
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect catalytic conversion of cellulose to liquid straight-chain alkanes†
Beau
Op de Beeck
a,
Michiel
Dusselier
ac,
Jan
Geboers
a,
Jensen
Holsbeek
a,
Eline
Morré
a,
Steffen
Oswald
b,
Lars
Giebeler
b and
Bert F.
Sels
*a
aCenter for Surface Chemistry and Catalysis, KU Leuven, Kasteelpark Arenberg 23, 3001 Heverlee, Belgium. E-mail: bert.sels@biw.kuleuven.be
bLeibniz-Institute for Solid State and Materials Research, Institute for Complex Materials, Helmholtzstr. 20, D-01069 Dresden, Germany
cChemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA
First published on 9th September 2014
Abstract
High yields of liquid straight-chain alkanes were obtained directly from cellulosic feedstock in a one-pot biphasic catalytic system. The catalytic reaction proceeds at elevated temperatures under hydrogen pressure in the presence of tungstosilicic acid, dissolved in the aqueous phase, and modified Ru/C, suspended in the organic phase. Tungstosilicic acid is primarily responsible for cellulose hydrolysis and dehydration steps, while the modified Ru/C selectively hydrogenates intermediates en route to the liquid alkanes. Under optimal conditions, microcrystalline cellulose is converted to 82% n-decane-soluble products, mainly n-hexane, within a few hours, with a minimum formation of gaseous and char products. The dominant route to the liquid alkanes proceeds via 5-hydroxymethylfurfural (HMF), whereas the more common pathway via sorbitol appears to be less efficient. High liquid alkane yields were possible through (i) selective conversion of cellulose to glucose and further to HMF by gradually heating the reactor, (ii) a proper hydrothermal modification of commercial Ru/C to tune its chemoselectivity to furan hydrogenation rather than glucose hydrogenation, and (iii) the use of a biphasic reaction system with optimal partitioning of the intermediates and catalytic reactions. The catalytic system is capable of converting subsequent batches of fresh cellulose, enabling accumulation of the liquid alkanes in the organic phase during subsequent runs. Its robustness is illustrated in the conversion of the raw (soft)wood sawdust.
Broader contextA novel one-pot catalytic approach is presented that is able to directly transform cellulose into straight-chain alkanes (mainly n-hexane). The carbon-based yields are high (up to 82%) and the process completes in less than 6 hours at only 493 K. The so produced and thus bio-derived light naphta fraction is an ideal green feedstock for existing processes that produce aromatics, gasoline or olefins. Considering the vast and cheap amounts of cellulosic residue and the absence of its pretreatment for this process, this catalytic one-pot approach seems highly promising en route to more sustainable chemicals and fuels. |
Introduction
Interest in lignocellulosic biomass as a renewable feedstock for fuels, chemicals and materials has increased tremendously in recent years.1–14 The high oxygen-to-carbon ratio of cellulosic biomass creates ample opportunities to produce chemicals and polymer building blocks with high chemical functionality, which cannot be produced as cheaply from fossil feedstock.15–17 5-hydroxymethylfurfural (HMF),18 (vinyl) glycolic acid,19,20 lactic acid21,22 and levulinic acid23,24 are four examples of such chemicals, for which synthesis directly from cellulose is under investigation. Targeting fuels with biomass feedstock primarily concentrates on depolymerization and defunctionalization strategies to produce molecules with high heating value like alkanes and aromatics.25–35 As the value of a fuel per tonnage is usually low, but the targeted volumes are enormous, process and energy cost should be kept to an absolute minimum. There are elaborate examples in literature describing the production of new generation biofuels from sugars, sugar alcohols or other platform molecules such as HMF and levulinic acid,3,10,25,30–33,35–41 but research on the direct route from low cost cellulose to alkanes is still in its infancy. Although high temperature hydropyrolytic routes from biomass towards mixtures of gasoline and other compounds are promising,42–49 there is room to improve the carbon efficiency to liquid alkanes. Due to its high natural abundance50 and uniform chemical structure with repeating C6 sugar units, cellulose should be the ideal precursor for selectively making C6 alkanes (and thus light naphtha) as C–C bond breaking and forming are not required. The main challenge is to selectively break C–O in presence of C–C bonds.The prospect of renewable n-hexane directly made from cellulose, as outlined in Scheme 1, is exciting as this alkane has many uses such as technical solvent,51 fuels and building block for chemicals. For use as a transportation fuel, viz. gasoline, n-hexane needs to be isomerized to branched hexanes with higher motor octane numbers (MON) like 2,2-dimethylbutane (MON 95).52 It is well known that, unlike C7+ alkanes, light (C4, C5 and C6) alkanes can be selectively isomerized with minimal cracking (e.g., in the Hysomer Process of Shell), and mixed in with gasoline.52,53 Since highly branched alkanes, possibly mixed with some ethers, constitute the environmentally most friendly gasoline,52 bio-based isomerized light naphtha (with or without ethers) may be an interesting option to improve the renewability of gasoline in short term. Besides fuel and solvent use, n-hexane may also serve as ideal feedstock for bio-benzene production54 and for bio-ethylene and propylene production via steam or catalytic cracking.55
 | ||
| Scheme 1 Biphasic catalytic conversion of cellulose into liquid alkanes like n-hexane, methylcyclopentane and n-pentane at elevated temperature under H2 pressure with modified Ru/C. | ||
During submission of this manuscript, the group of Tomishige reported the first selective one-pot conversion of cellulose to n-hexane using Ir-ReOx/SiO2 and H-ZSM-5.56 The reaction proceeds via the hydrolytic hydrogenation of cellulose to sorbitol, which is subsequently converted to n-hexane through consecutive hydrodeoxygenation cycles. Apart from this report, only multistep processes57 and the conversion of cellobiose36 (94.8% n-hexane yield) and methylcellulose58 into n-hexane (80% total yield) were demonstrated. The major obstacles for direct cellulose conversion are its poor solubility in conventional solvents and high chemical recalcitrance.59 These issues necessitate severe reaction conditions in terms of acidity and/or temperature, which can lead to unwanted side reactions. This paper reports a direct, fast and selective conversion of cellulose into liquid straight-chain alkanes, mainly n-hexane, by tuning the hydrogenation selectivity of a commercial Ru catalyst in a biphasic liquid system. The surface modification steers the reaction via a novel pathway, forming liquid alkanes through intermediate HMF (see Scheme 2).
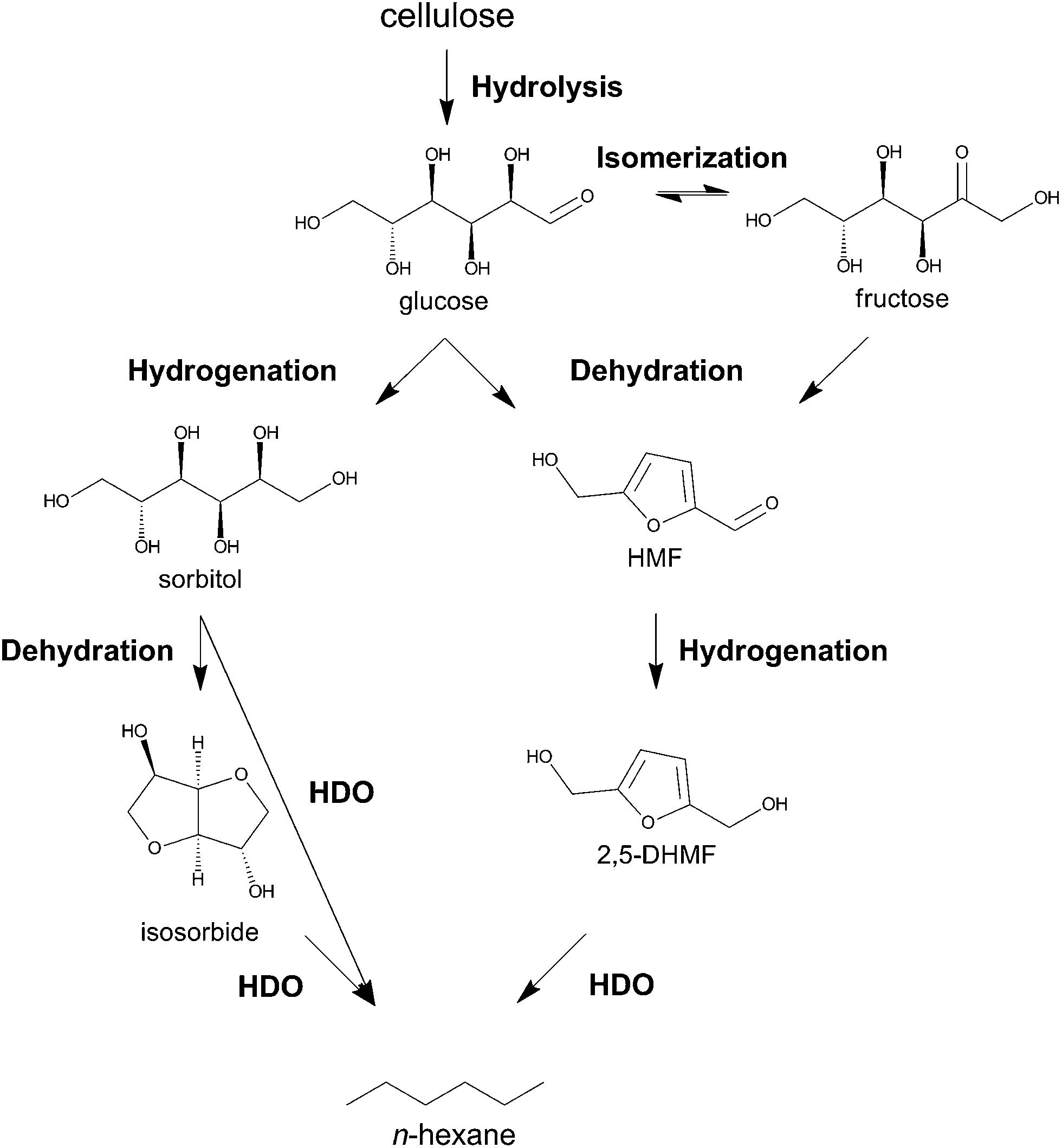 | ||
| Scheme 2 Two different pathways for the selective one-pot conversion of cellulose to n-hexane: the established sorbitol route (left hand-side) and the here proposed HMF route (right hand-side). Abbreviations: HDO, hydrodeoxygenation. 2,5-DHMF, 2,5-dihydroxymethylfuran. | ||
Experimental
A typical modification of commercial 5 wt% Ru/C proceeded as follows: Ru/C (1 g), tungstosilicic acid (TSA) hydrate (0.25 g) and water (40 ml) were loaded into a 100 ml stainless steel batch reactor (Parr Instruments Co.). The reactor was flushed with N2 and subsequently pressurized with 5 MPa H2. The mixture was stirred at 700 rpm and heated to 483 K at an average rate of 10 K min−1 and kept at this temperature for 1 h. The reactor was then cooled, depressurized and opened. The synthesized catalyst (htTSA(2)Ru/C) was filtered, thoroughly washed with distilled water and dried to constant weight.In a typical catalytic experiment, microcrystalline Avicel PH-101 cellulose (2 g), TSA hydrate (5 g), htTSA(2)Ru/C (0.5 g), water (20 ml) and n-decane (20 ml) were loaded into a 100 ml stainless steel batch reactor (Parr Instruments Co.). The reactor was flushed with N2 and subsequently pressurized with 5 MPa H2. The mixture was stirred at 700 rpm and heated to 493 K at an average rate of 12 K min−1 from room temperature to 423 K and further to 493 K at a fixed rate of 0.5 K min−1. The mixture was kept at 493 K for an additional 40 min. After reaction, the reactor was cooled, depressurized and opened. Samples were taken from both the water and n-decane phases and centrifuged before GC and TOC analysis. For determination of cellulose conversion and catalyst reuse experiments, centrifuged particles were added back to the reaction mixture. The reaction mixture was subsequently filtered, thoroughly washed and dried to constant weight.
