
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile hydrogen atom transfer to iron(III) imido radical complexes supported by a dianionic pentadentate ligand†
Denis M.
Spasyuk
a,
Stephanie H.
Carpenter
b,
Christos E.
Kefalidis
c,
Warren E.
Piers
*a,
Michael L.
Neidig
*b and
Laurent
Maron
*c
aUniversity of Calgary, Department of Chemistry, 2500 University Drive N.W., Calgary, Alberta, Canada T2N 1N4. E-mail: wpiers@ucalgary.ca
bDepartment of Chemistry, University of Rochester, Rochester, New York 14627, USA
cLPCNO, Université de Toulouse, INSA, UPS, CNRS, 135 avenue de Rangueil, F-31077 Toulouse, France
First published on 31st May 2016
Abstract
A dianionic tetrapodal pentadentate diborate ligand is introduced. This ligand forms a high spin neutral iron(II) complex that reacts with a variety of organoazides to yield transient Fe(III) imido radicals that are extremely potent hydrogen atom abstractors. The nature of these species is supported by full characterization of the Fe(III) amido products, kinetic studies, density functional computations and Mössbauer spectroscopy on the –C6H4-p-tBu substituted derivative.
Introduction
Tetrapodal pentadentate ligands can be used to provide a defined platform for small molecule activation and catalysis.1 One successful ligand design is the polypyridyl PY5 system first reported by the Stack2 and Feringa3 groups. This neutral ligand coordinates to metals from across the periodic table4 and its oxidative stability allows access to a range of metal oxidation states,5 a key feature for applications in catalysis.6 This ligand framework has been successfully employed in catalyst systems that generate dihydrogen7–10 or oxygen11–13 from water, using earth abundant metals such as molybdenum and cobalt.14 More recently, Gardinier has introduced a related ligand system, Pz4Py, incorporating pyrazolyl donors instead of pyridyl moieties.15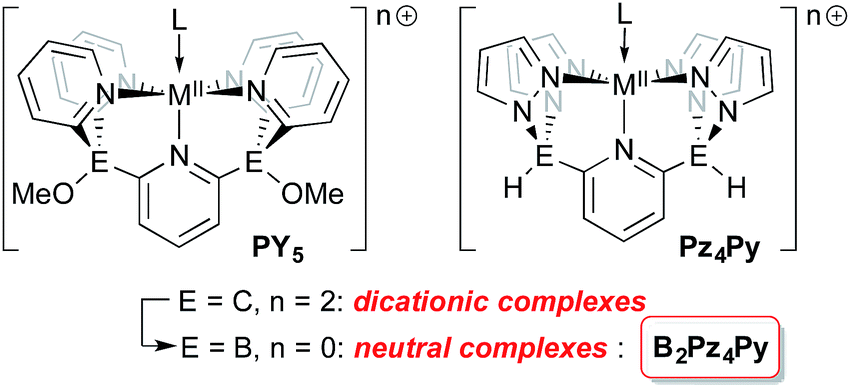
The overall neutral charge of the PY5 and Pz4Py ligands necessarily means that higher oxidation state intermediates bear positive charges and are fleeting, highly reactive species. Rendering ligands anionic16–18 may stabilize higher oxidation state intermediates so they can be studied in more detail. Dianionic renditions of these ligands should allow for the synthesis of neutral higher oxidation state compounds; the transposition of carbon for boron at the linking positions of the PY5 or Pz4Py ligands is an established way to introduce the negative charges in the form of borates.19 Here, we introduce the new dianionic ligand B2Pz4Py and its use in the synthesis of iron imido complexes that are strong hydrogen atom acceptors.
Results and discussion
Details concerning the synthetic assembly of the B2Pz4Py ligand framework are given in Fig. 1. The most convenient ligand precursor, the [B2Pz4LiPyH]2 dimer shown in Fig. 1, can be prepared in two steps starting from 2,6-bis(trimethylstannyl)pyridine20 and bromo-(dimethylamino)phenyl borane.21 Mixing these two reagents together in toluene and stirring at room temperature for 24 hours yields a pale yellow semi solid upon removal of volatiles. This 2,6-diborylpyridyl species is stabilized by the strongly π donating NMe2 groups, but does exhibit strong sensitivity to even weak donors and so is used without further purification in a reaction with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of freshly sublimed pyrazole and lithium pyrazolate. This leads to elimination of HNMe2 and production of gram quantities of [B2Pz4LiPyH]2, isolated in up to 81% yield as an air and water stable white solid. Crystals were obtained from THF/hexanes and the structure solution shows that the pyrazolyl nitrogens from opposing units coordinate a lithium counteranion in an approximately tetrahedral environment. The pyridine nitrogens are protonated to balance the second borate negative charge (Fig. 1). Metrical data within the structure are unremarkable and details can be found in the ESI (Fig. S1†).
:1 mixture of freshly sublimed pyrazole and lithium pyrazolate. This leads to elimination of HNMe2 and production of gram quantities of [B2Pz4LiPyH]2, isolated in up to 81% yield as an air and water stable white solid. Crystals were obtained from THF/hexanes and the structure solution shows that the pyrazolyl nitrogens from opposing units coordinate a lithium counteranion in an approximately tetrahedral environment. The pyridine nitrogens are protonated to balance the second borate negative charge (Fig. 1). Metrical data within the structure are unremarkable and details can be found in the ESI (Fig. S1†).
