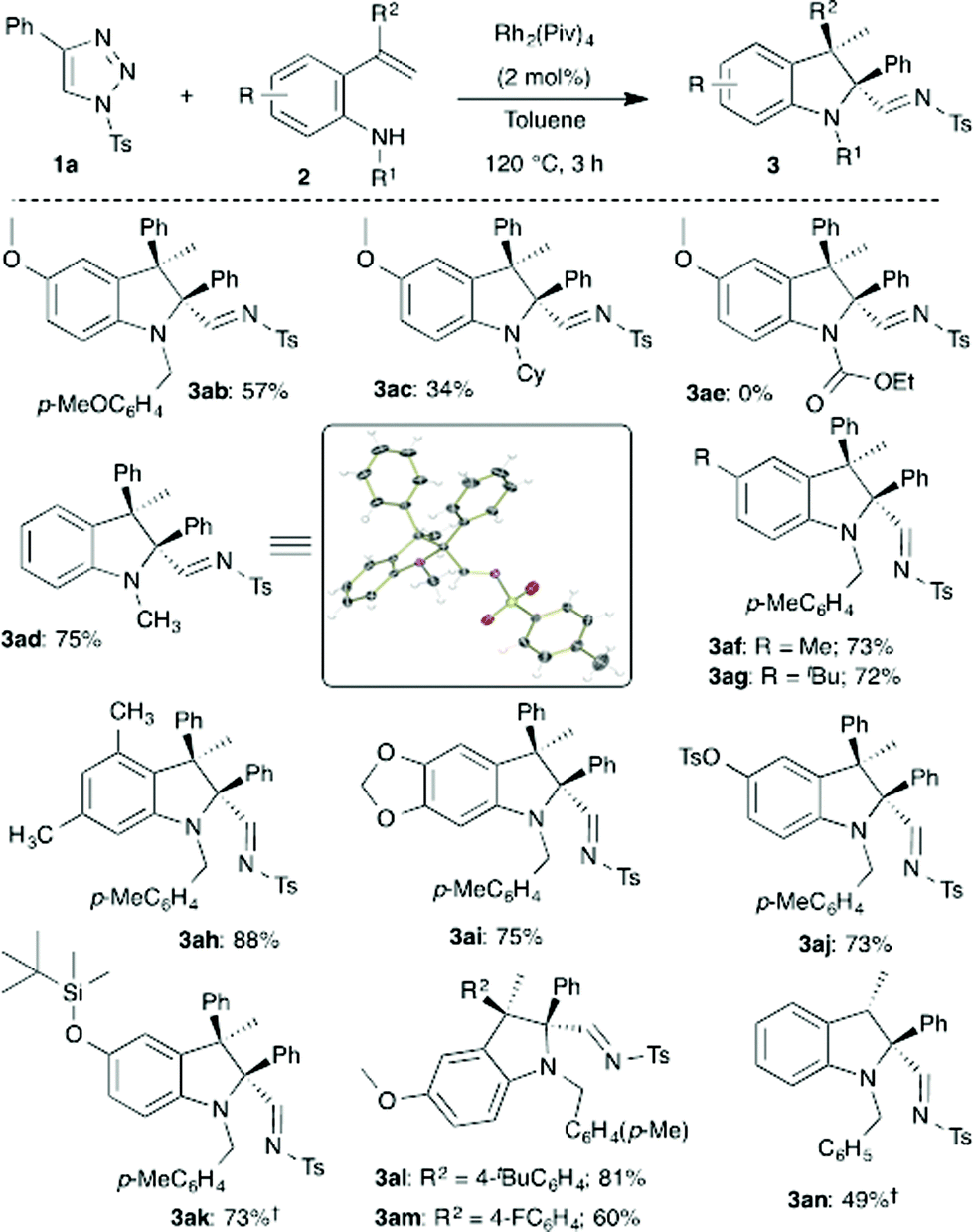 | ||
| Scheme 3 Rhodium catalyzed carbenylative amination: scope of the o-vinylanilines 2. †NMR yield. | ||
Diastereoselective formation of the tetrasubstituted indolines can be explained with the following plausible mechanism (Scheme 4a). The reaction starts with the generation of rhodium carbenoid A from the N-sulfonyl-1,2,3-triazole using the rhodium catalyst,9 which on subsequent reaction with the o-vinylaniline 2 would afford the ammonium ylide B. A 1,3-rhodium shift in the ammonium ylide B would produce the zwitterion C. Formation of indoline 3 from C could be rationalized with two pathways: (1) a rhodium promoted intramolecular metalloene15 reaction of C would generate the cyclized intermediate D with high diastereoselectivity, and a following protonation of rhodium species D would furnish the product 3 along with regeneration of the rhodium catalyst which continues the catalytic cycle. (2) Initial protonation of rhodium species C to provide enamine E followed by an intramolecular aza-ene reaction16 would afford the observed product 3. To investigate the feasible pathways, various attempts were made to isolate the enamine E. Unfortunately, all our attempts either resulted in no reaction or the formation of