Complete experimental procedures are provided in the ESI.†
Results and discussion
Rationale
From literature on n-hexane production from sugar and sugar-derived feedstock,31,32 and the recent one-pot approach from Tomishige and co-workers,56 one may deduce one major pathway, which proceeds via a combination of various reactions including hydrolysis, hydrogenation, dehydration and hydrodeoxygenation (HDO), catalyzed by bifunctional acid/redox catalytic systems (Scheme 2). There is general agreement that the route involves the initial formation of sorbitol as key intermediate towards n-hexane.32,36,60,61 However, we propose here an alternative pathway that runs through HMF. Deep HDO of HMF to n-hexane, e.g. by direct metal-catalyzed C–O hydrogenolysis or acid/metal-catalyzed dehydration/hydrogenation cycles,60,62 has not been demonstrated experimentally.The use of cellulose rather than sugar solutions significantly complicates the balance of reaction rates required for selective n-hexane formation. As fast cellulose hydrolysis generally requires strong acidic conditions or high temperatures, sorbitol produced from glucose may undergo rapid dehydration to sorbitan and isosorbide. As a remarkable stability of isosorbide in the presence of acid/redox catalysts at high temperatures was encountered,63 isosorbide formation may be a significant hurdle for the low energy conversion of cellulose to n-hexane. Isosorbide formation can be prevented as long as glucose dehydration to HMF is kinetically favored over glucose hydrogenation to sorbitol. Additionally, subsequent and fast hydrogenation of HMF to e.g., 2,5-dihydroxymethylfuran (2,5-DHMF, Scheme 2) and 2,5-dihydroxymethyltetrahydrofuran (2,5-DHMTHF) should be promoted to avoid HMF degradation into levulinic acid and humin (char). From that point on, a series of HDO cycles of the furanic species should ensue. A recent example of Buntara et al., showing a selective conversion of 2,5-DHMTHF to 1,6-hexanediol, underscores the potential of an HMF route that ultimately leads to n-hexane.64
The main challenge seems the integration of the acidic hydrolysis of cellulose with a selective hydro(deoxy)genation of acid-sensitive HMF in presence of glucose. We anticipate that these demands can be fulfilled by (i) compartmentalization of the acidity in an aqueous phase and redox activity in an organic phase and (ii) modification of the redox catalyst to increase its selectivity towards HMF hydrogenation instead of glucose hydrogenation (vide infra). The biphasic system is essential to extract acid-sensitive intermediates from the acidic aqueous phase into the organic phase, while the organic phase should be a favorable medium for hydrogenation and dehydration reactions due to a higher hydrogen solubility and more efficient dehydration in organic solvents, respectively. The important role of catalysis at water–oil interfaces in biomass conversion has been suggested in other work as well.65,66 The beneficial effect of a biphasic solvent system on the selective hydrogenation of HMF to 2,5-DHMTHF for instance has been investigated and confirmed by Alamillo et al.67 and Yang et al.68 Furthermore, glucose hydrogenation is suppressed in favor of HMF hydrogenation by the use of a hydrophobic hydrogenation catalyst, which predominantly resides in the organic phase. Since HMF traverses phase boundaries, the latter will result in selective hydrogenation of HMF, avoiding sorbitol formation.
Tuning the hydrogenation properties of Ru/C
Since Ru/C is commercially used to hydrogenate glucose to sorbitol,69 it seems at first sight an unlikely catalyst choice, but its selection here was primarily based on its high affinity for the organic phase (see ESI, Fig. S1†) and its commercial relevance.In order to suppress its glucose hydrogenation ability and favor HMF hydrogenation, Ru/C was modified. Modifying the chemoselectivity of metal redox catalysts is usually accomplished by adding promoters.70 Below, we show that the hydrogenation selectivity of commercial Ru/C is drastically changed in favor of HMF hydrogenation by hydrothermal treatment (ht) in presence of tungstosilicic acid (TSA, H4SiW12O40). The modification was carried out under H2 pressure (5 MPa at room temperature) at 483 K for 1 h in water in presence of varying TSA concentrations. Despite the harsh treatment, we barely noticed Ru leaching during the hot water treatment in presence of TSA: elemental analysis of the filtrate demonstrated the presence of 2.5 ppm Ru, corresponding to 0.3 wt% of the initial Ru content.
The change in hydrogenation selectivity was evidenced in a kinetic study. A first series of experiments with glucose was carried out in water in presence of unmodified Ru/C, ht-treated Ru/C and Ru/C ht-treated in a 2 and 135 mM TSA solution, denoted as Ru/C, htRu/C, htTSA(2)Ru/C and htTSA(135)Ru/C, respectively. The TSA loading on Ru/C after drying, studied by gravimetric analysis, correlates to the TSA concentration in the pretreatment mixture (Table S1†), in agreement with the strong adsorption of heteropoly acids on carbon supports.71–77 The final TSA modified catalysts htTSA(2)Ru/C and htTSA(135)Ru/C contain approximately 9 and 27 wt% TSA (on dry basis), respectively. The kinetic profiles are presented in Fig. 1. As expected, glucose is selectively converted to sorbitol by each catalyst. The data show a significant decrease in activity after modification with TSA, and this decrease is more pronounced with the higher TSA loading. Comparison of the initial conversion rates of htTSA(2)Ru/C and htTSA(135)Ru/C versus pristine Ru/C showed a remarkable three- and six-fold activity loss, respectively, whereas a hydrothermal treatment in absence of TSA only shows a minor impact on the hydrogenation activity.
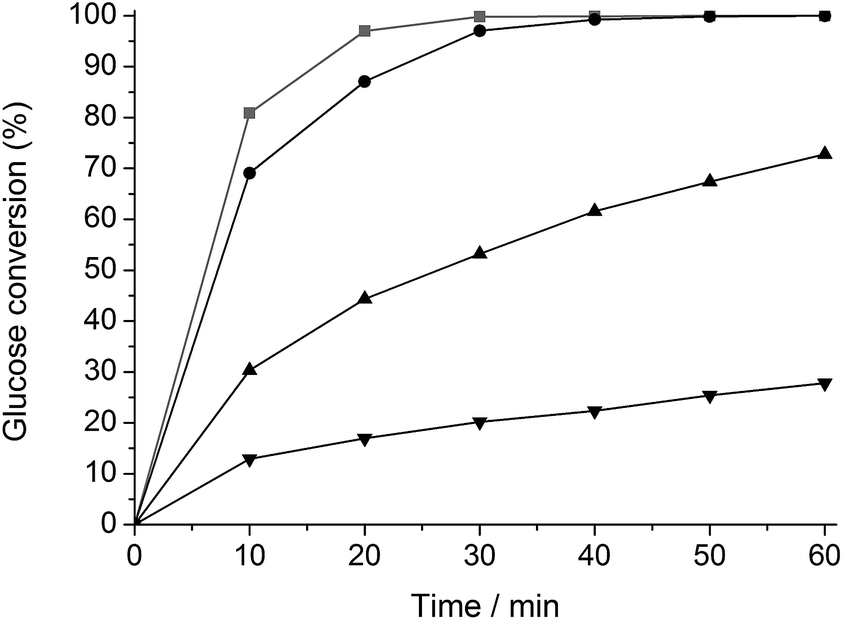 | ||
| Fig. 1 Glucose hydrogenation with Ru/C (gray, ■), htRu/C (black, ●), htTSA(2)Ru/C (black, ▲) and htTSA(135)Ru/C (black, ▼). Reaction conditions: 5.56 mmol glucose, 0.25 g Ru/C (content in the different catalysts), 50 ml H2O, 5 MPa H2, 373 K. | ||
A similar set of kinetic experiments was carried out for the hydrogenation of HMF (Fig. 2). All reactions formed 2,5-DHMF as main product. Interestingly, the hydrothermal modification of Ru/C both with and without TSA results in a hydrogenation activity increase.