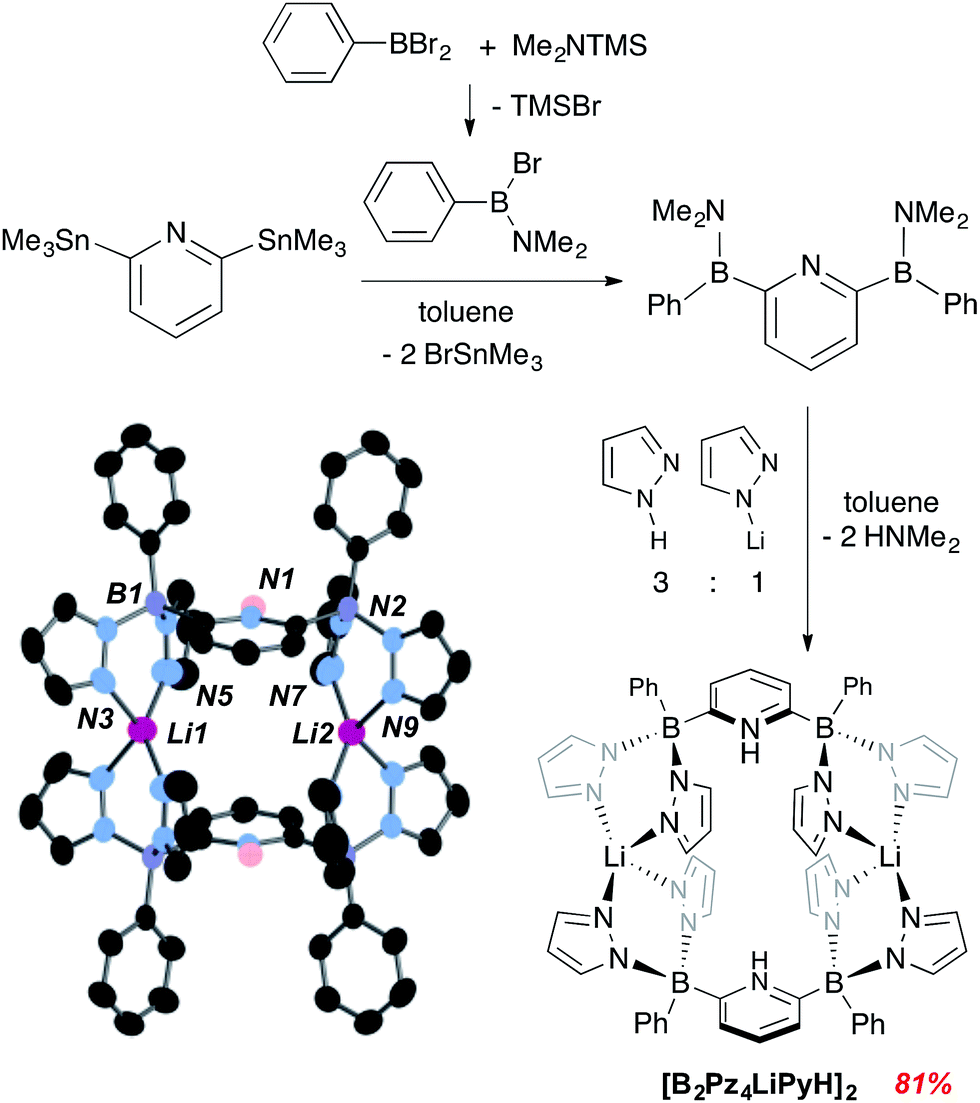 | ||
| Fig. 1 Synthesis and structure of dianionic pentadentate ligand precursor [B2Pz4LiPyH]2. Thermal ellipsoids are drawn to the 50% probability level. | ||
For attachment to iron, this ligand precursor was taken up in THF and deprotonated using LiOiPr; this solution of in situ generated dilithio salt was then transferred to a suspension of FeBr2 in THF (Scheme 1) to form the Fe(II) complex 1·THF. Yellow-green crystals of this air and moisture sensitive, paramagnetic compound were obtained, and its structure confirmed via X-ray crystallography (Fig. S2†). The ligand coordinates in a square-pyramidal array, while the THF ligand occupies the position trans to the pyridyl group to form an overall octahedral geometry. An Evan's method measurement yields a μeff of 5.64 B. M., consistent with a high spin (HS) electron configuration with 4 unpaired electrons (S = 2). The 80 K Mossbauer spectrum of 1·THF features a quadrupole doublet with parameters of δ = 1.13 mm s−1 and ΔEQ = 2.40 mm s−1, consistent with an S = 2 iron(II) species (Fig. S3† and Table 1).22–25 DFT computations using the B3PW91 functional indicate that the HS S = 2 configuration is 10.2 kcal mol−1 lower in energy than the low spin (LS) S = 0 configuration (Table S1†).
