
Efficient synthesis of 2-nitroimidazole derivatives and the bioreductive clinical candidate Evofosfamide (TH-302)†
Liam J.
O'Connor
ab,
Cindy
Cazares-Körner
ab,
Jaideep
Saha
a,
Charles N. G.
Evans
ab,
Michael R. L.
Stratford
b,
Ester M.
Hammond
*b and
Stuart J.
Conway
 *a
*a
aDepartment of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: stuart.conway@chem.ox.ac.uk
bCancer Research UK/MRC Oxford Institute for Radiation Oncology, Department of Oncology, University of Oxford, Old Road Campus Research Building, Oxford, OX3 7DQ, UK
First published on 10th July 2015
Abstract
Hypoxia, regions of low oxygen, occurs in a range of biological environments, and is involved in human diseases, most notably solid tumours. Exploiting the physiological differences arising from low oxygen conditions provides an opportunity for development of targeted therapies, through the use of bioreductive prodrugs, which are selectively activated in hypoxia. Herein, we describe an improved method for synthesising the most widely used bioreductive group, 2-nitroimidazole. The improved method is applied to an efficient synthesis of the anti-cancer drug Evofosfamide (TH-302), which is currently in Phase III clinical trials for treatment of a range of cancers.
Hypoxia, an inadequate supply of oxygen, is found in many biological contexts, including bacterial biofilms,1 plant root nodules,2,3 and human disease – most notably solid malignant tumours.4,5 Tumour hypoxia arises from the high metabolic demands of the solid tumour, which results in a disorganised vasculature, and poor oxygen supply to regions of the tumour. The presence of hypoxia in solid tumours correlates with an increased likelihood of tumour metastasis, resistance to all modalities of treatment, and consequently a poor patient prognosis.6 Hypoxic cells are resistant to radiotherapy because radiation-induced DNA damage relies on the generation of reactive oxygen species, and thus occurs in an oxygen-dependent manner. The disruption in tumour vasculature also results in poor delivery of chemotherapeutic agents, reducing the effectiveness of such drugs. Consequently, the hypoxic tumour volume comprises the most aggressive tumour fraction, which is also therapy resistant.6 Therefore, overcoming the challenge of effectively targeting these cells is critical to the successful treatment of solid tumours, and improvement in patient prognosis.
Despite these complications, the reduced oxygen levels present an opportunity to exploit the chemical difference in physiological conditions within regions of hypoxia. Bioreductive prodrugs can be used to target therapeutic compounds to regions of hypoxia, affording enhanced precision in therapeutic compound delivery.7 A number of chemical functionalities, which are selectively reduced under hypoxic conditions via enzymatic metabolism, have been employed in these prodrugs. The moieties used in such prodrugs include nitroaryl groups, quinones and N-oxides.7–12
The nitroimidazole group has been widely used in the development of bioreductive prodrugs and imaging agents. It has found application in anaerobic antibacterial treatments including metronidazole,13,14 in anti-parasitic drug candidates,15 and also in anti-cancer agents. A notable anti-cancer therapeutic that utilises this bioreductive principle is Evofosfamide (TH-302, Fig. 1).16 Under conditions of normal oxygen concentration (normoxia) the nitro group undergoes a process of futile cycling. In hypoxic conditions the nitro group is reduced, resulting in fragmentation to release the active drug. Consequently, increased DNA crosslinking occurs selectively in hypoxia. TH-302 is in advanced Phase II clinical trials for a variety of cancers (ClinicalTrials.gov Identifiers: NCT02093962; NCT01403610; NCT02402062; NCT00742963). It has also recently entered Phase III trials, in combination with other therapeutics, for treatment of metastatic pancreatic cancer and metastatic soft tissue sarcoma (ClinicalTrials.gov Identifiers: NCT01746979; NCT01440088).
