
Recent research progress on organoboron-based stimuli responsive materials
Zhiyu
Jia
 *,
Yue
Gao
,
Tongtong
Zhang
and
Nan
Wang
*
*,
Yue
Gao
,
Tongtong
Zhang
and
Nan
Wang
*
Key Laboratory of Cluster Science, Ministry of Education, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: jzy@bit.edu.cn; nanwang@bit.edu.cn
First published on 17th February 2026
Abstract
Organoboron-based stimuli-responsive materials have attracted intense research interest over the past decade owing to their broad utility in optoelectronics, chemical sensing, and smart devices. The boron center combines Lewis acidity, pronounced electron-accepting character, and synthetically tunable orbital hybridization, enabling boron-embedded scaffolds to undergo rapid and reversible structural or electronic transformations under optical, thermal, mechanical, or electrical stimuli. These perturbations modulate intra- and intermolecular interactions, producing pronounced changes in photophysical signature, charge-transport behavior, or mechanical response that can be exploited for real-time optical read-out or switchable device function. This review describes recent progress in boron-containing responsive systems ranging from small molecules and polymers to supramolecular assemblies, with emphasis on luminescent materials and representative advances of the last five years. Current challenges and future directions toward next-generation boron-based smart materials are also discussed.
 Zhiyu Jia | Zhiyu Jia is an associate professor at the School of Chemistry and Chemical Engineering, Beijing Institute of Technology (BIT). He received his BSc from Lanzhou University, MSc from Renmin University of China, and PhD from Universitat Autònoma de Barcelona. He then carried out postdoctoral research at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS). In 2017, Dr Jia started his independent career at BIT. His current research focuses on the design and synthesis of photo-functional materials and their applications in (photo)electrocatalysis and energy conversion. |
 Yue Gao | Yue Gao graduated from Yan’an University (China) with a BS degree in 2024. She formerly studied at BIT as an exchange student and is now a master's student under the guidance of Prof. Wang at Beijing Institute of Technology (China). Her research interests are broad, focusing mainly on stimuli-responsive boron-based materials and the photophysics of excited-state intramolecular proton-transfer (ESIPT) materials. |
 Tongtong Zhang | Tongtong Zhang began her studies at Qingdao University of Science and Technology and obtained a bachelor's degree in Applied Chemistry. During her master's program, she worked under Professor Wang Nan, conducting research on the stimulus-responsive properties of organic boron compounds, and received her master's degree in June 2025. |
 Nan Wang | Dr Nan Wang is an Associate Professor at the School of Chemistry and Chemical Engineering in Beijing Institute of Technology (BIT). She received her BSc from Nankai University in China, and MSc and PhD from Queen's University in Canada. Dr Wang worked as a postdoctoral fellow at University of Toronto Scarborough. In 2015, she started her independent career at BIT. Her research interests include organoboron based materials, luminescent transition metal complexes, and photochemistry. |
1. Introduction
Stimuli-responsive materials are widely recognized as “smart” systems owing to their ability to reversibly and instantaneously modulate their optical, electronic, or mechanical properties in response to external stimuli such as light,1 heat,2 mechanical force3 or chemical microenvironments.4,5 This dynamic behavior endows traditionally static materials with adaptive functionality, significantly broadening the scope of applications in drug delivery, catalysis, data storage, encryption and energy harvesting.6–9 Compared with their inorganic counterparts, organic stimuli-responsive materials offer distinct advantages for designing intelligent systems by virtue of their structural tunability, chemical diversity and facile processability. To date, extensive research has been reported, encompassing a wide range of materials from small molecules and supramolecular assemblies to polymeric architectures. Escalating performance demands now dictate that next-generation intelligent materials simultaneously deliver high quantum efficiencies, ultrafast response kinetics, synergistic multi-stimuli operability and impeccable biocompatibility.It is now well established that embedding main-group heteroatoms into π-scaffolds constitutes a powerful design platform for next-generation functional materials.10 By harnessing the distinct orbital symmetries, variable coordination numbers and unique local geometries inherent to main-group elements, unprecedented π-conjugated architectures with tailored photophysical and electronic characteristics become accessible. Among these elements, group-13 boron has attracted particular attention. The intrinsic electron deficiency of sp2-hybridized boron enforces trigonal-planar geometry and leaves an unoccupied p orbital coplanar with the surrounding π-system. This vacant orbital (i) mediates strong p–π* conjugation, enabling precise modulation of frontier molecular orbital energies, optical bandgaps and emission wavelengths; (ii) imparts high Lewis acidity to the boron center, permitting reversible coordination of intra- or intermolecular nucleophiles. Consequently, reversible switching between tri- and tetra-coordinate boron centers can be realized, affording pronounced and fully reversible changes in luminescence color, quantum yield and charge-transport behavior. These unique features have established boron-doped π-systems as versatile building blocks for stimuli-responsive materials, single-molecule sensors and multifunctional optoelectronic devices.10–12
Driven by the exponential expansion of organoboron materials chemistry and the intensifying quest for intelligent responsive systems, this review provides a critical survey of advances emerging since 2018 in boron-containing stimuli-responsive platforms, especially those exhibiting multi-stimuli responsiveness. The foundational analysis by Wang and co-workers has already codified the three prevailing operational paradigms: (1) reversible B←X bond formation/cleavage, (2) intramolecular manipulation of excited-state dynamics and (3) stimulus-directed intermolecular interactions.13 Consequently, the present article concentrates on materials that either extend these established motifs or adopt previously unreported switching pathways. The discussion is organized hierarchically according to molecular dimensionality: (1) small molecules, (2) macromolecules/polymers and (3) supramolecular assemblies. Each class is described with respect to its luminescence and/or physicochemical response, followed by an evaluation of their prospective deployments in sensing, bio-imaging, data encryption and adaptive optoelectronics. To avoid redundancy with existing literature, photochromic tetra-coordinate boranes,14 BODIPY-centric supramolecular constructs15 and boron-based systems whose responsiveness originates primarily from aggregation phenomena16 are therefore excluded from the present coverage.
2. Responsive systems based on boron-containing small molecules
Organoboron small molecules, especially triarylboranes, have long been mainstays in materials science because of their unique optoelectronic properties. As outlined above, boron exhibits a distinctive electronic signature that combines an unoccupied p orbital, moderate Lewis acidity and strictly trigonal planar geometry, rendering it an indispensable hub for luminescent π conjugated scaffolds. The vacant pπ–π* overlap lowers the LUMO level, narrows the band gap and delivers intense, readily tunable emission across the visible to near-infrared window. The same empty orbital provides an on-board binding site. Reversible coordination to Lewis bases or nucleophiles perturbs the electronic landscape in real time, enabling fluorescence switching without extra chromophores. Furthermore, the rigid, flat boron center locks adjacent aryl rings into a coplanar arrangement, suppressing vibrational relaxation and promoting aggregation-enhanced emission or through-space conjugation (Chart 1). Capitalizing on this principle, one of the earliest and most successful applications of organoboron “smart” materials has been anion sensing. Among Lewis-basic triggers, the fluoride ion is particularly important because of its roles in health care and environmental pollution.17,18 Pioneering design strategies fall into three paradigms: (1) fluoride binding switches the boron center between sp2 and sp3, interrupting π-conjugation and producing a blue-shift in absorption and/or emission together with fluorescence quenching; (2) the change in boron coordination redirects intramolecular energy-transfer pathways, yielding ratiometric blue- and red-shifted signals; and (3) fluoride coordination saturates the boron acceptor, suppressing intramolecular charge-transfer (ICT), through-space CT (TSCT) or long-range ICT, so that emission reverts to that of the local π-framework, usually accompanied by a blue-shifted and enhanced signal.19 Consequently, traditional triarylborane-based anion sensors operate predominantly in blue-shift or turn-off modes.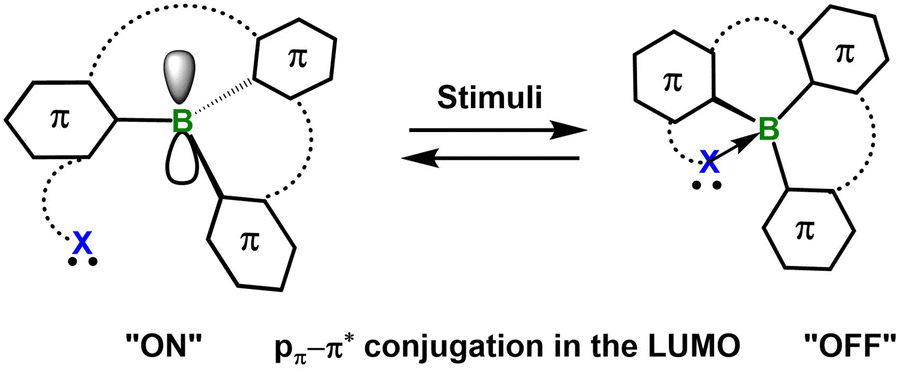 | ||
| Chart 1 Schematic showing reversible inter-/intramolecular Lewis pair coordination. | ||
Recently, Takeda et al. reported a fluoride-sensing strategy that breaks away from the traditional “blue-shift/quenching” framework. By appending the strongly electron-withdrawing dibenzo[a,j]phenazine (DBPHZ) to a nitrogen-bridged triarylborane (1,4-phenazaborine, PAzB) core, they constructed D–A–D architecture 1 (Fig. 1a).20 In this scaffold, PAzB plays a contradictory dual role as both a Lewis acid and an electron donor. Upon fluoride coordination the boron center converts from tri- to tetra-coordinate, the oxidation potential shifts much more negatively (ΔoxEonset = −1.53 V) than the reduction potential (ΔredEonset = −0.50 V), raising the HOMO level of the PAzB unit, while the LUMO remains localized on DBPHZ. Consequently, the HOMO–LUMO gap narrows and a new, low-energy CT absorption band appears at around 500 nm. Solution emission shifts progressively from 539 nm to 592–634 nm. In polystyrene (PS) or poly(methyl methacrylate) (PMMA) thin films, incremental fluoride doping tunes the emission from 465 nm to 684 nm with the photoluminescence quantum yield (ΦPL) as high as 0.20. Further fluoride doping yields a deep-red emission band centered at 621 nm (ΦPL = 0.06–0.07). This system affords an unprecedented red-shifted response of triarylboranes to fluoride stimuli through a novel “CT enhancement + bandgap narrowing” mechanism.
 | ||
| Fig. 1 (a) Chemical structure of a fluoride-sensor 1; (b) stepwise protonation of 2 with selected Brønsted acids and photographs of the solutions (left to right: 2-BAr4·OEt2, 2-OTf·HOTf, 2-OTf and 2-Cl) under ambient light and simultaneous 254/366 nm UV illumination; reproduced with permission from ref. 22. Copyright 2022, John Wiley & Sons, Inc. (c) Chemical structures of pH-responsive fluorophores 3 and 4; (d) aldehyde-functionalized amino-borane D–π–A dye 5 and the phosphine-oxide platform 6–9; (e) multi-color B←N based molecular switch 10; (f) switch 11: crystals under white light (top) and 365 nm UV lamp (bottom). Reproduced with permission from ref. 27. Copyright 2024, John Wiley & Sons, Inc. | ||
Beyond extensively studied fluoride-responsive systems, anion-tunable motifs are rapidly emerging as versatile additions to the stimuli responsive toolkit for water quality surveillance and biofluorescence imaging.21 A recent proof-of-concept from the Braunschweig group illustrates how a single molecular platform can deliver an entire color palette without any covalent redesign.22 Their doubly protonated, boron-doped thiazolothiazole core, stabilized by two cyclic alkyl(amino)carbene ligands, transduces subtle differences in counter-anion hydrogen-bond strength into a continuous, visible and fluorescence color sweep. As shown in Fig. 1b, the non-emissive compound 2 contains a cyclic alkyl(amino)carbene (CAAC)-stabilized “boron-doped” thiazolo[5,4-d]thiazole (TzTz) core. Systematic protonation with HCl, HOTf or [H(OEt2)2][BAr4] affords mono- or diprotonated species. The brightly emissive doubly protonated dications display pronounced colorimetric and fluorescence dependence on the nature of the hydrogen-bonded counter anions, spanning a broad spectral window. In dichloromethane (DCM), the absorption maximum shifts hypsochromically from 493 nm (X = Cl−) to 450 nm (BAr4·OEt2), while the emission moves from 580 nm to 539 nm and the ΦPL increases from 0.23 to 0.33. Theoretical calculations reveal a tunable boron-heterocycles → CAAC ICT characteristic for the S0 → S1 transition. Weakening the hydrogen bond enlarges the HOMO–LUMO gap by ∼0.2–0.3 eV, consistent with the observed blue-shift.
Using this model, a pair of tetra-coordinate boron heterocycles, 3 and 4, has been reported (Fig. 1c).23 While the neutral compounds are non-emissive, protonation of the methylamine donor triggers efficient blue photoluminescence, producing tetrahedral ammonium borates 3·HX and 4·HX that switch fluorescence “on” (3·HCl, λ = 488 nm, ΦPL = 0.71; 4·HCl, λ = 465 nm, ΦPL = 0.79), with emission wavelengths virtually independent of the counter-anion. Single-crystal X-ray analyses of both the neutral and cationic species reveal a saddle-to-planar conformational change upon protonation. Theoretical calculations indicate that photoinduced electron transfer (PET) quenching from the methylamine lone pair operates in the neutral state. Upon protonation, the HOMO−1 energy drops by ∼0.4 eV, thereby blocking PET and enabling a bright S1 → S0 transition.
When a Lewis base is incorporated into the molecular framework, a dynamic intramolecular B←X (X = O, N, S) coordination bond is established, which has become a cornerstone of stimuli-responsive luminogen design. In an earlier study, Wang et al.24 described the simple aldehyde-functionalized amino-borane 5, whose emission can be reversibly switched via intramolecular B←O bond cleavage and reformation under multiple external stimuli (Fig. 1d). The high chemical reactivity of the aldehyde group, however, compromises long-term stability and may limit practical deployment, prompting the need for further optimization. To this end, compounds 6–9 were devised in which the aldehyde is replaced by a triarylphosphine oxide unit.25 This modification markedly enhances chemical robustness while preserving the switchable ICT manifold. Systematic tuning of the boron Lewis acidity and/or the nucleophilicity of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety enables precise modulation of B←O bond strength: the carbazole-based congener 6 is too robust to undergo stimulus-induced rupture, whereas methoxy-substituted 8 and sterically encumbered 9 exhibit suitably weakened B←O interactions that can be reversibly disrupted by heat, water or hydrogen-bonding media. Consequently, 8 and 9 exhibit the most sensitive and fully reversible multi-state photophysical switching, with no detectable degradation over numerous cycles.
O moiety enables precise modulation of B←O bond strength: the carbazole-based congener 6 is too robust to undergo stimulus-induced rupture, whereas methoxy-substituted 8 and sterically encumbered 9 exhibit suitably weakened B←O interactions that can be reversibly disrupted by heat, water or hydrogen-bonding media. Consequently, 8 and 9 exhibit the most sensitive and fully reversible multi-state photophysical switching, with no detectable degradation over numerous cycles.
