
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon nanohorn-based electrolyte for dye-sensitized solar cells†
Fabian
Lodermeyer
a,
Rubén D.
Costa
*a,
Rubén
Casillas
a,
Florian T. U.
Kohler
b,
Peter
Wasserscheid
b,
Maurizio
Prato
c and
Dirk M.
Guldi
*a
aDepartment of Physical Chemistry and Pharmacy, Interdisciplinary Center for Molecular Materials, University of Erlangen-Nürnberg, Egerlandstr. 3, 91052, Erlangen, Germany. E-mail: ruben.costa@fau.de; dirk.guldi@fau.de
bDepartment of Chemical and Bioengineering, Reaction Engineering, University of Erlangen-Nürnberg, Egerlandstr. 3, 91058, Erlangen, Germany
cCenter of Excellence for Nanostructured Materials, INSTM, Unit of Trieste, Dipartimento di Scienze Chimiche e Farmaceutiche, Università degli Studi di Trieste, Piazzale Europa 1, 34127 Trieste, Italy
First published on 18th September 2014
Abstract
For the first time, carbon nanohorns were implemented into solid-state electrolytes for highly efficient solid-state and quasi-solid-state DSSCs. They feature an effective catalytic behavior towards the reduction of I3− and enhance the I3− diffusivity in the electrolyte. In a final device, solar cells with 7.84% efficiency at room temperature were achieved. As a matter of fact, this is the highest reported efficiency for nanocarbon-based electrolytes up to date.
Broader contextThe continuously growing world energy demand and the depletion of fossil fuels necessitate alternative and ecofriendly energy resources. Thereby, costs and efficiencies play a crucial role. In this context, dye-sensitized solar cells (DSSC) are a viable alternative to conventional solid-state semiconductor devices, owing to their low-cost productions from earth abundant materials. So far, benchmark efficiencies were obtained using liquid electrolyte based organic solvents, which bear, however, the risk of volatilization and toxicity. In contrast, ionic liquids (IL) exhibit negligible vapor pressure, a wide electrochemical window, and non-toxicity. To date, the major bottleneck is the low conductivity of ILs, which impacts the overall efficiency of DSSCs, and, which have triggered investigations towards enhancing ionic conductivity. Among these investigations, work on nanocarbons stand out, since they are earth abundant, feature high conductivity, and exhibit catalytic activity towards the reduction of I3−. In this work, carbon nanohorns (CNHs) were implemented into IL-based electrolytes, since they feature better miscibility with organic solvents and ILs compared to graphene and carbon nanotubes. Consequently, solar cells with 7.84% efficiency at room temperature were achieved, when CNHs were implemented into the electrolyte. |
Dye-sensitized solar cells (DSSCs) are a promising alternative to convert solar energy, especially when compared to conventional silicon-based technologies. In this context, material costs and facile fabrication processes are valuable assets.1 Typically, in n-type DSSCs, one or several dyes are anchored to a mesoporous semiconducting electrode, for example, TiO2 or ZnO. Upon light excitation of the dye, the latter injects electrons into the conduction band of the semiconductor electrode followed by the reduction of the oxidized dyes by the electrolyte. Under such conditions, the injected electrons perform work through an external load before they reduce the electrolyte at the platinum counter-electrode.2,3 Several decades of continuous optimizations resulted in state-of-the-art efficiencies in the range from 10 to 13%.4,5
Under operation, the electrolyte plays a key role in terms of fully functioning DSSCs. To this date, the I−/I3− redox couple is the most commonly used redox pair. Nevertheless, it suffers severe limitations including inherent corrosive nature, distinct absorption features in the visible region, and a prominent charge recombination arising from I3− ion pair formation at the electrode surface.6 Furthermore, its use in the form of liquid-based electrolytes impacts the long-term stability owing to solvent evaporation and I3− induced corrosion at electrode contacts. All of the aforementioned has fueled research on iodine-free and solid-state dye-sensitized solar cells (ssDSSCs).7 Notably, despite all efforts, ssDSSCs feature efficiencies of around 8%,8 which are much inferior compared to liquid-based electrolytes.
In light of the aforementioned, novel concepts for efficient iodine-free and solid-state electrolytes are currently among the major challenges in the field of ssDSSC. To the best of our knowledge, there are several approaches to replace liquid-based electrolytes without compromising the solar cell performance. Firstly, the design of novel photoanodes to enhance the penetration of the electrolyte into the pores is considered.9 As a very recent example, Ahn et al. reported a novel honeycomb TiO2 nanostructure for ssDSSCs yielding an efficiency of 7.4%.10 Secondly, iodine-free alternatives for ssDSSCs such as hole transport materials11 and conductive polymers12 are under investigation. Starting with the first report by Bach et al. in 1998,13 continuous improvements led to efficiencies of around 6% in, for example, spiro-OMeTAD-based ssDSSCs.11 Thirdly, the replacement of conventional dyes by perovskites in combination with spiro-OMeTAD has recently boosted the efficiencies to more than 15%.14 Finally, the use of carbonaceous materials – carbon black,15,16 single- and multi-walled carbon nanotubes,17,18 and several graphene derivatives19 – as additives to iodine-free and solid-state electrolytes is emerging.7,20 Among them, the benefits of implementing nanocarbons stand out given (i) better ionic diffusion throughout the electrolyte, (ii) catalytic activity towards the reduction of I3− to I−, (iii) dye regeneration to reach yields that are almost unity, and (iv) reduction of the internal device resistance by quasi-zero distances between oxidized dyes and the counter-electrode.16,21 To this end, using multi-walled carbon nanotubes based electrolytes 6.86% efficiencies evolved after continuous optimization steps.22 But, integrating nanocarbons into DSSCs by means of solution-based techniques is still the major bottleneck of this approach due to nanocarbon bundling in solution and poor nanocarbon solubility.
