
Copper-mediated oxidative difluoromethylation of terminal alkynes with TMSCF2H†
Sheng-Qing
Zhu
a,
Xiu-Hua
Xu
a and
Feng-Ling
Qing
*ab
aKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China. E-mail: flq@mail.sioc.ac.cn
bCollege of Chemistry, Chemical Engineering and Biotechnology, Donghua University, 2999 North Renmin Lu, Shanghai 201620, China
First published on 29th June 2015
Abstract
A copper-mediated oxidative difluoromethylation of terminal alkynes using nucleophilic TMSCF2H in the presence of an oxidant 9,10-phenanthraquinone was developed. This reaction provided a direct and efficient method for preparation of difluoromethylated alkynes.
Fluorinated alkynes have become a type of useful building blocks taking advantage of the unique properties of fluorine atoms and fluorine-containing groups.1 Among them, difluoromethylated alkynes (RC
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCF2H) are important intermediates for the preparation of valuable fluorinated compounds,2 because the CF2H group is considered isosteric and isopolar to a hydroxyl (OH)3 and it could act as a lipophilic hydrogen bond donor.4 Consequently, a number of methods have been developed for the synthesis of difluoromethylated alkynes. Fluorination of aldehydes with a nucleophilic fluorinating reagent is an orthodox route to difluoromethylated alkynes.5 However, this method suffers from harsh reaction conditions that are incompatible with many functional groups. These compounds could also be accessed through the transformation from other CF2-containing intermediates, such as gem-difluoromethylenated ketones,6 difluoromethylated vinyl iodides,7 and (phenylsulfonyl)difluoromethylated alkynes.8 Although these methods boast high levels of reactivity and site selectivity, the direct approaches to difluoromethylated alkynes are more attractive on account of the high atom and step economy.
CCF2H) are important intermediates for the preparation of valuable fluorinated compounds,2 because the CF2H group is considered isosteric and isopolar to a hydroxyl (OH)3 and it could act as a lipophilic hydrogen bond donor.4 Consequently, a number of methods have been developed for the synthesis of difluoromethylated alkynes. Fluorination of aldehydes with a nucleophilic fluorinating reagent is an orthodox route to difluoromethylated alkynes.5 However, this method suffers from harsh reaction conditions that are incompatible with many functional groups. These compounds could also be accessed through the transformation from other CF2-containing intermediates, such as gem-difluoromethylenated ketones,6 difluoromethylated vinyl iodides,7 and (phenylsulfonyl)difluoromethylated alkynes.8 Although these methods boast high levels of reactivity and site selectivity, the direct approaches to difluoromethylated alkynes are more attractive on account of the high atom and step economy.
While tremendous progress has been made recently in the direct synthesis of trifluoromethylated alkynes,9 the direct approaches to analogous difluoromethylated alkynes are less developed. The cross-coupling of 1-iodoalkynes with the thermally unstable reagent CuCF2H was developed by Burton and Hartgraves (Scheme 1a).10 This protocol required prefunctionalized terminal alkynes. Kitazume11a and Hu11b,c reported the difluoromethylation of the in situ generated lithium acetylides with difluorocarbene precursors (Scheme 1b). However, some sensitive functional groups would not be compatible with the alkynyl-Li and/or :CF2 used in these systems. Thus, it is highly desirable to develop new methods such as transition metal-mediated/catalyzed direct difluoromethylation of terminal alkynes for the preparation of difluoromethylated alkynes.