Based on the same amino-borane skeleton, Cao and co-workers developed a smart organoborane by condensing a Schiff base to give fluorescent probe 10 (Fig. 1e).26 The appended acylhydrazone acts as a hydrogen-bond acceptor that interacts with primary alcohols, cleaving the B←N bond and modulating the ICT manifold to produce multi-color emission. The steric demand of the alcohol governs hydrogen-bond strength: the less hindered the hydroxyl group, the more complete the B←N cleavage and the greater the population of tri-coordinate boron. At 0.01 mM and under 365 nm excitation, the 490 nm/600 nm emission intensity ratio increases in the order tert- < sec- < iso- < n-butanol, enabling naked-eye discrimination of the four isomers within seconds. This reagent-free protocol offers a safe and operationally simple alternative to the classical Lucas test.
Further modification of this system by introducing a sulfone unit afforded 11 (Fig. 1f),27 which exhibits full-color-tunable emission (456–610 nm) across multiple states via reversible B←N bond formation and conformational transitions. The transformation can be triggered by the crystallization solvent (DCM/MeOH → THF/EtOH), the water fraction in THF (0–99%), the PMMA doping level (0.1–50 wt%) or mechanical grinding. In the open form, ICT proceeds from the NMe2-phenyl donor to the vacant p orbital of the tri-coordinate boron (ΔE = 2.97 eV), affording short-wavelength emission. Closure of the B←N bond redirects ICT toward the sulfone-Schiff-base acceptor (ΔE = 1.74 eV) and produces a pronounced red-shift. Single-crystal analyses reveal systematic structural tightening across the blue → red series: B←N distance contracts from 4.28 to 1.67 Å, the rotation angle α decreases from 86.2° to 83.5°, and crystal density increases from 1.203 to 1.231 g cm−3. White light (CIE 0.31, 0.35) is observed when 85% aqueous THF generates nearly equal populations of open and closed forms, yielding dual emission at 467 and 560 nm (I467/I560 = 1.10) facilitated by Förster resonance energy transfer (FRET). Quantum yields of 92% in pure THF and 21% in the orange-emitting aggregate are obtained. Coating a 460 nm LED with 5 wt% 11@PMMA affords white electroluminescence with CIE coordinates of (0.33, 0.34).
Compared with the extensively investigated B←N-based stimuli-responsive systems, materials that employ dynamic B←chalcogen (O, S) dative bonds remain scarce. This paucity stems from the stronger electron-donating ability and better size-matching of nitrogen, which provides a moderately strong yet metastable B←N bond and thereby enables reversible on/off coordination switching at boron under external stimuli.28 To fill this gap, Yamaguchi and co-workers introduced a family of ortho-P(X)R2-substituted triarylborane D–π–A fluorophores (12–16, Fig. 2) in which the B←chalcogen bond dissociates in the excited state.29 Systematic variation of the R substituent and the heteroatom X (S or O) tunes the Lewis basicity of the PX moiety so that B–X cleavage occurs upon excitation, endowing the dyes with dual-emission characteristics and responsiveness to temperature and bulk viscosity. In cyclohexane, the PS derivative 14 undergoes almost complete photodissociation to emit at 437 nm with ΦPL = 0.53, whereas the PO analogue 13 remains predominantly coordinated and exhibits weaker, shorter-wavelength emission at 388 nm (ΦPL = 0.35). The extent of photodissociation is further modulated by solvent polarity: in acetonitrile, the long-wavelength band of 13 dominates and ΦPL increases to 0.64. These results establish that PS cleaves more readily than PO, and that the population of the emissive tri-coordinate species increases with solvent polarity.
 | ||
| Fig. 2 Triarylborane D–π–A fluorophores 12–20 reported by Yamaguchi and co-workers. | ||
Building on the same triarylborane π-scaffold, Yamaguchi and co-workers next replaced the PX moiety with a purely carbon-based Lewis base, namely an olefin, to examine how a weaker dative interaction would influence the photophysical behavior.30 They prepared D–π–A fluorophores 17 and 18, each bearing an alkenyl-bridged diarylboryl moiety (Fig. 2), and arranged them to form an intramolecular frustrated Lewis pair (FLP). Single-crystal X-ray analysis corroborates a modest olefin → borane contact that raises the LUMO and consequently blue-shifts both absorption and emission. Remarkably, the addition of an external Lewis base (tricyclohexylphosphine, 4-dimethylaminopyridine or tetrabutylammonium fluoride) does not generate a classical boron←base adduct. Instead, the FLP engages in 1,2-addition across the CC bond to deliver a zwitterionic species accompanied by pronounced spectral changes. The resulting adduct displays fully reversible thermochromism between 293 and 363 K. A tandem ring-closing metathesis/chemoselective hydrogenation sequence furnishes modular, air-stable scaffolds that serve as a versatile springboard for stimuli-responsive luminescent materials.
To further widen the stimuli-response window, the same group moved from the mono-boron FLP manifold to a diboron platform capable of sustaining two labile B–S contacts.31 The key design, 9,10-dihydro-9,10-diboraanthracene (DBA) derivatives 19 and 20 (Fig. 2), locks arylthiomethyl pendants at the ortho positions of the B-bound phenyl rings, affording five-membered B←S chelates that are isolable as cis- and trans-isomers (cis favored). Because the dative B←S bond is intrinsically weak, subtle electronic or steric changes in the aryl substituent translate into predictable modulation of binding strength and enable fully reversible cis–trans interconversion on the variable temperature (VT) NMR time-scale. TD-DFT studies indicate that both sulfur atoms dissociate in concert upon photoexcitation, generating a π-conjugated bis-borane excited state. This photodissociation event triggers a multi-emission manifold comprising: (1) residual fluorescence from the tetra-coordinate ground state (420–460 nm), (2) dominant emission from the tri-coordinate photoproduct (500–580 nm), (3) red-shifted ICT fluorescence from the relaxed excited geometry (∼570 nm), and (4) low-temperature phosphorescence (580–680 nm). Each channel can be selectively accessed by tuning temperature, solvent polarity, or excitation wavelength, exemplifying how diboron scaffolds can multiply the output modes of a single molecular switch.
Rigid aromatic boron doped D–π–A skeletons typically exhibit high emission efficiencies because of intense through-bond charge-transfer (TBCT) transitions. Most of the aforementioned stimuli-responsive organoboranes rely on the reversible formation/cleavage of a B–X dative bond, thereby interrupting TBCT and generating marked spectral changes. Beyond this conventional approach, an emerging alternative employs conformationally flexible, non-conjugated scaffolds embedding B←X interactions that modulate through-space charge-transfer (TSCT) transitions. Because external stimuli can precisely tune B←X bond strength, the TSCT manifold can be regulated at will, affording tailor-made photophysical properties that are decoupled from through-bond conjugation.32–34
Building on the prior eight-membered B/N Lewis pairs,34 Chen and co-workers extended the series to 21–28 (Fig. 3a), in which the balance of steric demand at boron (Tip, FMes or Mes) and ortho-substitution on nitrogen (ranging from diisopropyl to H) continuously tunes the B⋯N interaction from open-chain frustrated pairs to closed Lewis adducts.35 Highly congested congeners (21–26) exhibit long B⋯N separations (3.63–3.74 Å) and retain an FLP characteristic, whereas less shielded 27 and 28 adopt butterfly conformations with short B–N bonds [1.805(2) and 1.792(3) Å]. VT 11B NMR reveals that 27 equilibrates between sp2 and sp3 boron centers, while 28 exists exclusively as a fixed adduct, consistent with X-ray diffraction data. In solution, 27 emits at 485–571 nm, depending on solvent and temperature, from an open-chain excited state. This emission blue-shifts with increasing temperature, attributed to excited-state structural relaxation and reduced solvent stabilization. In the crystalline state, heating induces B–N bond cleavage, red-shifting the emission from 470 nm (closed form) to 540 nm (open form) and intensifying it, tracking a phase transition and conformational flip. Notably, 27 also exhibits highly sensitive solid-state photochromism (colorless → purple → colorless) under UV irradiation. The photoproduct rad-27 displays rare room-temperature phosphorescence (RTP) (τ = 232 ms, afterglow ≈1.3 s), exemplifying fluorescence-to-phosphorescence switching in a single dynamic B/N Lewis pair.
 | ||
| Fig. 3 Molecular structures of (a) eight-membered-ring B/N Lewis pairs 21–28; (b) dual-B/N-pair dimers 29–33; (c) ortho-functionalized donor-BMes2 stilbenes; (d) B/N Lewis pairs 38–40 with central eight-membered octacyclic scaffolds. | ||
Encouraged by this reversible single-pair photoswitching, the authors devised a “two-in-one” strategy, constructing eight-membered-ring dimers 29–33 that incorporate two B/N Lewis pairs within a single π-bridge (Fig. 3b).36 By tuning the steric hindrance of ortho-substituents on the central arylamino π-linkers, the electronic properties of the two LP subunits are precisely modulated. Consequently, 31 and 32 coexist with both sp2- and sp3-hybridized boron centers within a single molecule. VT fluorescence reveals an anti-correlated two-band profile: heating attenuates the 465 nm emission while a broad 590 nm band emerges, enabling continuous color tuning and delivering single-molecule white-light emission at 290 K (CIE 0.31, 0.33). This opposite trend is attributed to thermally induced conversion of boron centers from sp3 to sp2, as corroborated by VT 11B NMR measurements.
Beyond flexible scaffold engineering, incorporation of a photoactive unit offers a complementary and rapid route to light-triggered TSCT control. Building on the zwitterionic ladder stilbenes developed by Yamaguchi and co-workers,37 where a cascade cyclization locks the B and P centers into a rigid, charge-separated framework, Chen et al. chose a simpler and reversibly photo-responsive strategy. They leveraged the classical CC double bond as a photo-switchable core and prepared ortho-functionalized donor-BMes2 stilbenes 34–37 whose trans–cis isomerization modulates the B⋯N distance, and hence, the TSCT efficiency, thereby furnishing a concise molecular toolkit for photo-responsive organoboranes.38 A monotonic decrease in nitrogen steric bulk [34 (Ph, Ph) → 35 (Ph, Me) → 36 (Me, Me) → 37 (Me, H)] shortens the ground-state B⋯N separation, enhances photochemical conversion and tightens intramolecular constraints. Consequently, the cis population spans a complete activity spectrum: weak B/N coupling and rapid thermal back-reversion (cis-34/35), emission-ON switching via enhanced ICT (cis-36), or robust B←N bond formation that quenches fluorescence by an order of magnitude (cis-37, ΦFL 60% → 13%) and yields a long-lived bistable state requiring thermal reset. This steric hierarchy delivers fully reversible, fatigue-resistant photoswitching and provides a minimalist TSCT platform for next-generation photo-responsive organoboranes.
Complementing these light-driven switches, temperature-controlled conformational dynamics offer an alternative handle over TSCT. A recent design is provided by dibenzo-8-membered bridge-type scaffold donor–acceptor systems (38–40, Fig. 3d),39 in which a flexible eight-membered ring separates a triarylamine donor from a triarylborane acceptor, enabling temperature-dependent twisting that modulates TSCT. Each compound displays two emission bands: a solvent-insensitive local-excited (LE) peak at ∼390 nm and a strongly temperature-dependent CT band. For 38, the CT maximum shifts hypsochromically from 628 nm (150 K) to 490 nm (345 K), affording a 176 nm thermochromic window; comparable shifts are observed for 39 (176 nm) and 40 (145 nm). As temperature rises, the CT intensity increases while the LE signal remains constant, producing a visible color change from orange to blue. VT 1H NMR and femtosecond transient-absorption measurements attribute this behavior to a thermally driven twist-boat ↔ chair interconversion of the dibenzo-8-membered bridge that shortens the donor–acceptor distance and enhances electronic coupling. Additionally, 38 and 39 exhibit aggregation-induced emission (AIE), making them attractive for solid-state luminescent thermometers and other smart photonic applications.
In 2016, Hatakeyama et al. unveiled the short-range charge-transfer (SR-CT) concept of multi-resonance thermally activated delayed fluorescence (MR-TADF). By harnessing the counteracting resonance of boron and nitrogen, an alternant HOMO/LUMO arrangement can be achieved in the central phenyl ring (Fig. 4a),40 giving a tiny singlet–triplet gap that enables TADF while almost no excited-state geometry change occurs. While DOBNA,40 DABNA41 and their numerous boron-rich analogues continue to expand the emission palette and push device performance, the chemically addressable central boron atom inherent to these “star” cores now offers an additional, largely unexplored dimension. Recent studies demonstrate that external stimuli can trigger large, fatigue-resistant and fully reversible changes in emission color, intensity or excited-state lifetime, positioning MR-TADF frameworks as emerging platforms for next-generation stimuli-responsive photonics rather than mere high-efficiency emitters.
 | ||
| Fig. 4 (a) MR-TADF design principles and the molecular structures of DOBNA and DABNA; (b) reversible formation and dissociation of the B–O bond between 41 and p-BQ2−; reversible emission switching of the EFC device and photographs under daylight and UV light; reproduced with permission from ref. 42. Copyright 2024, Springer Nature. (c) Molecular structures of 42–47; (d) molecular structure of 48 and the cyclic interconversion of TPAPy-48, TPAPy-48A, and TPAPy-48AB triggered by external stimuli. Reproduced with permission from ref. 44. Copyright 2024, John Wiley & Sons, Inc. | ||
Zhang et al. demonstrated that utilizing the electrochemically reversible B←O dynamic coordination bond can precisely regulate the photophysical behavior of 41, which is a widely studied DABNA derivative.42 In this study, the boron-doped polycyclic aromatic hydrocarbon (PAH) 41 serves as an electron acceptor, while p-benzoquinone (p-BQ) functions as an electrochemically switchable electron donor. Upon reduction of p-BQ to p-BQ2−, a B–O coordination bond is formed at the boron center of 41 (Fig. 4b). The association constant increases from 210 ± 7 M−1 to (2.6 ± 0.6) × 104 M−1 upon reduction, resulting in complete quenching of the 488 nm emission and the emergence of a new, blue-shifted fluorescence band at 382 nm. Concurrently, the absorption maximum shifts from 467 nm to 350 and 367 nm. 11B NMR data reveal a chemical shift change of over 40 ppm, consistent with a sp2 → sp3 rehybridization of the boron atom, which is further supported by theoretical calculations. Femtosecond transient absorption spectroscopy indicates that, prior to coordination, the excited-state relaxation involves a 340 fs internal conversion, a 20.6 ps intersystem crossing, and a 5.82 ns radiative decay. After coordination, only a 265 fs vibrational cooling and a 1.83 ns blue fluorescence component remain. An electrofluorochromic (EFC) device based on this mechanism exhibits an on/off contrast ratio (Ion/Ioff) of 10.2, with 90% fluorescence quenching and recovery achieved within 0.4 s and 0.8 s, respectively. The device maintains over 80% contrast after 1000 switching cycles. This study introduces the first electrochemically controllable B–O dynamic covalent bond in B,N-doped PAHs.