In the current work, we present the implementation of carbon nanohorns (CNH) into solid-state electrolytes. CNHs are conical structures with diameters between 2 and 5 nm and a length from 30 to 50 nm. In solution, they tend to associate into roundish aggregates of around 100 nm (Fig. 1 and ESI† for further details).23 Notably, the interest in CNHs has recently been intensified given their metal-free synthesis, semiconductor character, and relatively easy suspendibility in organic solvents.24 CNHs have, however, never been tested inside DSSC electrolytes, despite recent reports on the benefits of CNH-based interlayers and CNH doping of TiO2-based photoanodes.24,25 In the present study, we have realized an iodine-free and highly-efficient ssDSSC based on a mixture of 1-ethyl-3-methylimidazolium iodide ([EMIM]I), several additives, and CNHs. After a series of optimization steps, our ssDSSCs feature 7.84% efficiency under room temperature conditions, which exceeds that seen in current state-of-the-art nanocarbon-based ssDSSCs.26
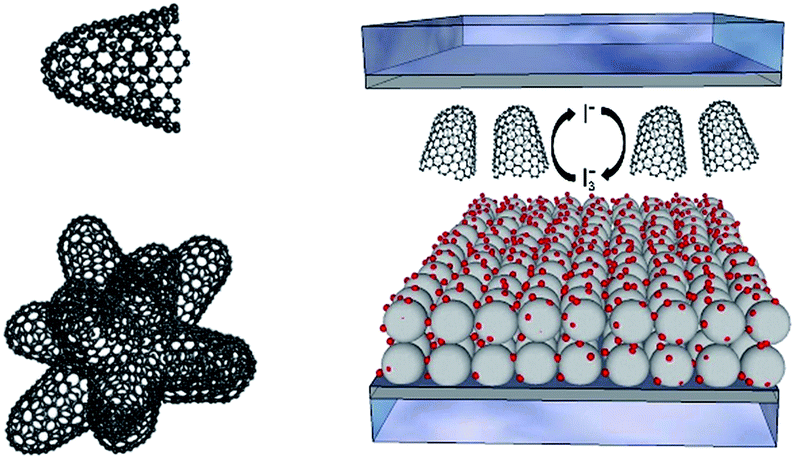 | ||
| Fig. 1 (Left) Representation of a CNH (top), and a bundle-like CNH aggregate present in solution (bottom). (Right) Schematic representation of a ssDSSC containing CNHs in the electrolyte. | ||
First, we elucidated the optimum [EMIM]I to CNH weight ratios for their application as solid-state electrolytes. In particular, different amounts of [EMIM]I were added to acetonitrile suspensions of CNHs (37 mg mL−1) and the solvent was slowly evaporated at 80 °C under vigorous stirring conditions until a viscous binary mixture was left – for more details see ESI.† [EMIM]I to CNH weight ratios of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 5:1, and 1:1 wt% were realized. Higher CNH amounts could not be achieved due to immiscibility. Electrolytes with 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM][BF4]) as crystal growth inhibitor and with 4-tert-butylpyridine (TBP) as well as guanidinium thiocyanate (GuSCN) as additives were also prepared by the same procedure – for more details see ESI.† [BMIM][BF4] was added to enhance the electrode coverage, whereas TBP and GuSCN assisted in preventing the back-electron-transfer processes and improving the dye regeneration, respectively.3
:1, 5:1, and 1:1 wt% were realized. Higher CNH amounts could not be achieved due to immiscibility. Electrolytes with 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM][BF4]) as crystal growth inhibitor and with 4-tert-butylpyridine (TBP) as well as guanidinium thiocyanate (GuSCN) as additives were also prepared by the same procedure – for more details see ESI.† [BMIM][BF4] was added to enhance the electrode coverage, whereas TBP and GuSCN assisted in preventing the back-electron-transfer processes and improving the dye regeneration, respectively.3
Differential scanning calorimetry (DSC) was employed to probe the effect of CNH addition on the transition temperatures, that is, from the solid to the liquid phase and vice versa, by heating up to 120 °C and subsequent cooling down to 0 °C (Fig. S4†). Upon heating, a similar solid-to-liquid transition at around 74 °C was noted for bare [EMIM]I and [EMIM]I/CNH mixtures. During cooling, [EMIM]I forms the solid phase at room temperature. In stark contrast, [EMIM]I/CNH mixtures show a liquid-to-solid phase transition, which onsets independently on the amount of CNHs at around 45 °C. A likely rationale implies that CNHs induce crystallization. As a consequence, the hysteresis temperature shift from the liquid to the solid phases is reduced from 54 to 29 °C for pure [EMIM]I and CNH containing mixtures. Addition of [BMIM][BF4] leads to featureless DSCs due to a melting point beyond 0 °C. Likewise, after addition of TBP and GuSCN the [EMIM]I/CNH mixtures are at room temperature best described as ionic liquids.