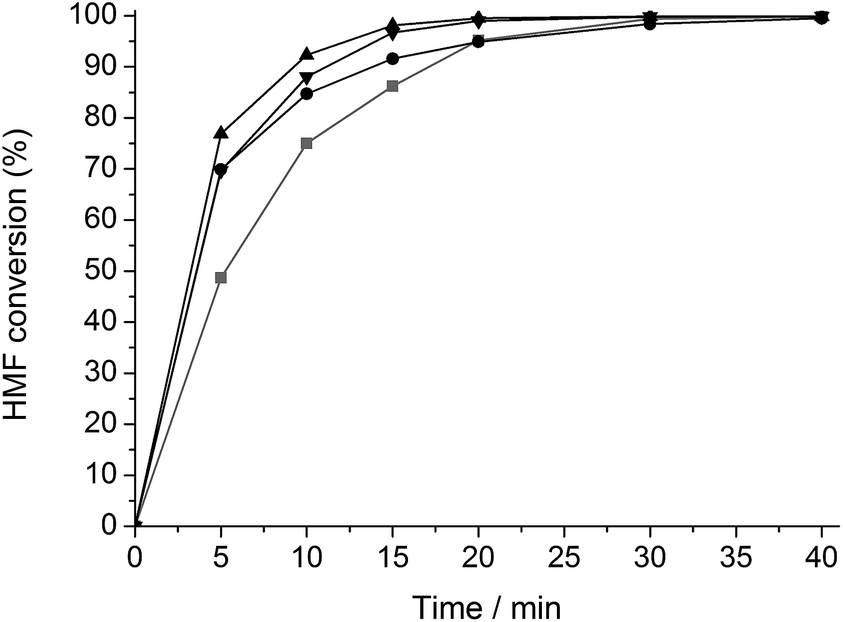 | ||
| Fig. 2 HMF hydrogenation with Ru/C (gray, ■), htRu/C (black, ●), htTSA(2)Ru/C (black, ▲) and htTSA(135)Ru/C (black, ▼). Reaction conditions: 5.56 mmol HMF, 0.25 g Ru/C (content in the different catalysts), 50 ml H2O, 5 MPa H2, 333 K. | ||
Although the fundamentals behind the selective modification of Ru/C with TSA are unclear, CO chemisorption (see ESI†) showed a decreased number of total active sites upon modification and this decrease correlates linearly with the initial glucose hydrogenation activity of the different catalysts (Fig. S2a†). Unless CO is selectively probing glucose adsorption sites, this observation is indicative of a structural change of Ru e.g. Ru sintering. Calculation of the turnover frequency (TOF, s−1) shows that modification with TSA has little impact on glucose hydrogenation, while there is a significant increase in the TOF (s−1) (calculated as mol converted HMF per mol surface Ru per second) of HMF hydrogenation (Fig. S2b†). The modified Ru surface thus seems to benefit the planar adsorption of HMF, with strongly adsorbed C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and parallel-oriented CO bonds,62 likely on atomically smoother Ru surfaces of the sintered Ru. Investigation of the physicochemical properties of the TSA-modified Ru/C catalyst is ongoing.
C and parallel-oriented CO bonds,62 likely on atomically smoother Ru surfaces of the sintered Ru. Investigation of the physicochemical properties of the TSA-modified Ru/C catalyst is ongoing.
Exploring cellulose to liquid alkanes conversion with modified Ru/C
The modified htTSA(135)Ru/C catalyst, with its altered hydrogenation selectivity, was used to explore the one-pot conversion of cellulose to liquid alkanes. A biphasic water/n-decane (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 vol%) solvent mixture was initially chosen. Microcrystalline cellulose was used and its conversion to liquid alkanes was initially tested with htTSA(135)Ru/C at temperatures ranging from 483 K to 503 K at 5 MPa H2 pressure. The reaction uses an additional amount of water-soluble TSA catalyst to accelerate cellulose hydrolysis. Unlike most inorganic solids like alumina and silica/alumina, TSA is a strong Brønsted acid, and most importantly, it shows a high selectivity to glucose during cellulose hydrolysis.63,75,78–82 The catalytic results are summarized in Table 1. The table includes the main reaction products found: n-hexane, methylcyclopentane (MCP), n-pentane, 2,5-DMTHF, 1-hexanol and some hexitols (sorbitol, mannitol and their anhydrides like sorbitan and isosorbide).
:50 vol%) solvent mixture was initially chosen. Microcrystalline cellulose was used and its conversion to liquid alkanes was initially tested with htTSA(135)Ru/C at temperatures ranging from 483 K to 503 K at 5 MPa H2 pressure. The reaction uses an additional amount of water-soluble TSA catalyst to accelerate cellulose hydrolysis. Unlike most inorganic solids like alumina and silica/alumina, TSA is a strong Brønsted acid, and most importantly, it shows a high selectivity to glucose during cellulose hydrolysis.63,75,78–82 The catalytic results are summarized in Table 1. The table includes the main reaction products found: n-hexane, methylcyclopentane (MCP), n-pentane, 2,5-DMTHF, 1-hexanol and some hexitols (sorbitol, mannitol and their anhydrides like sorbitan and isosorbide).
| Entry | [TSA] [mM] | T [K] | t [min] | Yieldd [%] | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n-decane | Water | Sum | ||||||||||||
| n-hex | MCP | n-pent | DMTHF | 1-hexol | Other | Sum | Hexitols | Other | Sum | |||||
| a Reaction conditions: Avicel PH-101 cellulose (2 g), TSA hydrate, htTSA(135)Ru/C (containing approx. 0.5 g of Ru/C), water (20 ml), n-decane (20 ml), initial H2 pressure at RT = 5 MPa. b Final temperature, reached by heating at an average rate of 12 K min−1 from RT to 423 K, and at a fixed rate of 0.5 K min−1 from 423 K onward (except for entry 1; reached by heating at an average rate of 17 K min−1 from RT to 423 K and 5.5 K min−1 from 423 K onward). c Reaction time at final temperature. d Yield (%) = (moles C in product/moles C in cellulose) × 100, products: n-hex = n-hexane; MCP = mainly methylcyclopentane and some cyclohexane; n-pent = n-pentane; DMTHF = 2,5-dimethyltetrahydrofuran; 1-hexol = 1-hexanol; hexitols = sorbitol, mannitol and their anhydrides (i.e. isosorbide). Other = sum of yields of other decane-soluble products like n-butane, 2-methyltetrahydrofuran and 2-methyltetrahydro-2H-pyran) and water-soluble products (like HMF, levulinic acid or 1,2-hexanediol). | ||||||||||||||
| 1 | 135 | 483 | 60 | 22.4 | 8.7 | 0.8 | 1.4 | 2.7 | 6.3 | 42.2 | 5.7 | 11.5 | 17.1 | 59.4 |
| 2 | 135 | 483 | 60 | 34.0 | 9.6 | 2.1 | 1.8 | 4.8 | 7.5 | 59.9 | 8.2 | 9.8 | 18.0 | 77.8 |
| 3 | 71 | 483 | 60 | 31.3 | 7.3 | 1.9 | 8.5 | 6.8 | 7.9 | 63.7 | 7.4 | 11.2 | 18.6 | 82.3 |
| 4 | 37 | 483 | 60 | 24.6 | 5.9 | 2.4 | 16.4 | 7.2 | 7.8 | 64.3 | 6.5 | 13.2 | 19.7 | 84.0 |
| 5 | 15 | 483 | 60 | 13.8 | 4.2 | 1.9 | 22.7 | 8.1 | 8.6 | 58.4 | 6.2 | 25.0 | 31.2 | 89.6 |
| 6 | 0 | 483 | 60 | 0.2 | 0.0 | 0.1 | 0.1 | 0.0 | 0.4 | 0.7 | 4.5 | 13.0 | 17.4 | 18.2 |
| 7 | 71 | 493 | 40 | 37.1 | 7.7 | 2.9 | 4.0 | 5.1 | 8.6 | 65.3 | 6.8 | 7.8 | 14.6 | 80.0 |
| 8 | 71 | 503 | 20 | 36.4 | 7.6 | 2.8 | 2.6 | 4.2 | 8.0 | 61.6 | 5.3 | 8.6 | 13.9 | 75.5 |
A first experiment, in presence of 135 mM of soluble TSA and htTSA(135)Ru/C (Table 1, entry 1), showed appreciable yields of n-decane-soluble products (42%, including 22% n-hexane yield), while only 6% hexitol yield was obtained in one hour at 483 K, reached by rapidly heating the reactor (see conditions in Table 1). This first result validates the concept of directly converting cellulose to liquid alkanes in the biphasic liquid conditions using hydrothermally TSA-treated Ru/C. The 41% carbon deficit indicates significant losses in form of gaseous and insoluble polymeric products. To minimize these side reactions, a slower heating rate of 0.5 K min−1 instead of 5.5 K min−1 was applied from 423 K onward (Table 1, entry 2). This stepwise heating protocol resulted in a notable yield increase of n-decane-soluble products to 60%, including 34% n-hexane and a 78% yield of identified liquid phase products. Insignificant amounts of gaseous products were detected in this experiment (mainly methane, see ESI†).
TSA in the aqueous phase plays a key role in the conversion of cellulose to liquid alkanes. As expected, low yields of liquid phase products are observed in absence of soluble TSA (Table 1, entry 6), since the acid is responsible for cellulose hydrolysis. An increase of TSA concentration from 15 to 135 mM considerably enhances the total liquid alkane yield, mainly at the expense of oxygenates like DMTHF and 1-hexanol (Table 1, entries 2–5). This yield increase is in line with the strong dehydration property of TSA, required to efficiently carry out series of bifunctional HDO reactions. Reactions with only TSA and no hydrogenation catalyst should obviously be avoided, as it leads to pronounced char formation.
