
Antimony alloying electrode for high-performance sodium removal: how to use a battery material not stable in aqueous media for saline water remediation†
Stefanie
Arnold
 ab,
Lei
Wang
ab,
Öznil
Budak
ab,
Mesut
Aslan
a,
Pattarachai
Srimuk
a and
Volker
Presser
*ab
ab,
Lei
Wang
ab,
Öznil
Budak
ab,
Mesut
Aslan
a,
Pattarachai
Srimuk
a and
Volker
Presser
*ab
aINM – Leibniz Institute for New Materials, 66123 Saarbrücken, Germany. E-mail: volker.presser@leibniz-inm.de
bDepartment of Materials Science and Engineering, Saarland University, 66123 Saarbrücken, Germany
First published on 25th November 2020
Abstract
Capacitive deionization (CDI) is based on ion electrosorption and has emerged as a promising desalination technology, for example, to obtain drinking water from brackish water. As a next-generation technology, battery desalination uses faradaic processes and, thereby, enables higher desalination capacities and remediation of feed water with high molar strength such as seawater. However, the full use of a large capacity of charge transfer processes is limited by the voltage window of water and the need to use electrode materials non-reactive towards the water. Using our multi-channel bi-electrolyte cell, we now introduce for the first time an alloying electrode for sodium removal in the context of water desalination. Separated by a ceramic sodium superionic conductor (NASICON) membrane, the antimony/carbon composite electrode accomplished sodium removal while chlorine removal is enabled via ion electrosorption with nanoporous carbon (activated carbon cloth). In a sodium-ion battery half-cell setup, the antimony/carbon composite electrode reaches an initial capacity of 714 mA h g−1 at a specific current of 200 mA g−1, which shows a slow but continuous degrading over the course of 80 cycles (426 mA h g−1 in 80th cycle). Our hybrid CDI cell provides a desalination capacity of an average of 294 mgNa gSb−1 (748 mgNaCl gSb−1) with a charge efficiency of ca. 74% in a 600 mM NaCl at a specific current of 200 mA g−1 and a voltage range of −2.0 V to +2.0 V.
1 Introduction
In the 21st century, due to the ever-increasing population and correspondingly the deterioration of the environment, obtaining clean potable water becomes a serious issue for sustaining livelihood, especially in arid regions.1 Large-scale efforts have been invested in developing desalination technology to address this issue. From simple distillation, multi-stage flash distillation,2 to reverse osmosis,3,4 many methods have been studied. However, many of these methods are expensive and require high energy input. Among them, capacitive deionization (CDI) is considered as an energy-efficient technology, compared to the pressure or filtration-based desalination technology.5 In CDI, ions are immobilized at the fluid/solid interface of nanoporous carbon by reversible ion-electrosorption.6 A typical CDI cell employs a pair of nanoporous carbon electrodes and a separator (open channel or porous dielectric material), to prevent short-circuiting.7 By applying a constant voltage or current, the salt ions present in feedwater migrate into the electrical double-layer (EDL) and, thus, remove salt from the water (ion-electrosorption). When the cell is discharged, the ions are released again, and the invested charge is recovered. The voltage applied to each electrode pair is typically around (slightly above) 1 V, depending on the overpotential of water splitting. Because of the low voltage, and because there is no need for other energy input such as high pressure or high temperature, CDI holds the promise of being a more energy-efficient method of desalinating water on a larger scale.7However, uncharged carbon nanopores are populated by both co-ions and counter-ions, that is, ions with the same or opposite charge compared to the electrode.8,9 Once the electrodes are polarized, co-ions will be released from the EDL at the same extent as counter-ion attraction takes place; only once the co-ion population has been depleted (at higher states-of-charge), permselective ion removal will be possible.10 Thereby, CDI remains limited to low molar concentrations (with an initially low number of co-ions present in carbon nanopores).11 This condition is only (partially) remedied when the pore diameter significantly falls below 1 nm.12 Also, the charge storage capacity intrinsically linked to EDL carbon electrodes is limited to about 0.1 F m−2 (typical values for the gravimetric capacitance of activated carbon is about 100–150 F g−1).13–15 Therefore, the desalination capacity of low-concentration brackish water with carbon electrodes is typically in the range of 10–30 mgNaCl gelectrode−1 (∼4–12 mgNa gelectrode−1).16
To overcome the desalination limitations of carbon-based CDI, Pasta et al. introduced the concept of the desalination battery built of sodium manganese oxide and silver electrodes to deionize seawater.17 Instead of using two faradaic electrodes, it is also possible to pair carbon with a charge-transfer electrode. For example, Lee et al. used sodium manganese oxide to capture sodium and carbon to electrosorb chloride.18 By doing so, the authors achieved a desalination capacity of 31 mgNaCl gelectrode−1 (∼12 mgNa gelectrode−1), while the carbon itself only provided 14 mgNaCl gelectrode−1 (∼6 mgNa gelectrode−1).18 Until now, mostly two kinds of faradaic materials have been applied for desalination: intercalation-type materials (e.g., sodium manganese oxides,19 nickel hexacyanoferrate,20 titanium disulfide21) and conversion-type materials (e.g., silver22), which show a desalination capacity of 85–115 mgNaCl gelectrode−1 (up to about 22 mgNa gelectrode−1).23–27
The selection of suitable sodium-removal electrode for electrochemical desalination is limited by their compatibility with water. Many new materials could be used for desalination, especially anode materials of sodium-ion batteries if the requirement to expose the electrode to the feedwater stream would be lifted. As a first step towards this goal, we introduced the use of organic solvent CDI in 2016 (ref. 28) and aqueous/organic bi-electrolyte CDI in 2018 (ref. 29). In the latter concept, a multi-channel system is used, and one of the electrodes, covered by an ion-exchange membrane, is operated in an organic electrolyte instead of an aqueous solution. Ion transfer into and out from the organic compartment is ensured by the ion-exchange membrane. Our initial design used a polymer-based ion-exchange membrane, so water cross-over was not fully eliminated. Still, the system allowed stable cell operation at voltages significantly above 1.2 V and resulting desalination capacities of up to 60 mgNaCl gelectrode−1 (∼24 mgNa gelectrode−1; at 2.4 V cell voltage).
Research at the water/energy nexus between battery desalination and seawater batteries is a rapidly growing field for stationary applications.30–33 Our present proof-of-concept now uses the bi-electrolyte concept and demonstrates the highly promising concept of combining a ceramic ion-exchange membrane (sodium superionic conductor: NASICON)34 with antimony (Sb) as a high-performance sodium-alloy electrode material. Antimony offers a very high theoretical capacity of 660 mA h g−1 when assuming the formation of Na3Sb.35 However, it readily reacts with water and the electrode potential for the alloying reactions is far outside of the stability window of aqueous electrolytes. Therefore, one would never consider it for use in conventional desalination batteries. In previous work by Pfeifer et al., the influence of different carbon additives in the antimony electrode on the electrical performance in sodium-ion batteries was investigated.36 The highest capacity (620 mA h g−1) and stability (capacity loss of 19% after 100 cycles) were shown by a composite mixture of 70 mass% nanoscale antimony with 20 mass% of carbon onions and 10% carboxymethyl cellulose binder.36 The low alloying reaction potential, generally lower than 1.0 V vs. Na+/Na, make alloy materials impossible to be adopted to conventional cells for electrochemical desalination because of the exposure to water.37 Due to that reason, we chose this promising electrode material for use in the bi-electrolyte desalination concept. In this multi-channel cell, Sb and carbon electrodes were in organic and aqueous electrolyte, respectively. The two electrodes were separated by a Na+ permeable membrane (NASICON). During the charging process, Na+ in the organic electrolyte was captured by the alloying reaction between Sb and Na+ in the organic compartment. To keep the charge neutrality in the organic compartment, Na+ from feed water would diffuse into organic electrolytes resulting in desalination. The initial performance of this cell in 600 mM NaCl is about 495 mgNa gSb−1 (excluding the first two preconditioning cycles), and the performance stabilizes at around 294 mgNa gSb−1 with a charge efficiency of about 74% in aqueous 600 mM NaCl for 40 operating cycles.