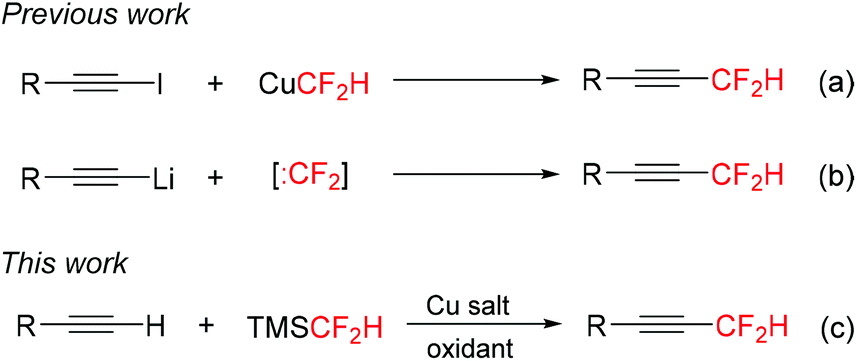 | ||
| Scheme 1 Direct approaches to difluoromethylated alkynes. | ||
Recently, we have developed a new type of trifluoromethylation reaction, oxidative trifluoromethylation, for the synthesis of a wide range of trifluoromethylated compounds.12 This protocol allows the direct installation of a trifluoromethyl group in place of C–H bonds of terminal alkynes with TMSCF3.9a–c Inspired by these results, we anticipated that a similar copper-mediated oxidative C–H difluoromethylation of terminal alkynes with TMSCF2H might be possible. However, this transformation is more challenging than trifluoromethylation of alkynes, because the Si–CF2H bond is more inert than the Si–CF3 bond13 and difluoromethyl copper complexes are less stable than trifluoromethyl copper complexes.14 Herein, we describe the copper-mediated oxidative difluoromethylation of terminal alkynes with TMSCF2H (Scheme 1c) on the basis of recent developments in copper-mediated difluoromethylation of arenes.15 This protocol avoids the use of prefunctionalized alkynes or the generation of alkynyl-Li intermediates.
Optimization of the reaction conditions was explored using phenylacetylene 1a as the model substrate (Table 1). Under the optimized conditions for copper mediated trifluoromethylation of terminal alkynes,9a the reaction of 1a with TMSCF2H (2.0 equiv.), CuI (1.0 equiv.), and 1,10-phenanthroline (phen, 1.0 equiv.) in the presence of KF (3.0 equiv.) under an air atmosphere in DMF at 100 °C provided dimer 3a as the major product, and none of the desired product 2a was observed (entry 1). Then, the reaction was investigated using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as the oxidant and t-BuOK as the initiator based on our previous work of oxidative cross-coupling reaction of terminal alkynes with α-silyldifluoromethylphosphonates.16 To our delight, the desired product 2a was formed in 32% yield (entry 2). Switching to other oxidants such as air, PhI(OAc)2, and Ag2CO3 led to a dramatic decrease in the reaction yield (entries 3–5). Considering the effectiveness of DDQ as the oxidant, several quinones, including 2,3,5,6-tetrachloro-1,4-benzoquinone (O-1), naphthoquinone (O-2), 3,4,5,6-tetrachloro-1,2-benzoquinone (O-3), and 9,10-phenanthraquinone (O-4) were examined (entries 6–9). 9,10-Phenanthraquinone (O-4) proved to be better than other oxidants, giving 2a in 43% yield (entry 9). Surprisingly, the yield of 2a was increased up to 66% in the absence of any ligand (entry 10). In our opinion, the solvent DMF might also act as a ligand to stabilize Cu–CF2H species.15b In fact, 2a was obtained in extremely low yields in other solvents such as DMSO or MeCN (entries 11 and 12). Different Cu salts, including CuBr, CuCN, and CuSCN, could also mediate this transformation, but none of them showed higher reactivity than CuI (entries 13–15). Finally, the yield reached 72% when the reaction was performed from 0 °C to rt (entry 16). It was noteworthy that both the amounts of CuI and t-BuOK were crucial to this transformation. None of 2a was produced when the amount of CuI was decreased from 1.0 equiv. to 0.5 equiv. (entry 17) or the amount of t-BuOK was decreased from 3.0 equiv. to 2.0 equiv. (entry 18).