Interestingly, reactions at higher temperatures require less acid (Table 1, compare entries 3, 7 and 8).83 For instance, by increasing the reaction temperature to 493 K, 71 mM TSA is sufficient to completely convert cellulose to 65% n-decane-soluble products of which more than half is n-hexane. The total product yield from both liquid phases accounts for 80% of the carbon balance. Based on the amount of TSA in the aqueous phase, and assuming cellulose hydrolysis, various acid-catalyzed rearrangements and dehydration steps to break C–O bonds en route to n-hexane, a catalytic turnover of about 12 can be estimated for each proton, showing a catalytic contribution of TSA.
The effect of modifying Ru/C with TSA, as predicted in the rationale of this contribution, is apparent from the experiments in Table 2. Unmodified Ru/C (Entry 8) led to significantly less alkane formation, while the hexitol yield considerably increased (from about 9 to 29%). Main compound in the hexitol fraction is isosorbide (with 18%), followed by sorbitan, isoidide and isomannide. This difference in product distribution is in line with the well-known glucose hydrogenation ability of commercial Ru/C. Hydrothermal treatment in absence of TSA (htRu/C, entry 7) partly decreases the hexitol fraction, but this decrease is not as efficient as with the TSA-modified Ru/C catalysts. Entries 1 to 6 illustrate the catalytic results with different htTSA(x)Ru/C catalysts, where x represents the TSA concentration during hydrothermal pretreatment (ranging from 2 to 135 mM). The highest carbon efficiency and liquid alkane yield, viz. 90% and 60% respectively, were obtained with the lowest TSA modification (entry 6).
| Entry | htTSA(x)Ru/Cb | Yieldc [%] | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n-decane | Water | Sum | ||||||||||
| n-hex | MCP | n-pent | DMTHF | 1-hexol | Other | Sum | Hexitols | Other | Sum | |||
|
a Reaction conditions as in Table 1, entry 7 (71 mM of TSA, slow heating, 40 min at 493 K).
b Catalyst notation depending on pretreatment conditions: ht, hydrothermal; x mM aqueous TSA solution during pretreatment.
c Yield (%) = (moles C in product/moles C in cellulose) × 100.
d Hexitol distribution (%): isosorbide:isomannide:sorbitan of 6.7:0.7:1.0.
e Hexitol distribution (%): isoidide:isosorbide:isomannide:sorbitan of 3.6:17.9:2.4:4.6.
|
||||||||||||
| 1 | htTSA(135)Ru/C | 37.1 | 7.7 | 2.9 | 4.0 | 5.1 | 8.6 | 65.3 | 6.8 | 7.8 | 14.6 | 80.0 |
| 2 | htTSA(71)Ru/C | 35.8 | 6.9 | 3.2 | 4.9 | 5.1 | 7.2 | 63.1 | 9.7 | 7.2 | 16.9 | 79.8 |
| 3 | htTSA(37)Ru/C | 35.9 | 6.7 | 3.6 | 5.6 | 5.1 | 6.6 | 63.3 | 9.0 | 8.8 | 17.8 | 80.9 |
| 4 | htTSA(15)Ru/C | 38.7 | 6.0 | 5.0 | 6.2 | 4.2 | 6.5 | 66.8 | 8.0 | 9.4 | 17.4 | 84.2 |
| 5 | htTSA(8)Ru/C | 39.7 | 4.4 | 8.1 | 7.8 | 4.2 | 6.5 | 70.6 | 9.9 | 8.5 | 18.3 | 88.9 |
| 6 | htTSA(2)Ru/C | 41.6 | 4.4 | 12.2 | 7.5 | 1.5 | 6.0 | 73.1 | 8.5d | 9.3 | 17.8 | 90.3 |
| 7 | htRu/C | 30.5 | 3.5 | 19.3 | 4.1 | 0.0 | 10.5 | 68.0 | 17.9 | 3.2 | 21.1 | 89.1 |
| 8 | Ru/C | 19.0 | 1.4 | 13.1 | 6.2 | 0.1 | 8.1 | 47.9 | 28.5e | 7.5 | 36.1 | 83.9 |
Reaction network study
The previous data displayed a wealth of intermediates and end-products, with the liquid alkanes being the desired ones in this work. To gain more insight into the reaction network, a systematic catalytic study was carried out by feeding the major reaction intermediates into the reactor under identical conditions. The data are collected in Table 3.| Entry | Feedstock | Modeb | Yieldc [%] | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n-decane | Water | Sum | |||||||||||
| n-hex | MCP/CH | n-pent | DMTHF | Hexold | Other | Sum | Hexitols | Other | Sum | ||||
| a Reaction conditions as in Table 2, entry 6 (71 mM of TSA, slow heating, 40 min at 493 K), added carbon amount of the substrate approximates the carbon amount in 2 g of Avicel PH-101 cellulose). b Reaction mode: batch or fed-batch. In fed-batch mode, 20 ml of the aqueous reaction mixture containing feedstock and TSA was fed into the reactor containing the htTSA(2)Ru/C catalyst in 20 ml n-decane between 423 K and 493 K (including the 40 min isothermal stage). c Yield (%) = (moles C in product/moles C in feedstock) × 100. d Hexol: 1- or 2-hexanol, as indicated between parentheses. e No TSA was added to the aqueous solution. f 5 MPa H2 pressure was introduced at 453 K. | |||||||||||||
| 1 | Sorbitol | Fed-batch | 4.3 | 1.0 | 4.9 | 0.6 | 0.1 (1) | 3.3 | 14.1 | 78.6 | 0.0 | 78.6 | 92.7 |
| 2e | <0.1 | 0.0 | 0.2 | 0.2 | 0.0 | 2.6 | 3.1 | 83.8 | 12.3 | 96.1 | 99.2 | ||
| 3 | Batch | 11.7 | 2.7 | 9.7 | 0.7 | 0.3 (1) | 5.0 | 30.1 | 59.9 | 4.4 | 64.3 | 94.4 | |
| 4 | Isosorbide | Batch | 8.7 | 1.0 | 5.8 | 0.5 | 0.2 (1) | 3.2 | 19.3 | 70.0 | 2.7 | 72.7 | 92.0 |
| 5 | Glucose | Fed-batch | 25.9 | 5.6 | 12.6 | 12.5 | 0.8 (1) | 8.8 | 65.1 | 0.0 | 8.8 | 8.8 | 73.9 |
| 6 | Batch | 44.4 | 7.9 | 16.5 | 3.6 | 0.1 (1) | 10.6 | 83.2 | 3.6 | 4.0 | 7.7 | 90.9 | |
| 7 | Fructose | Fed-batch | 23.9 | 3.7 | 12.9 | 12.4 | 0.2 (1) | 9.3 | 62.4 | 0.0 | 8.5 | 8.5 | 70.9 |
| 8 | Batch | 35.2 | 2.9 | 7.3 | 4.5 | 1.3 (1) | 10.7 | 62.0 | 2.8 | 6.5 | 9.2 | 71.2 | |
| 9 | HMF | Fed-batch | 16.7 | 2.0 | 3.9 | 22.7 | 2.7 (1) | 5.8 | 53.8 | 0.0 | 11.8 | 11.8 | 65.7 |
| 10 | Batch | 8.2 | 2.2 | 0.2 | 4.0 | 0.0 | 5.6 | 20.1 | 0.0 | 21.1 | 21.1 | 41.1 | |
| 11 | DMTHF | Batch | 82.9 | 0.0 | 0.4 | 7.8 | 0.0 | 1.2 | 92.4 | 0.0 | 2.7 | 2.7 | 95.0 |
| 12e | Batch | 7.2 | 0.0 | 0.1 | 62.7 | 9.2 (2) | 0.5 | 79.7 | 0.0 | 8.5 | 8.5 | 88.2 | |
| 13 | 2,5-Hexanediol | Batch | 74.1 | 0.0 | 0.5 | 6.8 | 0.2 (2) | 1.7 | 83.2 | 0.0 | 3.7 | 3.7 | 87.0 |
| 14 | 2,5-Hexanedione | Batch | 90.5 | 0.0 | 0.7 | 8.4 | 0.1 (2) | 1.9 | >100 | 0.0 | 3.2 | 3.2 | >100 |
| 15f | Batch | 69.2 | 1.4 | 0.4 | 16.4 | 0.2 (2) | 5.8 | 92.2 | 0.0 | 9.6 | 9.6 | >100 | |
| 16 | 1,2-Hexanediol | Batch | 39.7 | 0.0 | 52.2 | 0.0 | 0.1 (1) | 2.1 | 94.1 | 0.0 | 2.3 | 2.3 | 96.3 |
| 17 | 2-Hexanol | Batch | 93.9 | 0.0 | 0.1 | 0.0 | 0.0 | 0.9 | 94.9 | 0.0 | 1.2 | 1.2 | 96.1 |
| 18 | 1-Hexanol | Batch | 36.9 | 0.0 | 49.0 | 0.0 | 0.2 (1) | 0.8 | 87.0 | 0.0 | — | — | 87.0 |
In contrast to previously reported pathways to alkanes,32,60,84,85 sorbitol and isosorbide turned out to be fairly unreactive (see Table 3, entries 1–4): only 30% of sorbitol carbon (or 14% in fed-batch mode) and 19% of isosorbide carbon were converted into n-decane-soluble products. Sorbitol was mostly dehydrated to isosorbide (here also referred to as ‘hexitol’). These observations confirm a kinetically less favorable route from cellulose to alkanes via sorbitol. In line with our hypothesis, it predicts that hydrogenation of glucose should be slow compared to its dehydration to HMF in order to avoid yield loss to hexitols and their anhydrides. Interestingly, modification of Ru/C with TSA fulfills this particular role.