2 Experimental
2.1 Materials synthesis
The synthesis of antimony nanopowder was carried out by optimizing a synthesis route given in the literature.38 To obtain a high purity of antimony, an excess of sodium borohydride (NaBH4, 1.216 g, ≥98.0% purity, Sigma Aldrich) was suspended in 400 mL technical ethanol (>99.0% purity, Merck). Antimony chloride (SbCl3, 2.244 g, ≥99.0% purity, Sigma Aldrich) was dissolved in 100 mL absolute ethanol (>99.8% purity, Honeywell). The antimony chloride solution was added dropwise into the stirring solution of sodium borohydride in ethanol at room temperature within one hour. The resultant mixture was stirred for another hour at room temperature. To break up agglomerates, the reaction mixture was sonicated for 10 min after stirring (P120H, Elmasonic). After sedimentation of the antimony particles, they were filtered under vacuum, washed three times with ethanol and three times with deionized water. Antimony particles were dried in an oven at +80 °C for 4 h. NASICON powder with excess Na was synthesized in analogy to literature (via Na3.3Zr2Si2PO12)39 by using a solid-state reaction. As the precursor, 26.80 g NH4H2PO4 (98.0% purity, Alfa Aesar), 40.35 g Na2CO3 (100.0% purity, Alfa Aesar), 27.45 g SiO2 (particle size <50 nm, Aerosil O × 50) and 56.00 g ZrO2 (particle size 40 nm, Tosoh) were applied. They were homogenized in a tumbler mixer for first step dry with 10 mm zirconia balls for 1 h, and then for another 24 h as an ethanolic slurry (absolute ethanol) with 15 mass% solid content (without balls to avoid contamination). After evaporation of ethanol, the dry powder was calcinated in two steps: first, at +600 °C in the air for 4 h, followed by a second calcination step at +1150 °C in the air for 4 h. The calcinated powder was crushed by hand and dry milled with 10 mm diameter zirconia balls for 1 h in a tumbler mixer. After sieving, the powder was cold-isostatically pressed at 400 MPa in a cylindrical rubber mold to obtain a green body.The pressed body with a relative density of around 74% was sintered at +1100 °C for 10 h in an air atmosphere to obtain a NASICON cylinder with a relative density of 78%. Membrane discs of 0.5 ± 0.05 mm thickness and a diameter of 40 mm were cut using a saw with a diamond blade. The open pores in the membrane were filled with epoxy resin. More details about this synthesis procedure are given in previous work.34
2.2 Structural and chemical characterization
X-ray diffraction (XRD) measurements of the antimony and NASICON powder were performed with a D8 Discover diffractometer (Bruker AXS) with a copper X-ray source (Cu-Kα (λ = 1.5406 Å), 40 kV, 40 mA) and a Göbel mirror and a 1 mm point focus as optics. With a VANTEC-500 (Bruker AXS) two-dimensional X-ray detector positioned at 17° 2θ, 37° 2θ, 57° 2θ, and 97° 2θ with a measurement time of 1000 s per step, five frames were recorded.Scanning electron microscope (SEM) images were recorded with a field emission scanning electron microscope (JEOL JSM-7500F) operating at an acceleration voltage of 3 kV. The samples were dispersed in ethanol, drop casted multiple times on the carbon film sticky tape on the steel sample holder. The samples were dried under vacuum for 30 min and analyzed without the aid of an additional, conductive sputter coating.
Raman spectroscopy was carried out with a Renishaw inVia Microscope equipped with a neodymium-doped yttrium aluminum garnet laser with an excitation wavelength of 532 nm and a laser power of approximately 0.05 mW, a 2400 mm−1 grating, and a 50× objective lens with a numeric aperture of 0.75. Three to four different spots of each sample were recorded with five accumulations and 30 s exposure time.
Elemental analysis (CHNS) was performed with a Vario Micro Cube system from Elementar. The samples were each weighed in tin boats with the same amount of WO3 and pressed under air exclusion. The reduction temperature in the pipe represented +850 °C (combustion tube temperature: +1150 °C), and the device was calibrated through repeated measurements of sulfanilamide.
Quantitative analysis of elemental oxygen was performed by using a rapid OXY cube oxygen analyzer from Elementar at a pyrolysis temperature of +1450 °C. The samples were weight in silver boats and pressed under air exclusion. The system was calibrated by measurements of benzoic acid.
2.3 Electrode materials and preparation
The Sb/C electrodes were manufactured by mixing the active material of 70 mass% synthesized antimony nanopowder with 20 mass% carbon onions, produced from nanodiamond powder at a temperature of 1300 °C under vacuum (OLC1300-Va),40 and 10 mass% carboxymethyl cellulose as binder from a 3 mass% aqueous solution (CMC, degree of substitution = 0.7, molecular weight = 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Sigma Aldrich), by hand mixing for 40 min. First, the active material and the carbon were mixed and ground dry in a mortar. After adding isopropanol, the suspension was kept grinding until the isopropanol is completely vaporized. In the next step, this was repeated with ethanol. After that, the electrode material with a small amount of ethanol and water (ratio 1:1) kept stirring for 30 min. Finally, the CMC binder solution was added, and the electrode past kept stirring for another 1 hour. The suspension was stirred for several hours on a magnetic stirrer to obtain a homogeneous slurry. The obtained electrode slurries were doctor bladed on aluminum foil (Ranafoil, Toyo Aluminium), used as a current collector, with a thickness of 15 μm. The electrodes were initially dried at ambient conditions overnight. Then, they were dried further in a vacuum oven at +120 °C for 12 h. Subsequently, the electrodes were punched out with a 12 mm diameter and transferred into a vacuum oven. Finally, a vacuum drying step at +80 °C for 12 h was conducted to remove the remaining solvent. The resulting electrode thickness of the dried electrodes was 40–60 μm with a material loading of 1.4 ± 0.2 mg cm−2.
000 g mol−1, Sigma Aldrich), by hand mixing for 40 min. First, the active material and the carbon were mixed and ground dry in a mortar. After adding isopropanol, the suspension was kept grinding until the isopropanol is completely vaporized. In the next step, this was repeated with ethanol. After that, the electrode material with a small amount of ethanol and water (ratio 1:1) kept stirring for 30 min. Finally, the CMC binder solution was added, and the electrode past kept stirring for another 1 hour. The suspension was stirred for several hours on a magnetic stirrer to obtain a homogeneous slurry. The obtained electrode slurries were doctor bladed on aluminum foil (Ranafoil, Toyo Aluminium), used as a current collector, with a thickness of 15 μm. The electrodes were initially dried at ambient conditions overnight. Then, they were dried further in a vacuum oven at +120 °C for 12 h. Subsequently, the electrodes were punched out with a 12 mm diameter and transferred into a vacuum oven. Finally, a vacuum drying step at +80 °C for 12 h was conducted to remove the remaining solvent. The resulting electrode thickness of the dried electrodes was 40–60 μm with a material loading of 1.4 ± 0.2 mg cm−2.
To evaluate the performance of the carbon on the aqueous side, commercially available microporous activated carbon cloth (Kynol ACC-507-20) was investigated as a working electrode. These electrodes, showing thickness of 250 μm, were punched in 12 mm discs and can be operated as free-standing, binder-free electrodes. In aqueous half-cell setups, a free-standing oversized activated carbon powder electrode (YP-80F, Kuraray) function as a counter electrode. These electrodes were prepared by mixing 90 mass% of activated carbon and 10 mass% polytetrafluoroethylene (60 mass% dispersion in water, Sigma Aldrich) together with ethanol. The mixture was ground until a dough-like slurry was obtained, and then rolled to a thickness of about 600 μm. These electrodes were dried under vacuum at +80 °C and punched into circular plates with a diameter of 12 mm.
2.4 Cell preparation and electrochemical characterization
All cell parts were dried overnight at +80 °C and introduced into an argon-filled glovebox (MBraun Labmaster 130; O2 and H2O < 0.1 ppm). First, the 12 mm diameter working electrode was placed in the cell, followed by a 13 mm diameter vacuum dried compressed glass-fiber separator (GF/D, Whatman). The counter electrode was punched into circular plates with a diameter of 10 mm and placed on top of the separator. Before using sodium metal, the oxidized surface of sodium was removed to obtain a smooth surface to avoid inhomogeneity and impurities. The counter electrodes were pressed to a uniform thickness of approximately 1 mm. A copper foil current collector was placed on the backside of each counter electrode. The sodium reference electrode was placed on a 2 mm diameter compressed glass-fiber separator (GF/D, Whatman) in a cavity close to the working electrode/counter electrode stack and contacted with titanium wire. The other three holes were closed with PEEK-screws.