Information about the morphology of the [EMIM]I/CNH electrolytes came from scanning electron microscopy (SEM) measurements. Fig. S5† documents the impact of CNHs onto the features of the [EMIM]I matrix. For example, the homogenous [EMIM]I crystal arrangement is disrupted upon CNH addition. Now, [EMIM]I reveal grain boundaries, into which the CNHs are integrated. In the presence of [BMIM][BF4], TBP, or GuSCN, the low viscosity of the samples hampered a meaningful SEM analysis.
Next, the ionic diffusivity of the electrolytes was measured by means of linear sweep voltammetry (LSV). Here, the anodic and cathodic steady-state polarization diffusion current densities (JSS) of the electrolyte sandwiched between two fluorine-doped tin oxide (FTO) electrodes coated with platinum are monitored – i.e., FTO–Pt/electrolyte/Pt–FTO.17 Increasing the CNH content leads to higher JSS in [EMIM]I/CNH (Fig. S6†). The lowest JSS is, indeed, observed for pure [EMIM]I (E1). In contrast, the highest JSS evolves for the [EMIM]I/CNHs mixture with a 1:1 wt% ratio (E2). As a matter of fact, the same observation holds for electrolytes with [BMIM][BF4], TBP, and GuSCN, that is, [EMIM]I/[BMIM][BF4]/TBP/GuSCN with a weight ratio of 1:0.25:0.83:0.17 (E3) and with the optimized amount of CNHs [EMIM]I/CNHs/[BMIM][BF4]/TBP/GuSCN with weight ratios of 1:1:0.25:0.83:0.17 (E4). Please note that E2 and E4 feature almost the same JSS (Fig. S6†). From the latter we conclude that upon addition of CNHs to the electrolyte the ionic diffusivity increases. This is a crucial prerequisite for highly efficient dye regeneration under device operation conditions.
To corroborate our findings, the aforementioned FTO–Pt/electrolyte/Pt–FTO cell configuration was investigated by means of electrochemical impedance spectroscopy (EIS).27 This is meant to provide insights into the charge transfer resistance across the different interfaces. Fig. S7† depicts the Nyquist plots and the equivalent electrical circuits. Two semicircles are discernable each at a well distinct frequencies. Firstly, a 1000–10000 Hz semicircle in the Nyquist plot relates to the charge transfer resistance (RPt) and the corresponding Helmholtz capacitance (CPt) at the FTO–Pt/electrolyte interface. Secondly, the 0.1–100 Hz semicircle correlates with the resistance of the Warburg diffusion of the I−/I3− redox pair in the bulk electrolyte (Rdiff). With this data at hand, the effective diffusion coefficient (Ddiff) of the electrolyte can be calculated by using eqn (1):27
| Ddiff = (kBT)/(m2q2N[I3−]Rdiffδ) | (1) |
EIS analysis of the FTO–Pt/electrolyte/Pt–FTO cells confirms the two major assumptions made when implementing CNHs into the electrolyte, namely improving the catalytic reduction of I3− to I− and facilitating ionic diffusivity as noted from the LSV assays. The former is confirmed by lower RPt, which ultimately relates to the resistance to catalysis at the electrode interface. In particular, RPt gradually reduces from 190.47 to 44.84, 28.13, and to 7.38 Ω for [EMIM]I/CNH ratios of 1:0, 10:1, 5:1, and 1:1, respectively. Especially, the 26-fold reduction of RPt in E2 relative to E1 corroborates an improved catalytic behavior of CNHs towards the I3− reduction (Table S1 and Fig. S7†). Reassuring is the fact that the absence and the presence of CNHs in E3 and E4 reduces RPt from 167.33 to 41.21 Ω, respectively. Finally, we were also able to confirm the latter. As a matter of fact, CNHs in the electrolyte drastically results in increasing Ddiff, since Rdiff decreases from 2676.10 to 477.65, 205.63, and to 35.86 Ω for [EMIM]I/CNH ratios of 1:0, 10:1, 5:1, and 1:1, respectively (Table S1 and Fig. S7†) as well as from 781.04 to 31.39 Ω for E3 and E4, respectively. By using eqn (1), we confirm that Ddiff increases drastically from 7.55 × 10−12 to 0.42 × 10−10, 0.98 × 10−10, and to 5.63 × 10−10 m2 s−1 for [EMIM]I/CNH ratios of 1:0, 10:1, 5:1, and 1:1, respectively. Likewise, Ddiff increases from 0.26 × 10−10 m2 s−1 for E3 to 6.44 × 10−10 m2 s−1 for E4.
To prove the effects of the implementation of CNHs into the electrolyte on the device performance, we prepared TiO2-based ssDSSCs with cis-diisothiocyanato-bis(2,2′-bipyridyl-4,4′-dicarboxylato) ruthenium(II) bis(tetra-butylammonium) (N719) using electrolytes without CNHs (E1 and E3) and with CNHs (E2 and E4). Fig. 2 depicts the current–voltage (J–V) curves, while Table 1 summarizes the device performance features of ssDSSCs.