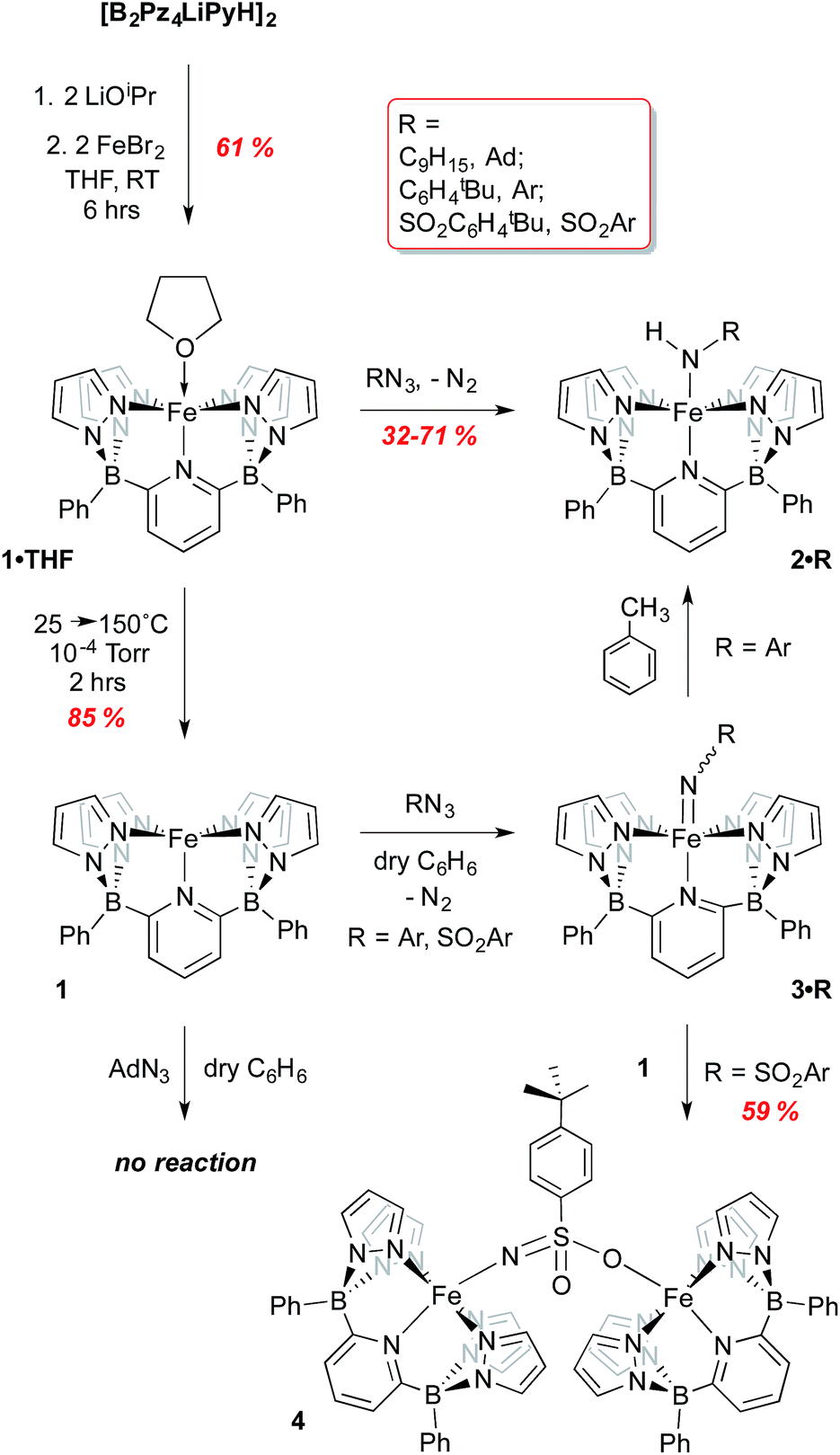 | ||
| Scheme 1 Synthesis of iron(II) complexes of [B2Pz4LiPyH]2 and their reactions with organoazides RN3 (R = adamantyl, C6H4tBu, SO2C6H4tBu). | ||
Low valent iron complexes can react with nitrene transfer agents to generate higher oxidation state imido derivatives.26–41 Complex 1·THF was thus reacted with organic azides, RN3, of varying electronic properties (R = adamantyl, C6H4-p-tBu and SO2C6H4-p-tBu) using dry benzene as a solvent in an attempt to generate isolable, neutral Fe(IV) imido derivatives. In each case, the product was the corresponding Fe(III) amido complex 2·R (Scheme 1), isolated in moderate yields. Amidos 2·R were all fully characterized, including via X-ray crystallography for 2·Ar (shown in Fig. 2A) and 2·Ad (Fig. S5†). All are paramagnetic with μeff values of 1.82–2.05 B. M., indicating a LS, S = 1/2 configuration, and exhibit sharp N–H stretches in the IR spectrum at 3262 (2·Ad), 3303 (2·Ar) and 3071 (2·SO2Ar) cm−1. EPR spectroscopy of 2·Ar further confirms that it is an S = 1/2 species (see ESI, Fig. S6†). In the UV-vis spectra, these amides exhibit low energy absorptions at 625 nm (2·Ad) and 829 nm (2·Ar). Qualitatively, the rate of the reaction was related to the nature of R; for adamantyl azide, the reaction required heating, while for the aryl and sulfonyl azides, gas evolution was immediate upon addition of the azide to solutions of 1·THF. For the aryl azide, an initially formed brown solution turned green over a couple of hours, suggesting the intermediacy of a transient species. We surmised that the path to amido products 2·R may involve the predicted Fe(IV) imido species, which then abstract a hydrogen atom from the liberated THF ligand to form the observed products; indeed, when THF-d8 is added, an N–D stretch in the IR spectrum at 2451 cm−1 for 2·Ar-d1 is observed, but full conversion to 2·Ar takes significantly longer and the sample is only partially deuterated (see below). The products of H atom abstraction from THF in this reaction were not definitively identified, but THF is the most likely source of hydrogen atoms in this system.