The cells were vacuum filled with the electrolyte. The preparation and handling of the electrolyte solvent and salt were conducted in an argon-filled glovebox (MBraun Labmaster 130, O2, H2O < 0.1 ppm). A 1 M sodium perchlorate (NaClO4, >99% purity, Alfa Aesar) solution in a solvent mixture of ethylene carbonate (EC, ≥99% purity, Sigma Aldrich) and dimethyl carbonate (DMC, ≥99% purity, Sigma Aldrich) in a 1:1 mass ratio with the addition of 5 mass% fluoroethylene carbonate (FEC, 99% purity, Sigma Aldrich) was used as an electrolyte. FEC is an additive for sodium-ion batteries used to improve the stability by increasing the solid electrolyte interface (SEI) stability, modifying the composition of the SEI layer, and preventing the decomposition of EC and DMC because the oxidation potential of FEC is higher than those of EC and DMC.42–46 The sodium salt for the electrolyte was dried under vacuum at +80 °C for 48 h. The electrolyte was examined via Karl-Fischer titration and was found to contain less than 25 ppm water.
For measurements in aqueous electrolyte, half-cells were prepared by using Kynol ACC-507-20 as a working electrode and YP-80F as the counter electrode, the electrodes were separated by a 13 mm diameter vacuum dried glass-fiber disc (GF/A, 210 μm thickness, Whatman) to avoid short-circuiting. We used aqueous 600 mM NaCl as the electrolyte. Graphite foil was used as a current collector, and the Ag/AgCl (3 M KCl E0 Ag/AgCl = 0.210 V vs. normal hydrogen electrode) electrode was employed as a reference electrode.
To determine the electrochemical behavior and electrochemical stability window of aqueous electrolyte, an S-value test, according to Xu et al.47 was conducted. The vertex potential started at 0 V and was increased with an incremental of 50 mV steps until the final potential of 1.2 V vs. Ag/AgCl was reached. These measurements were done with a scan rate of 1 mV s−1. For S-value calculation, the data at the 4th cycle is selected so that the electrode is conditioned at every vertex potential. For positive potential window opening, the S-values obtained from cyclic voltammetry were calculated by applying the eqn (1):
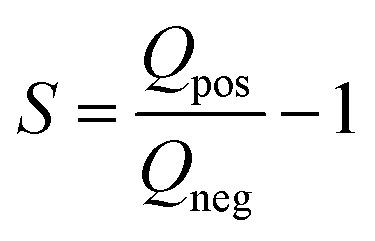 | (1) |
By integrating the positive and negative current vs. time of each cycle, the values for Qneg and Qpos are calculated.
Galvanostatic cycling with potential limitation (GCPL), cyclic voltammetry (CV), and performance measurements were carried out using a VMP3 multi-channel potentiostat/galvanostat from Bio-Logic. All obtained values for the capacity in Sb/C vs. sodium cells relate to the respective active mass (i.e., the total mass of antimony). All electrochemical measurements were carried out at a climate chamber (Binder) with a constant temperature of +25 ± 1 °C. The galvanostatic charge/discharge cycling with potential limitation (GCPL) experiments was recorded at voltages in the range of 0.1–2.0 V vs. Na+/Na. For all GCPLs in this work, a specific current of 200 mA g−1 was used. Rate performance measurements were conducted at different currents to get more information about the half-cell rate capability and stability at higher currents. The applied specific currents were 0.1 A g−1, 0.2 A g−1, 0.5 A g−1, 1.0 A g−1, 2.0 A g−1, 4.0 A g−1, 8.0 A g−1, and (again) 0.1 A g−1. All CV measurements were carried out with a scan rate of 0.1 mV s−1 in a potential window of 0.1–2.0 V vs. Na+/Na.
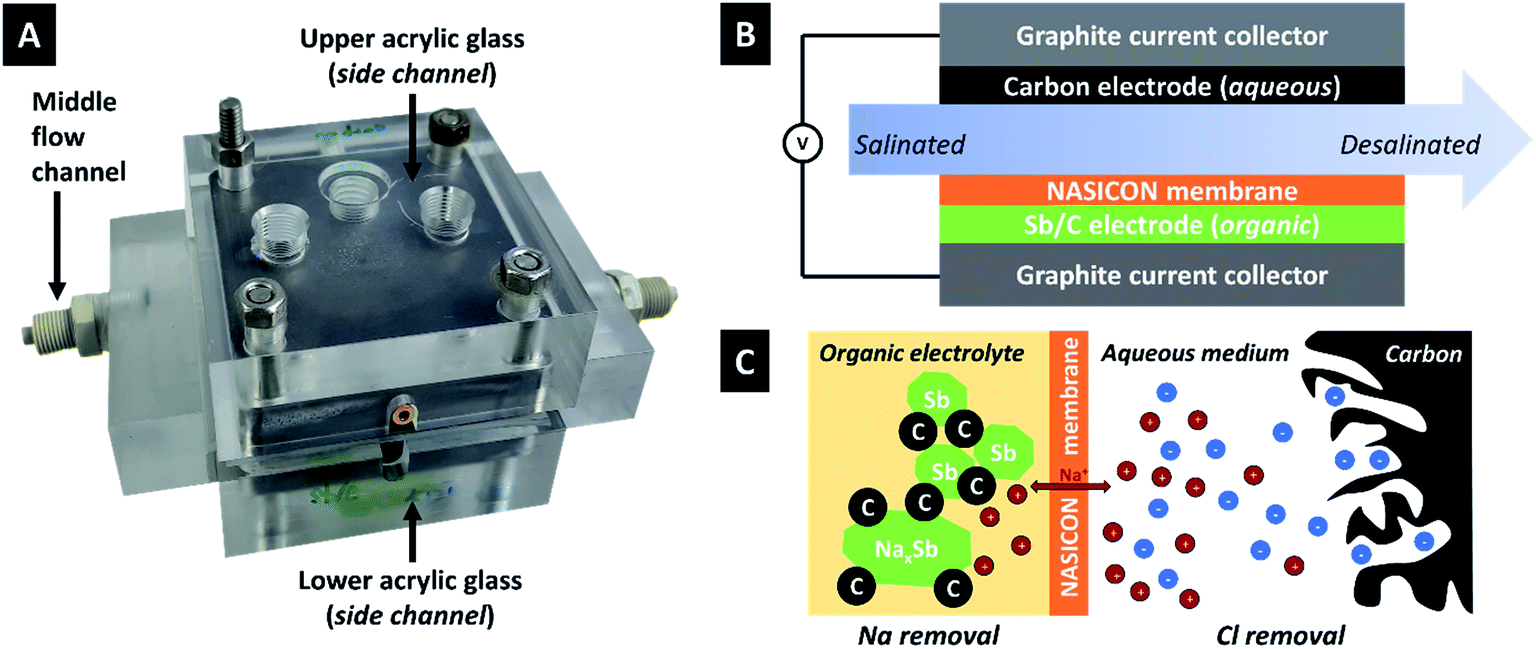 | ||
| Fig. 1 (A) Photograph and (B) schematic drawing of the desalination cell used for this work. (C) Schematic drawing of the desalination mechanism. | ||
Mass balancing between a carbon electrode and Sb/C electrode followed eqn (2):
| mSb/CQSb/C = mCQC | (2) |
The feed solution with 600 mM NaCl was prepared by dissolving NaCl (≥99.5% purity, Sigma Aldrich) in deionized water (Milli-Q). Using a 10 L reservoir, this aqueous electrolyte was pumped at an average of 1.1 mL min−1 into one side channel of the cell by a peristaltic pump (Masterflex, L/S Series). The electrolyte tank was continuously flushed with nitrogen gas to remove dissolved oxygen. The electrochemical test was carried out using a VSP3 multi-channel potentiostat/galvanostat from Bio-Logic. The conductivity- and pH-modules (Metrohm) are controlled by the Tiamo software. All electrochemical measurements were carried out in a climate chamber (Binder) with a constant temperature of +25 ± 1 °C. The GCPL experiments were recorded in the range of ±2.0 V vs. antimony.