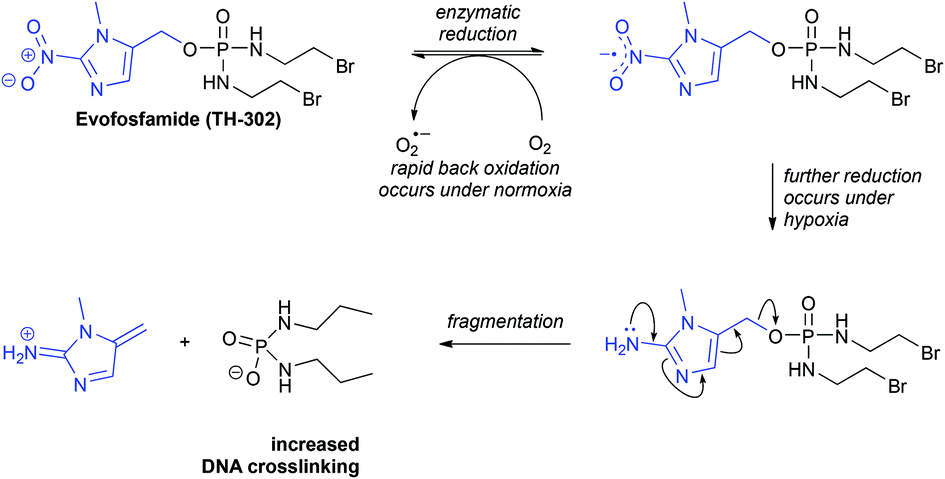 | ||
| Fig. 1 The 2-nitroimidazole-containing bioreductive prodrug Evofosfamide (TH-302) is activated under hypoxic conditions. Under conditions of normal oxygen concentration (normoxia) the nitro group undergoes a process of futile cycling. Under hypoxia, the nitro group is reduced resulting in fragmentation to release the active drug, which crosslinks DNA. | ||
Despite the frequent use of 2-nitroimidazole derivatives as bioreductive prodrugs, existing literature procedures17 for the synthesis of this group are capricious, time intensive, and afford low yields in our hands. There is, consequently, a need for a reliable, time efficient and higher-yielding synthesis of the key synthetic intermediates that can be used for the synthesis of bioreductive prodrugs. Here, we report a time efficient and repeatable synthesis of the aminoimidazole-based ester 4 in yields of 48–54% over three steps. This compound can readily be converted to the nitroimidazole 7. Methods to transform the ester (7) into a range of useful synthetic handles are described. Finally, the alcohol 8 is employed to complete an efficient synthesis of the bioreductive prodrug TH-302.
To synthesise the key 2-aminoimidazole intermediate 4, we initially investigated the procedure of Cavalleri et al.,18 but we were not successful in synthesising the N-methyl-β,β-diethoxyalanine ethyl ester required for this route. We therefore investigated a one-pot procedure developed by Matteucci et al., which was disclosed in the patent literature (Scheme 1A).17 Formylation of sarcosine methyl ester (1), at both the α-carbon and the secondary amine, was achieved by treatment with sodium hydride and ethyl formate to afford 2. The unwanted formyl group on the secondary amine was removed by heating under reflux in acidic EtOH to give the presumed intermediate 3. Subsequent treatment with cyanamide under reflux in aqueous acetate-buffered conditions furnished the desired 2-aminoimidazole (4) in yields that typically ranged from 10–25%, over five repeats of the reaction (n = 5). It should be noted that this procedure was reported on an 82 g scale, and is perhaps less suitable for the scale of 2–3 g, which we employed. Our investigations into an alternative reported synthesis19 were equally variable in the yield of the key intermediate (4).