product 3, formation of enamine E was not observed. Hence, at this point both the pathways are equally possible. As can be seen in Scheme 4a, the cis-diastereoselectivity possibly arises from an intramolecular ene-type reaction. To explain the observed stereochemistry, a plausible transition state model is proposed (Scheme 4b). The 1,3-rhodium shift or the formation of an enamine through intramolecular protonation could afford preferentially the Z-alkene.11 Subsequent cyclization through a six membered ene-type transition state F is highly favourable, where intramolecular interactions between [Rh] or H and the alkene would facilitate the cyclization and the cis-diastereoisomer could be obtained.
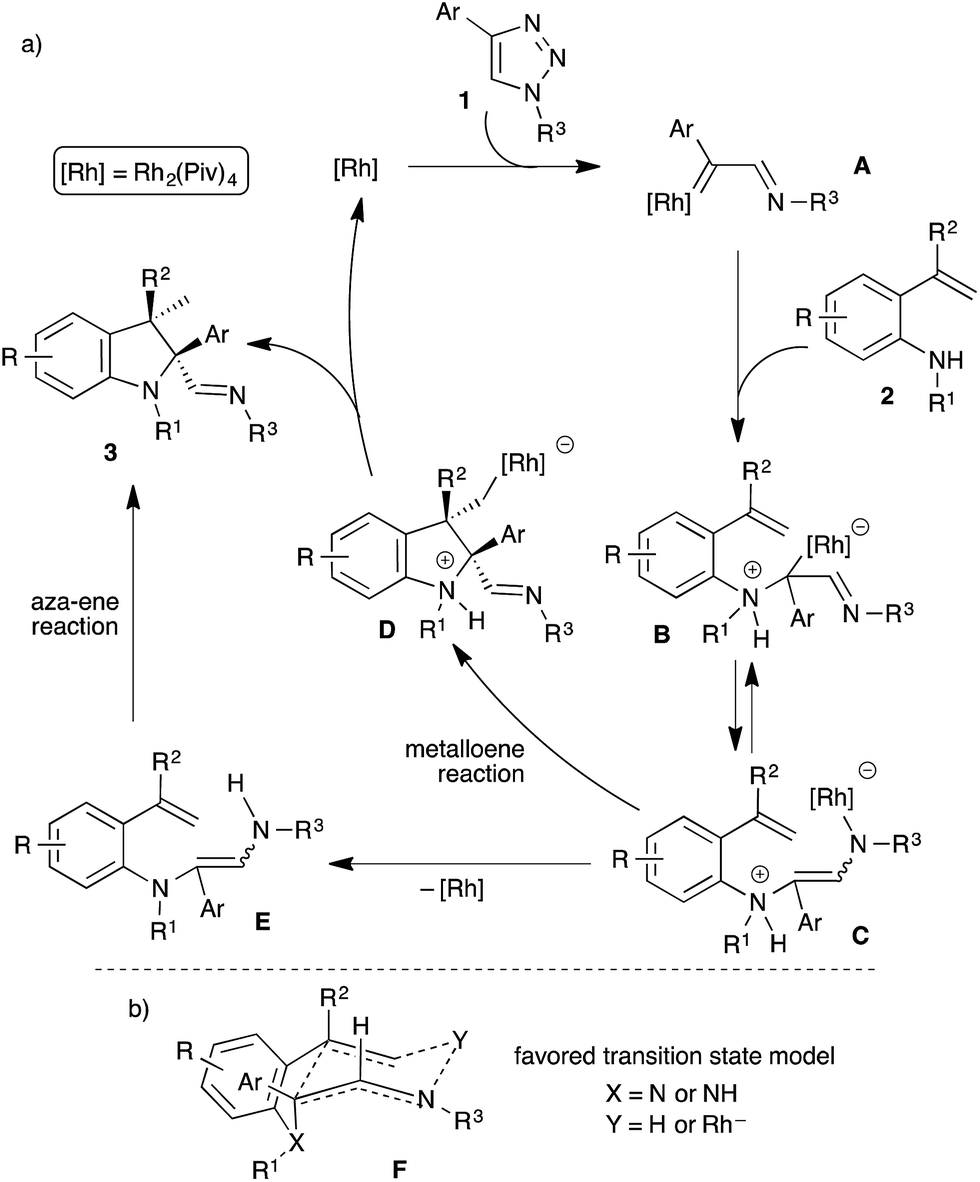 | ||
| Scheme 4 (a) Plausible mechanism; (b) proposed transition state for the ene-type reaction. | ||
After successful demonstration of the generality of the carbenylative amination, synthetic applications of the substituted indolines 3 were revealed via the manifestation of readily available functional groups. Reaction of 3aa with K2CO3 and methanol afforded the corresponding aldehyde 4a in 69% yield (Scheme 5). On the other hand, conversion of the imine moiety in 3aa to a sulfonamide (5a) was achieved through reduction