Another interesting example is DOBNA derivative 42–47 (Fig. 4c).43 These molecules exhibit distinct stimuli-responsive luminescence behaviors depending on the type of N-heterocyclic substituents attached to the DOBNA core. Specifically, when n-butyl-1H-1,2,4-triazolyl groups are introduced (compounds 42–44), these molecules can form dynamic dimeric structures in solution via weak intermolecular B←N coordination. This coordination is thermally reversible: upon heating, the dimers dissociate, leading to a significant enhancement in fluorescence intensity. For instance, the emission intensity of 42 increases by nearly six-fold as the temperature rises from 200 K to 320 K. VT 11B NMR spectroscopy further confirms this process, showing a transition from a tetra-coordinate boron environment (δ ∼ −4 ppm) to a tri-coordinate one (δ ∼ 39 ppm). This indicates that the dynamic dissociation of the B←N dative bond is central to the luminescence enhancement. Single-crystal X-ray diffraction analyses support these findings: 42–44 all adopt head-to-tail dimeric structures in the solid state, with B–N bond lengths of approximately 1.66–1.67 Å, which are significantly longer than typical aromatic B–N bonds (1.43–1.47 Å), suggesting a relatively weak and thermally labile coordination. In contrast, when n-butyl-1H-1,2,3-triazolyl groups (45–46) or other substituents such as pyridyl are used, the intermolecular B–N interactions are substantially diminished or absent. As a result, these compounds exhibit conventional thermal quenching behavior under the same conditions. This difference is primarily attributed to the greater steric hindrance and altered electronic properties of the 1,2,3-triazolyl group, which reduce the responsiveness of the B–N interaction to temperature changes.
Building on the same DOBNA core, Li and co-workers devised a reversible “capture-release” platform that switches RTP on and off through a dynamic B←N bond (Fig. 4d).44 Lewis-acidic 48 is first trapped by the Lewis-basic pyridiyl donor N,N-diphenyl-4-(pyridin-4-yl)aniline (TPAPy) to afford the adduct TPAPy-48, in which RTP is silenced. Exposure to HCl cleaves the B←N linkage, releases and re-aggregates 48, and restores a yellow after-glow (λem = 539 nm, τ = 154 ms) via FRET from 48 to the concomitantly formed protonated TPAPyH. Subsequent NH3 neutralization deprotonates TPAPyH, switches FRET off, and generates a green RTP (λem ≈ 529 nm, τ = 186 ms). The cycle-fluorescence “on”/RTP “off” (TPAPy-48) ↔ yellow RTP (TPAPy-48A) ↔ green RTP (TPAPy-48AB) can be repeated more than 8 times without spectral fatigue and can be triggered by grinding, heating (100 °C) or solvent, affording an exceptional fatigue-resistant, multi-stimuli RTP system that has been implemented in ink-jet printing and multi-level anti-counterfeiting films.
In addition to utilizing intermolecular B–X interactions to modulate photophysical properties for stimulus responsiveness, another widely adopted strategy involves harnessing noncovalent intermolecular forces, such as π–π stacking, restriction of intramolecular motions (also known as AIE), hydrogen bonding, crystal-to-amorphous transitions, and related interactions to construct stimuli-responsive boron-based systems. This approach has proven particularly effective in the design of mechanochromic luminescent (MCL) materials. Among these, tetracoordinate boron difluoride (BF2) complexes, including difluoroboron β-diketonates, diketoiminates, and diiminates, represent the most commonly employed structural motifs.45 Their MCL was first reported by Fraser and co-workers in 2007.46 A recent representative example was reported by Tanaka et al., who described a thienyl-substituted fused boron ketoiminate (compound 49 in Fig. 5).47 The thienyl ring extends π-conjugation and increases molecular planarity, thereby amplifying mechanochromic sensitivity. Compound 49 crystallizes in two polymorphs: a green-emissive phase (crystal A) and an orange-emissive phase (crystal B). Upon grinding, crystal A undergoes a single-step transition, with the emission maximum shifting from 500 nm to 487 nm and ΦPL dropping from 0.20 to 0.15. In contrast, crystal B displays multi-step luminescence changes: gentle tapping moves the emission from 566 nm to 529 nm (yellow-green), whereas prolonged grinding stabilizes it at 491 nm (yellow). This behavior arises from the progressive disruption of crystalline order and the formation of distinct emissive states, as confirmed by peak deconvolution and excited-state lifetime measurements. This study exemplifies how minor structural modifications can endow boron complexes with exquisite mechano-responsiveness and enable the extent of mechanical perturbation to be read out through well-defined luminescent signals, underscoring their promise as mechanosensors and smart materials.
 | ||
| Fig. 5 Molecular structure of 49 and photographs of polymorphs A and B under 365 nm UV irradiation before and after mechanical treatment; excitation wavelength 400 nm. Reproduced with permission from ref. 47. Copyright 2020, The Royal Society of Chemistry. | ||
Subsequently, the same group demonstrated that reversible single-molecule conformational change in a β-diketiminate boron complex is sufficient to evoke macroscopic vapochromic luminescence.48 As illustrated in Fig. 6a, they designed propeller-shaped pentaphenyl boron complex 50, whose central boron atom toggles between two low-energy conformations – planar and half-chair. This structural flexibility furnishes two pseudopolymorphs: the neat G phase (λem = 490 nm) emits green light through an intermolecular charge-transfer (CT) state, whereas the B phase, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cocrystal with a small aprotic volatile organic compound (VOC), gives blue emission (λem = 443 nm) because the CT manifold is disrupted. The two states interconvert quantitatively within seconds to tens of seconds at room temperature under VOC vapor, without heating or vacuum, and sustain their integrity over numerous cycles. Conformational self-selection directs the complex to adopt either a “solvent-cocrystal” or a “desolvated-pure” packing motif dictated by the surrounding vapor pressure. VOC ingress generates the B phase, whereas VOC egress restores the G phase. Each conformational switch thus translates into a crystal-structure reorganization that dictates the ultimate emission color. Multiscale characterization combining XRD, photoluminescence, NMR, and TD-DFT calculations corroborates that the reversible appearance and disappearance of the CT state underlies the chromic behavior. Importantly, only small, aprotic solvents (e.g., DCM) trigger the G → B transition, affording exceptional selectivity. This system offers a low-energy, recyclable platform for visual VOC detection.
:1 cocrystal with a small aprotic volatile organic compound (VOC), gives blue emission (λem = 443 nm) because the CT manifold is disrupted. The two states interconvert quantitatively within seconds to tens of seconds at room temperature under VOC vapor, without heating or vacuum, and sustain their integrity over numerous cycles. Conformational self-selection directs the complex to adopt either a “solvent-cocrystal” or a “desolvated-pure” packing motif dictated by the surrounding vapor pressure. VOC ingress generates the B phase, whereas VOC egress restores the G phase. Each conformational switch thus translates into a crystal-structure reorganization that dictates the ultimate emission color. Multiscale characterization combining XRD, photoluminescence, NMR, and TD-DFT calculations corroborates that the reversible appearance and disappearance of the CT state underlies the chromic behavior. Importantly, only small, aprotic solvents (e.g., DCM) trigger the G → B transition, affording exceptional selectivity. This system offers a low-energy, recyclable platform for visual VOC detection.
 | ||
| Fig. 6 (a) Schematic illustration showing the conformational change of compound 50 and its reversible vapochromic behavior. Reproduced with permission from ref. 48. Copyright 2021, John Wiley & Sons, Inc. (b) Molecular structures of 51–55 and photographs under UV irradiation (365 nm) before and after mechanical grinding. Reproduced with permission from ref. 49. Copyright 2025, John Wiley & Sons, Inc. (c) Chemical structure, dihedral angles and optical images of the different polymorphs of 56 taken under daylight. Reproduced with permission from ref. 50. Copyright 2019, American Chemical Society. (d) Molecular structure of the phthalimide–difluoroboron Schiff-base complexes 57 and 58. Photographs show their luminescent color and mechanochromic properties under UV irradiation. Reproduced with permission from ref. 53. Copyright 2023, American Chemical Society. | ||
A BF2-diketiminate-based dimeric series 51–54 was subsequently reported, in which the bridge topology was systematically varied (Fig. 6b).49 In dilute solution, the meta-linked isomer 52 retains monomer-like behavior (λem = 493 nm, ΦPL = 0.41), akin to the parent monomer 55. In contrast, the para-connected analogue 53 exhibits a symmetry-broken CT characteristic, resulting in red-shifted emission (519 nm), an enhanced ΦPL of 0.64, and a large Stokes shift of 5500 cm−1, accompanied by pronounced solvatochromism. The directly tethered derivative 54, featuring a freely rotatable interchromophoric C–C bond, displays the largest Stokes shift within the series (6900 cm−1) and uniquely shows reversible mechanochromism in the solid state: crystalline green emission (504 nm) switches to orange (557 nm) upon manual grinding, with full restoration of the original emission upon thermal annealing (150 °C for 10 min), as confirmed by DSC and powder XRD (PXRD) analysis. TD-DFT calculations attribute the spectral trends to through-space orbital overlap (ortho and direct dimers) or through-bond π-delocalization (para dimer), and further establish that torsion-dependent exciton coupling in 54 underlies its mechanochromic response.
Another example of a tetra-coordinate organoboron mechanochromic luminogen was reported by Fang and co-workers (Fig. 6c).50 Among three tetrahedral monoboron complexes bearing 8-hydroxyquinoline ligands, only the derivative with 5,7-diiodo-8-hydroxyquinoline as the chelating unit and hexylbenzene as the monodentate ligand (56) exhibits dual emission. The two bands are assigned to a LE and a relaxed-excited (RE) state whose relative intensities are governed by molecular conformation. Compound 56, whose conformational landscape is defined by the dihedral angles α and β between each phenyl ring and the rigid boron-chelated core, can be isolated as an amorphous green-emitting powder (P) or as three polymorphs: A (green), B (yellow) and C (red). These polymorphs undergo quantitative interconversion under mechanical or thermal stimuli. Grinding proceeds sequentially C → B → A → P, whereas heating (≈70 °C) regenerates the thermodynamically most-stable C form (Fig. 6c). In situ fluorescence, DSC and PXRD track the phase transformations, and VT photoluminescence shows that cooling selectively enhances the LE contribution while heating favors the RE bands. Consequently, a single ink formulation can write green, yellow or red patterns simply by adjusting the substrate temperature. Single-crystal analyses reveal that the dihedral angles between the chelating hydroxyquinoline plane and the two rotatable phenyl rings differ in each polymorph, modulating intermolecular C–H⋯π distances (3.7–3.9 Å) and lattice density (1.520–1.523 g cm−3). These structural changes collectively tune the LE/RE population ratio and hence the observed emission color.
Recently, tetra-coordinate boron Schiff-base complexes have gained momentum because of their outstanding potential in light-emitting materials, fluorescence sensing and related areas. Locking the CN bond and suppressing excited-state proton transfer curtail non-radiative decay, which in turn significantly boosts emission efficiency. Since their emission is dictated by an intramolecular rotation axis, they exhibit solution-phase sensitivity toward viscosity, temperature or pH and solid-state mechanochromism that can be reversibly cycled.51,52 A representative example is the work of Georgiev and co-workers,53 who prepared two positional isomers of a phthalimide–difluoroboron Schiff-base dye (Fig. 6d, 57, 4-substituted; 58, 3-substituted) and quantified how a single atom change in connectivity dictates both photophysical output and mechanical response. Single-crystal X-ray analysis revealed that relocating the imide nitrogen from C-4 to C-3 increases the ground-state B–N–C–C dihedral angle from 31.7° to 54.0° and raises the corresponding rotational barrier from 2.8 to 4.1 kcal mol−1. Consequently, the 3-substituted rotor 58 exhibits a photoluminescence quantum yield of 34–38% in solution, measured across solvents of different polarity, almost twice the yield of 4–21% recorded for 57. Time-resolved spectroscopy resolved two excited-state species that interconvert through a planarization-induced charge-transfer (PICT) pathway with an activation energy as low as 0.6 kcal mol−1 in 57, but 1.0 kcal mol−1 in the more hindered 58. In the solid state, only 57 displays reversible mechanochromism. Gentle grinding red-shifts emission from 468 nm to 510 nm (Δλ = 42 nm) and increases the ΦPL from 13% to 16.7%, whereas 58 merely brightens without spectral shift. The color change can be erased completely by brief exposure to DCM vapor, restoring the original crystal packing. These findings establish a quantitative structure–mechanochromism relationship that greater steric congestion suppresses rotation and enhances solution emission, whereas a lower planarization barrier is essential for mechanically gated solid-state switching, thus offering a readily accessible design principle for next-generation boron-based fluorophores destined for rewritable optical media, high-contrast mechanosensors and viscosity-mapping bio-imaging probes.
The boron Schiff-base platform has also been exploited to deliver controllable chiral emitters. Driven by the demand for high-gain circularly polarized luminescent (CPL) materials, systems that combine intense emission with stimuli-governed handedness are attracting sustained interest.54–56 In a representative study, Ikeshita, Imai and Tsuno et al.57 prepared bis(boron-difluoride) complexes 59 (phenylene bridge) and 60 (naphthylene bridge) anchored to a chiral salen ligand whose two BF2 planes are locked face-to-face by a 1,2-cyclohexyl linker (Fig. 7). Dynamic rotation about the CN axis generates (SC, SC, Ra, Ra) and (SC, SC, Sa, Sa) atropisomers whose equilibrium is tuned reversibly by temperature and solvent polarity. Both complexes act as dual-input CPL switches, but their signatures diverge markedly. Complex 59 shows fatigue-resistant (>102 cycles) sign inversion (glum +2.0 × 10−3 at 293 K → −1.8 × 10−3 at 213 K, λem fixed at 430 nm) and a linear glumvs. ET(30) span of 3.6 × 10−3 from toluene (+) to DMF (−), whereas 60 exhibits only monotonic intensity change (+1.3 → +0.4 × 10−3) without reversal. TD-DFT attributes the difference to mirror-image exciton couplets in 59 (ΔμS1 ≈ 3D) but weak- vs. strongly-positive couplets in 60, rendering the latter unidirectional. This study provides a rare instance of pure intramolecular-rotation-driven CPL sign flipping and underscores the boron-Schiff-base scaffold as a solution-processable platform for optical cryptography and multi-state molecular logic.
 | ||
| Fig. 7 Schematic illustration of CPL sign switching via dynamic axial chirality control of boron Schiff-base complexes 59 and 60. Reproduced with permission from ref. 57. Copyright 2024, John Wiley & Sons, Inc. | ||
As an important class of tetra-coordinate boron species, borondipyrromethylene (BODIPY) derivatives have been widely studied for their intriguing photophysical properties and are now ubiquitous in sensors, bio-imaging and electroluminescent devices. Unlike tri-coordinate organoboron luminogens, which often retain bright solid-state emission, the planar dipyrrin core of BODIPY favors strong π–π contacts that quench fluorescence and seemingly preclude mechanochromism. This limitation was recently overturned by Duan et al., who installed meso-aryl substituents to impose sufficient torsion within the aggregate (Fig. 8).58 Single crystals of compounds 61, 62 and 63 adopt slipped stacks with dipole-alignment angles of 72.2°, 76.5° and 83.8°, emitting red light at 632, 601 and 614 nm with photoluminescence quantum yields in the solid state of 32.2%, 9.2% and 13.0%, respectively. Gentle grinding fractures the crystals into micro-domains and hypsochromically shifts the emission 45–70 nm to the yellow (∼560 nm); further mechanical amorphisation disrupts the J-aggregate manifold to give an amorphous green powder (λem = 535 nm) whose absorption (498 nm) and lifetime (0.8 ns) mirror the monomer recorded in THF (520 nm, 0.9 ns). The red → yellow → green sequence is fully reversed by solvent vapor or thermal annealing, whereas the planar reference 64 exhibits only a 6 nm shift under identical stress. The study establishes that steric twist, rather than added bulk alone, can unlock large-amplitude mechanochromism in otherwise recalcitrant BODIPY solids.