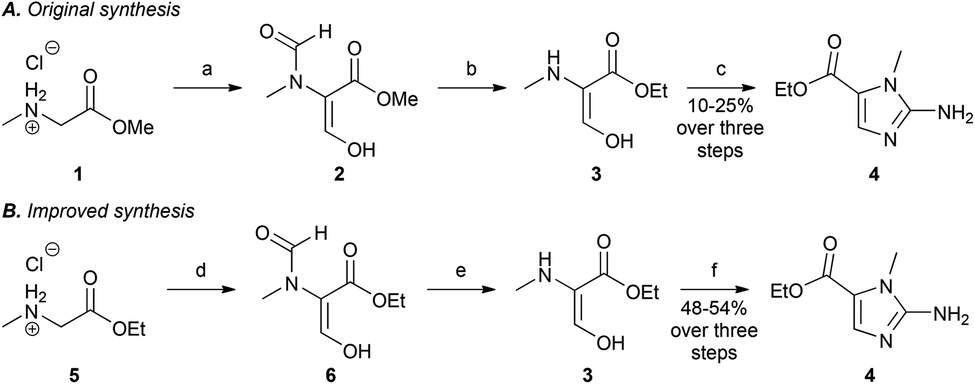 | ||
| Scheme 1 The original and optimised synthetic routes for the 2-aminoimidazole ester 4. A. Reagents and conditions: a. EtOCHO, NaH, 16 h; b. EtOH, conc. HCl, 90 °C, 2 h; c. AcOH (aq.), NaOAc, NH2CN, 100 °C, 1.5 h; 10–25% over three steps (n = 5). B. Reagents and conditions: d. EtOCHO, THF, NaH, 3 h; e. EtOH, conc. HCl, 90 °C, 2 h; f. EtOH, H2O, NH2CN, pH 3, 100 °C, 1.5 h; 48–54% over three steps (n = 5). | ||
When seeking to develop a more robust synthesis we identified the first step, in which the diformylated product (2) is furnished, as being especially capricious. In particular, the solubility of sarcosine methyl ester hydrochloride (1) in ethyl formate is moderate, and this step often failed to proceed, even when the starting material (1) was ground to a powder in an attempt to improve its solubility. We, therefore, hypothesised that the low yield of this reaction was linked to the poor solubility of 1 under the reaction conditions. To address this issue, THF was employed as co-solvent along with ethyl formate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to improve the solubility of the reactants. These modified conditions led to an improvement in the overall yield of the 2-aminoimidazole (4), ranging from 30–35%, over three steps (n = 3). In addition, THF as a co-solvent provided greater temperature control of the highly exothermic reaction, presumably resulting from improved heat transfer, and dilution of the reactants. A further improvement in yield was achieve by employing sarcosine ethyl ester hydrochloride (5) as the starting material (Scheme 1B), as this compound has improved solubility under the reaction conditions, compared to 1. We also noted that the initial formylation step was time intensive, requiring at least 16 h for completion. As the sarcosine ethyl ester (5) is more soluble than the methyl ester (1), we hypothesised that this would increase the reaction rate. This proved to be the case, and after a substantially shorter time period, of 3 h (on a 2 g scale), a milky yellow suspension formed suggesting the presence of the desired N-formylated enolate (6). Given the advantages of using the sarcosine ethyl ester hydrochloride (5), we proceeded with the synthesis using this starting material.
:1) to improve the solubility of the reactants. These modified conditions led to an improvement in the overall yield of the 2-aminoimidazole (4), ranging from 30–35%, over three steps (n = 3). In addition, THF as a co-solvent provided greater temperature control of the highly exothermic reaction, presumably resulting from improved heat transfer, and dilution of the reactants. A further improvement in yield was achieve by employing sarcosine ethyl ester hydrochloride (5) as the starting material (Scheme 1B), as this compound has improved solubility under the reaction conditions, compared to 1. We also noted that the initial formylation step was time intensive, requiring at least 16 h for completion. As the sarcosine ethyl ester (5) is more soluble than the methyl ester (1), we hypothesised that this would increase the reaction rate. This proved to be the case, and after a substantially shorter time period, of 3 h (on a 2 g scale), a milky yellow suspension formed suggesting the presence of the desired N-formylated enolate (6). Given the advantages of using the sarcosine ethyl ester hydrochloride (5), we proceeded with the synthesis using this starting material.