Selected key intermediates, which were analyzed in the previous experiments and are likely involved in the alternative HMF route, are glucose, fructose, HMF, 2,5-DMTHF, 2,5-hexanedione, 2,5-hexanediol, 1,2-hexanediol, 2-hexanol and 1-hexanol. Scheme 3 collects these chemicals in a tentative reaction network. Reactions with these molecules are presented in Table 3 (entries 5–18). Before giving a detailed description of the data and a network analysis, it can already be concluded from the data that high yields of n-decane soluble products were attained from all the above mentioned molecules, validating the proposed n-hexane pathway via HMF. Conversion of glucose and fructose with htTSA(2)Ru/C in presence of 71 mM TSA yields an insignificant amount of hexitols (in line with Fig. 1), while the alkane yield is highest for glucose (Table 3, entries 5–8). The formation of a significant amount of n-pentane is apparent. A fed-batch approach was used next to the batch reactions to imitate the gradual release of glucose from cellulose, as was done successfully in the direct conversion of glucose to ethylene glycol.86,87 The product distribution indeed changed with reactor type, the batch reactor systematically leading to higher alkane yields, in agreement with the preferred high contact time to form the alkane end-products. Indeed, test reactions with n-hexane and n-decane showed negligible conversion (data not shown), while longer reaction times provides higher liquid alkane yields, as will be demonstrated below.
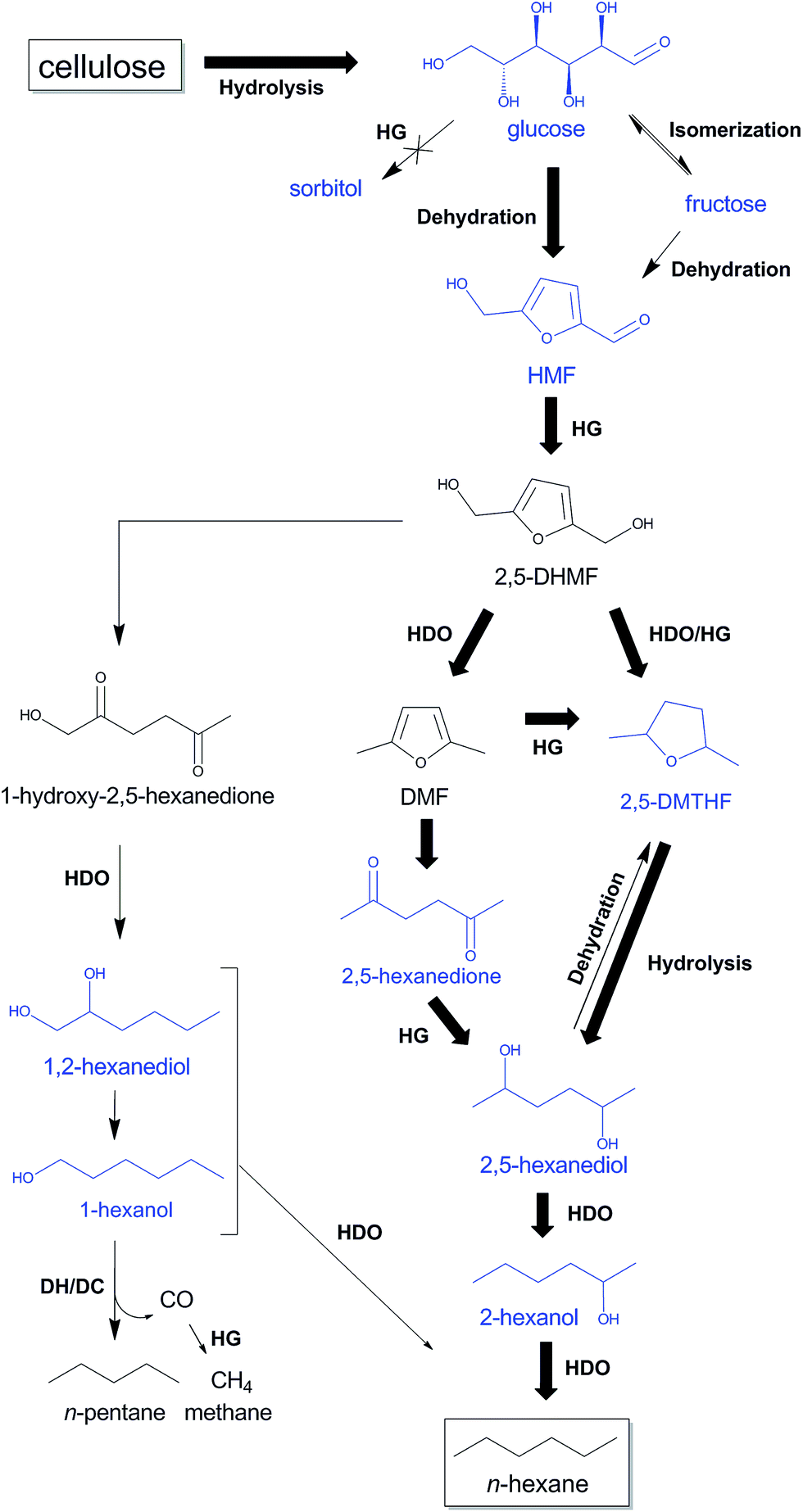 | ||
| Scheme 3 Proposed reaction pathways from cellulose to n-hexane and n-pentane through HMF with TSA and htTSA(2)Ru/C, partially based on Liu et al.,88 Alamillo et al.,67 Li et al.60 and Yang et al.92 Intermediates tested in this study are indicated in blue. The most selective reaction pathway from cellulose to n-hexane is indicated with bold arrows. HDO, hydrodeoxygenation; HG, hydrogenation; DH/DC, dehydrogenation/decarbonylation. | ||
Study of the HMF conversion, although the key molecule in the new n-hexane pathway, was somewhat problematic in batch mode due to its high reactivity (Table 3, entry 10). A low content of n-decane soluble products was observed and large quantities of polymers (char) and degradation products like levulinic acid were noticed. To obtain high liquid alkane yield from cellulose, gradual formation of HMF and subsequently fast HMF hydrogenation is thus important. Fed batch conversion of HMF is more efficient, yielding 22% alkanes and 23% 2,5-DMTHF (Table 3, entry 9), which will be further converted into alkanes upon longer reaction time. Indeed, reaction with 2,5-DMTHF shows an almost quantitative conversion to n-hexane, in agreement with our proposed HMF pathway (Table 3, entry 11). Presence of acidity in the aqueous phase is essential for the latter reaction. A similar reaction without TSA in the aqueous phase, results into a low 2,5-DMTHF conversion and showed additional formation of 2-hexanol (Table 3, entry 12). The acid-catalyzed ether bond hydrolysis of 2,5-DMTHF, likely first to 2,5-hexanediol, followed by a dehydration/hydrogenation to 2-hexanol, is thus an essential step en route to n-hexane (Table 3, entry 13 and Scheme 3). A suitable amount of acidity in the aqueous phase is thus crucial not only to hydrolyze cellulose to glucose and to dehydrate glucose to HMF, but also to achieve fast ring-opening hydrolysis of 2,5-DMTHF to 2,5-hexanediol. 2,5-Hexanediol is indeed very selectively converted to n-hexane, as demonstrated in entry 13 of Table 3.
2,5-Hexanedione was also occasionally analyzed in the cellulose experiments, especially at low contact time (see later). As this dione is reported to result from an acid-catalyzed ring opening hydrolysis/HDO of furans like MHMF (2-methyl-5-hydroxymethylfuran) and DMF (2,5-dimethylfuran),88,89 such reaction happens when ring hydrogenation is slower than hydrolysis. Interestingly, the occurrence of the reaction imposes no decrease of the n-hexane selectivity as 2,5-hexanedione is almost quantitatively converted into n-hexane in reaction conditions (Table 3, see entry 14). One may conclude at this point that 2,5-hexanediol, either resulting from 2,5-DMTHF or 2,5-hexanedione, is a crucial intermediate towards n-hexane formation.