with LAH. Next, a hypervalent iodine mediated cyclization17 of 5a was envisaged for the synthesis of a nitrogen based tricyclic compound. The reaction of sulfonamide 5a with (diacetoxyiodo)benzene (PIDA) in DCM at room temperature afforded imidazoindoline 6a in 68% yield over two steps, as a single diastereomer. The formation of 6a can be explained as due to oxidation of the tertiary amine in 5a with PIDA to provide an iminium ion followed by cyclization. The structure of 6a was unambiguously confirmed using single crystal X-ray analysis18 (see ESI†).
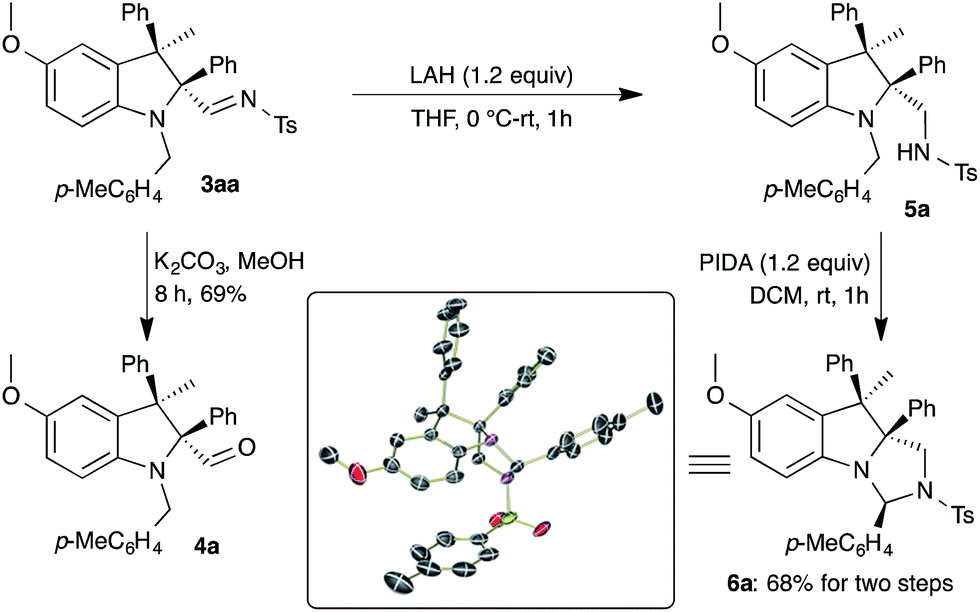 | ||
| Scheme 5 Synthesis of imidazoindoline 6a. | ||
The developed two-step protocol was subsequently applied to functionally different indolines 3. In general, all of the indolines that were examined afforded the imidazoindolines 6b–k in good yield under the two-step reduction and oxidative cyclization sequence (Scheme 6). It is important to note that the oxidative cyclization tolerates oxidation prone electron rich arene and thiophene substituents as well as electron deficient arenes, and led to the synthesis of the corresponding imidazoindolines 6d–j in good yield. Interestingly, replacement of N-Bn with N-Me also provided the imidazoindoline 6k in 69% yield.