The desalination capacity is calculated with eqn (3):
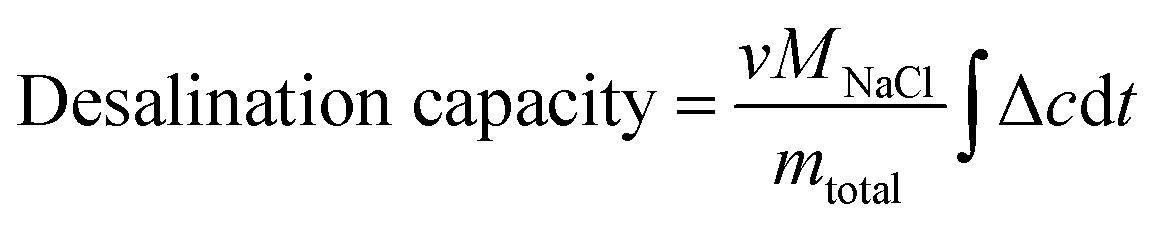 | (3) |
3 Results and discussion
3.1 Working principle of the Sb/C vs. activated carbon cloth cell
We chose an optimized Sb/C composite electrode material based on our previous work which surveyed an array of different carbon additives; a detailed report on the structural, chemical, and porosity-related properties of the used carbon onion/antimony composite electrodes is found in ref. 36.In this work, an Sb/C electrode is surrounded by an organic electrolyte and separated from the feedwater (aqueous 600 mM NaCl) by a ceramic cation-exchange membrane. During charging, 1 Sb atom reacts with (up to) 3 Na+-ions in a multi-stage alloying process. Na+ originates from the electrolyte solution on this side of the cell. This creates a concentration gradient of Na+ during charging and a lack of these ions in the electrolyte. The saltwater on the other side of the cell is rich in Na+ and the concentration gradient allows Na+ to move through the NASICON membrane, whereby sodium removal of the feedwater stream is accomplished. On the other side of the electrochemical desalination cell, chloride is removed from the saline solution via ion electrosorption at the fluid/solid interface. When discharging, the process would be reversed, and an increased amount of NaCl is released back into the effluent stream. Thereby, both the Sb and the carbon electrodes are both regenerated and ready for the next desalination cycle.
Data on the material characterization of antimony nanopowder and NASICON powder and membrane are given in ESI, Fig. S1 and S2.†
3.2 Electrochemical characterization
Cyclic voltammetry was carried out to characterize the redox behavior of the electrode material. Fig. 2A shows the typical CV of the first, fifth, and tenth cycle at a scan rate of 0.1 mV s−1 between 0.1 V and 2.0 V vs. Na+/Na. The redox peaks at around 1.2 V and 0.5 V vs. Na+/Na in the first cycle correspond to the SEI formation and conversion of crystalline Sb to NaxSb, respectively.49 The oxidation peak in the subsequent sodiation scan at a potential of 0.8 V vs. Na+/Na characterize the de-sodiation reaction of NaxSb alloy back to elemental amorphous antimony. In the 5th and 10th cycles, there are additional reduction peaks at the potential of 0.7 V and 0.45 V vs. Na+/Na. This is related to the reaction mechanism of the crystalline antimony, which reacts with the sodium to form amorphous compounds. NaSb further reacts with additional sodium to form crystalline Na3Sb. Between the first and the following cycle, we can see key differences. The oxidation peak, resulting from the de-sodiation reaction of the alloy, is shifting from 0.74 V vs. Na+/Na (1st cycle) over 0.87 V vs. Na+/Na (5th cycle) to 0.91 V vs. Na+/Na (10th cycle). These differences and the significant decrease in the current intensity after cycling can be attributed to the decomposition of the electrolyte, a change of reaction mechanism, and several changes in the involved structures.
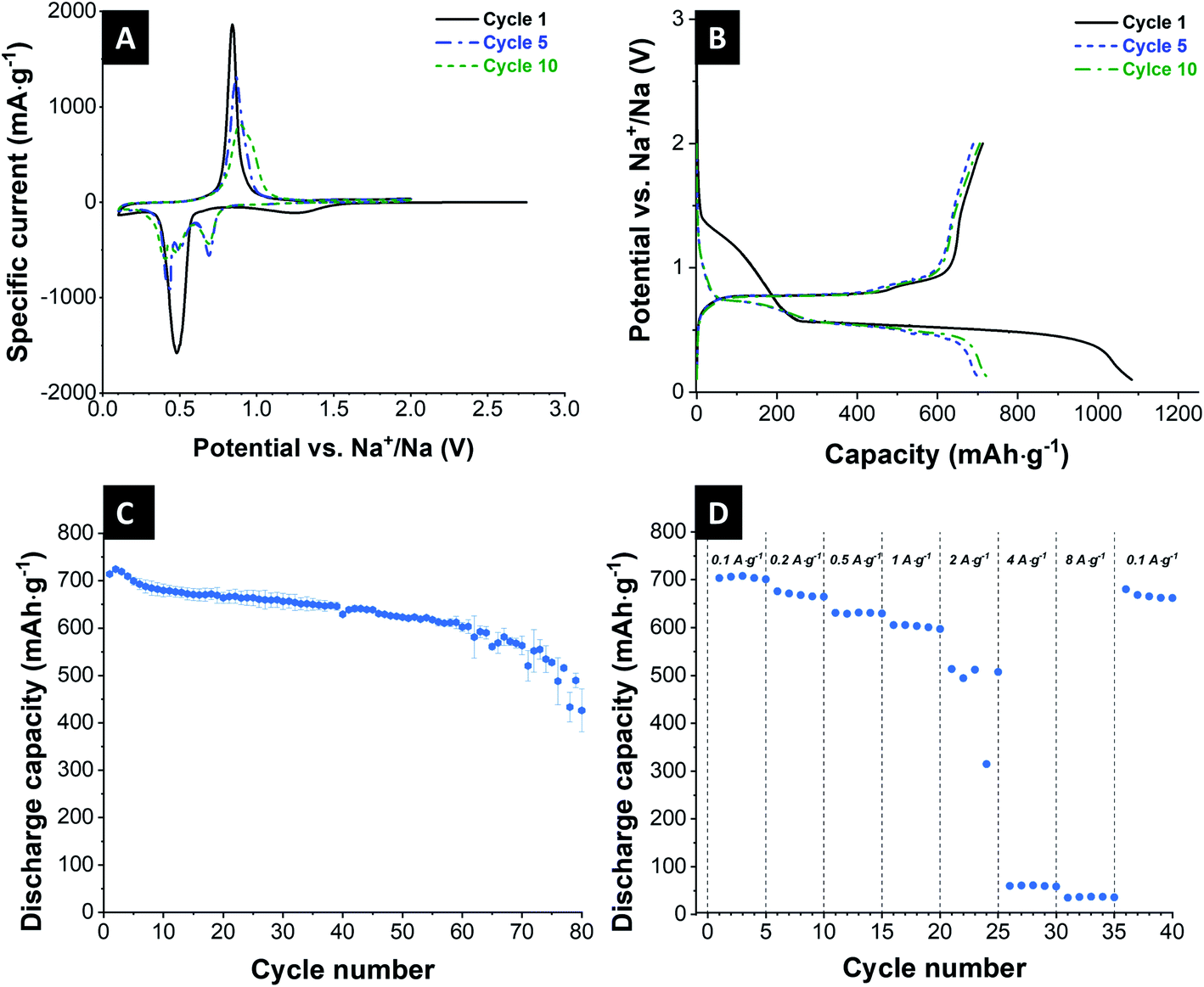 | ||
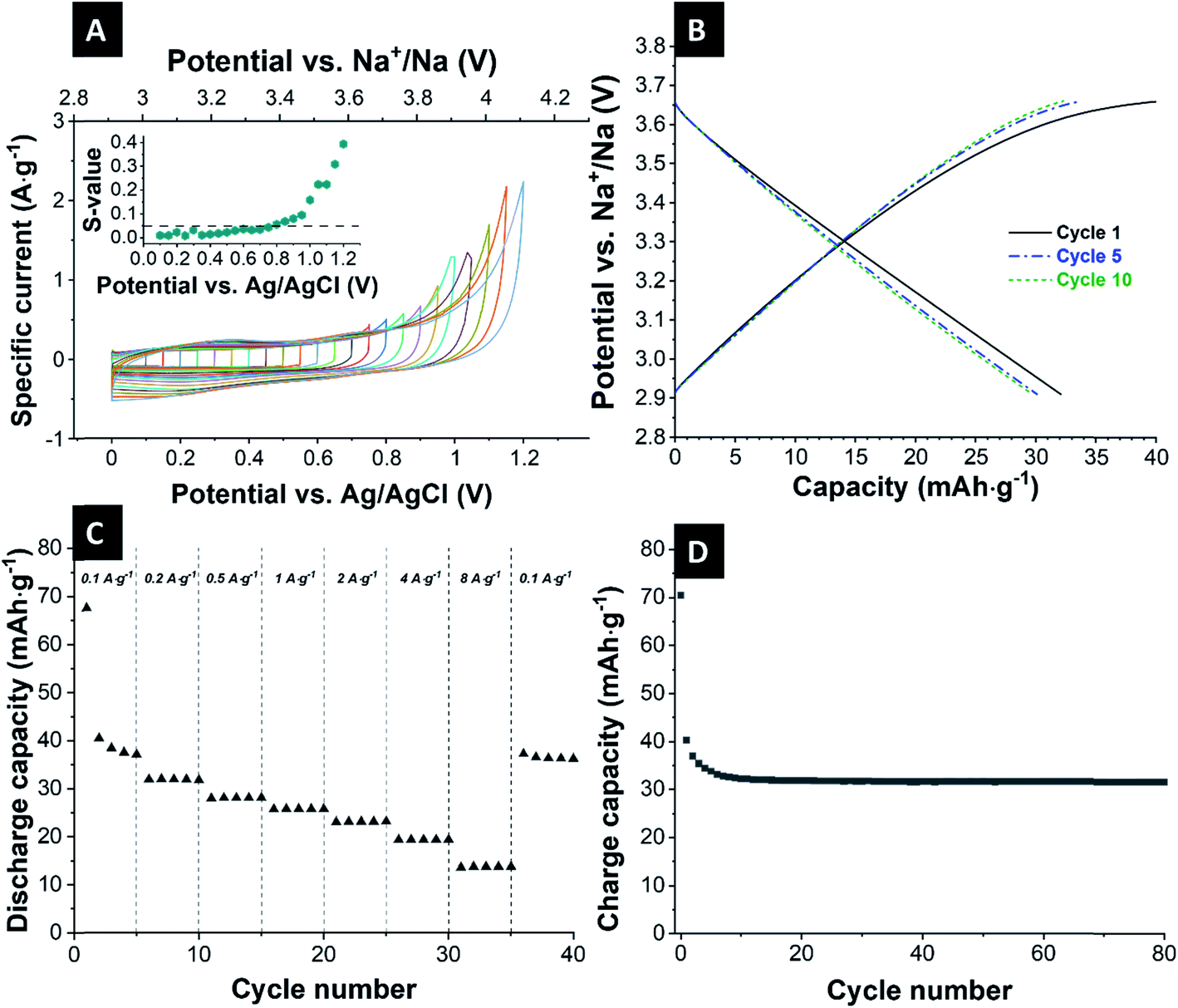
| Fig. 2 Electrochemical performance of the Sb/C electrode. (A) 1st, 5th, and 10th cyclic voltammogram at a scanning rate of 0.1 mV s−1 from 0.1 V up to 2.0 V vs. Na+/Na. (B) Galvanostatic charge and discharge profiles of the 1st, 5th, and 10th cycle at 200 mA g−1 between 0.1 V and 2.0 V vs. Na+/Na. (C) Galvanostatic charge/discharge cycling performance electrochemical stability at a specific current of 200 mA g−1. (D) Rate performance using galvanostatic charge/discharge cycling at different values for the specific current. | ||