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cu salt | Ligand | Oxidant | Solvent | Temperature | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), TMSCF2H (2.0 equiv.), Cu salt (1.0 equiv.), ligand (1.0 equiv.), oxidant (1.2 equiv.), t-BuOK (3.0 equiv.), DMF (2.0 mL), temperature, under Ar, 10 h. b Yields determined by 19F NMR spectroscopy using fluorobenzene as an internal standard. c KF was used as the initiator. d CuI (0.5 equiv.). e t-BuOK (2.0 equiv.). | ||||||
| 1c | CuI | Phen | Air | DMF | 100 °C | 0 |
| 2 | CuI | Phen | DDQ | DMF | −15 °C to rt | 32 |
| 3 | CuI | Phen | Air | DMF | −15 °C to rt | Trace |
| 4 | CuI | Phen | PhI(OAc)2 | DMF | −15 °C to rt | 5 |
| 5 | CuI | Phen | Ag2CO3 | DMF | −15 °C to rt | Trace |
| 6 | CuI | Phen | O-1 | DMF | −15 °C to rt | 34 |
| 7 | CuI | Phen | O-2 | DMF | −15 °C to rt | Trace |
| 8 | CuI | Phen | O-3 | DMF | −15 °C to rt | 16 |
| 9 | CuI | Phen | O-4 | DMF | −15 °C to rt | 43 |
| 10 | CuI | — | O-4 | DMF | −15 °C to rt | 66 |
| 11 | CuI | — | O-4 | DMSO | −15 °C to rt | Trace |
| 12 | CuI | — | O-4 | MeCN | −15 °C to rt | 4 |
| 13 | CuBr | — | O-4 | DMF | −15 °C to rt | 60 |
| 14 | CuCN | — | O-4 | DMF | −15 °C to rt | 34 |
| 15 | CuSCN | — | O-4 | DMF | −15 °C to rt | 48 |
| 16 | CuI | — | O-4 | DMF | 0 °C to rt | 72 |
| 17d | CuI | — | O-4 | DMF | 0 °C to rt | 0 |
| 18e | CuI | — | O-4 | DMF | 0 °C to rt | 0 |
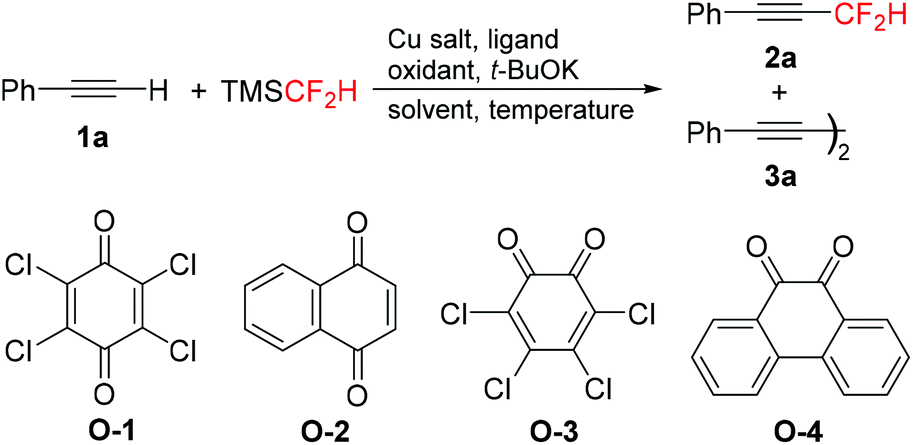
With the optimized reaction conditions in hand (Table 1, entry 16), we next investigated the substrate scope of copper-mediated oxidative difluoromethylation of terminal alkynes. A variety of aromatic and aliphatic alkynes 1 could be transformed into the corresponding difluoromethylated products 2 in moderate to good yields (Table 2). Both electron-rich and -deficient aryl alkynes were compatible in this reaction. In general, the electron-rich aryl alkynes (1b–1h) afforded slightly higher yields compared to the electron-deficient substrates (1i–1k). Many functionalities, including alkoxyl, amino, bromo, cyano, and ester groups were well-tolerated in the reaction. Importantly, substrate 1i bearing a ketone group exhibited moderate reactivity in this transformation, despite the potential for ketones to undergo competing addition of the difluoromethyl group to the carbonyl unit.17 The aliphatic alkynes 1l–1n were also effective to produce the desired products in moderate to good yields.