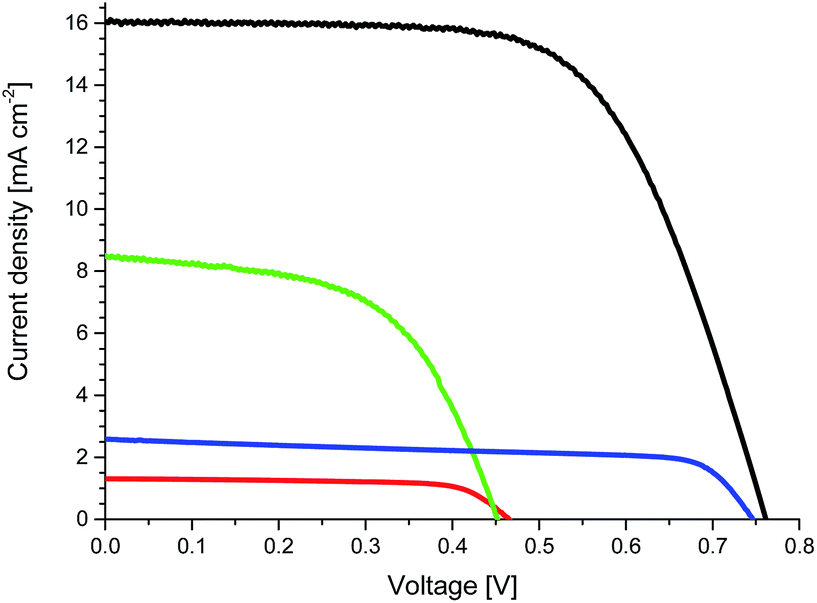 | ||
| Fig. 2 J–V curves of devices with E1 (red) and E2 (green) measured at 70 °C as well as devices with E3 (blue) and E4 (black) measured at room temperature. | ||
| Electrolyte | J SC [mA cm−2] | V OC [V] | FF | η [%] | R Pt [Ω] | R diff [Ω] | D diff [× 10−10 m2 s−1] |
|---|---|---|---|---|---|---|---|
| a Measured at 70 °C. b Measured at room temperature. c Measured between two Pt electrodes. d Measured with 10% error. | |||||||
| E1 | 1.27 | 0.47 | 0.71 | 0.42 | 190.47c | 1966.10 | 0.10 |
| E2 | 8.29 | 0.45 | 0.56 | 2.09 | 7.57 | 26.54 | 7.61 |
| E3 | 2.59 | 0.75 | 0.67 | 1.29 | 7.99 | 273.48 | 0.74 |
| E4 | 16.03 | 0.76 | 0.64 | 7.84 | 1.81 | 7.66 | 26.38 |
At room temperature, the initial efficiencies (η) of ssDSSCs with E1 and that with different CNH-amounts were all around 0.01% caused by low fill factors (FF) and low short-circuit current densities (JSC) (Fig. S8†). This fact is expected due to the solid-state nature of the electrolytes. Upon heating, the device performance starts to improve at temperatures close to the melting point of the electrolytes, plateauing at around 70 °C in good agreement with the DSC measurements. Here, the liquid phase enables, on one hand, a better electrode coverage and, on the other hand, a better ion diffusion throughout the electrolyte. Overall, higher FF and JSC evolve. Upon cooling to room temperature, the electrolytes remain in their liquid phase as seen in DSC measurements – vide supra – and impact the ssDSSC performance. Fig. S9† attests that at temperatures above the melting point, the efficiency is still at its maximum regardless of the number of heating–cooling cycles. Even more striking is the fact that after the first cycle, the high temperature maximum efficiency remains even at room temperature and only slowly decreases as a function of time (Fig. S10†).
Overall, upon increasing the CNH content inside [EMIM]I, the efficiency improves, owing to a boost in JSC (Table S2 and Fig. S8†). Among all of the electrolytes, devices with [EMIM]I/CNHs at a 1:1 wt% ratio (E2) show the best ssDSSC performance, in line with LSV and EIS measurements – vide supra. In details, E2 ssDSSCs feature open-circuit voltage (VOC) of 0.45 V, JSC of 8.29 mA cm−2, FF of 0.56, and η of 2.09% (Table 1 and Fig. 2). On the contrary, E1 ssDSSC show VOC of 0.47 V, JSC of 1.27 mA cm−2, FF of 0.71, and η of 0.42%. Mainly responsible for the 5-fold improvement in efficiency is a higher JSC with CNHs in the electrolyte.
Quite likely, the major bottleneck of the aforementioned electrolytes relates to their penetration into the mesoporous network. However, this aspect should not be applicable to E3 and E4 devices, in which the presence of additives such as [BMIM][BF4] transforms them into ionic liquids at room temperature – vide supra. To provide a reasonable comparison between the performances, devices with E1 and E2 were measured at 70 °C, whereas those with E3 and E4 were measured at room temperature. As expected from our previous results with E1 and E2 ssDSSCs, E3 ssDSSCs featured VOC of 0.74 V, JSC of 1.17 mA cm−2, FF 0.70, and η of 0.61%, whereas E4 ssDSSCs exhibited VOC of 0.76 V, JSC of 16.03 mA cm−2, FF of 0.64, and η of 7.84% (Table 1 and Fig. 2). Notably, both devices feature comparable VOC and FF, only JSC increases by a factor of nearly 6. This is also valid when comparing devices with E1 and E2 – Table 1. The increase in VOC of ssDSSCs with E3 and E4 compared with those with E1 and E2 is ascribed to the use of additives.28
Decisively, the latter was confirmed in EIS measurements with E1, E2, E3, and E4 devices (Table 1 and Fig. 3). Two findings should be highlighted. Firstly, a direct comparison between E3 and E4 devices demonstrates the catalytic behavior of CNHs in terms of reducing I3−, since RPt is lowered from 7.99 to 1.81 Ω for E3 and E4, respectively (Table 1 and Fig. 3). Due to the poor ionic diffusivity of E1, it was impossible to dissect RPt, since the interface between TiO2 and Pt is not distinguishable and, as a consequence, the frequencies in the Bode phase of both interfaces coalesce (Fig. S11†). Secondly, Rdiff drops from 1966.10 Ω for E1 to 26.54 Ω for E2 and similarly from 273.48 Ω for E3 to 7.66 Ω for E4. This prompts to an ion diffusivity, which increases upon CNH addition (Table 1). Here, E1 exhibits a Ddiff of 0.10 × 10−10 m2 s−1, while that of E2 increases to 7.61 × 10−10 m2 s−1 due to a lower charge transport resistance (Table 1). Among all of the electrolytes, E4 features the highest diffusion coefficient of 26.38 × 10−10 m2 s−1. It is safe to conclude at this point that the major impact of CNHs as an integrative part of the electrolytes is on the Warburg diffusion resistance and on the catalytic I3− reduction. The net result of the aforementioned is an appreciably increased dye regeneration rate, which leads to higher short-circuit current densities, and, in turn, to enhanced η. To demonstrate the lack of regeneration limits, E4 devices were measured at different illumination intensities (Fig. S12†). Given the linear relationship between light intensity and the JSC, on one hand, and an ordinate intercept of nearly zero, on the other hand, we postulate that the electrolyte effectively regenerates the oxidized dye even under high illumination conditions.