 | ||
| Fig. 8 Chemical structures of BODIPY derivatives 61–64. The inset photographs show single crystals of 61 (a), 62 (b), 63 (c) and 64 (d) after gentle grinding and strong grinding under a 365 nm UV lamp. Reproduced with permission from ref. 58. Copyright 2019, The Royal Society of Chemistry. | ||
In recent years, the tetracoordinate BF2-hydrazone (BODIHY) scaffold has attracted sustained interest because of its facile synthesis, tunable absorption/emission property, large Stokes shifts, aggregation-induced emission enhancement (AIEE) and so on, making it suitable for solid-state applications.59 Building on these advantages, Aprahamian et al. recently disclosed an unusual configuration-selective 1,2-BF2 shift that couples visible-light photoisomerization with spontaneous fluorescence turn-on (Fig. 9).60 The azo-BF2 photoswitch trans-65 (λabs = 593 nm) can be quantitatively converted to its cis isomer cis-65 (λabs = 508 nm) or reverted with 480 nm light; importantly, only trans-63 undergoes a room-temperature 1,2-BF2 shift (solid-state activation at 338 K) to yield orange-emitting BODIHY 66 (λem = 655 nm). DFT calculations reveal that an adjacent methoxy group is essential for this structural transformation, which lowers the activation barrier from ca. 40 to 25.3 kcal mol−1via lone-pair-assisted B←O attack, whereas the cis isomer is blocked by a 55.3 kcal mol−1 wall. Heating (≥ 373 K) closes the cycle by regenerating the hydrazone precursor 67, yielding a four-state, four-color loop (purple → red → orange → yellow). Using this cascade, the authors fabricated a cross-linked polydimethylsiloxane (PDMS) film that can be repeatedly patterned in micrometre-scale multi-color motifs with 650 nm light and 338 K heat, demonstrating a new class of programmable, multi-stimuli chromic materials for anticounterfeiting and optical recording.
 | ||
| Fig. 9 Reaction pathways with solution color changes for azo-BF2 derivatives mentioned in the study. Reproduced with permission from ref. 60. Copyright 2024, American Chemical Society. | ||
Beyond the widely studied tetra-coordinate BF2 luminophores, Yin and co-workers have demonstrated that sterically shielded tri-coordinate organoboranes can simultaneously furnish RTP and fatigue-resistant mechanochromism without resorting to tetra-coordinate BF2 motifs.61 Two donor–acceptor systems, 68 and 69, carrying bulky Mes* or 2,6-bis(trifluoromethyl)phenyl substituents, are air/moisture-stable and exhibit >92% ΦPL in solution (Fig. 10). In the crystalline phase, multiple C–H⋯π, C–F⋯π and C–H⋯F contacts (2.48–2.80 Å) promote aggregation-induced intersystem crossing, giving intense green phosphorescence (λ = 495–520 nm, τ = 52–62 µs) together with weak purple fluorescence (λ ≈ 400–416 nm, τ < 4 ns). Mechanical grinding disrupts long-range order and switches emission to fluorescence-rich purple. While 68 requires heating (150 °C, 20 min) to regenerate the original green emission, 69 shows ambient “self-recovery”, with the green phosphorescence returning within 4 h at 25 °C thanks to –CF3 enhanced self-crystallization. A 5 wt% 68 doping PMMA film yields pure white emission (CIE = (0.33, 0.34), ΦPL = 47%), and solution-processed single-molecule WOLEDs deliver white EL at CIE (0.31, 0.33) with EQE = 0.49%. These results establish heavily protected tri-coordinate organoboranes as a versatile platform for multi-stimuli-responsive solid-state lighting materials.
 | ||
| Fig. 10 Molecular structures of compounds 68 and 69, and their corresponding emission spectra and photographs under UV irradiation showing dual fluorescence-phosphorescence emission for white light emission and mechanochromism. Reproduced with permission from ref. 61. Copyright 2023, John Wiley & Sons, Inc. | ||
At the end of this section, selected examples exhibiting uncommon output profiles or distinctive operating mechanisms are highlighted. A notable example is triarylborane-decorated [2.2]paracyclophane 70, reported by Zhao and co-workers (Fig. 11).62 Contrary to the anticipated CPL activity,63 this pseudo-gem-substituted cyclophane displays unusual solid-state three-color MCL: green crystals (497 nm, ΦPL = 0.62) convert to a yellow amorphous phase (515 nm, ΦPL = 0.60) upon grinding and revert completely after annealing at 120 °C; solvent vapor exposure yields a denser polymorph that emits cyan light (473 nm, ΦPL = 0.87). The rigid, π–π-contact-free lattice suppresses aggregation caused quenching (ACQ) and progressively restricts excited-state planarization, blue-shifting emission while maintaining high brightness. Reversible acidochromic luminescence (ACL, TFA/TEA) further furnishes a blue state at 429 nm (ΦPL = 0.31), enabling four-color solid-state fluorescence switching from a single molecule.
 | ||
| Fig. 11 Molecular structure of 70 and photographs under UV irradiation after different treatments. Reproduced with permission from ref. 62. Copyright 2024, John Wiley & Sons, Inc. | ||
In 2022, Wagner and co-workers leveraged the intrinsic temperature sensitivity of the TADF manifold to develop a color-tuneable, room-temperature afterglow system (Fig. 12).64 Two laterally expanded 9,10-dimesityl-9,10-diboraanthracenes bearing lateral acenaphthylene or phenanthrene donors and weakly donating mesityl protecting groups 71 and 72 were rationally designed, thereby establishing an intramolecular charge-transfer framework. When dispersed in a rigid PMMA matrix, both chromophores exhibit intense and persistent afterglow emission. Photophysical study reveals that 71 delivers red afterglow centered at 614 nm with ΦPL of 3%, a mono-exponential lifetime of 0.42 s and a visually detectable duration of 5 s. In contrast, 72 produces blue-green afterglow at 531 nm, an outstanding ΦPL of 15%, bi-exponential lifetimes of 0.17 s and 0.93 s, and a prolonged emission lasting up to 25 s. Each spectrum consists of a high-energy TADF band and a low-energy RTP band; the relative contribution of the former increases monotonically with temperature, enabling precise chromatic tuning. Consequently, the afterglow of 71 shifts from red to orange-yellow, whereas that of 72 moves from green to blue-violet. Theoretical calculations indicate that ICT-derived states together with a dense manifold of nearby triplet levels create efficient intersystem-crossing pathways, while the T1 state exhibits exceptionally low radiative and non-radiative decay rates, ensuring a long-lived triplet reservoir. Moreover, intermediate-state-mediated reverse intersystem crossing permits pronounced TADF activation even when ΔE(S1–T1) exceeds 440 meV, thereby affording sensitive and reversible temperature control over both the color and intensity of the afterglow emission.
 | ||
| Fig. 12 Molecular structures of stimuli-responsive boron containing PAHs. | ||
Wang and co-workers subsequently retained the boron-doped anthracene core but pushed boron incorporation to its structural limit. In 2023 they reported acenes 73–76, in which four boron atoms are embedded within a single polycyclic framework – the highest boron density yet achieved in such scaffolds.65 Among them, 75 uniquely integrates mechanochromic and Lewis-base-responsive solid-state emission. In toluene it shows a record ΦPL of 90% (λem = 559 nm). Upon grinding, its loose lamella packing enables a 91 nm mechanochromic shift (576 → 605 nm) with high CIE contrast (green → red), the largest ever recorded for boron-doped PAHs.66 This dual-stimuli platform affords three bright, fatigue-free emission states and highlights the potential of high-level boron doping for next-generation responsive luminescent materials.
Another interesting study was reported by Yang and co-workers who reported the high-yield synthesis of a new class of polycyclic aromatic helicene molecules.67 By incorporating nitrogen–boron–nitrogen (NBN) units and seven-membered-ring defects, the photoluminescence performance of the materials was markedly enhanced. 77, a tetra-helicene compound bearing two NBN units, and a double helicene compound 78 containing two NBN-doped heptagons were prepared (Fig. 12). Both compounds exhibit outstanding ΦPL (99% for 77 and 65% for 78 in toluene), with extremely narrow full-width-at-half-maximum (FWHM) values of 24 nm and 22 nm. All derivatives show fluoride-ion responsiveness. Notably, in contrast to the fluorescence quenching usually observed for tri-coordinate boron compounds upon fluoride binding, helicene 78 undergoes a stepwise coordination with fluoride. Its emission successively shifts from the original green (522 nm) to orange (567 nm, 78-F1) and then to yellow (541 nm, 78-F2), while the ΦPL increases dramatically from 65% to 99% (78-F1) and 90% (78-F2). The process is fully reversible by addition of BF3·OEt2. Single-crystal X-ray diffraction and theoretical calculations reveal that fluoride coordination enhances intramolecular charge transfer, reduces the HOMO–LUMO gap, and alters molecular conformation, which are the key mechanisms enabling the luminescence tuning.
Recently, the Dou group further expanded the scope of boron-containing functional materials. By leveraging B/N- and B/O-PAHs, they constructed a novel family of boron-containing organic diradicaloids. Upon addition of a Lewis base, Lewis acid–base adducts are formed, enabling dynamic tuning of (anti)aromaticity and the diradical characteristic in organic diradicaloids, as well as the assembly of supramolecular diradicaloids.68,69 Intriguingly, their subsequent work demonstrated that B/N-type diradicaloids can also serve as effective photothermal materials.70 Two organic diradicaloids (Fig. 12, 79 and 80) featuring B/N-heterocycle fusion and enhanced quinoidal conjugation were designed for this purpose. These structural features confer increased open-shell characteristics, red-shifted NIR absorption (ca. 900 nm), and excellent ambient stability. Although the molecules lack conventional stimulus-responsive moieties, their nanoparticulate formulations exhibit robust photothermal behavior: NIR irradiation elicits efficient heat generation and tumor ablation. This reflects a functional photoresponse rather than a molecular-level structural transformation.
3. Responsive systems based on boron-containing polymers
The past decade has witnessed tremendous advances in the synthesis of main-group-element-containing polymers,71,72 especially boron-containing polymers,73–75 which have attracted significant research interest. This is largely due to their ability to combine intrinsic functionality with the practical advantages of macromolecular matrices – such as flexibility, low weight, and facile processability.76 By leveraging the empty p-orbital of the boron center and its highly electron-deficient nature, which enables the formation of dative bonds with Lewis bases, countless opportunities have emerged not only in catalysis but also in the development of stimuli-responsive, sensory, and supramolecular materials.77,78Boron can be incorporated into polymer frameworks via three well-established strategies: (1) direct integration of boron-centered units into the π-conjugated backbone to enhance electronic communication;79,80 (2) pendant attachment of boron-containing side chains that provide specific recognition or reactive sites;81 and (3) dynamic B–X cross-linking that leverages the reversible nature of B–X dative bonds to construct adaptable networks.82 In this section, recent representative advances in polymeric organoboron-responsive materials are surveyed according to these three categories. Given the vast number of studies in this field and the existence of several excellent review articles,16,71,73,83–85 only the most recent representative examples based on luminescent organoboron-containing polymers will be discussed. Macromolecules designed for self-healing and shape-memory applications are beyond the scope of this review.
In the first category, boron is directly embedded within the π-conjugated backbone. The low-lying vacant orbital of the boron center effectively extends the conjugation length, giving rise to distinct optoelectronic properties and pronounced environmental sensitivity. Additional functionalities can be readily introduced through controlled aggregation, side-chain engineering and/or integration with complementary building blocks,86 collectively conferring boron-containing π-conjugated polymers with high sensitivity and broad application potential. Tanaka et al. illustrated this concept with a series of donor–acceptor polymers 81–86 (Fig. 13a) prepared by alternating an O^N^O-chelate boron-fused azomethine (BAm) acceptor with a bithiophene (BT) donor whose alkyl side chains were systematically shortened from dodecyl to hydrogen.87 In dilute chloroform solution, progressive shortening of the BT-bound alkyl substituents from dodecyl (82) to hydrogen (86) red-shifted the emission maximum from ca. 632 nm to 683 nm while maintaining a high absolute ΦPL (0.27). Theoretical study shows that desubstitution reduces the inter-thiophene dihedral angle, leading to planarizing the backbone and narrowing the HOMO–LUMO gap. Spray-coated films of long-chain (≥hexyl) polymers retained ΦPL up to 0.33–0.42 at 635–644 nm due to sterically hindered π-stacking, whereas short-chain or non-alkylated analogues exhibited ACQ (ΦPL 0.02–0.07). Remarkably, DCM vapor annealing induced an unusual film-state “red-shift” response: 83 and 85 films exhibited an additional ca. 40 nm red-shift, pushing the emission to 685–709 nm in the near-infrared region with ΦPL preserved at 0.36 and virtually no loss in efficiency, which is attributed to vapor-induced side-chain swelling and backbone planarization rather than polymorphic transitions, and demonstrate that the magnitude of the response can be predictably tuned by the side-chain length. Building on this work, they further expanded the ligand platform to N^N^O-chelate BAm derivative monomers and incorporated the moderate donor fluorene (FL) to generate polymers 87–90 (Fig. 13b).88 These materials display luminescence both in solution and as films. Replacing the ligand oxygen with nitrogen endows the polymers with acid-responsive behavior: exposure to HCl vapor produces a reversible red-shift in photoluminescence that is erased by base vapor. Furthermore, the boron chromophore is highly sensitive to solvent polarity. Bilayer films of 87 and poly(n-butyl methacrylate) (PBMA) undergo chloroform-vapor-induced interfacial mixing that gradually blue-shifts the emission (orange → yellow → green), providing a built-in optical read-out for evaluating the vapor permeability of protective membranes.
 | ||
| Fig. 13 (a) Structures of boron-fused azomethine (BAm) conjugated copolymers 81–86 and their film photoluminescence (PL) spectra; films were prepared by spray-coating followed by annealing. (b) Design of nitrogen-substituted BAm polymers 87–90 and PL spectra of 87/PBMA bilayer films before and after chloroform-vapour annealing, together with PL spectra of 87/PBMA dispersion films and photographs of the solvent-vapour permeability test. Reproduced with permission from ref. 88. Copyright 2023, American Chemical Society. (c) Molecular design of 91 and 92 and photographs of a drop-cast 91 film before and after piercing with a syringe needle. Reproduced with permission from ref. 89. Copyright 2024, American Chemical Society. (d) Molecular structure of polymer 93 and its PL spectra (1.0 × 10−5 M per repeating unit, THF/buffer pH 9, 9:1 v/v) recorded in the presence of 0–1000 equiv. D-(−)-fructose. Reproduced with permission from ref. 90. Copyright 2024, American Chemical Society. | ||
As discussed in the previous section, β-diketiminate boron complexes have been well documented for their MCL properties. Considering the superior advantages of conjugated polymers in optoelectronic properties, developing conjugated polymer based MCL materials is of great importance. Thus, embedding a boron diketiminate moiety into the main chain offers a straightforward route to MCL conjugated polymers. To this end, conjugated polymers 91 and 92, prepared by directly alternating a boron pyridylenolate complex acceptor with a BT donor were designed (Fig. 13c).89 This direct linkage effectively improves inter-chain interactions and environmental sensitivity. Polymer 91 exhibits bright luminescence at 574 nm (ΦPL = 34%) in dilute DCM. The spin-coated film is red-shifted to 591 nm (ΦPL = 4.3%) due to planarization and aggregation. Critically, both powder and thin-film samples show irreversible MCL: gentle grinding blue-shifts the emission from 641 nm to 623 nm and changes the color from red to orange, while ΦPL increases from 2.8% to 5.3%. The orange signal persists for >2 weeks even after 80 °C thermal annealing. Single-crystal and TD-DFT analyses reveal that the BF2 substituent affords strong π–π stacking; mechanical disruption of this packing decreases the non-radiative rate and produces the observed blue-shift and quantum-yield enhancement. In contrast, the BPh2 analogue 92 shows negligible mechano-response because of steric hindrance and weak intermolecular interactions. Exploiting these features, the authors fabricated <200 µm-thick films of 91. Puncture with a 20 G needle creates a permanent orange spot around the hole, realizing for the first time a shear-force “write-and-memory” function in a conjugated-polymer film and offering a high-spatial-resolution optical recording material for stress mapping and security tagging.