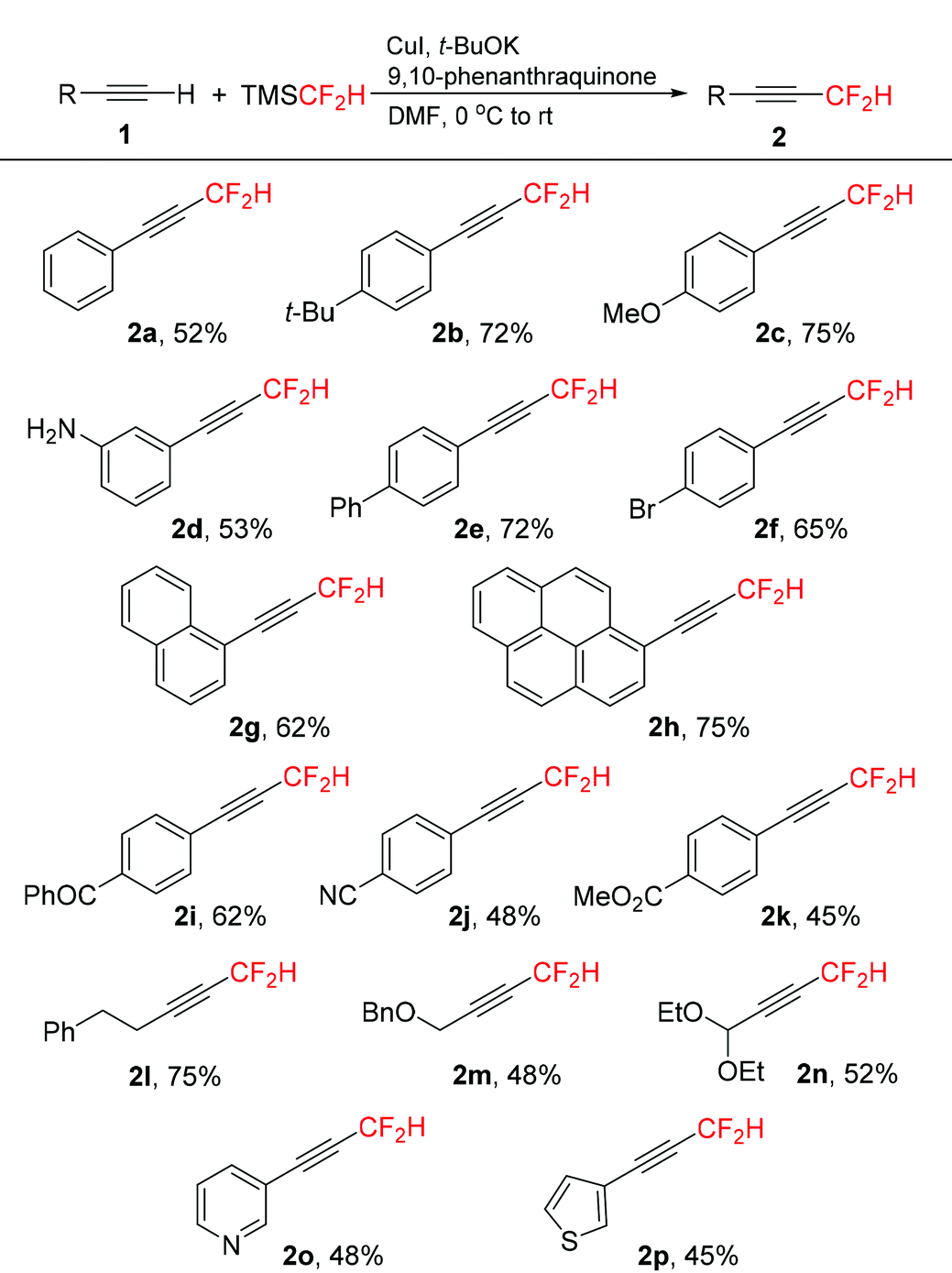
Notably, heteroaryl alkynes 1o and 1p derived from pyridine and thiophene proceeded smoothly to give products 2o and 2p respectively in moderate yields.
This direct difluoromethylation protocol could also be applied for complex molecules, such as estrone and glucofuranose derivatives 1q and 1r (Scheme 2). The corresponding difluoromethylated alkynes 2q and 2r were isolated in moderate to good yields. These results showed that this protocol can be applicable to “late-stage difluoromethylation” of medicinally relevant compounds.