The cycling stability of the Sb/C electrode was tested for 80 cycles at a specific current of 200 mA h g−1 in a voltage range between 0.1–2.0 V vs. Na+/Na. The corresponding galvanostatic charge and discharge curves are displayed in Fig. 2B, which show typical characteristics of the antimony electrodes.38,49,50 The different plateaus correspond to the redox reactions associated with Na+ alloying/de-alloying. The initial sodiation curve exhibits a long plateau at 0.4 V vs. Na+/Na, which can, besides the alloying products, be assigned to the formation of SEI films on the electrode.51 In the following cycles, it continuously shows the plateaus according to the alloying reaction from Sb to amorphous NaxSb to cubic and hexagonal Na3Sb mixture to hexagonal Na3Sb.52 Subsequent to the 2nd cycle, there are no significant changes in the shape of the curves. Only a small loss of capacity is visible in the discharge curves. The results of these galvanostatic curves confirm the findings received from cyclic voltammetry.
The galvanostatic curves display a potential drift during the electrochemical measurements of the cell. The plateau of the charge curve shifts in the direction of lower capacity, while the plateau of the discharge curve shifts partially in the direction of higher capacity. This characterizes an overvoltage in the cell, and the capacity is less affected compared to the plateau position (ESI, Fig. S3A†). The reasons for this shift may include low kinetics, inhomogeneities of the electrode, and undesired side reactions to a change in the reaction mechanism according to previous works on alloying electrodes.38,53–55
The cycling stability is depicted in Fig. 2C. The initial capacity has a value of 714 mA h g−1. As usual for sodium-ion batteries, there is a small increase of the capacity over the course of the first cycles. The cells slowly degrade continuously over the course of the 80 cycles, especially toward the higher cycle numbers. The degradation occurred in all cells, but at different rates, which resulted in an increased error bar seen at higher cycle numbers in Fig. 2C. After 80 cycles, Sb/carbon electrode reaches a capacity of 426 mA h g−1, which corresponds to a value of 60% of the initial capacity.
The rate capability of these Sb/C electrodes is evaluated at a varying specific current from 0.1 A g−1 to 8 A g−1 (Fig. 2D). Up to a specific current of 1 A g−1, the electrodes show stable electrochemical behavior with a capacity of higher than 600 mA h g−1 (0.1 A g−1: 708 mA h g−1; 0.2 A g−1: 668 mA h g−1; 0.5 A g−1: 632 mA h g−1; 1 A g−1: 603 mA h g−1). At a specific current of 2 A g−1 and higher, the cells lost most of their charge storage capacity. When the specific current is again reduced to 0.1 A g−1, the cell nearly reaches its initial capacity but degrades toward the 5th cycle.
 | ||
| Fig. 3 Characterization of the activated carbon cloth electrode in 600 mM NaCl with an oversized activated carbon counter-electrode. (A) Voltammetric potential window opening experiments were recorded at a scan rate of 0.1 mV s−1 (inset: S-value vs. vertex voltage plot). (B) Galvanostatic charge–discharge profiles of the 1st, 5th, and 10th cycle at 200 mA g−1 of the activated carbon cloth electrode between 0 V and 0.75 V vs. Ag/AgCl, which corresponds with 2.91 V and 3.66 V vs. Na+/Na. (C) Rate performance of the activated carbon cloth electrode from galvanostatic charge/discharge cycling at different values for the specific current. (D) Galvanostatic charge/discharge cycling performance electrochemical stability at a specific current of 200 mA g−1. | ||
The obtained galvanostatic charge and discharge data (Fig. 3B) agree with the data from cyclic voltammetry. As expected for a nearly perfect electrical double-layer capacitor, the galvanostatic profiles show no plateaus, and the electrode delivers an initial capacity of 40 mA h g−1. Over the next cycles, the capacity is stabilized at 32 mA h g−1 (≈146 F g−1) for 80 cycles (Fig. 3C).
Fig. 3D shows the reversible capacities at various discharge/charge rates. The system retained a capacity of 36/32/27/24/23/19/14 mA h g−1 at a specific current of 0.1/0.2/0.5/1.0/2.0/4.0/8.0 A g−1. During running different values of specific current, stable values for the capacities can be obtained, even for high currents of 8 A g−1, which is the advantage of the ion-electrosorption. Only at the very first cycle, one can see that the cell needs one cycle for stabilization. When the specific current is returned to 0.1 A g−1, the cell nearly reaches its initial capacity and shows good stability.