When the embedded boron center is tri-coordinate, the sp2 ↔ sp3 coordination change familiar from small-molecule systems can be translated into macromolecular stimuli responsiveness. For example, via Migita–Kosugi–Stille copolymerization, tri-coordinate cyclic benzoxaborin was incorporated directly into a π-conjugated backbone alternated with a bithiophene donor, yielding polymer 93 (Fig. 13d).90 At pH 9 (THF/buffer, 9:1 v/v), 93 binds D-(−)-fructose in a reversible 1:1 fashion:titration (0 → 1000 equiv.) red-shifts absorption from 505 to 525 nm and converts emission from blue (ca. 505 nm) to green (ca. 525 nm), while the absolute ΦPL increases from 51% to 59% (K = 1.18 × 103 M−1). DFT calculations reveal that diol coordination converts boron from tri- to tetra-coordinate, elevating the HOMO and narrowing the band gap; enhanced rigidity lowers the non-radiative rate from 7.2 × 108 to 5.1 × 108 s−1, simultaneously color-switching and brightening the emission. Glucose or sucrose, which lack cis-1,2-diols, induce <2 nm shifts, confirming selectivity. Leveraging this binding-induced color switch, an assay for urinary glucose was demonstrated: addition of 50 mg dL−1 glucose to a 93 solution produces a naked-eye color change from blue-green to green within seconds, providing a proof-of-concept for instrument-free, point-of-care glycosuria detection.
Besides tetra-coordinate boron centers, tri-coordinate boron moieties have also been widely used in the main chain conjugated polymers. Jäkle and co-workers systematically engineered the regiochemistry of boron attachment (2- vs. 3-position of thiophene) and the electronic characteristic of the protecting groups (2,4,6-tri-tBu-phenyl vs. 2,4,6-tri-CF3-phenyl) to construct two classes of alternating borane–terthiophene conjugated polymers 94–97 (Fig. 14a),91 elucidating how regioisomerism governs optoelectronic and stimuli-responsive behavior. Polymers in which boron is anchored at the 2-position (94 and 95) exhibit strong p–π conjugation, narrow optical band gaps (2.25–2.38 eV), high fluorescence quantum yields (up to 32% for 95) and pronounced positive solvatochromism, with emission red-shifts >20 nm in polar solvents, indicating significant intramolecular charge transfer in the excited state. Reversible luminescence modulation is achieved upon Lewis-base coordination: partial pyridine complexation quenches emission, whereas addition of BF3·OEt2 scavenges the base and restores fluorescence, affording an “off–on” switching response. In contrast, 3-linked analogues (96 and 97) suffer from cross-conjugation that disrupts π-delocalization, resulting in wider band gaps (∼2.70 eV), blue-shifted emission, lower quantum yields (<10%) and weaker solvent sensitivity. These trends are corroborated by DFT calculations, which reveal that 2-linked systems exhibit lower-lying LUMOs that facilitate ICT, whereas 3-linked models exhibit symmetry-forbidden or weakened ICT transitions.
 | ||
| Fig. 14 (a) Chemical structures of tri-coordinate boron conjugation polymers 94–97. The photographs show the polymer solutions under ambient light (top) and UV irradiation at 365 nm (bottom). Reproduced with permission from ref. 91. Copyright 2023, John Wiley & Sons, Inc. (b) Representative π-conjugated BN-containing organic polymers (Mes = 2,4,6-trimethylphenyl; Tip = 2,4,6-triisopropylphenyl) and the target p-conjugated organic polymer 98; (c) photoluminescence of 98 recorded in various solvents and in a PMMA matrix. (d) Solid-state emission profiles of 98 measured under different environments. Reproduced with permission from ref. 94. Copyright 2025, John Wiley & Sons, Inc. | ||
Beyond the established boron-containing π-conjugated polymers that rely on B–C linkages, the strategic insertion of >BN< fragments into π-conjugated scaffolds has emerged as a versatile route to new materials.92 Until the past few years, the deliberate incorporation of >BN< motifs into conjugated polymer backbones remained largely unexplored.93 To introduce a permanent dipole along the backbone, recently Helten and co-workers synthesized an unprecedented poly(p-phenylene iminoborane) 98 featuring a strictly alternating BN sequence via AB-type polymerization of an N- and B-terminated monomer (Fig. 14b).94 Monodisperse oligomers containing up to three BN units were prepared and fully characterized. The regioregular BN backbone endows both polymer and oligomers with pronounced solution fluorescence, large Stokes shifts and AIEE, distinguishing them from earlier BBNN-sequenced congeners. As a PMMA film, 98 exhibits a single, narrow, intense blue-emission band with a fluorescence quantum yield of 65% (Fig. 14c). Mechanical grinding red-shifts and broadens this band reversibly, demonstrating clear mechanochromic behavior (Fig. 14d). This response opens opportunities for modulation by diverse stimuli—solvent polarity, viscosity, temperature and mechanical forces alike.
Compared with conjugated polymers that incorporate boron into the main chain, boron moieties are more frequently appended as side chains because of the inherently simpler synthetic protocols. This architectural choice allows the responsive boron fragment to retain its intrinsic sensitivity toward external stimuli. Additionally, multiple functionalities can be further introduced via co-monomers incorporation. Consequently, the corresponding polymers are expected to exhibit amplified and/or multi-modal responses to external stimuli through the emergence of intermolecular interactions during aggregation or self-assembly processes.
In general, borane-functionalized polymers are prepared by two strategies: (1) direct polymerization of pre-functionalized boron-containing monomers and (2) post-polymerization modification that attaches the borane to a reactive side chain. The first route demands careful selection of the borane structure and the polymerization conditions to prevent decomposition of the boron functionality or contamination by unreacted precursors and side products. To date, several robust methods have been established for polymerizing a variety of boron-decorated monomers, including reversible-deactivation radical polymerization of boron-functionalized styrenes,95 Pd-catalyzed vinyl-addition polymerization,96 and ring-opening copolymerization of epoxides with functional anhydrides.97 These techniques enable tri- and tetra-coordinate borane monomers to be converted into homopolymers,98 random copolymers,99 and block copolymers100 (Fig. 15a).
 | ||
| Fig. 15 (a) Selected examples of poly(triarylborane)s and poly(borate)s obtained by direct polymerization. (b) Molecular structure of the block copolymer PNIPAM-b-PBM 99 and a schematic illustration of the dual-responsive fluoride sensor based on 99 micelles in DMF/THF (99:1 v/v). Reproduced with permission from ref. 101. Copyright 2013, American Chemical Society. (c) Molecular structures of polymers 100–103 and their corresponding monomers M1–M4. | ||
For example, Jäkle et al. incorporated electron-deficient triarylboranes into a reversible addition–fragmentation chain transfer (RAFT) controlled radical system to synthesize the well-defined, narrow-dispersity amphiphilic block copolymer PNIPAM-b-PBM 99 (Fig. 15b).101 Taking advantage of the strong Lewis acid–base interaction between the empty boron p-orbital and F−, they achieved 0.1 ppm, second-scale fluorescence quenching in THF with a 10-fold sensitivity increase over the monomer. In DMF/THF mixtures, F− binding triggers charge inversion that spontaneously disassembles the block copolymer micelles, accompanied by a sharp drop in turbidity and complete emission turn-off, enabling naked-eye dual-signal detection. As of this writing, the same group further realized the controlled polymerization of pre-formed triarylborane-functionalized norbornenes through a ring-opening metathesis polymerization (ROMP) method (Fig. 15c).102 Their work demonstrates that via this general approach highly Lewis acidic borane moieties can be introduced into polymers which reach B(C6F5)3 level. Under mild conditions, both homo- and copolymers (100–103) can be obtained. Systematic variation of the aryl substituents and linker steric bulk enables synergistic electronic–steric tuning: the most electron-deficient monomer M2 exhibits 83% of the Lewis acidity of B(C6F5)3, and its homopolymer 101 displays hierarchical, fully reversible coordination in solution. Its 11B NMR shifts from 62 ppm to 8.7 ppm upon exposure to acetonitrile (weak base) and collapses to ≈0 ppm with one equiv. of pyridine (strong base), indicating the quantitative conversion to tetra-coordinate mode. Subsequent addition of BF3·OEt2 quantitatively liberates the free borane, affording a robust “capture-release” cycle. Homopolymer 100 emits at 496 nm in DCM (ΦPL = 15%) and undergoes a 37 nm hypsochromic shift in thin films. Its emission is partially quenched by pyridine coordination, but is restored when BF3·OEt2 scavenges the base, realizing an “off–on” luminescence switch. In contrast, homopolymer 103, bearing a less electron-deficient boron center, shows inertness toward base coordination, emitting deep-blue light at 427 nm with high air/moisture tolerance. This study represents the first polymeric framework that unites tunable super-electrophilicity, reversible host–guest chemistry, and emission modulation on a single ROMP scaffold, offering a versatile route to smart dynamic optoelectronic materials.
Beyond triarylborane-appended stimuli-responsive polymers, electron-deficient borinic acid moieties have emerged as promising monomers for constructing smart polymeric materials. In 2017, Wan and co-workers reported a class of electron-deficient borinic acid polymers (PBAs, 104, Fig. 16) synthesized via RAFT polymerization, achieving well-controlled molecular weights and narrow dispersity.103 These PBAs exhibited upper critical solution temperature (UCST) type thermo-responsive behavior, with phase-transition temperatures tunable from 20 to 100 °C in DMSO by adjusting the water content (0–2.5 vol%). More importantly, 104 demonstrated sensitive fluorescence recognition toward F−, Alizarin Red S (ARS), and 8-hydroxyquinoline (HQ), attributed to the formation of a highly emissive boron quinolate complex via dative N → B interaction. Building on this foundation, the same group further advanced the system by revealing that the borinic acid moiety itself can function as a polymerization-induced emission (PIE) luminogen, eliminating the need for external fluorophores or coordinative emission.104 Through RAFT polymerisation of non-emissive borinic-acid monomers, intrinsically luminescent PBAs 105 were directly obtained (Fig. 16), exhibiting blue emission (λ ≈ 349 nm for 105-Et) despite their non-emissive monomers and model compounds. DFT calculations revealed that as the number of repeat units increased, the HOMO–LUMO energy gap narrowed, supporting a through-space conjugation mechanism responsible for the observed PIE. These PBAs retained their thermo-responsive behavior (UCST ≈ 34–90 °C depending on substituent and solvent composition) and chemical sensitivity toward F−, ARS, and HQ, with fluorescence quenching or enhancement depending on the analyte. Together, these studies map an “all-in-one” roadmap that merges multi-responsiveness with intrinsic emission, propelling PBAs into smart luminescent materials.
 | ||
| Fig. 16 Borinic acid polymers 104 and 105 with different substituents and their stimuli responsive mechanism toward fluoride ions, ARS and HQ. Reproduced with permission from ref. 104. Copyright 2024, Elsevier. | ||
Alternatively, boron moieties can be introduced into the side chains via post-functionalization strategies. This approach offers significant advantages, most notably the ability to access a diverse range of functional materials from a single precursor polymer.81,105 It enables convenient access to polymers bearing triarylboranes, boronic acids and esters, luminescent tetra-coordinate organoboron hydroxyquinolates, as well as tetra-arylborates with weak or strong coordination abilities.106 For example, Jäkle and coworkers developed well-defined borane-end-functionalized PS via atom transfer radical polymerization (ATRP) combined with a silicon–boron exchange reaction. The resulting polymers can be readily functionalized into various end groups, such as boronic acids and triarylboranes, which are capable of reversibly interacting with pyridine bases or water to form supramolecular structures through Lewis acid–base interactions or boroxine linages. This strategy offers new avenues for the development of advanced stimuli-responsive soft materials.107 Besides static covalent linkages, boron moieties can be installed in the side chain through dynamic covalent bonds that are kinetically stable under ambient conditions yet become labile upon heating, pH jump, light or mechanical stress.108 This “two-birds-with-one-stone” tactic simultaneously installs the luminescent center and the stimulus-responsive site, and has therefore gained momentum.
Among the dynamic linkages, B–O bonds are particularly attractive because boron readily forms reversible complexes with Lewis bases and nucleophiles. Boronic acids, boronic esters and boroxines have consequently become routine building blocks for adaptive polymers. Moreover, aryl-boronic acids and their esters were recently found to display bright phosphorescence in rigid matrices or under mechanical stimulation, extending the scope to stimuli-responsive RTP materials.109–111 A representative example was reported by Li and co-workers.112 An AIE-active phosphor 1,1′:3′,1″-terphenyl-5′-boronic acid (DPP-BOH, 106, Fig. 17a) carrying a single boronic acid group was synthesised via Suzuki–Miyaura coupling and exhibited lifetimes of 5.5 s and 2.2 s in solution and solid under ultralow temperature. By dehydrative condensation with poly(vinyl alcohol) (PVA), 106 was grafted onto the polymer backbone through dynamic B–O bonds, producing a flexible film that displays an ultralong RTP lifetime of 2.43 s and a ΦPL of 7.51% at room temperature. The boron unit fulfils a dual role: it acts as the phosphorescent chromophore and, through covalent cross-linking, suppresses molecular motion and rigidifies the matrix. The film is reversibly modulated by water and heat: water disrupts the PVA hydrogen-bond network and quenches RTP, whereas heating removes water and restores the rigid environment, recovering the afterglow. Doping the film with fluorescein or rhodamine B further enables yellow (1.60 s) and red (1.90 s) afterglow via efficient triplet-to-singlet FRET, while retaining water/heat responsiveness. This fully aqueous-processable platform paves the way for eco-friendly, printable and multi-color smart phosphorescent materials in anti-counterfeiting, information encryption and bio-imaging.
 | ||
| Fig. 17 (a) Synthetic route to 106-PVA and reversible thermal-/water-gated modulation of its intermolecular interactions. Water-fumed 106-PVA films were heated for 15 min at 30–80 °C (10 °C intervals) and cooled to room temperature, showing temperature-dependent RTP spectra and decay traces together with corresponding photographs. Reproduced with permission from ref. 112. Copyright 2022, Springer Nature. (b) Chemical structures of boronic acids 107 and 108 and schematic illustration of the preparation of their PVA films. (c) Emission photographs of the 1% 107-PVA@5% 108-PVA (1:1) film in the dry and wet states under 365 nm UV irradiation and after ceasing irradiation, respectively; and (d) an anti-counterfeiting demonstration of the bifunctional doped film. Reproduced with permission from ref. 113. Copyright 2024, John Wiley & Sons, Inc. | ||
Spurred by this ready attainment of second-scale RTP lifetimes via boron–PVA dehydration chemistry, ultra-long room-temperature phosphorescence (URTP) materials have rapidly come to the fore. Zhang et al. developed all-aqueous, multi-stimuli-responsive URTP polymer films by incorporating a naphthalene-based pyridinium bromide (107) and a viologen photochromic arylboronic acid (108) into PVA (Fig. 17b).113 The 1%-107-PVA film emits cyan light at 507 nm (τ = 9.2 ns) with an RTP lifetime of 1.34 s and an 8 s visible afterglow; the corresponding results for the 5%-107-PVA film were 513 nm, 9.6 ns, 1.11 s and ∼7 s. The 5%-108-PVA film is non-phosphorescent but turns blue (613 nm radical absorption) within 1 min of 365 nm irradiation and fades instantly upon wetting. Blending 107 and 108 into a single matrix produces films that integrate >6 s afterglow, reversible photochromism and water responsiveness: 1%-107-PVA@5%-108-PVA-1-1 shows 490/515 nm dual emission, 6.8 ns fluorescence, 1.16 s RTP and 7 s afterglow. After 5 min irradiation the film turns blue, the lifetime drops to 0.80 s and afterglow shortens to 3 s, while wetting red-shifts emission to 538 nm and completely quenches RTP, with full reversibility upon drying. The system enables an eight-level, reversible anti-counterfeiting cycle on paper or cloth, offering a green, printable and multi-level information-encryption platform (Fig. 17c and d).