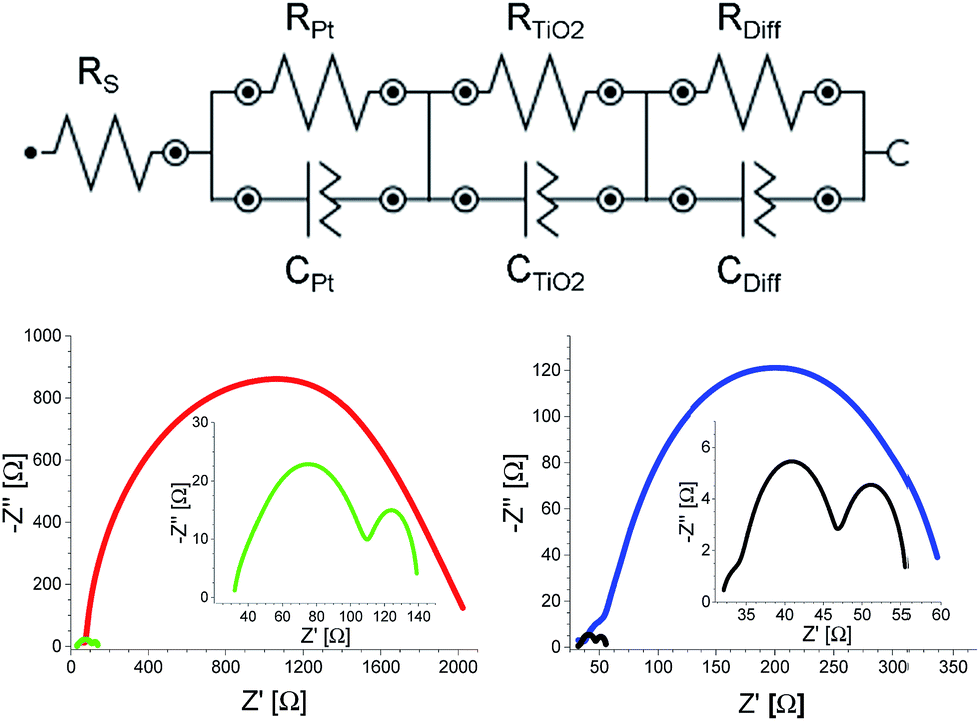 | ||
| Fig. 3 Upper part: equivalent circuit for EIS analysis. Left bottom part: Nyquist plots of devices with E1 (red) and E2 (green) measured at 70 °C. Right bottom part: Nyquist plots of devices with E3 (blue) and E4 (black) measured at room temperature. | ||
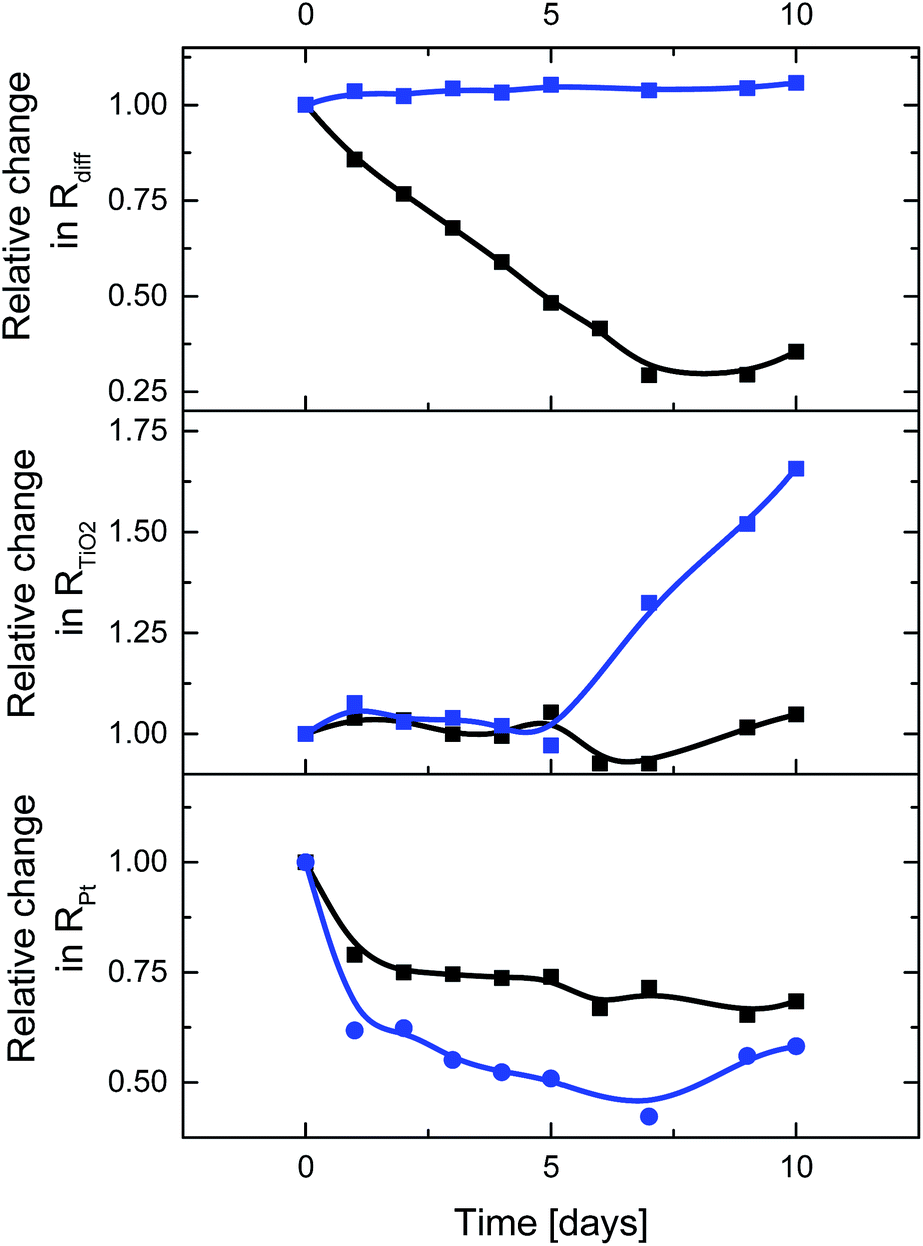
Finally, interactions between CNHs and the electrolyte were probed in time-dependent J–V and EIS measurements of DSSCs with E3 and E4 – Fig. 4 and 5. As time progresses, ssDSSCs with E3 and E4 feature nearly similar VOC, but JSC decreases for ssDSSCs with E3 by nearly 75%. In contrast, JSC of devices with E4 show a loss of only 9%. This is likely to relate to the dye degradation via, for example, substitution of the SCN− groups by I− when reacting with either I3− or polyiodide, which are formed in close proximity to the electrode surface.29,30 This impacts the overall dye regeneration. Negligible JSC decrease for E4 indicates, however, that CNHs support efficient electrolyte regeneration and, in turn, prevent dye degradation.
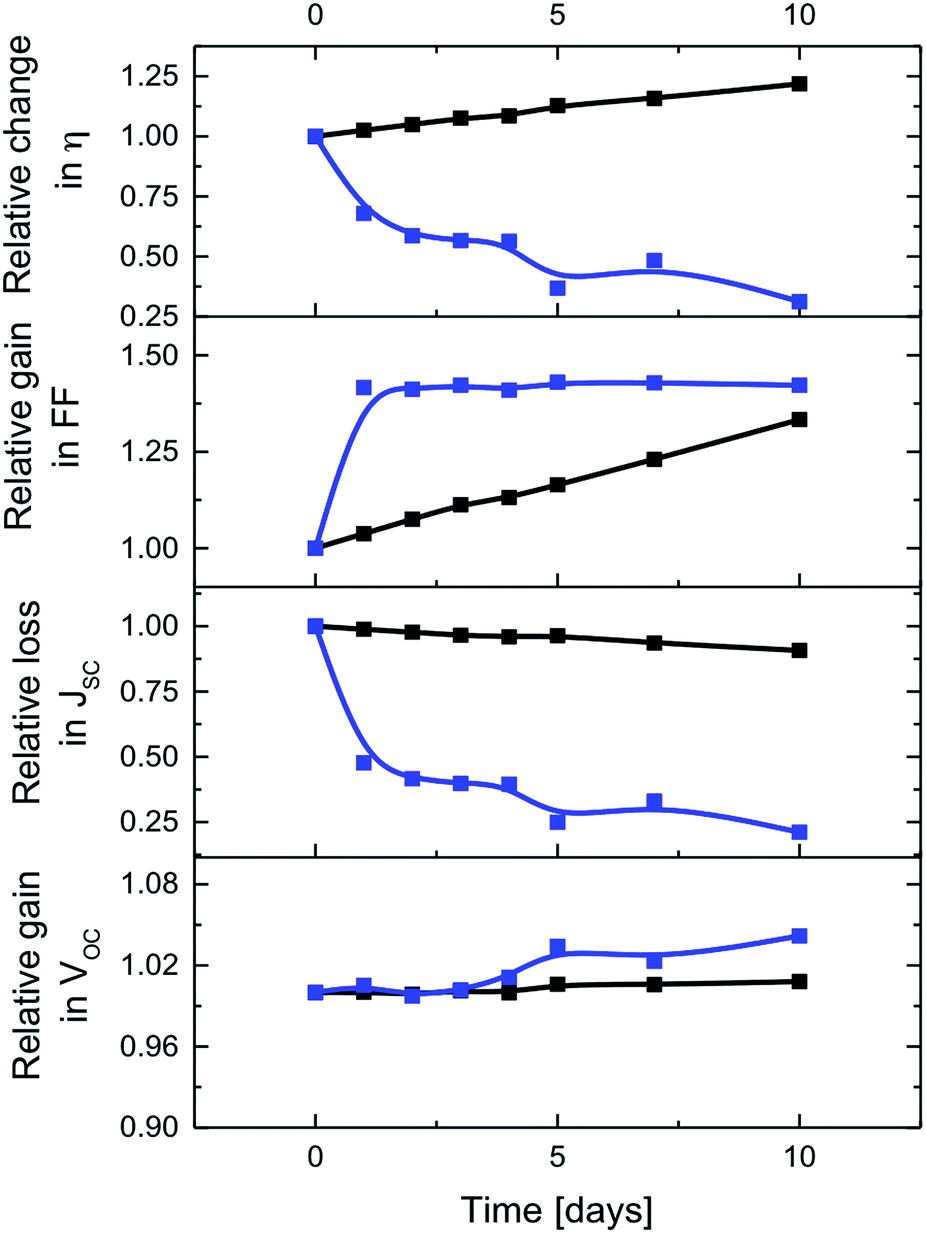 | ||
| Fig. 4 Time-dependent changes of the figures-of-merit for ssDSSCs with E3 (blue) and E4 (black). | ||
 | ||
| Fig. 5 Time-dependent changes of the figures-of-merit from EIS measurement of ssDSSCs with E3 (blue) and E4 (black). | ||
Interesting is the gradual increase of the FF in ssDSSCs with E4, while for ssDSSCs with E3 the FF raises only during the first day while remaining constant afterwards – Fig. 4. We rule out, however, a better electrolyte coverage of the dye-sensitized mesoporous network, owing to improved penetration as the reason for the continuous increase of the FF of ssDSSCs with E4.
To shed light onto this finding, we performed EIS analysis. Rdiff of ssDSSCs with E3 is not affected over time, while ssDSSCs with E4 exhibit a 71% decrease in Rdiff – Fig. 5. Similarly, RPt drops considerably after the first day, but remains from there on constant. A better contact between the electrolyte and the counter electrode is likely to be responsible for this