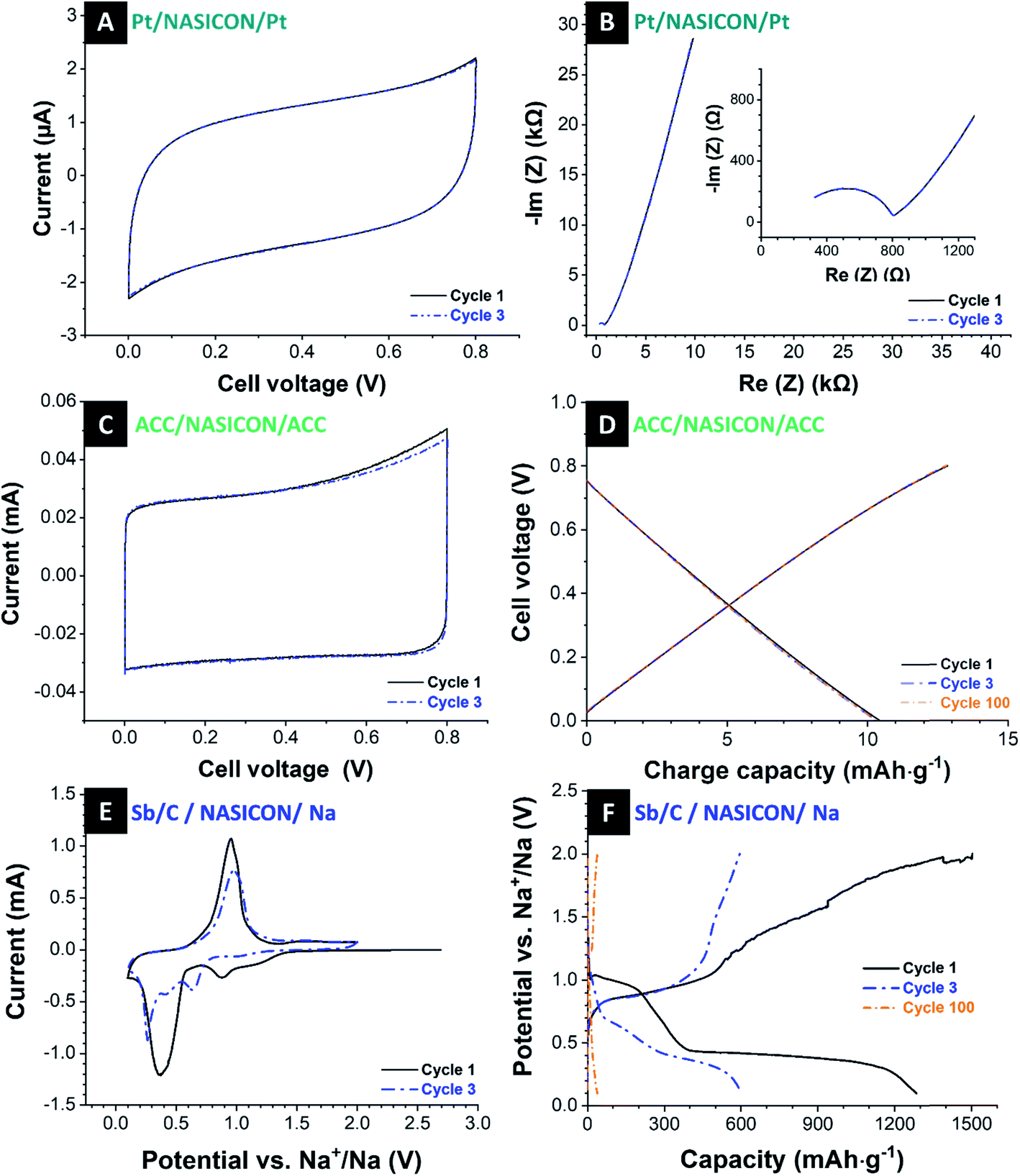 | ||
| Fig. 4 (A and B) Electrochemical characterization of the platinum-sputtered NASICON membrane: (A) cyclic voltammograms recorded at 5 mV s−1 in the potential range of 0.0–0.8 V, (B) Nyquist plots. (C and D) Electrochemical characterization of activated carbon cloth with a NASICON separator: (C) cyclic voltammograms recorded at 0.1 mV s−1 in the potential range of 0.0–0.8 V, (D) galvanostatic charge and discharge curves of the 1st, 3rd, and 100th cycle. (E and F) Electrochemical characterization of the Sb/C electrode with a NASICON separator paired with a Na-disk counter electrode: (E) first and third cyclic voltammogram recorded with 0.1 mV s−1 within the potential range from 0.1 V up to 2.0 V vs. Na+/Na, (F) galvanostatic charge and discharge curves of the 1st, 3rd, and 100th cycle. | ||
Electrochemical impedance spectroscopy (EIS) at a voltage amplitude of 100 mV was conducted using a frequency sweep of 100 mHz to 1 MHz to characterize the NASICON membrane conductivity. Fig. 4B shows the Nyquist plot with a semicircle in the medium-to-high frequency region. In the impedance spectra at high frequencies, the semicircle indicates the grain resistance of the membrane, whereas, at medium to low frequencies, the semicircle represents the grain boundary resistance. These resistors are characterized by a single semicircle as the total resistance of the membrane.39,58 At low frequencies, the diffusion phenomena can be observed. Here, the near-straight line in the low-frequency range corresponds with the interface components (electrode polarization).59
Using these data, we can calculate the effective ionic conductivity. The latter is given by the theoretical intersections of the low-frequency ends of the semicircle with the Z′ axis.
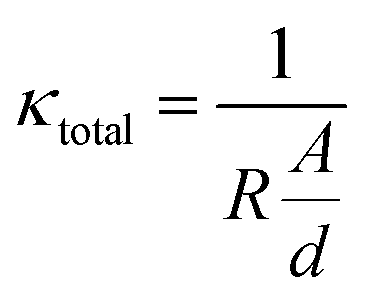 | (4) |
To characterize sodium transport across the ceramic NASICON membrane in an aqueous electrolyte, we paired the NASICON disc with two activated carbon cloth electrodes in 600 mM NaCl electrolyte and sandwich on the NASICON membrane. The first and third cyclic voltammograms at a scan rate of 0.1 mV s−1 are given in Fig. 4C within a cell voltage of 0.0 V to 0.8 V. Cyclic voltammograms at higher rates are provided in ESI, Fig. S4B.† We see highly rectangular profiles, which are typical for an ideal electrical double-layer capacitor.62 Additionally, the cell voltage of carbon cloth in the semi-solid cell is significantly lower than in a traditional cell. This is possibly due to the high contact resistance between carbon and NASICON interface as well as low ionic conductivity of the membrane.
The galvanostatic charge and discharge profiles of activated carbon cloth are shown in Fig. 4D. At a specific current of 200 mA g−1 within a voltage range of 0.0–0.8 V, there is a typical linear relation between charge and cell voltage in alignment with a nearly perfect capacitive behavior. There are no significant changes in charge/discharge capacity for 100 cycles (ESI, Fig. S5A and C†). However, considering the rate handling of carbon cloth (ESI, Fig. S5E†), the cell cannot provide fast charge/discharge as compared to the conventional cell with a glass fiber separator due to the fact that the sodium transport kinetics are much slower than that in glass fiber separator; a comparison thereof is shown in ESI, Fig. S3B.†
As in the next step, we used the Sb/C electrode as the working electrode and the pure sodium electrode as a counter electrode with the NASICON membrane as a separator. We used an organic electrolyte of 1 M NaClO4 in EC/DMC + 5% FEC and a scan rate of 0.1 mV s−1 within the range from 0.1–2.0 V vs. Na+/Na. The obtained cyclic voltammograms with the NASICON membrane (Fig. 4E) are similar to those with just a porous separator (Fig. 2A) with the characteristic peaks for the alloy reactions of antimony with sodium. This indicates that the comparatively thick NASICON electrode can transport Na+ to the extent or at speed required for the alloy reactions to take place. Cyclic voltammograms at higher rates are provided in ESI, Fig. S4C.† Conspicuous is the broader shape of the obtained cyclic voltammograms with the NASICON membrane compared with the same cell setup with glass fiber separator. In general, the breadth of peaks depends on the limiting stage of processes like the velocity of mass or electron transfer and diffusion and chemical processes before or after electron transfer. So, in this current case, the broader peaks may be an indicator of lower transport rates of the ions in the cell because of the thick NASICON membrane.
Fig. 4F presents the galvanostatic charge and discharge profiles of Sb/C electrode vs. Na+/Na with a NASICON membrane as the separator, at a specific current of 200 mA g−1 and a voltage range of 0.1–2.0 V. The initial charge curve exhibits the similar a long plateau at 0.4 V which represents the SEI formation. In addition, the discharge curve reaches beyond 1285 mA h g−1 of the corresponding charge curve. It seems that the sodium ions in the NASICON block the channels at times, and the cell cannot be discharged as easily. After the first cycle, this phenomenon is vanishing. During the next cycles, one sees the plateaus at 0.7–1.0 V during sodium-ion removal, which can be related to the reaction of Na3Sb alloys back to crystalline antimony. The observations from the galvanostatic charge and discharge curves are consistent with the information obtained from cyclic voltammetry.
The cycling stability is presented in ESI, Fig. S5B.† The performance is characterized by a continuous loss of capacity in each cycle. The cells exhibit a capacity of 643 mA h g−1 in the second cycle and show a capacity of about 453 mA h g−1 after only 10 cycles, which correspond to a loss of capacity of 30%. In the 50th cycle, the cells still reach a capacity of approximately 100 mA h g−1, and after the 80 cycles, only a low capacity of 44 mA h g−1 is delivered, which corresponds to a total capacity loss of 93% over 80 cycles. The coulombic efficiency, shown in ESI, Fig. S5D,† shows constant values of 96–97%. Compared to the cell with porous separator, the rate capability shows a lower capacity and stability at all rates (ESI, Fig. S5F†) as the sodium ion diffusion through the element-specific channels of the NASICON membrane is limited by the thick membrane. Yet, the ceramic NASICON membrane works sufficiently well to provide the antimony electrode with a sufficient supply of sodium for the alloying reaction.