A similar ring opening hydrolysis, followed by dehydration/hydrogenation, occurs with 2,5-DHMF, mainly forming 1-hydroxy-2,5-hexanedione.67,88,89 Accordingly, this intermediate is prone to convert ultimately to 1- and 2-hexanol through a family of diols such as 1,2-hexanediol (see Scheme 3) in our harsher reaction conditions. To better understand the reactivity and reaction pathways of the primary alcohol, a series of catalytic experiments was carried out with 1- and 2-hexanol and 1,2-hexanediol. The data are reported in Table 3, entries 16 to 18. Whereas 2-hexanol nearly quantitatively converts to n-hexane, 1,2-hexanediol and 1-hexanol yield remarkably lower n-hexane amounts (40% and 37%, respectively). Surprising amounts of n-pentane (53% and 49%) were obtained instead. The n-pentane is thus formed via C–C splitting of a primary alcohol under the reaction conditions, likely proceeding through a sequential dehydrogenation/decarbonylation reaction mechanism on the modified Ru/C.60 This reaction should form CO as by-product, which was indeed analyzed (as methane) in gas phase analysis in equimolar amounts with n-pentane (Fig. S12–S13 in ESI†). Since formation of 1-hexanol entities entails a significant loss of carbon yield in the liquid alkane fraction, ring opening of 2,5-DHMF should be delayed in favor of C–O hydrogenolysis and ring-hydrogenation. Besides n-pentane and n-hexane, the liquid alkane fraction also contains significant amounts of methylcyclopentane (MCP). The presumable formation route proceeds through the acid-catalysed Piancatelli rearrangement from 2,5-DHMF or MHMF,90 but this suggestion needs further confirmation. The reaction has been reported in the formation of cyclopentanone from furfural in presence of NiCu based catalysts under reducing conditions in water.91
Optimizing the cellulose to n-hexane reaction
Knowledge of the reaction network indicates that a minimum amount of redox catalyst htTSA(2)Ru/C is necessary to achieve efficient conversion of cellulose to n-hexane (Table 4, entry 1–3). Otherwise, char formation from HMF and water solubles like diols and triols will form, decreasing the content of n-decane solubles. Interestingly, reducing the original amount of catalyst twofold did not result in a significant change in total product yield, indicating that the hydrogenation activity in the biphasic system is still sufficient. A fourfold reduction of htTSA(2)Ru/C causes a drop in total carbon yield in the organic phase.| Entry | [TSA] [mM] | Ru/Cb [g] | Waterc (vol%) | Yieldd [%] | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n-decane | Water | Sum | ||||||||||||
| n-hex | MCP/CH | n-pent | DMTHF | 1-hexol | Other | Sum | Hexitols | Other | Sum | |||||
| a Reaction conditions as in Table 2 entry 6 (71 mM of TSA, slow heating, 40 min at 493 K) except for different water/n-decane ratios and catalyst amount. b Amount of Ru/C added in htTSA(2)Ru/C form. c Volume percentage of water, total volume = 40 ml. d Yield (%) = (moles C in product/moles C in cellulose) × 100. e Recycling: run 1 (2 g cellulose). f Run 2 (total of 4 g cellulose). g Run 3 (total of 6 g cellulose). | ||||||||||||||
| 1 | 71 | 0.48 | 50 | 41.6 | 4.4 | 12.2 | 7.5 | 1.5 | 6.0 | 73.1 | 8.5 | 9.3 | 17.8 | 90.3 |
| 2 | 71 | 0.24 | 50 | 40.7 | 8.0 | 3.4 | 6.7 | 6.7 | 8.3 | 73.8 | 7.1 | 8.8 | 16.0 | 89.8 |
| 3 | 71 | 0.12 | 50 | 26.3 | 10.8 | 1.1 | 4.2 | 4.6 | 12.5 | 59.5 | 1.8 | 14.5 | 16.3 | 75.8 |
| 4e | 49 | 0.24 | 75 | 42.4 | 6.9 | 5.6 | 2.5 | 3.3 | 7.1 | 67.8 | 6.3 | — | — | — |
| 5f | 49 | 0.24 | 75 | 36.7 | 7.2 | 3.5 | 3.0 | 4.8 | 8.7 | 63.9 | 3.1 | — | — | — |
| 6g | 49 | 0.24 | 75 | 29.2 | 7.0 | 2.6 | 4.3 | 5.9 | 9.3 | 58.3 | 1.8 | 12.8 | 14.7 | 72.9 |
In previous experiments the reaction mixture was analysed at a fixed reaction time, showing in some occasions significant amounts of intermediates like 1-hexanol and 2,5-DMTHF in the sampling mixture (Table 4, entry 4). As these molecules ultimately lead to n-hexane and n-pentane according to the results of Table 3, prolongation of the contact time is an obvious option to further increase the liquid alkane yields (see for instance an HPLC analysis of aqueous phase at various reaction times, Fig. S9†). Fig. 3 plots the product distribution and total cellulose conversion in function of time (with an indication of the reaction temperature at each time). The plotted data indeed confirm the increase in liquid alkane yield from cellulose with time. During the reactor heating stage between 423 and 453 K, the conversion of cellulose proceeds rapidly, reaching 40% and 90% at 30 and 60 minutes, respectively. During this interval, 2,5-DMTHF, glucose, some hexitols and their anhydrides and other water-solubles including HMF, 2,5-hexanedione and 1,2-hexanediol were formed (Fig. S9†). The accumulation of glucose and low hexitol (sorbitol, sorbitan and isosorbide) yield are indicative of the reduced glucose hydrogenation activity of the TSA-modified Ru/C, in agreement with the data presented in Fig. 1. The inability of this catalyst to rapidly hydrogenate glucose opens up a fast cascade route, involving glucose to HMF conversion, which is mainly hydrogenated to 2,5-DMTHF and some DMF was analyzed as well. The water phase contains a family of alcohols, mainly the most stable primary alcohols like 1-hexanol and 1,2-hexanediol, and also the secondary alcohols like 2-hexanol. After 1.5 h contact time, when the reactor temperature is in the range of 453–473 K, n-hexane is formed in expense of the reactive secondary alcohols through dehydration/hydrogenation cycles,60 while the primary alcohols remain largely untouched. 2,5-DMTHF and 1-hexanol are abundantly present, while also MCP is mainly formed in this period. Conversion of 2,5-DMTHF and the alcohols continues with longer reaction times (and increasing reaction temperature up to 493 K) until they are almost completely converted. At this temperature, n-pentane is formed, while 1-hexanol is completely converted. Thus, after a short reaction time of about 6 hours, high n-decane soluble product yields (about 82% based on carbon) and C5–C6 alkane yields (up to 75%, including 52% n-hexane) were obtained. In these conditions, the catalytic turnover based on surface Ru atoms can be estimated at about 200, assuming the consumption of 7H2 molecules for n-hexane production per glucose unit (and thus 7-metal catalyzed turnovers). The deficiency in the mass balance is due to some insoluble products (gas and solid, about 7% total estimated yield), while 11% carbon is present in the water phase as hexitols and some oligomeric products. Note that the hexitols were already formed very early in the reaction (after 30 min), but largely survived the reaction conditions, again proving the importance of the novel HMF route and differentiating the current biphasic system with the known pathways via sorbitol.
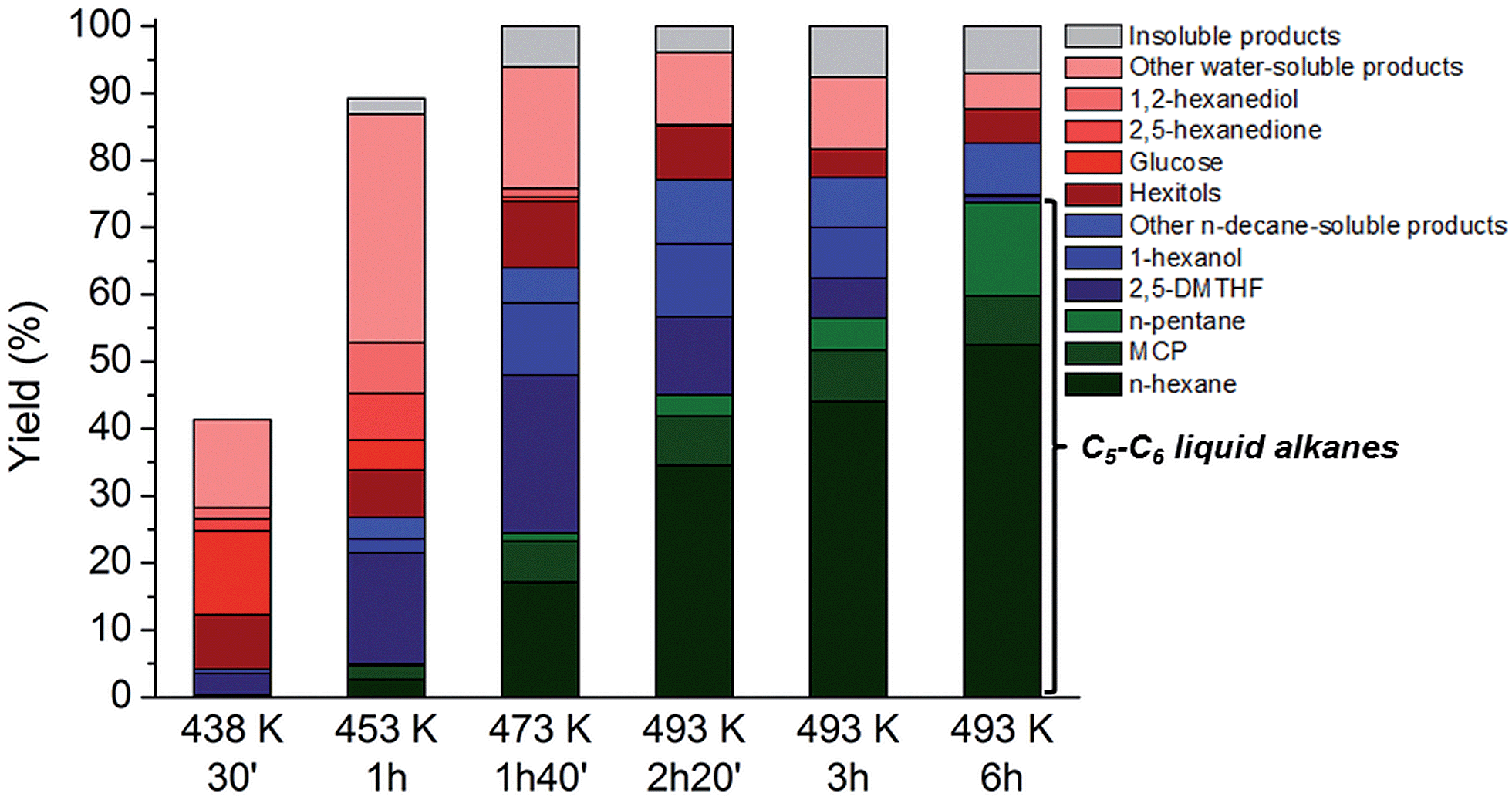 | ||
| Fig. 3 Conversion of microcrystalline cellulose to liquid alkanes with TSA and htTSA(2)Ru/C in function of time and temperature. Reaction conditions: Table 4, entry 4. Yield insoluble products (%) = cellulose conversion (%) − total yield dissolved products (%). | ||
Process robustness: converting real wood feedstock and catalyst and reuse
The direct conversion of softwood sawdust (from a local sawmill) to n-hexane was investigated in the aforementioned optimal reaction condition to assess the robustness of the catalytic system. Softwood was deliberately chosen here due to its high polyhexose content93 (here: 58%). Apart from cutting, no other pretreatment of the wood sample was foreseen as to omit biomass pretreatment costs and energy. Irrespective of that, an appreciable C5–C6 alkane yield of almost 60%, including approximately 40% n-hexane, was attained at full conversion of the polysaccharide component.Besides the use of real lignocellulosic feedstock, multiple catalyst reuse is of vital importance to a heterogeneous process and thus two types of reuse strategies were put forward to test the resilience and durability of the TSA-modified catalyst. At first, the catalyst was recovered from the reaction medium by filtration, washed and re-suspended in a fresh reaction medium after drying. The results of two such consecutive recycling runs are summarized in Fig. 4. Some loss of catalytic activity was noticed, which could originate from catalyst loss during filtration. The possibility of reusing both the heterogeneous htTSA(2)Ru/C catalyst and the soluble TSA co-catalyst in two successive runs was also investigated, by adding fresh cellulose to the batch reactor after each run and starting a new reaction, while accumulating the products. The results of this recycling are presented in Table 4 in entries 4 to 6, with yields based on the total amount of cellulosic carbon added. The catalytic system is acceptably reusable, not withstanding the harsh reaction conditions and high concentrations of products potentially inhibiting active metal sites. A small decrease in alkane and hexitol yield and an increase in oxygenate yield was monitored.