To further improve the synthesis, we speculated that the conditions previously employed to form 2-aminoimidazole 4, which involved heating under reflux in aqueous acetate-buffered conditions, would likely result in ethyl ester hydrolysis, depreciating the overall reaction yield. In addition, we speculated that a large excess of cyanamide would force the equilibrium towards the desired product. Therefore, the conditions for formation of the 2-aminoimidazole (4) were modified, such that intermediate (3) was treated with excess cyanamide under reflux in 70% aqueous EtOH (v/v). To minimise ester hydrolysis, the reaction solution was adjusted to pH 3–4 using 2 M aqueous NaOH. The product (4) was isolated by concentration in vacuo followed by adjustment of the pH to 8–9 with solid K2CO3, and filtration of the precipitate (Scheme 1B). This method resulted in reproducible yields of 48–54% (n = 5) for compound 4, representing a yield of approximately 80% per step, and an improvement on the previously employed methods, which gave yields of 10–25%.
With the key 2-aminoimidazole intermediate (4) in hand, we sought to introduce the nitro functionality via diazotisation of the amino group (Scheme 2). Using the procedure reported by Matteucci et al.,17 which employed a 4.8 M aqueous solution of NaNO2, we found yields of the 2-nitroimidazole product (7) were typically 10–20% lower than the reported value of 62%. This procedure was reported on a 37 g scale, and the conditions used seem to be less suitable for more moderate scales of 2–5 g. To minimise the volume of water in the reaction, we used a saturated aqueous solution of NaNO2, and increased the volume of AcOH in the reaction mixture from 50% to 66% (v/v). These modified conditions reliably afforded compound 7 in a yield of 72% (n = 3), and seem optimal for the introduction of the nitro group on a moderate scale.
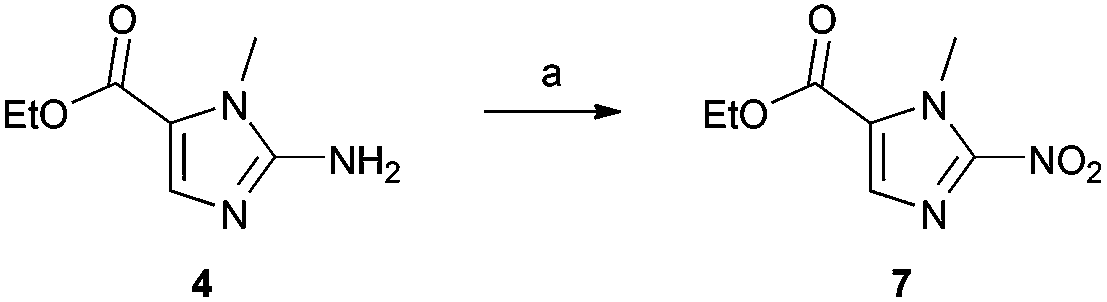 | ||
| Scheme 2 Diazotisation and nitration of 4. Reagents and conditions: AcOH, NaNO2 (aq), rt, 4 h, 72%. | ||
The 2-nitroimidazole ester 7 represents a versatile synthetic intermediate, which can be transformed into a number of useful compounds that possess suitable chemical handles for attachment to a range of complementary functionalities. Here we include procedures for synthesis of the alcohol (8), carboxyl (9), chloride (10), and carbaldehyde (11) derivatives (Scheme 3).18,20 Each of these intermediates is a useful synthetic substrate for the development of bioreductive prodrugs or dyes.