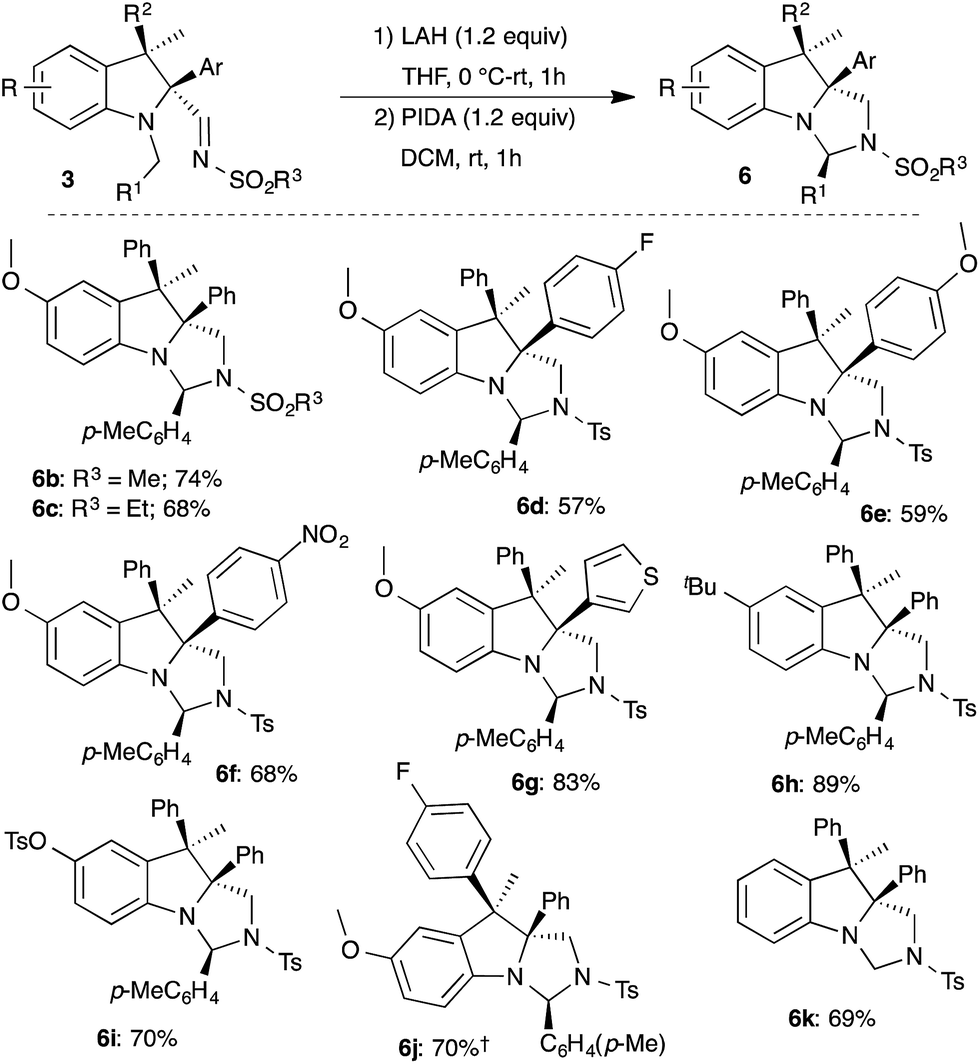 | ||
| Scheme 6 Synthesis of imidazoindolines 6. †1H NMR yield. | ||
Conclusions
In conclusion, an efficient rhodium catalyzed formal carbenylative amination of ortho-vinylanilines with iminocarbenes derived from N-sulfonyl-1,2,3-triazoles has been accomplished for the diastereoselective synthesis of indolines possessing two contiguous tetrasubstituted carbon centers. The reaction affords excellent cis-diastereoselectivity through the initial formation of a N-ylide, followed by trapping with unactivated alkenes via an ene-type reaction with a well-organized transition state. The present method tolerates various reactive functional groups, which allows the synthesis of various indoline derivatives in good to excellent yield. The developed transformation was further extended to the successful synthesis of tricyclic compounds, imidazoindolines, through reduction and hypervalent iodine mediated oxidative cyclization.Acknowledgements
We thank the Department of Science and Technology (DST), New Delhi, India (Project No. SR/S1/OC-48/2012) for funding this work. DY thanks IITM and ACSR thanks CSIR for fellowships. We also thank Mr Ramkumar for single crystal analysis support.Notes and references
- (a) E. Fattorusso and O. Taglialatela-Scafati, Modern Alkaloids: Structure, Isolation, Synthesis, and Biology, 2008 Search PubMed; (b) D. Crich and A. Banerjee, Acc. Chem. Res., 2007, 40, 151–161 CrossRef CAS PubMed; (c) M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS PubMed.