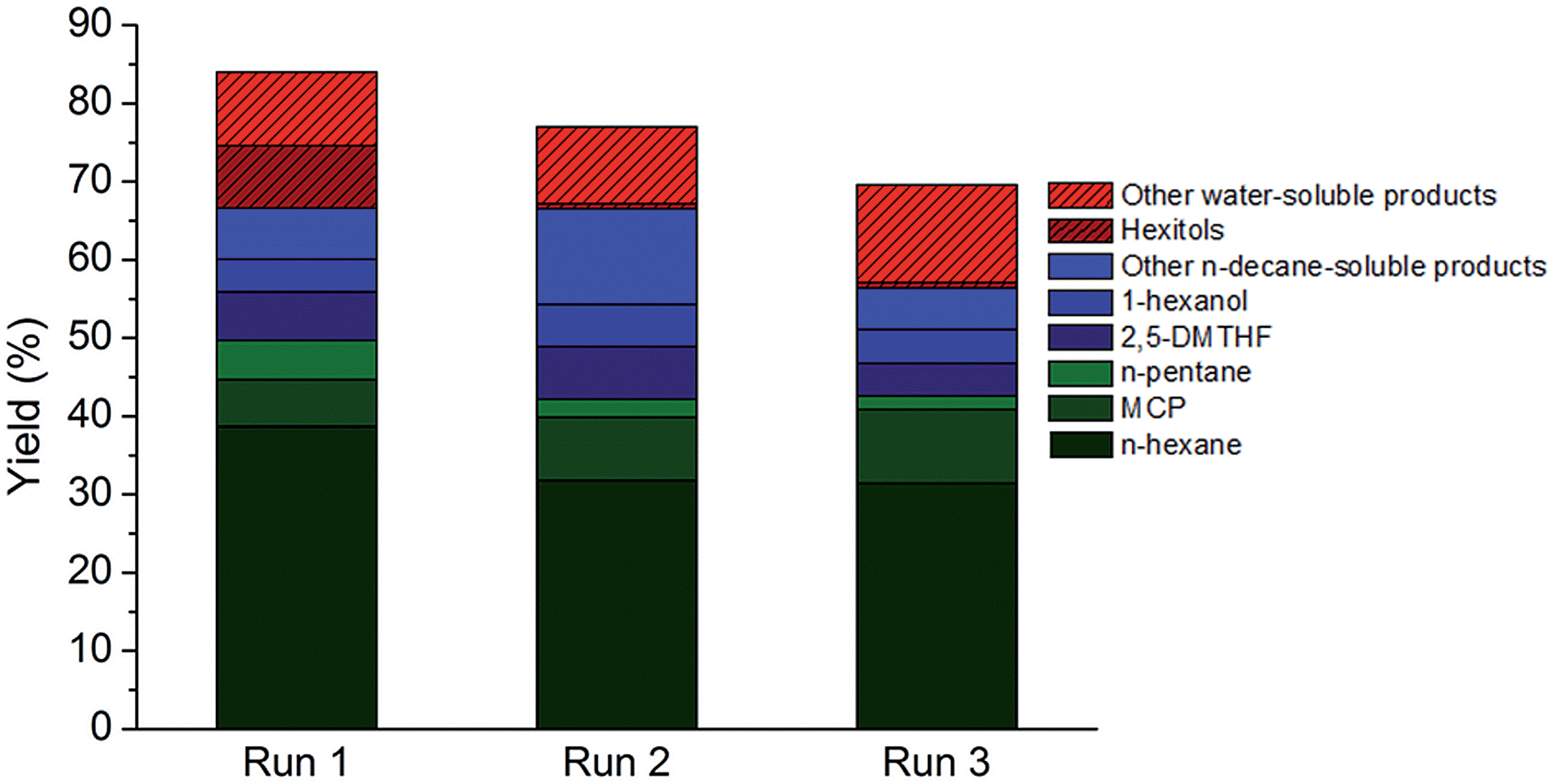 | ||
| Fig. 4 Reuse of the htTSA(15)Ru/C catalyst in 3 consecutive runs in the reaction conditions of Table 2, entry 4 (non-optimal conditions). | ||
Conclusions
This contribution has demonstrated the feasibility of a one-pot conversion of cellulose to alkanes. In contrast to recently reported hydroprocessing processes, this biphasic liquid approach at moderate temperatures mainly produces straight-chain alkanes with n-hexane and n-pentane as major components. The process allows an easy recuperation of alkanes, floating on top of a separate water phase, while hydrogen selectivity is high as almost no gaseous products are formed. A thorough reaction network study showed the dominant pathway, which deviates from the currently accepted sorbitol-to-alkane route.36,56,60 Instead, the major pathway proceeded via hydrolysis of cellulose to glucose, followed by dehydration into HMF. The latter needs to be hydrogenated quickly and leads to 2,5-DHMF and subsequently, via ring hydrogenation/hydrogenolysis, into 2,5-DMTHF. This cyclic ether is selectively converted into n-hexane via consecutive ring-opening hydrolysis and dehydration/hydrogenation cycles. Contribution of CO hydrogenation/hydrogenolysis of HMF to the methyl-furans, DMF and MHMF, followed by furan ring opening constitutes a productive parallel pathway to n-hexane through 2,5-hexanedione. The ring-opening of 2,5-DHMF on the other hand leads to the formation of linear primary alcohols such as 1-hexanol, and this path leads to a mixture of n-hexane, n-pentane and methane. The fast hydrogenation of 2,5DHMF to 2,5-DMTHF or hydrogenolysis to DMF or MHMF is thus an essential step in the cellulose-to-n-hexane reaction. MCP is proposed to be the result of a rearrangement reaction of 2,5-DHMF, but requires further confirmation.
The critical elements of the presented catalytic system are: (i) the use of a biphasic reaction solvent system - with redox activity in the organic phase and acidity in the aqueous phase - to partition reactive intermediates and to provide the best conditions for the different reactions to occur; (ii) controlled reactor heating to gradually release glucose and to form HMF in the right temperature zone to avoid their degradation; and (iii) modification of the hydrogenation selectivity of commercial Ru/C to steer the reaction from glucose to HMF hydrogenation to avoid sorbitol formation.
The catalytic system proved appreciably reusable and was applicable on raw softwood sawdust (almost 40% n-hexane yield). Future improvement in n-hexane yield is envisioned through a more selective formation of 2,5-DMTHF (or HMMF and DMF) to circumvent the n-pentane production. Identification of the modifying role of TSA on Ru/C, optimization of the stability of the catalytic biphasic system and decreasing the carbon content in the water phase are several focus points for future research.
Acknowledgements
This work was carried out within the EU FP7 project BIOCORE that is supported by the European Commission through the Seventh Framework Program for Research and Technical development under contract no FP7-241566. B.O.d.B. acknowledges the European Commission for his doctoral fellowship. Mia Tielen is acknowledged for her technical assistance. M.D. acknowledges the Research Foundation – Flanders (FWO) for postdoctoral funding and the Belgian American Educational Foundation (BAEF) for his honorary fellowship. B.F.S acknowledges IAP financing (Belspo). The authors would like to kindly thank Jeroen Snelders and Christophe M. Courtin (Laboratory of Food Chemistry and Biochemistry & Leuven Food Science and Nutrition Research Centre (LFoRCe), KU Leuven, Kasteelpark Arenberg 20, 3001 Heverlee, Belgium) for their support in the determination of the composition of the softwood sample.Notes and references
- S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS.
- J. A. Geboers, S. Van de Vyver, R. Ooms, B. Op de Beeck, P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 714–726 Search PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- M. Rose and R. Palkovits, Macromol. Rapid Commun., 2011, 32, 1299–1311 CrossRef CAS PubMed.
- Q. Zhang and F. Jérôme, ChemSusChem, 2013, 6, 2042–2044 CrossRef CAS PubMed.
- R. Carrasquillo-Flores, M. Käldström, F. Schüth, J. A. Dumesic and R. Rinaldi, ACS Catal., 2013, 3, 993–997 CrossRef CAS.
- N. Meine, R. Rinaldi and F. Schüth, ChemSusChem, 2012, 5, 1449–1454 CrossRef CAS PubMed.
- G. Gliozzi, A. Innorta, A. Mancini, R. Bortolo, C. Perego, M. Ricci and F. Cavani, Appl. Catal., B, 2014, 145, 24–33 CrossRef CAS PubMed.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed.
- Y. Liu, C. Luo and H. Liu, Angew. Chem., Int. Ed., 2012, 51, 3249–3253 CrossRef CAS PubMed.
- H. Kobayashi and A. Fukuoka, Green Chem., 2013, 15, 1740–1763 RSC.
- M. Yabushita, H. Kobayashi and A. Fukuoka, Appl. Catal., B, 2014, 145, 1–9 CrossRef CAS PubMed.
- M. Dusselier, M. Mascal and B. F. Sels, Top. Curr. Chem., 2014, 353, 1–40 CrossRef.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- T. Wang, M. W. Nolte and B. H. Shanks, Green Chem., 2014, 16, 548–572 RSC.
- J. Zhang, X. Liu, M. Sun, X. Ma and Y. Han, ACS Catal., 2012, 2, 1698–1702 CrossRef CAS.