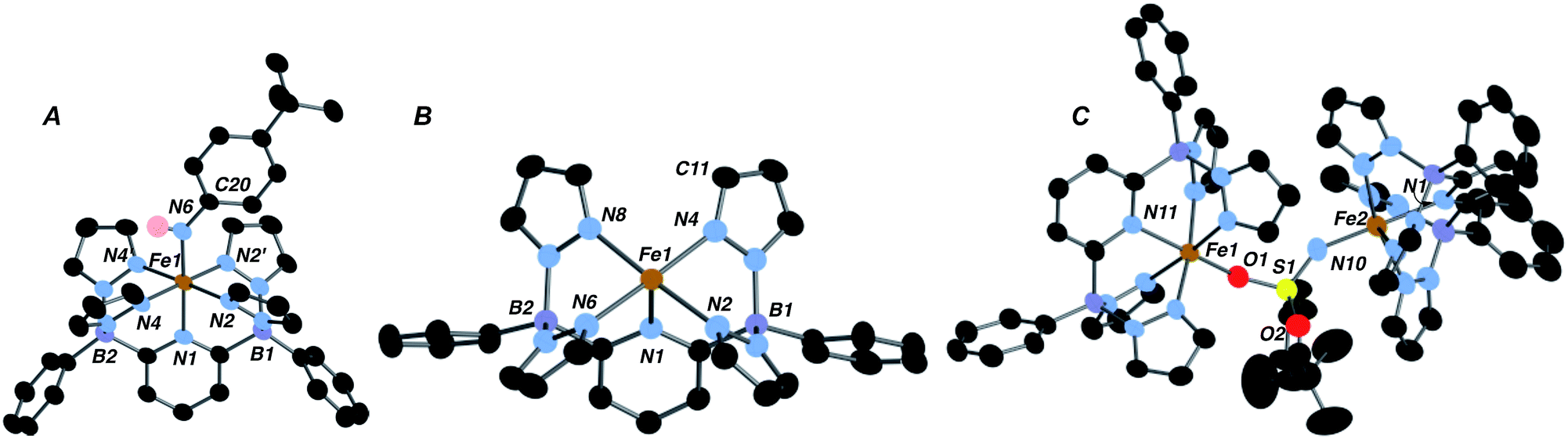 | ||
| Fig. 2 Molecular structures (50% ellipsoids) of compounds 2·Ar (A), 1 (B) and 4 (C). Selected metrical parameters (distances in Å, angles in °) for 2·Ar: Fe(1)–N(1), 1.994(3); Fe(1)–N(2), 1.982(2); Fe(1)–N(4), 2.002(2); Fe(1)–N(6), 1.869(3); N(2)–Fe(1)–N(6), 91.97(11); N(4)–Fe(1)–N(6), 89.45(11); N(1)–Fe(1)–N(6), 178.34(16); Fe(1)–N(6)–C(20), 139.0(3). The aryl amido ligand plane lies along the plane bisecting the N(2)–Fe(1)–N(2′) and N(4)–Fe(1)–N(4′) angles. Selected metrical parameters for 1: Fe(1)–N(1), 2.098(2); Fe(1)–N(2), 2.159(2); Fe(1)–N(4), 2.101(2); Fe(1)–N(6), 2.130(2); Fe(1)–N(8), 2.089(2). Selected metrical parameters for 4: Fe(1)–O(2), 1.913(3); Fe(1)–N(11), 2.111(4); S(1)–O(1), 1.460(3); S(1)–O(2), 1.513(3); S(1)–N(10), 1.503(4); Fe(2)–N(10), 1.909(4); Fe(2)–N(1), 2.144(4); Fe(1)–O(2)–S(1), 151.0(2); Fe(2)–N(10)–S(1), 148.6(3). | ||
We therefore hypothesized that removal of hydrogen atom donors from the system would be necessary in order to observe the putative Fe(IV) imido compounds. Heating solid samples of isolated 1·THF to 150 °C under high vacuum for a few hours resulted in removal of the ligated THF, forming THF-free 1 in excellent yield. Recrystallization from very dry benzene/pentane mixtures allowed for determination of its molecular structure, which is shown in Fig. 2B. The geometry at iron is square pyramidal, and the complex retains the HS, S = 2, configuration found in 1·THF, with a μeff of 5.33 B. M. DFT computations reproduce the experimental structure accurately and indicate the HS configuration is 14.4 kcal mol−1 more stable than the LS configuration in this 5 coordinate derivative (Table S2†). The “naked” 5-coordinate complex is somewhat unusual, but it should be noted that in the crystal structure, the complex packs in dimeric units in which C(11) comes into close contact with the iron center of the adjacent molecule (Fe(1)⋯C(11) = 3.282 Å, Fig. S7†). In solution, however, this vacant coordination site is available for reaction with Lewis bases and other reagents.
With THF-free 1 in hand, we examined its reactions with the three azides RN3 in hydrogen atom transfer (HAT) inert solvents such as benzene or bromobenzene (refer back to Scheme 1). It was necessary to rigorously exclude polar solvents and impurities to avoid producing amides 2; indeed, even working up the reactions in hexanes, which likely contain tertiary C–H bonds, led to the amido products and thus it was necessary to employ pure, dry pentane or Me3SiOSiMe3 to prevent formation of the amido complexes. Again, the facility of the reaction between 1 and the azides was dependent on the nature of the azide R substituent. Surprisingly, for adamantyl azide, no reaction was observed with 1, even upon heating to 60 °C in bromobenzene. Indeed, there was no visual or spectroscopic (UV-vis) evidence for interaction of the AdN3 reagent with 1. Only when a species capable of contributing a hydrogen atom (THF or toluene) was introduced did the reaction proceed, then giving 2·Ad directly. This observation suggests that an Fe imido complex is not readily formed for this azide and that reaction of 1 with this bulky azide requires the aid of a hydrogen atom donor in order to proceed.
In contrast, reaction with the sulfonyl azide N3SO2Ar was rapid at room temperature, evidenced by vigorous gas evolution upon dropwise addition of the azide to solutions of 1. The product isolated and characterized by X-ray crystallography was the dinuclear Fe(III)–Fe(III) species 4, the molecular structure of which is shown in Fig. 2C, along with selected metrical data. The Fe(1)–O(2) and Fe(2)–N(10) distances are consistent with Fe(III)-element single bonds, while the S(1)–N(10) distance of 1.503(4) Å is indicative of some multiple bonding in this linkage. Compound 4 likely forms through the rapid formation of an Fe(IV) imido, 3·SO2Ar, which rapidly picks up a second equivalent of Fe(II) 1 to give the observed dinuclear species (Scheme 1).42 Attempts to observe 3·SO2Ar by using an “inverse addition” strategy, or by treating 4 with an excess of azide reagent met with failure and only 4 was produced. When isolated samples of 4 were treated with an excess of THF at room temperature, a 1:1 mixture of 2·SO2Ar and 1·THF was produced rapidly.
However, when the aryl azide tBuC6H4N3 was employed in reaction with 1, evidence for formation of 3·Ar was observed. Rapid evolution of N2 was accompanied by a deepening of the light green color of 1 to a brown suspension from which dark green, solid 3·Ar could be isolated in 67% yield. The product is paramagnetic with a μeff value of 2.87 B. M., indicative of an S = 1 spin state, as determined via the Evans method on freshly prepared samples. Unfortunately, despite several attempts, crystals of this crucial material could not be obtained, likely due to its propensity to convert to amido complex 2·Ar. Consequently, 3·Ar was examined through DFT studies and the computed structure was obtained.