Recently, the boron moiety has also been used to construct 3D network polymer systems. For example, Ren et al. exploited the tri-coordinate binding mode of boron to construct a spatially networked polymer and reported a class of conjugated porous polymers (B-CPPs, 109–112, Fig. 18) featuring unprotected, high-density boron centers.114 Through B/Sn exchange polycondensation, commercially available BBr3 was directly incorporated into thiophene-bridged backbones, forming a three-dimensional rigid network that achieves the high Lewis acid density and strong p–π* conjugation. The resulting network polymers emit solid-state fluorescence tunable from 580 to 660 nm (maximum ΦPL = 8.5%, lifetime ca. 1 ns) and exhibit Brunauer–Emmett–Teller (BET) surface areas up to 649 m2 g−1; 11B MAS NMR δiso values of 44–46 ppm confirm strong electronic communication between tri-coordinate boron centers. Upon exposure to triethylamine or pyridine vapor, polymer fluorescence is sequentially quenched within 600 s. For 109, its pyridine uptake reaches 570 mg g−1, forming stable Lewis acid–base adducts and raising the thermal decomposition temperature of the adduct by 19 °C. This work is the first to introduce sterically unhindered boron centers into CPPs, combining high surface area, strong Lewis acidity, and reversible amine responsiveness.
 | ||
| Fig. 18 B-CPP polymers 109–112, and the proposed PL quenching mechanism of the B-CPPs. The inserted photograph shows the photoluminescence of 109 toward pyridine. Reproduced with permission from ref. 114. Copyright 2022, American Chemical Society. | ||
As outlined in the preceding section, the reversible B–O bond has made boronic acids and boroxines privileged building blocks for dynamic covalent polymers. Beyond their incorporation as side-chain motifs, these units can be used to construct macroscopic networks. Boronic acids condense rapidly with 1,2- or 1,3-diols to give boronic esters, whereas boroxines are generated by the dehydrative trimerization of boronic acids. These two orthogonal reactions furnish a versatile platform for dynamic covalent networks whose bond exchange can be modulated facilely by pH or ethanol concentration,115,116 enabling the design of a wide range of smart materials.117,118 For example, Feng and coworkers integrate dynamic boronic ester bonds into main-chain cholesteric liquid-crystal elastomers (CLCEs) to create intelligent photonic materials which exhibit unprecedented stimulus-responsive programmability and autonomous self-healing properties (Fig. 19a).119 In this study, they designed and synthesized a boron-containing dithiol, 2,2′-(1,4-phenylene)-bis[4-mercaptan-1,3,2-dioxaborolane] (BDB), in which the 1,3,2-dioxaborolane rings undergo facile B–O bond scission and re-formation under mild conditions, thereby enabling continuous topological rearrangement of the polymer network. This BDB monomer was formulated together with the commercial mesogen RM257, the chiral dopant LC756, the flexible spacer EDDET and the tetra-functional thiol cross-linker PETMP. The mixture was processed by a two-stage thiol-acrylate Michael addition followed by UV photopolymerization to yield free-standing CLCE films ca. 500 µm thick. Upon mechanical stretching, the CLCE films exhibit vivid and reversible mechanochromic behavior across the entire visible spectrum, with the reflection peak linearly blue-shifting from 659 nm to 468 nm as strain increases from 0% to 120%. Furthermore, by fixing a 60% uniaxial strain at 80 °C for 4 h, the authors programmed a red-reflecting CLCE into a green, monodomain actuator that retains both the new color and elongated length upon release. Upon cyclic heating/cooling between 25 °C and 100 °C, the film reversibly contracts and expands, lifting a 15 g load (≈300× its own weight) with a work density of 20.33 kJ m−3, thereby demonstrating simultaneous 4D color-shape actuation.
 | ||
| Fig. 19 (a) Molecular structures used for fabricating CLCEs. (b) The CLCE films reflecting red, green, and blue colors with a chiral dopant content of 4.5, 6.0, and 7.5 wt% of the total diacrylate monomers, respectively. Multi-colored CLCE films were prepared by pouring different CLCE precursors onto one single substrate (scale bar = 5 mm). (c) Photographs of a red-reflecting CLCE film being mechanically stretched. Reproduced with permission from ref. 119. Copyright 2022, John Wiley & Sons, Inc. (d) Synthesis route of 113 and 114. Reproduced with permission from ref. 121. Copyright 2024, John Wiley & Sons, Inc. | ||
However, considering the intrinsic susceptibility of both boronic esters and boroxines to hydrolysis or alcoholysis, in most cases their practical applications have been limited.120 To address this drawback, an effective strategy has been developed that introduces Lewis-basic atoms (N, O) as intra- or intermolecular electron donors. Coordination to the vacant p-orbital of boron converts the atom from sp2 to sp3, changes its geometry from planar to tetrahedral, and endows the network with additional responsiveness while simultaneously suppressing hydrolytic degradation. For example, He et al. constructed a new class of boron-containing hyperbranched polysiloxane stimuli-responsive luminescent polymers 113 and 114 (Fig. 19d).121 A cyclic boronic ester crosslinker-2,2′-(1,4-phenylene)-bis[4-mercaptan-1,3,2-dioxaborolane] (BDB)-was embedded into the network as a tetracoordinate boron node; the resulting B←N or B←OC interactions generate tetrahedral sp3 boron centers that act both as dynamically reversible B←X switches and as emissive/recognition sites. This dithiol-functionalized boronic ester was co-polymerized with multivinyl siloxane via hydrosilylation followed by self-cross-linking, affording free-standing 3D network films whose cross-linking density can be precisely tuned by varying the BDB feed (0–50 mol%). Systematic variation of solvent polarity and aromaticity shifts the emission of 113 continuously from blue (EtOH, 418 nm) to orange-yellow (Hacac, 588 nm) while maintaining a ΦPL of 9.15%—the highest value reported for red delayed-fluorescence unconventional polymers. Transient absorption and electrostatic-potential calculations reveal that n⋯π and hydrogen-bonding interactions between the solvent and the network equalize surface electrostatic potential (ESP) distributions, reduce ΔEST (0.068 eV) and amplify the reverse intersystem crossing (RISC) process, producing micro-second delayed fluorescence. Benefiting from the high surface area and Lewis-acidic boron sites of the boronic ester rings, the network film exhibits a colorimetric blue-to-green transition together with a ratiometric fluorescence increase at 503 nm toward F− in the 5–100 µM range, giving a detection limit as low as 5.33 µM with no interference from Cl−, Br− or NO3− at ten-fold higher concentrations.
4. Responsive systems based on boron-containing supramolecules
Boron-based supramolecules have evolved from structural curiosities into a versatile materials paradigm that seamlessly integrates molecular recognition, stimuli-responsiveness and cooperative function. In this section we showcase the most recent and representative architectures to illustrate the field; readers seeking a comprehensive survey are directed to the review by Jäkle and co-worker.16An efficient strategy for tailoring the structure and functionality of materials in their aggregated states involves the use of sophisticated supramolecular assembly methods. When aggregation is driven by complementary intermolecular interactions among preorganized monomeric units, it can lead to the formation of stable supramolecular nanostructures with defined dimensions and directional order, even in solution. These supramolecular constructs are inherently dynamic, enabling the development of stimuli-responsive soft materials such as gels, elastomers, and liquid crystals.122,123 Notably, the assembly of planar molecules through π–π stacking, along with other directional non-covalent forces, such as hydrogen bonding, halogen bonding, metal–metal interactions, electrostatics, and hydrophobic effects, has been extensively employed to engineer advanced supramolecular materials.
Assemblies with long-range order, constructed from boron-based building blocks, are now in the spotlight. Their distinctive molecular scaffolds and unusual photophysics open the door to previously inaccessible material functions. For example, triarylborane involving functional materials have received significant attention over the past few years due to their wide range of potential applications.124 Capitalizing on the outstanding luminescence of triarylborane, Han et al. constructed a tris-NHC ligand with a triarylborane core and assembled the isostructural hexacarbene cages with the metal–metal interactions (metal = Ag, Au).125 As shown in Fig. 20a, the Au complex 115 displays vivid solvatochromism: emission maxima shift stepwise from ≈450 nm in non-polar DCE/DCM/THF to ≈550 nm in polar DMSO/DMF/MeCN, indicative of a donor–acceptor CT characteristic. UV-Vis and fluorescence titrations show that the boron centers bind F− quantitatively. Addition of BF3·Et2O removes F− and restores emission, thus enabling fully reversible ON/OFF/ON fluorescence switching.
 | ||
| Fig. 20 (a) The triarylborane-based, discrete metal–carbene supramolecular cage 115 exhibits pronounced solvatochromic fluorescence and undergoes fluoride-selective quenching in both UV-vis absorption and emission titrations with TBAF. Reproduced with permission from ref. 125. Copyright 2023, The Royal Society of Chemistry. (b) UV-Vis and fluorescence titrations reveal that the aggregated boron-doped PAH 116 undergoes photo-dissociation upon DMAP binding; this process is reversibly modulated by TFA concentration, as visualized by color/fluorescence changes and schematized for its pH-responsive self-assembly in aqueous media. Reproduced with permission from ref. 126. Copyright 2022, The Royal Society of Chemistry. | ||
Recently, planarizable boron-doped PAHs have been advanced into aqueous-compatible, fully fused π-scaffolds that exploit a single B–N coordination toggle for multimodal fluorescence switching. Yamaguchi and co-workers appended hydrophilic chains to a rigid, all-sp2-carbon-bridged core, furnishing amphiphile 116 that quantitatively self-assembles into 4-nm bilayer H-aggregates in DMSO/H2O (Ke = 7.2 × 105 M−1, λabs = 510 nm) (Fig. 20b).126 The embedded boron center retains sufficient Lewis acidity to form a labile B←N adduct 116·DMAP that is non-emissive in the ground state yet undergoes quantitative photo-dissociation in the excited state, instantly restoring bright red emission (λem = 640 nm). Protonation of DMAP with TFA reversibly regenerates the tri-coordinate borane, re-establishing the H-aggregate and its 510 nm absorption within seconds. This acid/base/light-controlled, three-state cycle is fully reversible under physiologically relevant conditions.
As described in our discussion of small molecules, the boron atoms can play the role of Lewis acids and thus can form LP adducts with nitrogen-based Lewis bases. Building on this concept, numerous small-molecule-based stimuli-responsive systems have been reported. Using the same principle, boron–nitrogen LPs can also be assembled into supramolecular materials, such as macrocycles formed via B←N dative bonds. Indeed, such studies date back to 1991, when Niedenzu et al. reported the crystal structure of a macrocyclic (triazolyl)dimethylborane.127 Expanding on this foundation, Severin et al. have since broadened the structural scope to include a diverse array of motifs whose metrics and functions can be predictably tuned by modulating the electronic nature of the boron acceptor and/or the nitrogen donor.128 Moreover, Himmel et al. disclosed the first examples of highly charged cationic cyclophanes containing diboranyl units that exhibit distinctive redox behavior; in these systems, electron-rich diboranes and N-heteroarenes self-assemble into tetracationic diboranyl cyclophanes, wherein spontaneous electron transfer from the B–B bond to the heterocycle generates redox-active, electronically coupled macrocycles, heralding a new family of boron-based supramolecular redox materials.129
This conceptual foundation has recently been extended beyond traditional motifs toward highly symmetric, self-healing B←N macrocycles with engineered porosity and excited-state dynamics. For instance, Fei and co-workers have demonstrated a paradigm of stimuli-responsive supramolecular materials in which a single B←N dative bond acts as the sole actuator (Fig. 21a).130 Their “functional-molecular-wall” strategy (Fig. 21b) furnishes the quasi-pentagonal pillar macrocycle 117, which undergoes fully reversible, second-scale monomer ↔ pentamer interconversion between 303 and 403 K, driven by cleavage or re-formation of the B←N linkage as evidenced by temperature-dependent NMR/UV-vis/fluorescence titrations. The same bond is acid/base-switchable: protonation of the pyridyl-N with TFA disrupts the dative contact to generate the pyridinium salt, whereas addition of Et3N restores the pentamer, yielding naked-eye color switching (orange ↔ green) over >5 cycles without fatigue. Upon 477 nm photo-excitation, the macrocycle undergoes excited-state B←N dissociation, releasing monomeric boron PAH that competes radiatively with the intact pentamer and produces dual fluorescence (423 nm and 501 nm) plus micro-second-scale delayed fluorescence (τDF = 7.8 µs for pentamer, 11.8 µs for monomer). Lowering the temperature to 77 K freezes bond scission and simultaneously turns on millisecond phosphorescence (629 ms), providing a rare example of synchronized fluorescence–phosphorescence from a B←N assembly. The solid-state material is permanently porous (BET 554 m2 g−1) and exhibits ideal-adsorbed-solution-theory-predicted reverse C2H6/C2H4 selectivity of 1.61–1.75 at 298 K, completing a triple-responsive (thermal, pH, optical) platform that operates in both solution and the solid state.