trend. After five days a considerable RTiO2 increase sets in for devices with E3. The latter is attributed to the SCN−/I− exchange of the dye and/or the I3− induced corrosion of the TiO2 surface.29 For devices with E4, RTiO2 remains constant. Here, sufficient electrolyte regeneration and considerably reduced exposure of I3− to the electrode evoke long-term stability of DSSCs.
Taking all of the aforementioned into account, it is not surprising that the efficiency of ssDSSCs with E3 drops to 37%, while that of E4 increases by 22% from its initial value after 10 days. This clearly highlights the superior performance of CNHs-based electrolytes in terms of efficiency and stability.
Finally, it is important to point out that our findings leave the question unanswered on how CNHs improve the electrolyte features over time. To rationalize the time dependence we carried out Raman experiments of E4 extracted from different devices after 1, 2, 3, 4, and 10 days of operation – Fig. S13.† Raman results, namely the ratio intensification between the D and G bands from 1.08 to 1.21 for 0 and 10 days of exposure of the CNHs to the electrolyte, respectively, infer a gradual increase of defects. In other words, the trends for Rdiff and D/G ratio go hand-in-hand – Fig. 5 and S13,† from which conclude that the benefits upon adding CNHs correlate with the reaction between CNHs and I3−/I−. Here, defect loaded CNHs facilitate the diffusivity of the electrolyte and/or even reduce the I3− and polyiodide concentrations at the electrode/dye surface. Both effects are synergetic and lead to a gain in FF, which prompts to a suppression of electron recombinations with the electrolyte,31 and stable JSC due to the lack of both dye and electrolyte degradation. Still an open question is if the use of CNHs is self-limiting in terms of degradation or if, on the contrary, defects act as catalytic centers to further enhance the electrolyte features as our time-dependent assays suggest.
Conclusions