- (a) A. Ramirez and S. Garcia-Rubio, Curr. Med. Chem., 2003, 10, 1891–1915 CrossRef CAS PubMed; (b) J. Liu, J. Zhu, L. Tang, W. Wen, S. Lv and R. Yu, World J. Microbiol. Biotechnol., 2014, 30, 175–180 CrossRef CAS PubMed; (c) M. Serasanambati, S. R. Chilakapati, P. K. Manikonda, J. R. Kanala and D. R. Chilakapati, Nat. Prod. Res., 2014, 29, 484–490 CrossRef PubMed; (d) J. Chen, Y. Qu, D. Wang, P. Peng, H. Cai, Y. Gao, Z. Chen and B. Cai, Molecules, 2014, 19, 4395–4408 CrossRef PubMed.
- D. Zhang, H. Song and Y. Qin, Acc. Chem. Res., 2011, 44, 447–457 CrossRef CAS PubMed.
- (a) Q. Cai and S.-L. You, Org. Lett., 2012, 14, 3040–3043 CrossRef CAS PubMed; (b) S. Kirchberg, R. Frohlich and A. Studer, Angew. Chem., Int. Ed., 2009, 48, 4235–4238 CrossRef CAS PubMed; (c) S. Beaumont, V. Pons, P. Retailleau, R. H. Dodd and P. Dauban, Angew. Chem., Int. Ed., 2010, 49, 1634–1637 CrossRef CAS PubMed.
- (a) A. Minatti and S. L. Buchwald, Org. Lett., 2008, 10, 2721–2724 CrossRef CAS PubMed; (b) A. B. Dounay, P. G. Humphreys, L. E. Overman and A. D. Wrobleski, J. Am. Chem. Soc., 2008, 130, 5368–5377 CrossRef CAS PubMed; (c) Y. Yasui, T. Kinugawa and Y. Takemoto, Chem. Commun., 2009, 4275 RSC; (d) T. Watanabe, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2008, 10, 1759–1762 CrossRef CAS PubMed; (e) D. M. Schultz and J. P. Wolfe, Org. Lett., 2010, 12, 1028–1031 CrossRef CAS PubMed.