3.3 Desalination performance
Electrochemical desalination was performed in a desalination cell described elsewhere.29,34 The Sb/C electrode was surrounded by 1 M NaClO4 in EC/DMC + 5% FEC electrolyte and separated from the feedwater stream by a ceramic NASICON membrane with 300 μm thickness. Oversized activated carbon cloth is used as a counter electrode. As feedwater, aqueous 600 mM NaCl is fed to the cell with a peristaltic pump. To be sure that the electrolyte is oxygen-free, we constantly flushed the electrolyte with N2 gas throughout the desalination experiment. As a specific current of 200 mA g−1 was applied to the cell with the cut-off cell voltage of ±2.0 V, the concentration profile behaves like in a regular capacitive deionization cell. In the first cycle, the concentration of NaCl has decreased with the amplitude of about 5 mM when the cell is charged. After 80 min of charging, the cell is rested for 2 min and discharged, one can see the increasing of NaCl concentration, indicating the de-sodiation of Sb/C electrode (Fig. 5A). After 40 cycles of operation, the amplitude of decreased NaCl concentration is significantly lower than that of the first cycle (Fig. 5B). This is due to the intrinsic mechanism of sodium-alloying in the Sb electrode. The charge capacity of our desalination cell is about 669 mA h gSb−1 at the first cycle after conditioning cycles and decays to 360 mA h gSb−1 after 40 cycles (Fig. 5C). The corresponding desalination capacity of the first cycle is 495 mgNa gSb−1 and stabilizes at around 250 mgNa gSb−1 (corresponds to 635 mgNaCl gSb−1 and 1116 mgNa mmembrane−2) with the charge efficiency of about 74% for 40 cycles (Fig. 5D).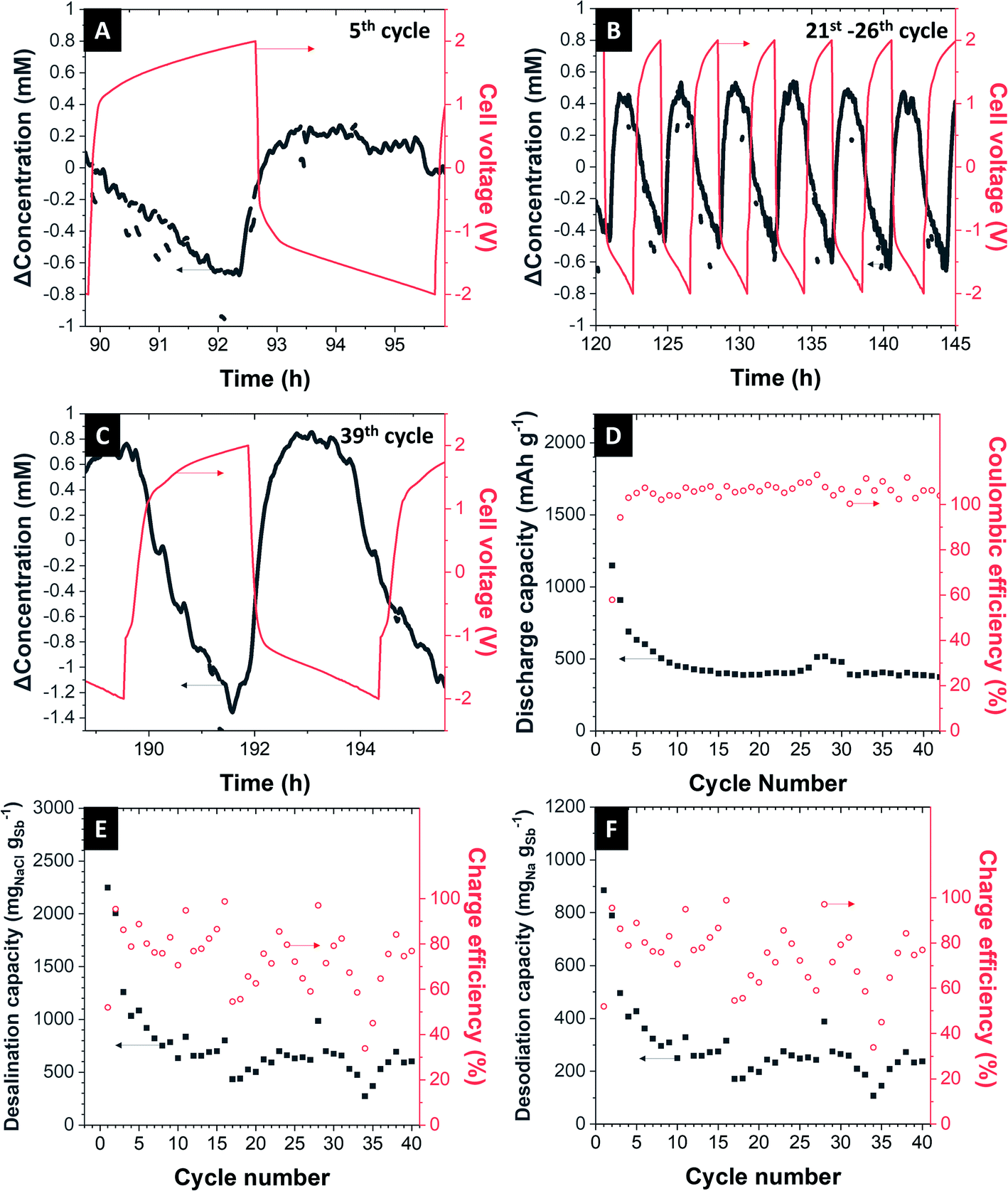 | ||
| Fig. 5 Performance of the electrochemical desalination cell. (A–C) The change of concentration and voltage profile at the 5th cycle, 21st to 26th cycles and the 39th cycle. (D) The charge capacity and the corresponding values of the coulombic efficiency. (E) The desalination capacity (mgNaCl gSb−1) and charge efficiency. (F) The desalination capacity (mgNa gSb−1) and charge efficiency. | ||
Reasons for the low charge efficiency could be, among others, the side reactions (like oxidation of carbon or splitting of water), the imperfect shape, and properties of the NASICON membrane, which prevents the ions from being transported in the fastest and easiest way, which also results in poorer kinetics. These factors also will contribute towards the decay of performance seen during continued cell operation.
In comparison to other systems (charge-transfer materials),33 which successfully desalinate with different mechanisms like an ion electrosorption (3–27 mgNaCl gelectrode−1; 1–11 mgNa gelectrode−1),63 insertion of different materials and redox couples (6–140 mgNaCl gelectrode−1; 2–55 mgNa gelectrode−1),27 or conversion type (17–115 mgNaCl gelectrode−1; 7–22 mgNa gelectrode−1),23,24 the system described in this work shows very promising values (Table 1).