The computed structures of the LS (S = 0), HS (S = 2) and S = 1 spin states of 3·Ar indicate that the last is the most stable by 12.1 kcal mol−1 over the LS configuration (Tables S3 and S4†). The structure is similar in geometry to that of Fe(III) amido complex 2·Ar (Fig. 3), but exhibits a much shorter Fe–N distance of 1.696 Å, in line with other Fe imido complexes,26,43,44 and a wider Fe–N–Cipso angle of 143.33°. The N–Cipso distance of 1.349 Å, coupled with alternation of long–short–long bond distances within the aryl ring suggest the NAr ligand is best described an imido based radical;37 indeed, the highest energy SOMO orbital shows significant localization on the imido nitrogen (Fig. 3, inset) and the calculated spin density population (Fig. S8†) is also in agreement with this description.
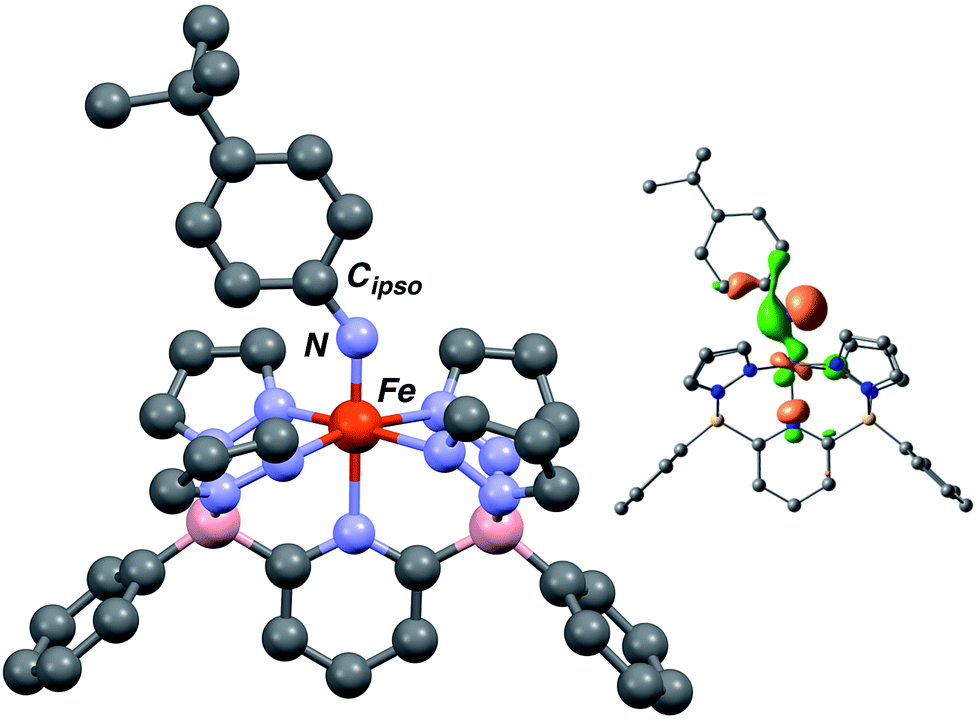 | ||
| Fig. 3 Computed structure of 3·Ar at the B3PW91 level of theory. Selected bond distances and angles (Å): Fe–N, 1.696 Å; Fe–N–Cipso, 143.33°. Depiction of the highest energy SOMO orbital (upper right). | ||
Mössbauer spectroscopy of 3·Ar was performed to evaluate the oxidation state of this species. While the Mössbauer spectrum of 3·Ar at 80 K is broadened, the spectrum at 5 K (Fig. 4B) is well-resolved. It contains a major quadrupole doublet (red component, 80% of all iron) with parameters of δ = 0.20 mm s−1 and ΔEQ = 1.96 mm s−1 as well as two additional iron species (see Fig. 4 caption), including one that is consistent with the presence of 2·Ar, likely formed from 3·Ar during transport and handling of the sample. Spin-quantitated EPR analysis further supports this minor component as being an S = 1/2 species (see ESI†). Importantly, the isomer shift of the major species corresponding to 3·Ar is similar to that for 2·Ar (an iron(III) species), suggestive of similar oxidation states of iron in both complexes. In addition, previously reported iron(IV) imido species typically have low isomer shifts: the iron(IV)-tosylamido species of Que and co-workers32 (δ = 0.02 mm s−1), the iron(IV)-imido corrolazine complex of Goldberg et al.45 (δ = −0.05 mm s−1), the iron(IV) pincer imido reported recently by Mindiola et al. (δ = −0.09 mm s−1)46 and the novel carbene-stabilized bis-imido iron(IV) complexes of Deng (δ = −0.29 to −0.52 mm s−1)47 are pertinent examples. Thus, the Mössbauer data is most consistent with an iron(III) description for 3·Ar. This assignment was further corroborated by computing the Mössbauer data using methodology developed by Neese et al.;48 as can be seen in Table 1, the computed values are in broad agreement with the experimental values for both 2·Ar and 3·Ar, taking into account the limitations of such calculations.