 | ||
| Fig. 21 (a) The reversible cycling of 117 and monomer: the pentameric ring closes from, and reopens to, pyridinium salt pyBNH+ and monomer pyBN-p. (b) Conceptual illustration of a pillar-like macrocycle based on the B←N bonds and design strategy for the “functional molecular wall” synthetic protocol. Reproduced with permission from ref. 130. Copyright 2025, American Chemical Society. | ||
Building on these advances in discrete B←N macrocycles, we turned our attention to linear, main-chain systems that translate bond-cleavage events into macroscopic solution properties. As depicted in Fig. 22, Tian and co-workers simply mix a bis-boronic ester (B2) with a telechelic 1-phenyl-imidazole-terminated PDMS (N21) to generate the main-chain supramolecular polymer 118 through reversible B←N dative bonds.131 Incremental addition of TFA triggers instantaneous turbidity and a ca. 8-fold drop in specific viscosity (20.4 → 2.5 mL g−1), signaling acid-induced protonation of the imidazole nitrogen and rupture of the dative linkages. Subsequent titration with TEA fully restores optical clarity and viscosity (Fig. 22b). The inset confirms >5 TFA/TEA cycles without fatigue. Orthogonal control at the boron center is showcased in Fig. 22c: tetrabutylammonium fluoride (TBAF) sequesters the Lewis-acidic boron via fluoride coordination, again cleaving the B←N bridges and lowering viscosity, whereas boron trifluoride etherate (BTE) strips the bound fluoride and quantitatively rebuilds the polymer, returning the solution to its high-viscosity state. The inset evidences ≥3 consecutive TBAF/BTE cycles with identical on/off amplitudes. Thus, the same macromolecule delivers triple-addressable (acid/base, fluoride/Lewis acid, thermal) responsiveness within seconds at room temperature.
 | ||
| Fig. 22 (a) Structural formulae and schematic model of B2 and N21, illustrating the built-in stimuli responsiveness of 118 that is programmed through dative B←N linkages: independent fluoride binding/release at boron (B2) and protonation/deprotonation at nitrogen (N21). (b) Changes in the intrinsic viscosity ([η]) of 118 (50 mg mL−1, CHCl3, 298 K) upon sequential addition of TFA and TEA; the inset traces the cycling behavior for N21 protonation/deprotonation. (c) Changes in [η] of 118 (50 mg mL−1, CHCl3, 298 K) during alternating treatment with TBAF and BTE; the inset depicts the reversible fluoride capture/release cycle at B2. Reproduced with permission from ref. 131. Copyright 2025, The Royal Society of Chemistry. | ||
While the foregoing examples illustrate how B←N bond scission in solution can reversibly modulate viscoelastic properties, the question remains whether the same dative linkage can orchestrate more complex, multi-modal responses in the solid state. In an elegant showcase of crystal-engineering-directed reactivity, Hecke and co-workers utilized a single organoboron Lewis pair to access two distinct, visible-light-driven single-crystal-to-single-crystal (SCSC) photopolymer-ization pathways (Fig. 23a).132 They designed a 1:2 adduct (para-cyano-triphenylboroxine: 4-(1-naphthylvinyl)pyridine) (NPE) stitched by B←N dative bonds and isolated it as solvent-free needles monomer-1 or toluene-solvated blocks monomer-2. The solvent-free monomer-1 presents an unusual, non-Schmidt geometry in which the vinyl group of one ligand sits parallel to a naphthalene CC bond of a neighbor (d ≈ 3.72 Å). Visible-light irradiation therefore triggers a rare olefin–arene [2+2] cycloaddition that stitches the molecules into a syn-[3]-ladderane backbone (119, 37% conversion). In contrast, monomer-2 incorporates toluene in the lattice; the guest templated a face-to-face arrangement of two olefinic bonds (d ≈ 3.52 Å) that satisfies the topochemical criterion for a conventional [2+2] reaction, enabling quantitative (100%) SCSC photopolymerization to the cyclobutane-linked polymer 120 within seconds. Both processes are accompanied by ultrafast macroscopic photomechanical motions (jumping, bending or cracking) and are accompanied by bright green RTP (τ = 8–15 µs), underscoring the combined light-to-mechanical and light-to-emissive energy transduction accessible through B←N-coordinated organoboron crystals.
 | ||
| Fig. 23 (a) Chemical structure of monomers-1 and monomer-2, and synthesis of polymer 119 and 120via [2+2] SCSC. Reproduced with permission from ref. 132. Copyright 2024, American Chemical Society. (b) Synthesis of polymer 121via the visible light induced [2+2] SCSC. Reproduced with permission from ref. 133. Copyright 2024, John Wiley & Sons, Inc. | ||
On the other hand, based on a similar concept, Wei and co-workers constructed a B←N-coordinated host crystal 121 using a methoxycarbonyl functionalized triphenylboroxine core and NPE ligands (Fig. 23b).133 Upon irradiation with visible light (447 nm), the crystal undergoes a [2+2] photodimerization reaction in a SCSC manner. The reaction reaches 100% conversion within 9 min and triggers the release of ∼80% of the encapsulated guest molecules (e.g., ethyl acetate and n-hexane). Moreover, 121 crystals exhibit a solvent-induced allosteric effect: exposure to different solvents (e.g. toluene, alkanes) enables reversible structural transformation and selective guest capture/release. The release efficiency is strongly correlated with the boiling point and molecular structure of the guest. For instance, n-pentane is released with 80.3% efficiency, whereas cyclohexane shows only 15.8%, demonstrating remarkable molecular recognition and controlled-release capabilities. Solid-state photophysics further reveal a blue-shifted emission (495 → 449 nm) and enhanced ΦPL (1.35 → 8.6%), offering an optical reporter for the release process. The dynamic B←N scaffold additionally enables solvent-induced allosteric exchange, generating four single-alkane-guest crystals (n-Pen, CP, n-Hex, CH) whose release efficiencies scale inversely with guest boiling point and molecular bulk. By integrating photo-reactivity, structural robustness, and guest tunability, the authors establish a mild and efficient light-driven release platform, offering significant potential for smart supramolecular materials, molecular delivery systems, and light-responsive devices. Compared with the Hecke's systems that exploit similar B←N and [2+2] chemistry but pursue rapid (seconds) photomechanical motion and ladderane polymer formation, the 121 platform emphasizes mild-light-triggered guest ejection and solvent-tunable selectivity rather than macroscopic actuation, thereby complementing the growing family of multifunctional organoboron smart crystals.
Concluding remarks
Over the past decade, the unique combination of Lewis acidity and reversible sp2 ↔ sp3 rehybridization at boron has established organoboron scaffolds as a versatile platform for stimuli-responsive materials. Early demonstrations of simple fluorescence on/off switching have evolved into multi-color, ratiometric and RTP outputs that report optical, redox or mechanical events with molecular resolution. This review has charted this progression from small-molecule probes through π-conjugated polymers to supramolecular crystals, and has distilled design rules that link the vacant p-orbital of boron to quantifiable macroscopic responses. Collectively, these advances position organoboron platforms at the threshold of industrial uptake, provided the following challenges are overcome.Three persistent bottlenecks continue to hinder large-scale deployment: (1) the intrinsic air- and moisture-sensitivity of electrophilic boron centers, (2) synthetic routes that are either lengthy or give low isolated yields, and (3) a historical focus on single-stimulus rather than multi-stimuli responses. Recent studies have delivered pragmatic solutions. Bulky mesityl or Tip substituents, together with intramolecular B←X (X = O, N, S) dative locks, kinetically stabilize boron centers, affording chromophores that withstand boiling water or 100 °C air for weeks without measurable decomposition. Streamlined synthetic toolkits, such as catalytic C–H borylation, one-pot tandem cyclizations and solid-state BF2 exchange, consistently afford gram-scale quantities of B/N-, B/O- or B/S-doped scaffolds in isolated yields exceeding 70%. Mechanistically, the field has advanced beyond simple anion sensing. Contemporary platforms exploit TSCT, MR-TADF, frustrated Lewis-pair reactivity and multi-boron cooperativity to deliver reversible red-shifts, white-light emission, RTP and diradicaloid states triggered by light, heat, pH, mechanical force or redox inputs. These capabilities have already enabled single-molecule multimodal sensors, anti-counterfeiting inks and electrically switchable OLED pixels.
Looking forward, three challenges must be addressed to position organoboron materials as robust components in next-generation intelligent systems. First, multi-stimuli orthogonality must be encoded at the molecular level so that optical, electrical and mechanical inputs can be processed simultaneously with minimal crosstalk. This requires design heuristics that integrate orthogonal binding sites and independent output channels within a single B-centered motif. Second, scalable and environmentally benign processing routes, such as aqueous-compatible synthesis, melt-extrusion of boron-containing polymers and vapor-phase deposition of supramolecular films, must be developed to advance from milligram-scale demonstrations to large-scale manufacturing. Third, the well-established stimuli-responsive properties of small molecules must be systematically translated into macromolecular architectures; this will necessitate coordinated experimental campaigns and multiscale theoretical studies coupled with real-time analytical characterization. Achieving these targets will accelerate the discovery of durable, low-cost and sustainable boron platforms for the trillion-sensor era.
Conflicts of interest
There are no conflicts to declare.Data availability
All data discussed in this review, including figures and charts, have been sourced from previously published literature, with permissions for reuse obtained from the respective copyright holders where required.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant 22171024) and the Analytical and Testing Center of Beijing Institute of Technology.References
- A. Díaz-Moscoso and P. Ballester, Chem. Commun., 2017, 53, 4635–4652 RSC
.
- J. Cui, J. E. Kwon, H.-J. Kim, D. R. Whang and S. Y. Park, ACS Appl. Mater. Interfaces, 2017, 9, 2883–2890 CrossRef CAS PubMed
- M. E. McFadden, R. W. Barber, A. C. Overholts and M. J. Robb, Chem. Sci., 2023, 14, 10041–10067 RSC
- Y. Wang, Y.-M. Zhang and S. X.-A. Zhang, Acc. Chem. Res., 2021, 54, 2216–2226 CrossRef CAS PubMed
- L. Huang, C. Qian and Z. Ma, Chem. – Eur. J., 2020, 26, 11914–11930 CrossRef CAS PubMed
- Y. Wang, M. S. Shim, N. S. Levinson, H.-W. Sung and Y. Xia, Adv. Funct. Mater., 2014, 24, 4206–4220 CrossRef CAS PubMed
- J.-Y. Li, J.-C. Wang, J.-H. Zhou, K. Zhao, S.-H. Liu, X.-J. Zhao, S.-L. Han, L.-Y. Yao and J.-K. Sun, Angew. Chem., Int. Ed., 2025, 64, e202509941 CrossRef CAS PubMed
- Y. Zhang, S. Zhang, C. Liang, J. Shi and L. Ji, Adv. Mater., 2023, 35, 2302732 CrossRef CAS PubMed
- B. H. Miller, H. Liu and M. Kolle, Nat. Mater., 2022, 21, 1014–1018 CrossRef CAS PubMed
- S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CrossRef CAS
- D. Hu, R. Huang and Y. Fang, Precis. Chem., 2025, 3, 10–26 CrossRef CAS PubMed
- C. Jin, X. Yang, W. Zhao, Y. Zhao, Z. Wang and J. Tan, Coord. Chem. Rev., 2024, 513, 215892 CrossRef CAS
- S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC
- J. Wang, M. Li, H. Gao, L. Xie, X. Zheng, G. Liu, T. Wu, L. Lin and L. Liu, Smart Mol., 2024, 2, e20240041 CrossRef CAS PubMed
- F.-Z. Li, J.-F. Yin and G.-C. Kuang, Coord. Chem. Rev., 2021, 448, 214157 CrossRef CAS
- D. Shimoyama and F. Jäkle, Aggregate, 2022, 3, e149 Search PubMed
- T. W. Hudnall, C.-W. Chiu and F. P. Gabbaï, Acc. Chem. Res., 2009, 42, 388–397 Search PubMed
- C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed
- Y. Takeda, ChemPhysChem, 2025, 26, e202400894 CrossRef CAS PubMed
- N. Aota, R. Nakagawa, L. E. de Sousa, N. Tohnai, S. Minakata, P. de Silva and Y. Takeda, Angew. Chem., Int. Ed., 2024, 63, e202405158 CrossRef CAS PubMed