In summary, we have successfully implemented for the first time CNHs into ssDSSC electrolytes. Thereby, ssDSSCs without CNHs exhibited rather poor performance of 0.42%, which was improved up to 2.09% upon the addition of CNHs. As such, CNHs inside solid-state electrolytes enhance JSC, whereas VOC and FF remained almost unchanged. We were able to attribute the boost in JSC to a better ionic diffusivity in the electrolyte as well as to a more effective catalytic reduction of I3− by CNHs. This enables effective dye regeneration due to a quasi-zero distance between oxidized dyes and the counter electrode. Optimized devices with [BMIM][BF4], TBP, and GuSCN as additives feature efficiencies of 7.84% with CNHs and only 0.61% without CNHs. Striking is the long-term stability when CNHs are used, an affect that led to better diffusivity of the electrolyte and to an effective suppression of dye degradation. Here, a reaction between CNHs and I3−/I− not only in fresh devices but also as time progresses seems to evolve as a likely rationale. This notion is supported by the correlation between CNH defects and the increase in FF and stable JSC. Ongoing work focuses on transient absorption spectroscopy to pinpoint the individual steps in the regeneration processes under device operation conditions as well as to provide more insights into the reaction mechanism between the CNHs and the redox couple. Finally, it is important to point out that the application of an alternative iodide source, which exhibits a better initial performance, could lead to even higher efficiencies.Acknowledgements
F.L. R.D.C. and R.C. acknowledge the EAM cluster in the frame of the DFG excellence programs for their support. D.M.G. acknowledges the DFG, ECRC, and the ZMP for financial and intellectual support. M.P. thanks the Italian Ministry of Education MIUR (cofin Prot. 2010N3T9M4 and Firb RBAP11C58Y), EU (Carinhyph).Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- L. E. Polander, A. Yella, B. F. E. Curchod, N. Ashari Astani, J. Teuscher, R. Scopelliti, P. Gao, S. Mathew, J.-E. Moser, I. Tavernelli, U. Rothlisberger, M. Grätzel, M. K. Nazeeruddin and J. Frey, Angew. Chem., Int. Ed., 2013, 52, 8731–8735 CrossRef CAS PubMed.