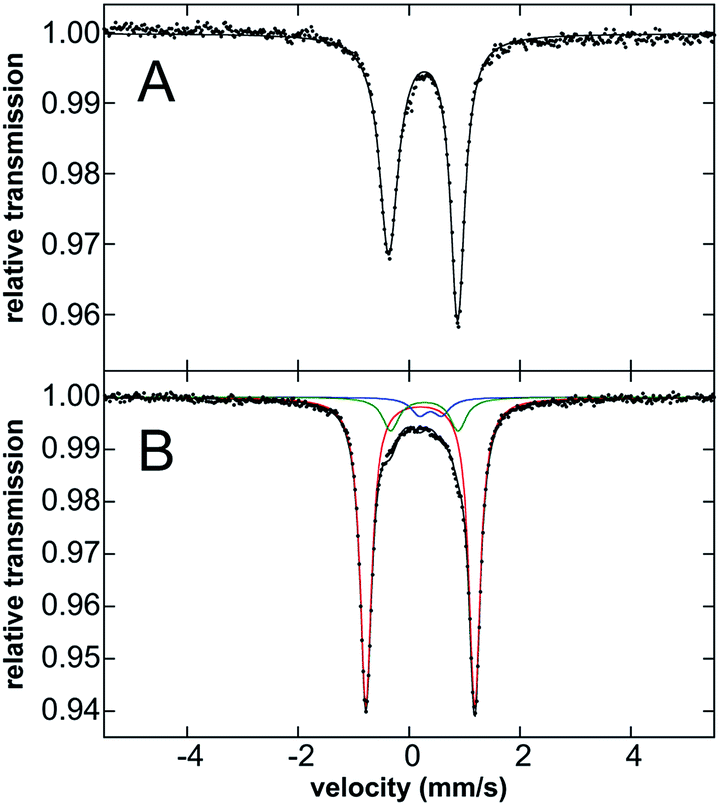 | ||
| Fig. 4 80 K Mössbauer spectra of solid (A) 2·Ar collected at 80 K and (B) 3·Ar collected at 5 K. The two minor impurities in the 3·Ar sample have Mossbauer parameters of δ = 0.26 mm s−1, ΔEQ = 1.24 mm s−1 (green, 13% of total iron) and δ = 0.39 mm s−1, ΔEQ = 0.40 mm s−1 (blue, 7% of total iron). | ||
Taken together, the above data suggests the nature of 3·Ar is best described as a LS Fe(III) (d5, S = 1/2) iron center bonded to an S = 1/2 imido radical species. Such imido radical formulations have been proposed in other systems, both for iron37 and cobalt.49–52 In 3·Ar, the two spins reside in orthogonal orbitals and so an S = 1 system is the result. This description also accounts for why this compound so readily converts to 2·Ar in the presence of even weak hydrogen atom donors, such as toluene (the Bond Dissociation Energy, BDE, for the benzylic C–H bond of toluene is ≈89 kcal mol−1).53 While the lower energy SOMO is essentially metal-based (see Fig. S8, ESI†), the orbital associated with the HAT reactivity is imido nitrogen-based and optimally oriented for accepting a hydrogen atom donor (Fig. 3, inset). Experiments are also consistent with this picture. Quantitative analysis of the rate of the reaction of 3·Ar with varying amounts of toluene (20–300 equivalents) was conveniently done by following the reactions by UV-vis spectroscopy at room temperature (Fig. S9, ESI†). The process is shown to be first order in [toluene], suggesting a bimolecular HAT reaction involving the benzylic toluene hydrogens as the source of the amido hydrogen in 2·Ar. From the plot of kobsvs. [toluene] (Fig. S10 and S11, ESI†), a second order rate constant of 1.60(5) × 10−3 M−1 s−1 is obtained. This is comparable to that of 9.1 ± 0.9 × 10−3 M−1 s−1 observed in the bimolecular HAT reaction between a 4 coordinate beta-diketiminato supported Fe(III) imido and 1,4-cyclohexadiene at −51 °C.36 While comparison of these two systems is tempered by the different conditions under which the rates were measured, we note that the lower coordinate iron imido is unreactive towards toluene, indicating qualitatively that the present system is highly reactive towards even weak hydrogen atom donors in comparison. Indeed, a transition state for HAT from toluene to 3·Ar was located and exhibits a near linear N–H–C vector with a low barrier of 6.4 kcal mol−1 for hydrogen transfer (Fig. S12 and Table S5†). The computed homolytic BDE for the N–H bond in 2·Ar was found to be 91.3 kcal mol−1, slightly greater than values found in Holland's beta-diketiminato amido complexes,36 and contributes to the exothermicity of HAT to the imido radical ligand in 3·Ar.
Since the product of HAT 2·Ar is paramagnetic, quantification of the NH vs. ND isotopologues was challenging; furthermore, we found by IR spectroscopy that the major isotopologue produced from reaction of 3·Ar with deuterated THF or toluene was in fact the NH isotopologue, suggesting that even the small amount of residual protons in the deuterated solvent are able to compete in these HAT reactions. Nonetheless, by integrating the N–H vs. the N–D stretching bands in the IR spectrum of a sample of 2·Ar-dn (n = 0, 1) generated by gentle heating of 3·Ar in THF-d8 (0.23% THF-d7), a kH/kD of 567 ± 16 was estimated. This experiment was repeated three times and a similar result was observed when toluene-d8 was employed. The calculated intensities of the N–H vs. N–D stretching frequencies indicate that they are of a similar magnitude, with the N–D stretch being less intense by a factor of 1.8; even with this correction a kH/kD of ≈315 is obtained. While certainly unusual, we note that KIEs of this magnitude are a hallmark of the HAT reaction. For example Holland et al. report a value of 105 ± 28 in the HAT from cyclohexadiene to a beta-diketiminato ligand supported Fe(III) imido complex.36 This value was obtained by taking the ratio of measured rate constants obtained using undeuterated and fully deuterated cyclohexadiene substrates and does not account for the possibility of competitive H atom transfer from residual protons in the deuterated substrate; it therefore may represent a low estimate of the true kH/kD in this system. Using a more accurate method, Meyer et al. report a “colossal” kH/kD of 439 ± 8 in the transfer of a hydrogen atom from N to a substrate in an osmium amide/imide system.54 Thus, such reactions do feature very large KIE, and while the value obtained for the conversion of 3·Ar to 2·Ar here likely has a large error associated with it, the key point is that it is much larger than a conventional KIE. We calculate a kH/kD of 5.82 by substituting D for H in the transition state calculation; clearly, the experimental value is much greater than that and supports a HAT mechanism with a substantial tunneling component during the transfer of the hydrogen atom.