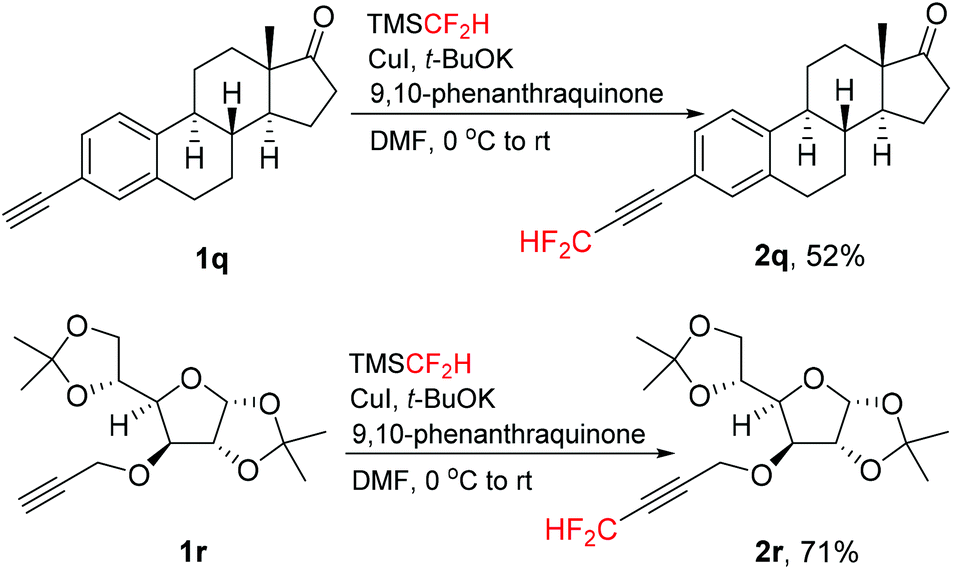 | ||
| Scheme 2 Direct difluoromethylation of estrone and glucofuranose derivatives. | ||
To understand the reaction mechanism, 19F NMR spectroscopy was used to track the reaction (see the ESI†). When TMSCF2H, CuI, and t-BuOK were combined, 19F NMR confirmed the generation of both CuCF2H (resonates at δ = −110.8 ppm, d, J = 45.3 Hz) and Cu(CF2H)2− (resonates at δ = −116.9 ppm, d, J = 44.2 Hz).14,15a After the alkyne was added, a new fluorine-containing intermediate was formed (resonates at δ = −115.9 ppm, d, J = 42.7 Hz). Finally, the desired difluoromethylated product was detected after the addition of oxidant O-4. On the basis of these experimental results, a plausible mechanism for copper-mediated difluoromethylation of terminal alkynes is shown in Scheme 3. The difluoromethylcopper species were firstly generated and then reacted with alkyne 1 to give intermediate A. Subsequently, intermediate A was oxidized to high-valent copper complexes, which finally underwent the reductive elimination to afford product 2.
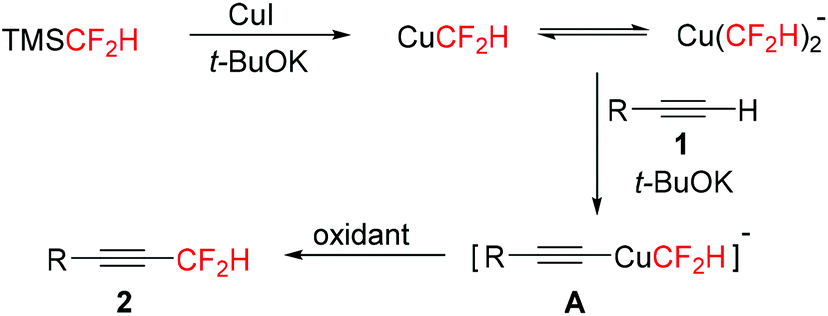 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed an efficient copper-mediated oxidative difluoromethylation of terminal alkynes with easily accessible TMSCF2H. This protocol provides a direct route to the difluoromethylated alkynes from alkynes. As difluoromethylated alkynes are important intermediates for the preparation of other fluorine-containing compounds, this methodology will be potentially useful in pharmaceutical, agrochemical, and material fields.Acknowledgements
We thank the National Natural Science Foundation of China (21421002, 21332010, 21272036) and the National Basic Research Program of China (2012CB21600) for funding this work.Notes and references
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (d) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (e) M. Cametti, B. Crousse, P. Metrangolo, R. Milani and G. Resnati, Chem. Soc. Rev., 2012, 41, 31 RSC; (f) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed.