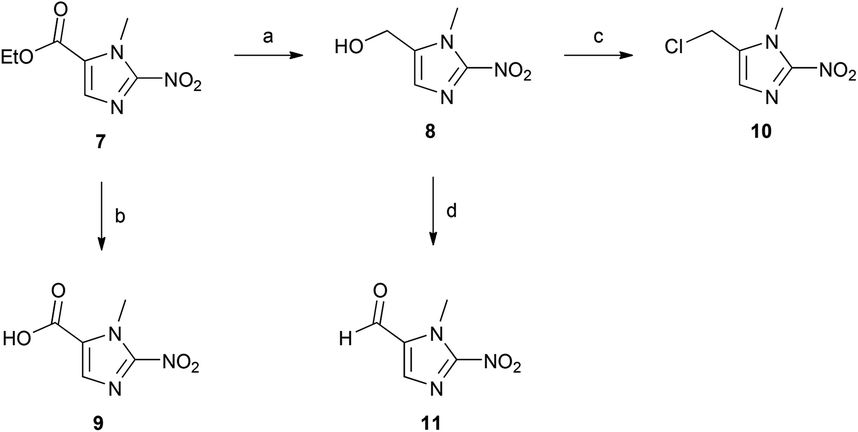 | ||
| Scheme 3 The introduction of versatile synthetic handles on to the 2-nitroimidazole ring. Reagents and conditions: a. NaBH4, THF, MeOH, 0 °C → rt, 2 h, 66%; b. NaOH (aq.), rt, 2 h, 95%; c. SOCl2, pyridine, CH2Cl2, 0 °C → rt, 2 h, 66%; d. MnO2, acetone, CH2Cl2, 3 d, 52%. | ||
To demonstrate the utility of our improved synthetic route, we applied it to the synthesis of the Phase III clinical candidate, and bioreductive prodrug, Evofosfamide (TH-302, Scheme 4).
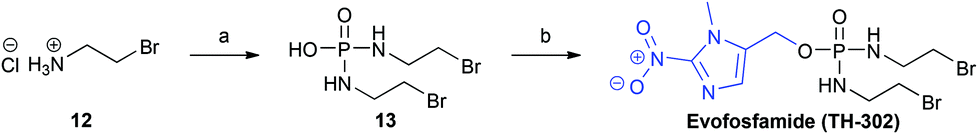 | ||
| Scheme 4 Synthesis of Evofosfamide (TH-302). Reagents and conditions: a POCl3, Et3N, CH2Cl2, −10 °C; b. PPh3, DIAD, 8, THF, 0 °C → rt, 3 h, 62%. | ||
The bromoisophosphoramide mustard intermediate (Br-IPM, 13), was synthesised from the corresponding 2-bromoethylamine hydrobromide salt (12), employing the method reported by Duan et al.16 Subsequent treatment of (13) with the 2-nitroimidazole alcohol (8) under Mitsunobu conditions,21 afforded Evofosfamide (TH-302) in 62% yield, which compares well with the previously reported synthesis (48% yield).16
In conclusion, we have developed a high yielding, rapid and reliable synthesis of the 2-aminoimidazole ester 4, which is a key intermediate in the synthesis of many bioreductive prodrugs. We have optimised the nitration conditions for this compound, and demonstrated that the 2-nitroimidazole-based ester 7 can be transformed into a range of synthetically useful derivatives. This work represents a comprehensive and optimised set of synthetic procedures for the synthesis of 2-nitroimidazole derivatives, which are essential components for developing bioreductive prodrugs and dyes. We have demonstrated the utility of our new methodology by applying it in the efficient synthesis of the bioreductive prodrug clinical candidate Evofosfamide (TH-302).
SJC, EMH and LJOC thank MRC for the award of a studentship to LJOC. SJC, EMH and CC-K thank Cancer Research UK for the award of a studentship to CC-K. SJC, EMH and CNGE thank Cancer Research UK and EPSRC for the award of a studentship to CNGE, through the CR-UK and EPSRC Cancer Imaging Centre in Oxford. SJC and JS thank the European Commission for the award of a Marie Curie Fellowship to JS (PIIF-GA-2012-331327, HYPOXPROBE). EMH thanks Cancer Research UK for research funding. SJC thanks St Hugh's College, Oxford, for research funding.