- (a) F. Brucelle and P. Renaud, J. Org. Chem., 2013, 78, 6245–6252 CrossRef CAS PubMed; (b) R. Viswanathan, C. R. Smith, E. N. Prabhakaran and J. N. Johnston, J. Org. Chem., 2008, 73, 3040–3046 CrossRef CAS PubMed; (c) D. L. J. Clive, J. Peng, S. P. Fletcher, V. E. Ziffle and D. Wingert, J. Org. Chem., 2008, 73, 2330–2344 CrossRef CAS PubMed; (d) H. Fuwa and M. Sasaki, Org. Lett., 2007, 9, 3347–3350 CrossRef CAS PubMed.
- (a) C. D. Gilmore, K. M. Allan and B. M. Stoltz, J. Am. Chem. Soc., 2008, 130, 1558–1559 CrossRef CAS PubMed; (b) Z. Wang, W. Wan, H. Jiang and J. Hao, J. Org. Chem., 2007, 72, 9364–9367 CrossRef CAS PubMed; (c) J. R. Dunetz and R. L. Danheiser, J. Am. Chem. Soc., 2005, 127, 5776–5777 CrossRef CAS PubMed.
- (a) C. Jing, D. Xing and W. Hu, Chem. Commun., 2014, 50, 951–953 RSC; (b) L. Jiang, R. Xu, Z. Kang, Y. Feng, F. Sun and W. Hu, J. Org. Chem., 2014, 79, 8440–8446 CrossRef CAS PubMed.
- (a) P. Anbarasan, D. Yadagiri and S. Rajasekar, Synthesis, 2014, 46, 3004–3023 CrossRef CAS; (b) H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151–5162 RSC.
- (a) T. Miura, Y. Fujimoto, Y. Funakoshi and M. Murakami, Angew. Chem., Int. Ed., 2015, 54, 9967–9970 CrossRef CAS PubMed; (b) Y. Yang, M.-B. Zhou, X.-H. Ouyang, R. Pi, R.-J. Song and J.-H. Li, Angew. Chem., Int. Ed., 2015, 54, 6595–6599 CrossRef CAS PubMed; (c) Y. Jiang, X.-Y. Tang and M. Shi, Chem. Commun., 2015, 51, 2122–2125 RSC; (d) Y. Wang, X. Lei and Y. Tang, Chem. Commun., 2015, 51, 4507–4510 RSC; (e) J. Pospech, R. Ferraccioli, H. Neumann and M. Beller, Chem.–Asian J., 2015, 10, 2624–2630 CrossRef CAS PubMed; (f) Y. Shi, X. Yu and C.-Y. Li, Eur. J. Org. Chem., 2015, 6429–6433 CrossRef CAS; (g) J. He, Z. Man, Y. Shi and C.-Y. Li, J. Org. Chem., 2015, 80, 4816–4823 CrossRef CAS PubMed; (h) B. Seo, W. H. Jeon, J. Kim, S. Kim and P. H. Lee, J. Org. Chem., 2015, 80, 722–732 CrossRef CAS PubMed; (i) S. Shin, Y. Park, C.-E. Kim, J.-Y. Son and P. H. Lee, J. Org. Chem., 2015, 80, 5859–5869 CrossRef CAS PubMed; (j) V. N. G. Lindsay, H. M. F. Viart and R. Sarpong, J. Am. Chem. Soc., 2015, 137, 8368–8371 CrossRef CAS PubMed; (k) M.-H. Shen, Y.-P. Pan, Z.-H. Jia, X.-T. Ren, P. Zhang and H.-D. Xu, Org. Biomol. Chem., 2015, 13, 4851–4854 RSC; (l) D. J. Jung, H. J. Jeon, J. H. Lee and S.-g. Lee, Org. Lett., 2015, 17, 3498–3501 CrossRef CAS PubMed; (m) E. Lee, T. Ryu, E. Shin, J.-Y. Son, W. Choi and P. H. Lee, Org. Lett., 2015, 17, 2470–2473 CrossRef CAS PubMed; (n) T. Miura, Y. Funakoshi, Y. Fujimoto, J. Nakahashi and M. Murakami, Org. Lett., 2015, 17, 2454–2457 CrossRef CAS PubMed; (o) H.