Unlike Betley's imido radical system,37,40 which also abstracts hydrogen atoms from suitable donors, we do not observe significant N–C bond formation via a radical-rebound process. Thus, our system does not mediate C–H bond amination, and we detect less that 4% of the Ar(PhCH2)NH amine product that would be expected if this were occurring in the reaction of 3·Ar with toluene. Following the intrinsic reaction coordinates from the TS for hydrogen atom transfer (Fig. S12†) leads to the full dissociation of the benzyl radical and there is no clear driving force for any rebound of the formed radical. Every attempt to rebound the radical leads back to the C–H activation TS so that it seems that there is no energetically accessible TS leading to C–N bond formation. We speculate that the change from a LS configuration in 2·Ar to a HS configuration in the Fe(II) amine product may play a role in the low level of C–N bond formation in this system.
Conclusions
In conclusion, we have generated and characterized a 6 coordinate Fe(III) imido radical via reaction of a 5 coordinate Fe(II) species with an aryl azide. It appears that the affinity of the imido group in 3·Ar for hydrogen atoms is quite high relative to previously reported, lower coordinate systems,36,37 an observation which may be related to the higher coordination number and anionic nature of the ligand in 3·Ar, which pushes significant spin density onto the imido nitrogen. In combination with the sterically modest (i.e., no ortho groups on the aryl substituent) and bent para-tBu N-aryl group, this effect results in an imido radical ligand that exhibits powerful hydrogen atom acceptor ability.Acknowledgements
Funding for this work was provided by NSERC of Canada (Discovery Grant) the Canada Research Chair secretariat (Tier I CRC 2013–2020) to W. E. P., the National Science Foundation (CHE-1454370) to M. L. N. and by an Alfred P. Sloan Research Fellowship to M. L. N. The Alexander von Humboldt Foundation (W. E. P. and L. M.) and the Chinese academy of Science (L. M.) are acknowledged for financial support. L. M. is member of the Institut Universitaire de France. L. M. and W. P. also acknowledge CNRS for a bilateral grant through the PICS program. CalMip is acknowledeged for a generous grant of computing time. D. M. S. thanks the University of Calgary for an Eyes High Postdoctoral Fellowship. Prof. Hans-Jörg Himmel of the Ruprecht-Karls-Universität Heidelberg is acknowledged for helpful discussions.Notes and references
- A. Grohmann, Dalton Trans., 2010, 39, 1432–1440 RSC.
- R. T. Jonas and T. D. P. Stack, J. Am. Chem. Soc., 1997, 119, 8566–8567 CrossRef CAS.
- M. E. de Vries, M. R. La Crois, G. Roelfes, H. Kooijman, A. L. Spek, R. L. Hage and B. Feringa, Chem. Commun., 1997, 1549–1550 RSC.
- R. J. M. Klein Gebbink, R. T. Jonas, C. R. Goldsmith and T. D. P. Stack, Inorg. Chem., 2002, 41, 4633–4641 CrossRef CAS PubMed.
- C. R. Goldsmith, R. T. Jonas, A. P. Cole and T. D. P. Stack, Inorg. Chem., 2002, 41, 4642–4652 CrossRef CAS PubMed.
- D. Z. Zee, T. Chantarojsiri, J. R. Long and C. J. Chang, Acc. Chem. Res., 2015, 48, 2027–2036 CrossRef CAS PubMed.
- J. P. Bigi, T. E. Hanna, W. H. Harman, A. Chang and C. J. Chang, Chem. Commun., 2010, 46, 958–960 RSC.
- Y. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 9212–9215 CrossRef CAS PubMed.
- Y. Sun, J. Sun, J. R. Long, P. Yang and C. J. Chang, Chem. Sci., 2013, 4, 118–124 RSC.
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, J. Borau-Garcia and C. P. Berlinguette, Chem. Commun., 2011, 47, 4249–4251 RSC.
- D. J. Wasylenko, R. D. Palmer, E. Schott and C. P. Berlinguette, Chem. Commun., 2012, 48, 2107–2109 RSC.
- D. W. Crandell, S. Ghosh, C. P. Berlinguette and M.-H. Baik, ChemSusChem, 2015, 8, 844–852 CrossRef CAS PubMed.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- T. J. Morin, B. Bennett, S. V. Lindeman and J. R. Gardinier, Inorg. Chem., 2008, 47, 7468–7470 CrossRef CAS PubMed.
- L. Tong, Y. Wang, L. Duan, Y. Xu, X. Cheng, A. Fischer, M. S. G. Ahlquist and L. Sun, Inorg. Chem., 2012, 51, 3388–3398 CrossRef CAS PubMed.
- K. J. Young, M. K. Takase and G. W. Brudvig, Inorg. Chem., 2013, 52, 7615–7622 CrossRef CAS PubMed.
- I. Siewert and J. Gałęzowska, Chem.–Eur. J., 2015, 21, 2780–2784 CrossRef CAS PubMed.
- S. Trofimenko, Scorpionates: The Coordination Chemistry of Polypyrazolylborate Ligands, Imperial College Press, London, 1999 Search PubMed.
- U. S. Schubert and C. Eschbaumer, Org. Lett., 1999, 1, 1027–1029 CrossRef CAS.
- P. A. Barfield, M. F. Lappert and J. Lee, J. Chem. Soc. A, 1968, 554–559 RSC.
- P. Gütlich, E. Bill and A. E. Trautwein, Mössbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications, Springer-Verlag, Berlin, Heidelberg, 2011 Search PubMed.
- S. C. Bart, K. Chłopek, E. Bill, M. W. Bouwkamp, E. Lobkovsky, F. Neese, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 13901–13912 CrossRef CAS PubMed.