Notes and references
- T. Rossignol, C. Ding, A. Guida, C. d'Enfert, D. G. Higgins and G. Butler, Eukaryotic Cell, 2009, 8, 550–559 CrossRef CAS PubMed.
- B. A. Smit, D. S. Neuman and M. L. Stachowiak, Plant Physiol., 1990, 92, 1021–1028 CrossRef CAS PubMed.
- M. C. Drew, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1997, 48, 223–250 CrossRef CAS PubMed.
- G. L. Semenza, F. Agani, D. Feldser, N. Iyer, L. Kotch, E. Laughner and A. Yu, Adv. Exp. Med. Biol., 2000, 475, 123–130 CrossRef CAS.
- M. Höckel and P. Vaupel, J. Natl. Cancer Inst., 2001, 93, 266–276 CrossRef PubMed.
- J. M. Brown and W. R. Wilson, Nat. Rev. Cancer, 2004, 4, 437–447 CrossRef CAS PubMed.
- W. R. Wilson and M. P. Hay, Nat. Rev. Cancer, 2011, 11, 393–410 CrossRef CAS PubMed.
- E. A. Blanche, L. Maskell, M. A. Colucci, J. L. Whatmore and C. J. Moody, Tetrahedron, 2009, 65, 4894–4903 CrossRef CAS PubMed.
- M. A. Colucci, C. J. Moody and G. D. Couch, Org. Biomol. Chem., 2008, 6, 637–656 CAS.
- T. Fryatt, H. I. Pettersson, W. T. Gardipee, K. C. Bray, S. J. Green, A. Slawin, H. D. Beall and C. J. Moody, Bioorg. Med. Chem., 2004, 12, 1667–1687 CrossRef CAS PubMed.
- T. Fryatt, D. T. Goroski, Z. D. Nilson, C. J. Moody and H. D. Beall, Bioorg. Med. Chem. Lett., 1999, 9, 2195–2198 CrossRef CAS.
- C. Cazares-Korner, I. M. Pires, I. D. Swallow, S. C. Grayer, L. J. O'Connor, M. M. Olcina, M. Christlieb, S. J. Conway and E. M. Hammond, ACS Chem. Biol., 2013, 8, 1451–1459 CrossRef CAS PubMed.
- G. Asato and B. G, J. Med. Chem., 1972, 15, 1086–1088 CrossRef CAS.
- F. P. Tally, V. L. Sutter and S. M. Finegold, Antimicrob. Agents Chemother., 1975, 7, 672–675 CrossRef CAS.
- W. J. Ross and W. B. Jamieson, J. Med. Chem., 1975, 18, 158–161 CrossRef CAS.
- J.-X. Duan, H. Jiao, J. Kaizerman, T. Stanton, J. W. Evans, L. Lan, G. Lorente, M. Banica, D. Jung, J. Wang, H. Ma, X. Li, Z. Yang, R. M. Hoffman, W. S. Ammons, C. P. Hart and M. Matteucci, J. Med. Chem., 2008, 51, 2412–2420 CrossRef CAS PubMed.
- M. Matteucci, J.-X. Duan, H. Jiao, J. Kaizerman, S. Ammons and M. H. Hopkins, US Patent Office, WO 2007/002931 A2, 2007 Search PubMed.
- B. Cavalleri, R. Ballotta and G. Lancini, J. Heterocycl. Chem., 1972, 9, 979–984 CrossRef CAS PubMed.
- M. Matteucci, P. Rao and J.-X. Duan, US Patent Office, WO 2004/087075 A2, 2004 Search PubMed.
- B. Cavalleri, R. Ballotta, V. Arioli and G. Lancini, J. Med. Chem., 1973, 16, 557–560 CrossRef CAS.
- O. Mitsunobu, Synthesis, 1981, 1–28 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, 1H, 13C and 31P NMR spectra, HPLC purity details for TH-302. See DOI: 10.1039/c5qo00211g |
| This journal is © the Partner Organisations 2015 |