- (a) M. Kuwabara, K. Fukunishi, M. Nomura and H. Yamanaka, J. Fluorine Chem., 1988, 41, 227 CrossRef CAS; (b) B. C. Hamper, J. Fluorine Chem., 1990, 48, 123 CrossRef CAS; (c) H. Yamanaka, K. Tamura, K. Funabiki, K. Fukunishi and T. Ishihara, J. Fluorine Chem., 1992, 57, 177 CrossRef CAS; (d) K. Tamura, T. Ishihara and H. Yamanaka, J. Fluorine Chem., 1994, 68, 25 CrossRef CAS; (e) B. C. Hamper, K. L. Leschinsky, S. S. Massey, C. L. Bell, L. H. Brannigan and S. D. Prosch, J. Agric. Food Chem., 1995, 43, 219 CrossRef CAS; (f) J. Chae, T. Konno, M. Kanda, T. Ishihara and H. Yamanaka, J. Fluorine Chem., 2003, 120, 185 CrossRef CAS; (g) B. C. Hamper and K. L. Leschinsky, J. Heterocycl. Chem., 2003, 40, 575 CrossRef CAS PubMed; (h) T. Konno, J. Chae, T. Ishihara and H. Yamanaka, Tetrahedron, 2004, 60, 11695 CrossRef CAS PubMed; (i) T. Konno, J. Chae, T. Ishihara and H. Yamanaka, J. Org. Chem., 2004, 69, 8258 CrossRef CAS PubMed; (j) T. Konno, J. Chae, T. Tanaka, T. Ishihara and H. Yamanaka, Chem. Commun., 2004, 690 RSC; (k) T. Konno, J. Chae, T. Miyabe and T. Ishihara, J. Org. Chem., 2005, 70, 10172 CrossRef CAS PubMed; (l) T. Konno, J. Chae, T. Tanaka, T. Ishihara and H. Yamanaka, J. Fluorine Chem., 2006, 127, 36 CrossRef CAS PubMed; (m) T. Konno, K. Taku, S. Yamada, K. Moriyasu and T. Ishihara, Org. Biomol. Chem., 2009, 7, 1167 RSC; (n) P. Bannwarth, A. Valleix, D. Grée and R. Grée, J. Org. Chem., 2009, 74, 4646 CrossRef CAS PubMed; (o) T. Konno, R. Kinugawa, A. Morigaki and T. Ishihara, J. Org. Chem., 2009, 74, 8456 CrossRef CAS PubMed; (p) B. Duda, S. N. Tverdomed and G.-V. Roschenthaler, Org. Biomol. Chem., 2011, 9, 8228 RSC; (q) B. Duda, S. N. Tverdomed, B. I. Ionin and G.-V. Röschenthaler, Eur. J. Org. Chem., 2012, 3684 CrossRef CAS PubMed; (r) B. Duda, S. N. Tverdomed and G.-V. Roschenthaler, RSC Adv., 2012, 2, 9135 RSC; (s) C. Alonso, M. González, M. Fuertes, G. Rubiales, J. M. Ezpeleta and F. Palacios, J. Org. Chem., 2013, 78, 3858 CrossRef CAS PubMed; (t) B. Duda, S. N. Tverdomed, B. I. Ionin and G.-V. Röschenthaler, Eur. J. Org. Chem., 2014, 3757 CrossRef CAS PubMed; (u) B. Duda, S. N. Tverdomed, B. S. Bassil and G.-V. Röschenthaler, Tetrahedron, 2014, 70, 8084 CrossRef CAS PubMed.
- (a) G. K. S. Prakash, M. Mandal, S. Schweizer, N. A. Petasis and G. A. Olah, J. Org. Chem., 2002, 67, 3718 CrossRef CAS PubMed; (b) Y. Li and J. Hu, Angew. Chem., Int. Ed., 2005, 44, 5882 CrossRef CAS PubMed.
- J. A. Erickson and J. I. McLoughlin, J. Org. Chem., 1995, 60, 1626 CrossRef CAS.
- (a) W. R. Hasek, W. C. Smith and V. A. Engelhardt, J. Am. Chem. Soc., 1960, 82, 543 CrossRef CAS; (b) W. J. Middleton, J. Org. Chem., 1975, 40, 574 CrossRef CAS; (c) M. Argentini, R. Weinreich, R. Oberti and L. Ungaretti, J. Fluorine Chem., 1986, 32, 239 CrossRef CAS; (d) J. T. Welch, Tetrahedron, 1987, 43, 3123 CrossRef CAS.