- D. Benito-Garagorri, L. G. Alves, M. Puchberger, K. Mereiter, L. F. Veiros, M. J. Calhorda, M. D. Carvalho, L. P. Ferreira, M. Godinho and K. Kirchner, Organometallics, 2009, 28, 6902–6914 CrossRef CAS.
- L. R. Widger, Y. Jiang, M. A. Siegler, D. Kumar, R. Latifi, S. P. de Visser, G. N. L. Jameson and D. P. Goldberg, Inorg. Chem., 2013, 52, 10467–10480 CrossRef CAS PubMed.
- S. D. Brown, T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2003, 125, 322–323 CrossRef CAS PubMed.
- M. P. Jensen, M. P. Mehn and L. Que, Angew. Chem., Int. Ed., 2003, 42, 4357–4360 CrossRef CAS PubMed.
- S. D. Brown and J. C. Peters, J. Am. Chem. Soc., 2005, 127, 1913–1923 CrossRef CAS PubMed.
- R. L. Lucas, D. R. Powell and A. S. Borovik, J. Am. Chem. Soc., 2005, 127, 11596–11597 CrossRef CAS PubMed.
- S. C. Bart, E. Lobkovsky, E. Bill and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 5302–5303 CrossRef CAS PubMed.
- N. A. Eckert, S. Vaddadi, S. Stoian, R. J. Lachicotte, T. R. Cundari and P. L. Holland, Angew. Chem., Int. Ed., 2006, 45, 6868–6871 CrossRef CAS PubMed.
- E. J. Klinker, T. A. Jackson, M. P. Jensen, A. Stubna, G. Juhász, E. L. Bominaar, E. Münck and L. Que, Angew. Chem., Int. Ed., 2006, 45, 7394–7397 CrossRef CAS PubMed.
- C. M. Thomas, N. P. Mankad and J. C. Peters, J. Am. Chem. Soc., 2006, 128, 4956–4957 CrossRef CAS PubMed.
- I. Nieto, F. Ding, R. P. Bontchev, H. Wang and J. M. Smith, J. Am. Chem. Soc., 2008, 130, 2716–2717 CrossRef CAS PubMed.
- A. C. Bowman, C. Milsmann, E. Bill, Z. R. Turner, E. Lobkovsky, S. DeBeer, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2011, 133, 17353–17369 CrossRef CAS PubMed.
- R. E. Cowley, N. A. Eckert, S. Vaddadi, T. M. Figg, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2011, 133, 9796–9811 CrossRef CAS PubMed.
- E. R. King, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 4917–4923 CrossRef CAS PubMed.
- M.-E. Moret and J. C. Peters, Angew. Chem., Int. Ed., 2011, 50, 2063–2067 CrossRef CAS PubMed.
- R. E. Cowley and P. L. Holland, Inorg. Chem., 2012, 51, 8352–8361 CrossRef CAS PubMed.
- E. T. Hennessy, R. Y. Liu, D. A. Iovan, R. A. Duncan and T. A. Betley, Chem. Sci., 2014, 5, 1526–1532 RSC.
- J. Xiao and L. Deng, Dalton Trans., 2013, 42, 5607–5610 RSC.
- J. A. Bellow, M. Yousif, A. C. Cabelof, R. L. Lord and S. Groysman, Organometallics, 2015, 34, 2917–2923 CrossRef CAS.
- R. E. Cowley, N. J. DeYonker, N. A. Eckert, T. R. Cundari, S. DeBeer, E. Bill, X. Ottenwaelder, C. Flaschenriem and P. L. Holland, Inorg. Chem., 2010, 49, 6172–6187 CrossRef CAS PubMed.
- A. K. Verma, T. N. Nazif, C. Achim and S. C. Lee, J. Am. Chem. Soc., 2000, 122, 11013–11014 CrossRef CAS.
- P. Leeladee, G. N. L. Jameson, M. A. Siegler, D. Kumar, S. P. de Visser and D. P. Goldberg, Inorg. Chem., 2013, 52, 4668–4682 CrossRef CAS PubMed.
- K. Searles, S. Fortier, M. M. Khusniyarov, P. J. Carroll, J. Sutter, K. Meyer, D. J. Mindiola and K. G. Caulton, Angew. Chem., Int. Ed., 2014, 53, 14139–14143 CrossRef CAS PubMed.
- L. Wang, L. Hu, H. Zhang, H. Chen and L. Deng, J. Am. Chem. Soc., 2015, 137, 14196–14207 CrossRef CAS PubMed.
- M. Römelt, S. Ye and F. Neese, Inorg. Chem., 2009, 48, 784–785 CrossRef PubMed.
- V. Lyaskovskyy, A. I. O. Suarez, H. Lu, H. Jiang, X. P. Zhang and B. de Bruin, J. Am. Chem. Soc., 2011, 133, 12264–12273 CrossRef CAS PubMed.
- A. I. Olivos Suarez, H. Jiang, X. P. Zhang and B. de Bruin, Dalton Trans., 2011, 40, 5697–5705 RSC.
- A. I. O. Suarez, V. Lyaskovskyy, J. N. H. Reek, J. I. van der Vlugt and B. de Bruin, Angew. Chem., Int. Ed., 2013, 52, 12510–12529 CrossRef CAS PubMed.
- M. Goswami, V. Lyaskovskyy, S. R. Domingos, W. J. Buma, S. Woutersen, O. Troeppner, I. Ivanović-Burmazović, H. Lu, X. Cui, X. P. Zhang, E. J. Reijerse, S. DeBeer, M. M. van Schooneveld, F. F. Pfaff, K. Ray and B. de Bruin, J. Am. Chem. Soc., 2015, 137, 5468–5479 CrossRef CAS PubMed.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- M. H. V. Huynh and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13138–13141 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization data, experimental and computational details. Crystallographic data for [B2Pz4LiPyH]2, 1·THF, 1, 2·Ar, 2·Ad and 4. CCDC 1439938 and 1439242–1439246. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc01433j |
| This journal is © The Royal Society of Chemistry 2016 |