- M. Dusselier, P. Van Wouwe, S. De Smet, R. De Clercq, L. Verbelen, P. Van Puyvelde, F. E. Du Prez and B. F. Sels, ACS Catal., 2013, 3, 1786–1800 CrossRef CAS.
- M. Dusselier and B. F. Sels, Top. Curr. Chem., 2014, 353, 85–125 CrossRef.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 CAS.
- R. Weingarten, W. C. Conner and G. W. Huber, Energy Environ. Sci., 2012, 5, 7559–7574 CAS.
- S. Van de Vyver, J. Thomas, J. Geboers, S. Keyzer, M. Smet, W. Dehaen, P. A. Jacobs and B. F. Sels, Energy Environ. Sci., 2011, 4, 3601–3610 CAS.
- A. Corma, O. de la Torre and M. Renz, Energy Environ. Sci., 2012, 5, 6328–6344 CAS.
- A. Corma, O. de la Torre, M. Renz and N. Villandier, Angew. Chem., Int. Ed., 2011, 50, 2375–2378 CrossRef CAS PubMed.
- G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184–7201 CrossRef CAS PubMed.
- J. Han, S. M. Sen, D. M. Alonso, J. A. Dumesic and C. T. Maravelias, Green Chem., 2014, 16, 653–661 RSC.
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 CAS.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gärtner and J. A. Dumesic, Science, 2008, 322, 417–421 CrossRef CAS PubMed.
- G. W. Huber, J. Chheda, C. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed.
- G. W. Huber, R. D. Cortright and J. A. Dumesic, Angew. Chem., Int. Ed., 2004, 43, 1549–1551 CrossRef CAS PubMed.
- M. Mascal, S. Dutta and I. Gandarias, Angew. Chem., Int. Ed., 2014, 53, 1854–1857 CrossRef CAS PubMed.
- M. Mascal and E. B. Nikitin, Angew. Chem., Int. Ed., 2008, 47, 7924–7926 CrossRef CAS PubMed.
- J.-P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed., 2010, 49, 4479–4483 CrossRef CAS PubMed.
- K. Chen, M. Tamura, Z. Yuan, Y. Nakagawa and K. Tomishige, ChemSusChem, 2013, 6, 613–621 CrossRef CAS PubMed.
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef CAS PubMed.
- G. W. Huber and J. A. Dumesic, Catal. Today, 2006, 111, 119–132 CrossRef CAS PubMed.
- G. Li, N. Li, J. Yang, L. Li, A. Wang, X. Wang, Y. Cong and T. Zhang, Green Chem., 2014, 16, 594–599 RSC.
- G. Li, N. Li, S. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, Chem. Commun., 2013, 49, 5727–5729 RSC.
- J. Q. Bond, A. A. Upadhye, H. Olcay, G. A. Tompsett, J. Jae, R. Xing, D. M. Alonso, D. Wang, T. Zhang, R. Kumar, A. Foster, S. M. Sen, C. T. Maravelias, R. Malina, S. R. H. Barrett, R. Lobo, C. E. Wyman, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2014, 7, 1500–1523 CAS.
- V. K. Venkatakrishnan, J. C. Degenstein, A. D. Smeltz, W. N. Delgass, R. Agrawal and F. H. Ribeiro, Green Chem., 2014, 16, 792–802 RSC.
- R. Agrawal and N. R. Singh, AIChE J., 2009, 55, 1898–1905 CrossRef CAS.
- N. R. Singh, W. N. Delgass, F. H. Ribeiro and R. Agrawal, Environ. Sci. Technol., 2010, 44, 5298–5305 CrossRef CAS PubMed.
- T. L. Marker, L. G. Felix and M. B. Linck, US 2010/0251600 A1, 2010.
- R. Agrawal and N. R. Singh, US 8,217,210 B2, 2012.
- R. Agrawal and N. R. Singh, US 2009/0084666 A1, 2009.
- R. Agrawal, M. Agrawal and N. R. Singh, US 2009/0082604 A1, 2009.
- R. Agrawal, M. Agrawal and N. R. Singh, US 8,217,211 B2, 2012.
- B. Kamm, Angew. Chem., Int. Ed., 2007, 46, 5056–5058 CrossRef CAS PubMed.
- N. K. Jorgensen and K. H. Cohr, Scand. J. Work, Environ. Health, 1981, 7, 157–168 CAS.
- G. Martino, P. Courty and C. Marcilly, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger and J. Weitkamp, Wiley-VCH, Weinheim, Germany, 1997, vol. 4, ch. 3, pp. 1801–2122 Search PubMed.
- H. W. Kouwenhoven and W. C. Van Zijill Langhout, Chem. Eng. Prog., 1971, 67, 65–70 CAS.
- W. C. Buss and T. R. Hughes, US4435283 A, 1984.
- N. Rahimi and R. Karimzadeh, Appl. Catal., A, 2011, 398, 1–17 CrossRef CAS PubMed.
- S. Liu, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2014, 2, 1819–1827 CrossRef CAS.
- J. M. Robinson, US5516960 A, 1996.
- L. L. Adduci, M. P. McLaughlin, T. A. Bender, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2014, 53, 1646–1649 CrossRef CAS PubMed.
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS PubMed.
- N. Li and G. W. Huber, J. Catal., 2010, 270, 48–59 CrossRef CAS PubMed.
- R. M. West, M. H. Tucker, D. J. Braden and J. A. Dumesic, Catal. Commun., 2009, 10, 1743–1746 CrossRef CAS PubMed.
- Y. Nakagawa, M. Tamura and K. Tomishige, ACS Catal., 2013, 3, 2655–2668 CrossRef CAS.
- B. Op de Beeck, J. Geboers, S. Van de Vyver, J. Van Lishout, J. Snelders, W. J. J. Huijgen, C. M. Courtin, P. A. Jacobs and B. F. Sels, ChemSusChem, 2013, 6, 199–208 CrossRef CAS PubMed.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS PubMed.
- S. Crossley, J. Faria, M. Shen and D. E. Resasco, Science, 2010, 327, 68–72 CrossRef CAS PubMed.
- P. A. Zapata, J. Faria, M. P. Ruiz, R. E. Jentoft and D. E. Resasco, J. Am. Chem. Soc., 2012, 134, 8570–8578 CrossRef CAS PubMed.
- R. Alamillo, M. Tucker, M. Chia, Y. Pagan-Torres and J. Dumesic, Green Chem., 2012, 14, 1413–1419 RSC.
- Y. Yang, Z. Du, J. Ma, F. Lu, J. Zhang and J. Xu, ChemSusChem, 2014, 7, 1352–1356 CrossRef CAS PubMed.
- H. Schiweck, A. Bär, R. Vogel, E. Schwarz, M. Kunz, C. Dusautois, A. Clement, C. Lefranc, B. Lüssem, M. Moser and S. Peters, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, Weinheim, 2000 Search PubMed.
- V. Ponec, Appl. Catal., A, 1997, 149, 27–48 CrossRef CAS.
- L. R. Pizzio, C. V. Cáceres and M. N. Blanco, J. Colloid Interface Sci., 1997, 190, 318–326 CrossRef CAS.
- Y. Izumi, R. Hasebe and K. Urabe, J. Catal., 1983, 84, 402–409 CrossRef CAS.
- M. E. Chimienti, L. R. Pizzio, C. V. Cáceres and M. N. Blanco, Appl. Catal., A, 2001, 208, 7–19 CrossRef CAS.
- J.-P. Tessonnier, S. Goubert-Renaudin, S. Alia, Y. Yan and M. A. Barteau, Langmuir, 2012, 29, 393–402 CrossRef PubMed.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS PubMed.
- M. A. Schwegler, P. Vinke, M. van der Eijk and H. van Bekkum, Appl. Catal., A, 1992, 80, 41–57 CrossRef CAS.
- S.-J. Yang, X.-X. Du, L. He and J.-T. Sun, J. Zhejiang Univ., Sci., B, 2005, 6, 373 CrossRef PubMed.
- J. Geboers, S. Van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577–3579 RSC.
- R. Palkovits, K. Tajvidi, A. M. Ruppert and J. Procelewska, Chem. Commun., 2011, 47, 576–578 RSC.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Green Chem., 2011, 13, 2167–2174 RSC.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Chem. Commun., 2011, 47, 5590–5592 RSC.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS PubMed.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS PubMed.
- R. R. Davda, J. W. Shabaker, G. W. Huber, R. D. Cortright and J. A. Dumesic, Appl. Catal., B, 2005, 56, 171–186 CrossRef CAS PubMed.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- R. Ooms, M. Dusselier, J. A. Geboers, B. Op de Beeck, R. Verhaeven, E. Gobechiya, J. A. Martens, A. Redl and B. F. Sels, Green Chem., 2014, 16, 695–707 RSC.
- G. Zhao, M. Zheng, J. Zhang, A. Wang and T. Zhang, Ind. Eng. Chem. Res., 2013, 52, 9566–9572 CrossRef CAS.
- F. Liu, M. Audemar, K. De Oliveira Vigier, J.-M. Clacens, F. De Campo and F. Jérôme, ChemSusChem, 2014, 7, 2089–2093 CrossRef CAS PubMed.
- V. Schiavo, G. Descotes and J. Mentech, Bull. Soc. Chim. Fr., 1991, 128, 704–711 Search PubMed.
- G. Piancatelli, A. Scettri and S. Barbadoro, Tetrahedron Lett., 1976, 17, 3555–3558 CrossRef.
- Y. Yang, Z. Du, Y. Huang, F. Lu, F. Wang, J. Gao and J. Xu, Green Chem., 2013, 15, 1932–1940 RSC.
- W. Yang and A. Sen, ChemSusChem, 2010, 3, 597–603 CrossRef CAS PubMed.
- D. Fengel and G. Wegener, Wood: chemistry, ultrastructure, reactions, W. de Gruyter, Berlin, 1983 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ee01523a |
| This journal is © The Royal Society of Chemistry 2015 |