- M. K. Goshisht and N. Tripathi, J. Mater. Chem. C, 2021, 9, 9820–9850 Search PubMed
- S. Hagspiel, F. Fantuzzi, M. Arrowsmith, A. Gärtner, M. Fest, J. Weiser, B. Engels, H. Helten and H. Braunschweig, Chem. – Eur. J., 2022, 28, e202201398 CrossRef CAS PubMed
- A. E. R. Watson, P. D. Boyle, P. J. Ragogna and J. B. Gilroy, Chem. Sci., 2025, 16, 2258–2264 RSC
- Y.-G. Shi, J.-W. Wang, H. Li, G.-F. Hu, X. Li, S. K. Mellerup, N. Wang, T. Peng and S. Wang, Chem. Sci., 2018, 9, 1902–1911 RSC
- Y. Wang, K. Liu, P. Chen, X. Yin, T. Peng, J. Iqbal and N. Wang, J. Mater. Chem. C, 2022, 10, 10981–10987 Search PubMed
- Y. Sun, H. Ding, M. Tang, J. Wen, S. Yue, Y. Peng, L. Zheng, Y. Shi and Q. Cao, Anal. Chem., 2023, 95, 5594–5600 Search PubMed
- Y. Sun, X. Cai, W. He, X. Ji, L. Zheng, Y. Shi and Q. Cao, Adv. Opt. Mater., 2024, 12, 2401445 Search PubMed
- J. Dong, L. Chen and D.-T. Yang, Chem. Sci., 2025, 16, 9577–9603 Search PubMed
- M. Kawashiro, T. Mori, M. Ito, N. Ando and S. Yamaguchi, Angew. Chem., Int. Ed., 2023, 62, e202303725 Search PubMed
- R. Oshimizu, N. Ando and S. Yamaguchi, Angew. Chem., Int. Ed., 2022, 61, e202209394 CrossRef CAS PubMed
- H. Narita, A. Virovets, H.-W. Lerner, M. Wagner and S. Yamaguchi, Chem. Sci., 2025, 16, 8764–8771 Search PubMed
- X. Y. Liu, D. R. Bai and S. Wang, Angew. Chem., Int. Ed., 2006, 45, 5475–5478 Search PubMed
- H. Pan, G.-L. Fu, Y.-H. Zhao and C.-H. Zhao, Org. Lett., 2011, 13, 4830–4833 Search PubMed
- Q. Hou, L. Liu, S. K. Mellerup, N. Wang, T. Peng, P. Chen and S. Wang, Org. Lett., 2018, 20, 6467–6470 CrossRef CAS PubMed
- Y. Shi, Y. Zeng, P. Kucheryavy, X. Yin, K. Zhang, G. Meng, J. Chen, Q. Zhu, N. Wang, X. Zheng, F. Jäkle and P. Chen, Angew. Chem., Int. Ed., 2022, 61, e202213615 CrossRef CAS PubMed
- Y. Shi, C. Li, H. Ma, Z. Cao, K. Liu, X. Yin, N. Wang and P. Chen, Org. Lett., 2022, 24, 5497–5502 CrossRef CAS PubMed
- A. Fukazawa, E. Yamaguchi, E. Ito, H. Yamada, J. Wang, S. Irle and S. Yamaguchi, Organometallics, 2011, 30, 3870–3879 CrossRef CAS
- G. Ji, N. Wang, X. Yin and P. Chen, Org. Lett., 2020, 22, 5758–5762 Search PubMed
- K. Zhang, G. Ji, N. Zhang, N. Wang, X. Yin, Q. Li and P. Chen, J. Mater. Chem. C, 2021, 9, 13851–13859 RSC
- H. Hirai, K. Nakajima, S. Nakatsuka, K. Shiren, J. Ni, S. Nomura, T. Ikuta and T. Hatakeyama, Angew. Chem., Int. Ed., 2015, 54, 13581–13585 CrossRef CAS PubMed
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed
- B. Yang, Y.-M. Zhang, C. Wang, C. Gu, C. Li, H. Yin, Y. Yan, G. Yang and S. X.-A. Zhang, Nat. Commun., 2024, 15, 5166 CrossRef CAS PubMed
- G. Meng, T. Peng, Y. Shi, H. Li, X. Wang, X. Yin, D.-T. Yang, S. Wang and N. Wang, J. Mater. Chem. C, 2020, 8, 7749–7754 RSC
- L. Tu, Y. Chen, X. Song, W. Jiang, Y. Xie and Z. Li, Angew. Chem., Int. Ed., 2024, 63, e202402865 CrossRef CAS PubMed
- P.-Z. Chen, L.-Y. Niu, Y.-Z. Chen and Q.-Z. Yang, Coord. Chem. Rev., 2017, 350, 196–216 CrossRef CAS
- G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 8942–8943 CrossRef CAS PubMed
- S. Saotome, K. Suenaga, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2020, 4, 1781–1788 RSC
- S. Ito, M. Yaegashi, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2021, 27, 9302–9312 CrossRef CAS PubMed
- S. Ito, Y. Sakai and K. Tanaka, Chem. – Eur. J., 2025, 31, e202500238 CrossRef CAS PubMed
- Y. Qi, N. Ding, Z. Wang, L. Xu and Y. Fang, ACS Appl. Mater. Interfaces, 2019, 11, 8676–8684 CrossRef CAS PubMed
- M. Ibarra-Rodríguez, B. M. Muñoz-Flores, R. Chan-Navarro, N. Waksman, A. Saucedo-Yañez, M. Sánchez and V. M. Jiménez-Pérez, Opt. Mater., 2019, 89, 123–131 CrossRef
- J. Wang, Q. Meng, Y. Yang, S. Zhong, R. Zhang, Y. Fang, Y. Gao and X. Cui, ACS Sens., 2022, 7, 2521–2536 Search PubMed
- D. Yordanov, R. Smolka, K. Nakashima, S.-I. Hirashima, Y. Matsushima, M. Vala, J. Krajčovič, M. Weiter, T. Miura and A. Georgiev, J. Org. Chem., 2023, 88, 17206–17214 CrossRef CAS PubMed
- B. H. Jhun, S. Y. Park and Y. You, Dyes Pigm., 2023, 208, 110786 Search PubMed
- Y.-L. Li, H.-L. Wang, Z.-H. Zhu, Y.-F. Wang, F.-P. Liang and H.-H. Zou, Nat. Commun., 2024, 15, 2896 CrossRef CAS PubMed
- J. Gong and X. Zhang, Coord. Chem. Rev., 2022, 453, 214329 Search PubMed
- M. Ikeshita, A. Kuroda, S. Suzuki, Y. Imai and T. Tsuno, ChemPhotoChem, 2024, 8, e202400110 Search PubMed
- C. Duan, Y. Zhou, G.-G. Shan, Y. Chen, W. Zhao, D. Yuan, L. Zeng, X. Huang and G. Niu, J. Mater. Chem. C, 2019, 7, 3471–3478 RSC
- A. N. Bismillah and I. Aprahamian, J. Phys. Org. Chem., 2023, 36, e4485 Search PubMed
- Q. Qi, S. Huang, X. Liu and I. Aprahamian, J. Am. Chem. Soc., 2024, 146, 6471–6475 CrossRef CAS PubMed
- M. Liu, H. Hao, G. Liao, C. Li, K. Liu, N. Wang, Q. Niu and X. Yin, Adv. Opt. Mater., 2023, 11, 2202918 CrossRef CAS
- L.-F. Guo, M. Wang and C.-H. Zhao, Chem. – Eur. J., 2024, 30, e202402287 CrossRef CAS PubMed
- M.-Y. Zhang, X. Liang, D.-N. Ni, D.-H. Liu, Q. Peng and C.-H. Zhao, Org. Lett., 2021, 23, 2–7 CrossRef CAS PubMed
- J. Jovaišaitė, S. Kirschner, S. Raišys, G. Kreiza, P. Baronas, S. Juršėnas and M. Wagner, Angew. Chem., Int. Ed., 2023, 62, e202215071 CrossRef PubMed
- C. Chen, Y. Guo, Z. Chang, K. Müllen and X.-Y. Wang, Nat. Commun., 2024, 15, 8555 CrossRef CAS PubMed
- H. Huang, Y. Zhou, Y. Wang, X. Cao, C. Han, G. Liu, Z. Xu, C. Zhan, H. Hu, Y. Peng, P. Yan and D. Cao, J. Mater. Chem. A, 2020, 8, 22023–22031 RSC
- D. Tan, J. Dong, T. Ma, Q. Feng, S. Wang and D.-T. Yang, Angew. Chem., Int. Ed., 2023, 62, e202304711 CrossRef CAS PubMed
- J. Guo, Y. Yang, C. Dou and Y. Wang, J. Am. Chem. Soc., 2021, 143, 18272–18279 CrossRef CAS PubMed
- L. Yuan, J. Guo, Y. Yang, K. Ye, C. Dou and Y. Wang, CCS Chem., 2023, 5, 876–884 CrossRef CAS
- Z. Li, H. Wen, X. Xiang, S. Sheng, T. Zhang, K. Ye, Z. Xie and C. Dou, Sci. China: Chem., 2026, 69, 339–346 CrossRef
- F. Vidal and F. Jäkle, Angew. Chem., Int. Ed., 2019, 58, 5846–5870 CrossRef CAS PubMed
- E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817–829 CrossRef CAS PubMed
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS PubMed
- H. Helten, Chem. – Eur. J., 2016, 22, 12972–12982 CrossRef CAS PubMed
- U. Yolsal, T. A. R. Horton, M. Wang and M. P. Shaver, Prog. Polym. Sci., 2020, 111, 101313 CrossRef CAS
- S. Ito, M. Gon, K. Tanaka and Y. Chujo, Polym. Chem., 2021, 12, 6372–6380 RSC
- Z.-H. Zhao, C.-H. Li and J.-L. Zuo, SmartMat, 2023, 4, e1187 CrossRef CAS
- B. Chen and F. Jäkle, Angew. Chem., Int. Ed., 2024, 63, e202313379 CrossRef CAS PubMed
- F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 CrossRef
- X. Yin, J. Liu and F. Jäkle, Chem. – Eur. J., 2021, 27, 2973–2986 Search PubMed
- Y. Qin, G. Cheng, O. Achara, K. Parab and F. Jäkle, Macromolecules, 2004, 37, 7123–7131 CrossRef CAS
- T. Patranika, K. Märker, S. Paul, A. J. Naylor, J. Mindemark, K. Edström and G. Hernández, ACS Appl. Polym. Mater., 2024, 6, 12429–12440 CrossRef CAS
- C. He, J. Dong, C. Xu and X. Pan, ACS Polym. Au, 2023, 3, 5–27 CrossRef CAS
- H. L. van de Wouw and R. S. Klausen, J. Org. Chem., 2019, 84, 1117–1125 CrossRef CAS PubMed
- K. Tanaka and Y. Chujo, Polym. J., 2020, 52, 555–566 CrossRef CAS
- T. Lei, J.-Y. Wang and J. Pei, Acc. Chem. Res., 2014, 47, 1117–1126 CrossRef CAS PubMed
- S. Ohtani, N. Yamada, M. Gon, K. Tanaka and Y. Chujo, Polym. Chem., 2021, 12, 2752–2759 RSC
- M. Kanjo, M. Gon and K. Tanaka, ACS Appl. Mater. Interfaces, 2023, 15, 31927–31934 CrossRef CAS PubMed
- Y. Aoyama, S. Ito and K. Tanaka, Macromolecules, 2024, 57, 6559–6567 CrossRef CAS
- S. Morimoto, K. Tanimura, M. Gon, T. Suematsu, K. Okamoto, H. Watanabe, H. Taka, H. Kita and K. Tanaka, Macromolecules, 2024, 57, 6531–6539 CrossRef CAS
- A. F. Alahmadi, X. Yin, R. A. Lalancette and F. Jäkle, Chem. – Eur. J., 2023, 29, e202203619 CrossRef CAS PubMed
- C. R. McConnell and S.-Y. Liu, Chem. Soc. Rev., 2019, 48, 3436–3453 RSC
- A. W. Baggett, F. Guo, B. Li, S.-Y. Liu and F. Jäkle, Angew. Chem., Int. Ed., 2015, 54, 11191–11195 CrossRef CAS PubMed
- J. Chorbacher, J. Klopf, A. Friedrich, M. Fest, J. S. Schneider, B. Engels and H. Helten, Angew. Chem., Int. Ed., 2025, 64, e202416088 CrossRef CAS PubMed
- H. Kuhtz, F. Cheng, S. Schwedler, L. Böhling, A. Brockhinke, L. Weber, K. Parab and F. Jäkle, ACS Macro Lett., 2012, 1, 555–559 CrossRef CAS PubMed
- E. A. Latif, J. D. Hilgar and N. A. Romero, J. Am. Chem. Soc., 2024, 146, 24764–24769 CrossRef CAS PubMed
- F. Vidal, S. Smith and C. K. Williams, J. Am. Chem. Soc., 2023, 145, 13888–13900 CrossRef CAS PubMed
- F. Cheng and F. Jäkle, Chem. Commun., 2010, 46, 3717–3719 RSC
- Y. Qin, V. Sukul, D. Pagakos, C. Cui and F. Jäkle, Macromolecules, 2005, 38, 8987–8990 CrossRef CAS
- P. O. Shipman, C. Cui, P. Lupinska, R. A. Lalancette, J. B. Sheridan and F. Jäkle, ACS Macro Lett., 2013, 2, 1056–1060 CrossRef CAS PubMed
- F. Cheng, E. M. Bonder and F. Jäkle, J. Am. Chem. Soc., 2013, 135, 17286–17289 CrossRef CAS PubMed
- J. McQuade, F. Vidal, M. Noor, L. Anand, R. A. Lalancette and F. Jäkle, ChemistryEurope, 2025, e202500327 Search PubMed
- W.-M. Wan, S.-S. Li, D.-M. Liu, X.-H. Lv and X.-L. Sun, Macromolecules, 2017, 50, 6872–6879 CrossRef CAS
- X.-Y. Zheng, T. Li, H.-W. Cai, X.-H. Wang, X.-L. Sun and W.-M. Wan, Polymer, 2024, 300, 126996 CrossRef CAS
- Y. Qin, G. Cheng, A. Sundararaman and F. Jäkle, J. Am. Chem. Soc., 2002, 124, 12672–12673 CrossRef CAS PubMed
-
F. Jäkle, in New Polymeric Materials Based on Element-Blocks, ed. Y. Chujo, Springer Singapore, Singapore, 2019, pp. 59–76 DOI:10.1007/978-981-13-2889-3_4
- Y. Qin, C. Cui and F. Jäkle, Macromolecules, 2007, 40, 1413–1420 CrossRef CAS
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed
- J. Yang, Z. Ren, Z. Xie, Y. Liu, C. Wang, Y. Xie, Q. Peng, B. Xu, W. Tian, F. Zhang, Z. Chi, Q. Li and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 880–884 CrossRef CAS PubMed
- J. Yang, Z. Ren, Z. Xie, Y. Liu, C. Wang, Y. Xie, Q. Peng, B. Xu, W. Tian, F. Zhang, Z. Chi, Q. Li and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 880–884 CrossRef CAS PubMed
- R. Tian, S. Gao, K. Li and C. Lu, Nat. Commun., 2023, 14, 4720 CrossRef CAS PubMed
- D. Li, Y. Yang, J. Yang, M. Fang, B. Z. Tang and Z. Li, Nat. Commun., 2022, 13, 347 CrossRef CAS PubMed
- X. Ma, T.-J. Ye, H.-J. Yang, G.-K. Ling, D. Wang, J.-S. Li, P. Gu, L.-J. Xu, T.-L. Sheng, F.-R. Lin, R.-F. Shen and Q. Zhang, Aggregate, 2024, 5, e579 CrossRef CAS
- C. Xue, M. Peng, Z. Zhang, X. Han, Q. Wang, C. Li, H. Liu, T. Li, N. Yu and Y. Ren, Macromolecules, 2022, 55, 3850–3859 CrossRef CAS
- L. He, D. E. Fullenkamp, J. G. Rivera and P. B. Messersmith, Chem. Commun., 2011, 47, 7497–7499 RSC
- M. Piest, X. Zhang, J. Trinidad and J. F. J. Engbersen, Soft Matter, 2011, 7, 11111–11118 RSC
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 CrossRef CAS
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed
- J. Ma, Y. Yang, C. Valenzuela, X. Zhang, L. Wang and W. Feng, Angew. Chem., Int. Ed., 2022, 61, e202116219 CrossRef CAS PubMed
- X. Zhang, S. Wang, Z. Jiang, Y. Li and X. Jing, J. Am. Chem. Soc., 2020, 142, 21852–21860 CrossRef CAS PubMed
- Y. He, Y. Qiao, Z. Li, W. Feng, Y. Zhao, W. Tian, B. Zhong Tang and H. Yan, Angew. Chem., Int. Ed., 2024, 63, e202413425 CrossRef CAS PubMed
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed
- J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 RSC
- Y.-F. Zhang, Y.-W. Zhang, X. Li, L.-Y. Sun and Y.-F. Han, Chem. Commun., 2023, 59, 2291–2294 RSC
- H. Narita, H. Choi, M. Ito, N. Ando, S. Ogi and S. Yamaguchi, Chem. Sci., 2022, 13, 1484–1491 RSC
- C. P. Brock, A. L. Companion, L. D. Kock and K. Niedenzu, Inorg. Chem., 1991, 30, 784–789 CrossRef CAS
- N. Christinat, R. Scopelliti and K. Severin, J. Org. Chem., 2007, 72, 2192–2200 CrossRef CAS PubMed
- A. Widera, E. Filbeck, H. Wadepohl, E. Kaifer and H.-J. Himmel, Chem. – Eur. J., 2020, 26, 3435–3440 CrossRef CAS PubMed
- Y. Li, J. Chen, W. Jiao, J. Ma, Y. Chen, J. Xu, Z. Liang, S. Dong, X. Chen, M. Heeney and Z. Fei, J. Am. Chem. Soc., 2025, 147, 33963–33975 CrossRef CAS PubMed
- X. Ji, Z. Wang, W. Yuan, Z. Gao and W. Tian, Chem. Commun., 2025, 61, 5447–5450 RSC
- S. Bhandary, M. Beliš, R. Shukla, L. Bourda, A. M. Kaczmarek and K. Van Hecke, J. Am. Chem. Soc., 2024, 146, 8659–8667 CrossRef CAS PubMed
- J. Xu, T. Wang, S. Deng, W. Lai, Y. Shi, Y. Zhao, F. Huang and P. Wei, Angew. Chem., Int. Ed., 2024, 63, e202411880 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2026 |