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- A. El-Shafei, M. Hussain, A. Atiq, A. Islam and L. Han, J. Mater. Chem., 2012, 22, 24048–24056 RSC.
- H. Tian and L. Sun, J. Mater. Chem., 2011, 21, 10592–10601 RSC.
- L. J. Brennan, M. T. Byrne, M. Bari and Y. K. Gun'ko, Adv. Energy Mater., 2011, 1, 472–485 CrossRef CAS.
- D. K. Roh, W. S. Chi, H. Jeon, S. J. Kim and J. H. Kim, Adv. Funct. Mater., 2013, 24, 379–386 CrossRef.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- S. H. Ahn, W. S. Chi, D. J. Kim, S. Y. Heo and J. H. Kim, Adv. Funct. Mater., 2013, 23, 3901–3908 CrossRef CAS.
- J. Burschka, F. Kessler, M. K. Nazeeruddin and M. Grätzel, Chem. Mater., 2013, 25, 2986–2990 CrossRef CAS.
- J. Kim, J. K. Koh, B. Kim, S. H. Ahn, H. Ahn, D. Y. Ryu, J. H. Kim and E. Kim, Adv. Funct. Mater., 2011, 21, 4633–4639 CrossRef CAS.
- U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissörtel, J. Salbeck, H. Spreitzer and M. Grätzel, Nature, 1998, 395, 583–585 CrossRef CAS PubMed.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed.
- C.-P. Lee, P.-Y. Chen, R. Vittal and K.-C. Ho, J. Mater. Chem., 2010, 20, 2356–2361 RSC.
- B.-X. Lei, W.-J. Fang, Y.-F. Hou, J.-Y. Liao, D.-B. Kuang and C.-Y. Su, J. Photochem. Photobiol., A, 2010, 216, 8–14 CrossRef CAS PubMed.
- Y.-H. Chang, P.-Y. Lin, S.-R. Huang, K.-Y. Liu and K.-F. Lin, J. Mater. Chem., 2012, 22, 15592–15598 RSC.
- C.-P. Lee, L.-Y. Lin, P.-Y. Chen, R. Vittal and K.-C. Ho, J. Mater. Chem., 2010, 20, 3619–3625 RSC.
- M. S. Akhtar, S. Kwon, F. J. Stadler and O. B. Yang, Nanoscale, 2013, 5, 5403–5411 RSC.
- R. D. Costa, F. Lodermeyer, R. Casillas and D. M. Guldi, Energy Environ. Sci., 2014, 7, 1281–1296 CAS.
- V. M. Mohan, K. Murakami, A. Kono and M. Shimomura, J. Mater. Chem. A, 2013, 1, 7399–7407 CAS.
- Y.-C. Wang, K.-C. Huang, R.-X. Dong, C.-T. Liu, C.-C. Wang, K.-C. Ho and J.-J. Lin, J. Mater. Chem., 2012, 22, 6982–6989 RSC.
- D. M. Guldi and N. Martín, Carbon Nanotubes and Related Structures: Synthesis, Characterization, Functionalization, and Applications, Wiley, 2010 Search PubMed.
- R. Casillas, F. Lodermeyer, R. D. Costa, M. Prato and D. M. Guldi, Adv. Energy Mater., 2014, 4, 1301577 Search PubMed.
- R. D. Costa, S. Feihl, A. Kahnt, S. Gambhir, D. L. Officer, G. G. Wallace, M. I. Lucio, M. A. Herrero, E. Vázquez, Z. Syrgiannis, M. Prato and D. M. Guldi, Adv. Mater., 2013, 25, 6513–6518 CrossRef CAS PubMed.
- R. D. Costa, F. Lodermeyer, R. Casillas and D. M. Guldi, Energy Environ. Sci., 2014, 7, 1281–1296 CAS.
- M. Adachi, M. Sakamoto, J. Jiu, Y. Ogata and S. Isoda, J. Phys. Chem. B, 2006, 110, 13872–13880 CrossRef CAS PubMed.
- M. K. Nazeeruddin, A. Kay, E. Müller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- H. Greijer, J. Lindgren and A. Hagfeldt, J. Phys. Chem. B, 2001, 105, 6314–6320 CrossRef CAS.
- J. R. Jennings, Y. Liu and Q. Wang, J. Phys. Chem. C, 2011, 115, 15109–15120 CAS.
- S. Y. Huang, G. Schlichthörl, A. J. Nozik, M. Grätzel and A. J. Frank, J. Phys. Chem. B, 1997, 5647, 2576–2582 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ee02037e |
| This journal is © The Royal Society of Chemistry 2015 |