- (a) T. Konno, A. Morigaki, K. Ninomiya, T. Miyabe and T. Ishihara, Synthesis, 2008, 564 CrossRef CAS PubMed; (b) S. N. Tverdomed, J. Kolanowski, E. Lork and G.-V. Röschenthaler, Tetrahedron, 2011, 67, 3887 CrossRef CAS PubMed.
- (a) K. Funabiki, T. Ishihara and H. Yamanaka, J. Fluorine Chem., 1995, 71, 5 CrossRef CAS; (b) T. Konno, J. Chae, M. Kanda, G. Nagai, K. Tamura, T. Ishihara and H. Yamanaka, Tetrahedron, 2003, 59, 7571 CrossRef CAS; (c) T. Konno, K. Moriyasu and T. Ishihara, Synthesis, 2009, 1087 CrossRef CAS PubMed; (d) T. Konno, M. Kishi, T. Ishihara and S. Yamada, Tetrahedron, 2014, 70, 2455 CrossRef CAS PubMed.
- J. Zhu, F. Wang, W. Huang, Y. Zhao, W. Ye and J. Hu, Synlett, 2011, 899 Search PubMed.
- (a) L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2010, 132, 7262 CrossRef CAS PubMed; (b) X. Jiang, L. Chu and F.-L. Qing, J. Org. Chem., 2012, 77, 1251 CrossRef CAS PubMed; (c) K. Zhang, X.-L. Qiu, Y. Huang and F.-L. Qing, Eur. J. Org. Chem., 2012, 58 CrossRef PubMed; (d) Z. Weng, H. Li, W. He, L.-F. Yao, J. Tan, J. Chen, Y. Yuan and K.-W. Huang, Tetrahedron, 2012, 68, 2527 CrossRef CAS PubMed; (e) D.-F. Luo, J. Xu, Y. Fu and Q.-X. Guo, Tetrahedron Lett., 2012, 53, 2769 CrossRef CAS PubMed; (f) N. Aiguabella, C. del Pozo, X. Verdaguer, S. Fustero and A. Riera, Angew. Chem., Int. Ed., 2013, 52, 5355 CrossRef CAS PubMed; (g) N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., Int. Ed., 2014, 53, 539 CrossRef CAS PubMed; (h) X. Li, J. Zhao, L. Zhang, M. Hu, L. Wang and J. Hu, Org. Lett., 2015, 17, 298 CrossRef CAS PubMed.
- D. J. Burton and G. A. Hartgraves, J. Fluorine Chem., 2007, 128, 1198 CrossRef CAS PubMed.
- (a) T. Konno and T. Kitazume, Chem. Commun., 1996, 2227 RSC; (b) W. Zhang, F. Wang and J. Hu, Org. Lett., 2009, 11, 2109 CrossRef CAS PubMed; (c) F. Wang, W. Huang and J. Hu, Chin. J. Chem., 2011, 29, 2717 CrossRef CAS PubMed.
- L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513 CrossRef CAS PubMed.
- T. Hagiwara and T. Fuchikami, Synlett, 1995, 717 CrossRef CAS PubMed.
- R. Eujen, B. Hoge and D. J. Brauer, J. Organomet. Chem., 1996, 519, 7 CrossRef CAS.
- (a) P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5524 CrossRef CAS PubMed; (b) G. K. S. Prakash, S. K. Ganesh, J.-P. Jones, A. Kulkarni, K. Masood, J. K. Swabeck and G. A. Olah, Angew. Chem., Int. Ed., 2012, 51, 12090 CrossRef CAS PubMed; (c) X.-L. Jiang, Z.-H. Chen, X.-H. Xu and F.-L. Qing, Org. Chem. Front., 2014, 1, 774 RSC; (d) C. Matheis, K. Jouvin and L. J. Goossen, Org. Lett., 2014, 16, 5984 CrossRef CAS PubMed.
- X. Jiang, L. Chu and F.-L. Qing, Org. Lett., 2012, 14, 2870 CrossRef CAS PubMed.
- Y. Zhao, W. Huang, J. Zheng and J. Hu, Org. Lett., 2011, 13, 5342 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00186b |
| This journal is © the Partner Organisations 2015 |