
Copper-mediated oxidative difluoromethylation of terminal alkynes with TMSCF2H†
Abstract
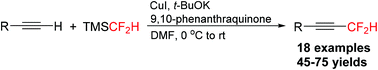
A copper-mediated oxidative difluoromethylation of terminal alkynes using nucleophilic TMSCF2H in the presence of an oxidant 9,10-phenanthraquinone was developed. This reaction provided a direct and efficient method for preparation of difluoromethylated alkynes.
- This article is part of the themed collection: Celebrating 70 Years of Shanghai Institute of Organic Chemistry
 Please wait while we load your content...
Please wait while we load your content...