-D. Xu, K. Xu, H. Zhou, Z.-H. Jia, H. Wu, X.-L. Lu and M.-H. Shen, Synthesis, 2015, 47, 641–646 CrossRef CAS; (p) Y. Jiang, R. Sun, Q. Wang, X.-Y. Tang and M. Shi, Chem. Commun., 2015, 51, 16968–16971 RSC; (q) S. Chuprakov, S. W. Kwok, L. Zhang, L. Lercher and V. V. Fokin, J. Am. Chem. Soc., 2009, 131, 18034–18035 CrossRef CAS PubMed; (r) N. Grimster, L. Zhang and V. V. Fokin, J. Am. Chem. Soc., 2010, 132, 2510–2511 CrossRef CAS PubMed; (s) J. C. Culhane and V. V. Fokin, Org. Lett., 2011, 13, 4578–4580 CrossRef CAS PubMed; (t) M. Zibinsky and V. V. Fokin, Org. Lett., 2011, 13, 4870–4872 CrossRef CAS PubMed; (u) J. S. Alford and H. M. L. Davies, Org. Lett., 2012, 14, 6020–6023 CrossRef CAS PubMed.
- S. Chuprakov, B. T. Worrell, N. Selander, R. K. Sit and V. V. Fokin, J. Am. Chem. Soc., 2014, 136, 195–202 CrossRef CAS PubMed.
- D. J. Lee and E. J. Yoo, Org. Lett., 2015, 17, 1830–1833 CrossRef CAS PubMed.
- (a) S. Rajasekar, D. Yadagiri and P. Anbarasan, Chem.–Eur. J., 2015, 21, 17079–17084 CrossRef CAS PubMed; (b) D. Yadagiri and P. Anbarasan, Chem. Sci., 2015, 6, 5847–5852 RSC; (c) D. Yadagiri and P. Anbarasan, Chem.–Eur. J., 2013, 19, 15115–15119 CrossRef CAS PubMed; (d) D. Yadagiri and P. Anbarasan, Org. Lett., 2014, 16, 2510–2513 CrossRef CAS PubMed; (e) S. Rajasekar and P. Anbarasan, J. Org. Chem., 2014, 79, 8428–8434 CrossRef CAS PubMed.
- CCDC 1438964† contains supplementary crystallographic data for the compound 3ad.
- (a) W. Oppolzer, Angew. Chem., Int. Ed., 1989, 28, 38–52 CrossRef; (b) T. J. J. Muller, in Comprehensive Organic Synthesis II, Elsevier, Amsterdam, 2nd edn, 2014, pp. 1–65 Search PubMed.
- (a) W. Tan, B.-X. Du, X. Li, X. Zhu, F. Shi and S.-J. Tu, J. Org. Chem., 2014, 79, 4635–4643 CrossRef CAS PubMed; (b) W. Adam and O. Krebs, Chem. Rev., 2003, 103, 4131–4146 CrossRef CAS PubMed; (c) J.-H. Zhang, M.-X. Wang and Z.-T. Huang, Tetrahedron Lett., 1998, 39, 9237–9240 CrossRef CAS; (d) J.-H. Zhang, M.-X. Wang and Z.-T. Huang, J. Chem. Res., Synop., 1998, 486–487 RSC.
- (a) R. Samanta, K. Matcha and A. P. Antonchick, Eur. J. Org. Chem., 2013, 5769–5804 CrossRef CAS; (b) F. V. Singh and T. Wirth, Chem.–Asian J., 2014, 9, 950–971 CrossRef CAS PubMed.
- CCDC 1438963† contains supplementary crystallographic data for the compound 6a.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1438963 and 1438964. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc01075j |
| This journal is © The Royal Society of Chemistry 2016 |