| System | Electrode | Desalination capacity mgNaCl gelectrode−1/mgNa gelectrode−1 | Charge efficiency (%) | Energy consumption | Cell voltage (V) | Electrolyte | Reference |
|---|---|---|---|---|---|---|---|
| Faradaic deionization | Na2−xMn5O10/Ag/AgCl | n.a. | n.a. | 0.29 W h L−1 | ∼0.55/0.19 | 600 mM NaCl | 17 |
| Capacitive deionization | Nanoporous carbon | 7/3 | ∼85% | n.a. | 1.4/1.2 V | 5 mM | 69 |
| Capacitive deionization | Nanoporous carbon | 13/ | 86% | n.a. | 1.2 V | 1000 M NaCl | 70 |
| Faradaic deionization | Ti3C2-MXene | 13 ± 2/5 | n.a. | n.a. | 1.2 V | 5 mM NaCl | 71 |
| Faradaic deionization | Bi/NaTi2(PO4)3 | 83/ | n.a. | n.a. | 0.75/0.55 V | 600 M NaCl | 25 |
| Multi-channel membrane capacitive deionization | Activated carbon cloth | 56/33 | ∼82% | ∼20 kT | 0.0/−1.2 V | Side channel: 1000 M, middle channel: 5 mM | 64 |
| Membrane capacitive deionization (aqueous/organic bi-electrolyte) | Nanoporous carbon | 64 ± 4/25 ± 2 | 95%. | 14–27 kT | 2.4 V | 5 mM NaCl | 29 |
| Faradaic deionization | MoS2-CNT | 25/10 | 80% | 24.6 kT | 0.8/0.0 V | 500 M NaCl | 72 |
| Hybrid capacitive deionization | Na4Mn9O18/nanoporous carbon | 31/12 | n.a. | n.a. | 0.8/0.0 V | 1000 M NaCl | 18 |
| Hybrid capacitive deionization (aqueous/organic bi-electrolyte) | Sb–C/nanoporous carbon | 748 mg NaCl g Sb −1 , 294 mg Na g Sb −1 | 74% | 16 kT (0.0005 W h L −1 ) |
+2.0/−2.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) V V
|
600 M NaCl | This work |
For membrane capacitive deionization (MCDI), a value of 22 kT is often reported.8,29 The first desalination battery achieved a value of 0.29 W h L−1 for energy consumption.17 Kim et al. report for a multi-channel membrane capacitive deionization an energy consumption of 20 kT.64 The energy consumption of the cell configuration in this work is calculated to be 16 kT or 39 kJ mol−1NaCl after the first cycles. This value corresponds to the energy per processed water of 0.0005 W h L−1, which is also comparable to traditional methods like reverse osmosis (0.5–2 W h L−1),65 multistage flash distillation (20–30 W h L−1),66 multi-effect distillation (15–20 W h L−1),67 freezing methods (6–8 W h L−1).68 Compared to other faradaic materials (including intercalation materials), conversion materials, redox electrolytes, and zinc–air desalination, alloying with the aid of NASICON deliver energy consumption lower than others. A direct comparison, however, is complicated by different experimental settings, such as flow rate, cell volume, and the extent of salt reduction.27
4 Conclusion
This study demonstrates a desalination cell by using antimony as a sodium-alloying material, which is a promising and high-performing electrode in sodium-ion batteries. By applying a bi-electrolyte system (organic and aqueous part), the cell can provide stable desalination performance at higher maximum cell voltage than the conventional capacitive deionization technology allows (1.0–1.2 V). In an optimized desalination cell configuration with a middle channel for the inflowing feed stream and a selective sodium permeable membrane (NASICON), the Sb/C electrodes exhibit an improved electrochemical performance with a discharge capacity of initially 669 mA h g−1 (395 mA h g−1 after 40 cycles) at a specific current of 200 mA g−1 and a voltage range of −2.0 V to +2.0 V, which results in a desalination capacity on the average of 294 mgNa gSb−1 with a charge efficiency of about 74% in aqueous 600 mM NaCl. Future work will have to further optimize the cell design, and further reduction of the ceramic membrane thickness will allow the faster operation of the desalination battery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. W. acknowledges funding from the Chinese Scholarship Council (CSC) via award number 201906260277. We thank Eduard Arzt (INM) for his continuing support. Further, we thank the INM Service Group Chemical Analytics for chemical analysis and Samantha Husmann (INM) for technical support and discussions.References
- N. C. Darre and G. S. Toor, Curr. Pollut. Rep., 2018, 4, 104–111 CrossRef CAS.
- A. D. Khawaji, I. K. Kutubkhanah and J.-M. Wie, Desalination, 2008, 221, 47–69 CrossRef CAS.
- A. Matin, Z. Khan, S. M. J. Zaidi and M. C. Boyce, Desalination, 2011, 281, 1–16 CrossRef CAS.
- Y.-y. Lu, Y.-d. Hu, D.-m. Xu and L.-y. Wu, J. Membr. Sci., 2006, 282, 7–13 CrossRef CAS.
- A. Rich, Y. Mandri, N. Bendaoud, D. Mangin, S. Abderafi, C. Bebon, N. Semlali, J.-P. Klein, T. Bounahmidi, A. Bouhaouss and S. Veesler, Desalin. Water Treat., 2010, 13, 120–127 CrossRef CAS.
- S. Porada, R. Zhao, A. van der Wal, V. Presser and P. M. Biesheuvel, Prog. Mater. Sci., 2013, 58, 1388–1442 CrossRef CAS.
- M. E. Suss, S. Porada, X. Sun, P. M. Biesheuvel, J. Yoon and V. Presser, Energy Environ. Sci., 2015, 8, 2296–2319 RSC.
- R. Zhao, S. Porada, P. M. Biesheuvel and A. van der Wal, Desalination, 2013, 330, 35–41 CrossRef CAS.
- S. Porada, L. Zhang and J. E. Dykstra, Desalination, 2020, 488, 114383 CrossRef CAS.
- P. M. Biesheuvel, S. Porada, M. Levi and M. Z. Bazant, J. Solid State Electrochem., 2014, 18, 1365–1376 CrossRef CAS.
- C. Prehal, C. Koczwara, H. Amenitsch, V. Presser and O. Paris, Nat. Commun., 2018, 9, 4145 CrossRef CAS PubMed.
- S. Bi, Y. Zhang, L. Cervini, T. Mo, J. M. Griffin, V. Presser and G. Feng, Sustainable Energy Fuels, 2020, 4, 1285–1295 RSC.
- N. Jäckel, P. Simon, Y. Gogotsi and V. Presser, ACS Energy Lett., 2016, 1, 1262–1265 CrossRef.
- F. Stoeckli and T. A. Centeno, Phys. Chem. Chem. Phys., 2012, 14, 11589–11591 RSC.
- H. Ji, X. Zhao, Z. Qiao, J. Jung, Y. Zhu, Y. Lu, L. L. Zhang, A. H. MacDonald and R. S. Ruoff, Nat. Commun., 2014, 5, 3317 CrossRef PubMed.
- Z.-H. Huang, Z. Yang, F. Kang and M. Inagaki, J. Mater. Chem. A, 2017, 5, 470–496 RSC.
- M. Pasta, C. D. Wessells, Y. Cui and F. La Mantia, Nano Lett., 2012, 12, 839–843 CrossRef CAS PubMed.
- J. Lee, S. Kim, C. Kim and J. Yoon, Energy Environ. Sci., 2014, 7, 3683–3689 RSC.
- P. Srimuk, J. Lee, Ö. Budak, J. Choi, M. Chen, G. Feng, C. Prehal and V. Presser, Langmuir, 2018, 34, 13132–13143 CrossRef CAS PubMed.
- S. Porada, A. Shrivastava, P. Bukowska, P. M. Biesheuvel and K. C. Smith, Electrochim. Acta, 2017, 255, 369–378 CrossRef CAS.
- P. Srimuk, J. Lee, A. Tolosa, C. Kim, M. Aslan and V. Presser, Chem. Mater., 2017, 29, 9964–9973 CrossRef CAS.
- E. Grygolowicz-Pawlak, M. Sohail, M. Pawlak, B. Neel, A. Shvarev, R. de Marco and E. Bakker, Anal. Chem., 2012, 84, 6158–6165 CrossRef CAS PubMed.
- P. Srimuk, S. Husmann and V. Presser, RSC Adv., 2019, 9, 14849–14858 RSC.
- J. Ahn, J. Lee, S. Kim, C. Kim, J. Lee, P. M. Biesheuvel and J. Yoon, Desalination, 2020, 476, 114216 CrossRef CAS.
- D.-H. Nam and K.-S. Choi, J. Am. Chem. Soc., 2017, 139, 11055–11063 CrossRef CAS PubMed.
- F. Yu, L. Wang, Y. Wang, X. Shen, Y. Cheng and J. Ma, J. Mater. Chem. A, 2019, 7, 15999–16027 RSC.
- P. Srimuk, X. Su, J. Yoon, D. Aurbach and V. Presser, Nat. Rev. Mater., 2020, 5, 517–538 CrossRef CAS.
- S. Porada, G. Feng, M. E. Suss and V. Presser, RSC Adv., 2016, 6, 5865–5870 RSC.
- C. Kim, P. Srimuk, J. Lee and V. Presser, Desalination, 2018, 443, 56–61 CrossRef CAS.
- Y. Kim, H. Kim, S. Park, I. Seo and Y. Kim, Electrochim. Acta, 2016, 191, 1–7 CrossRef CAS.
- Y. Kim, G.-T. Kim, S. Jeong, X. Dou, C. Geng, Y. Kim and S. Passerini, Energy Storage Materials, 2019, 16, 56–64 CrossRef.
- N. Kim, J.-S. Park, A. M. Harzandi, K. Kishor, M. Ligaray, K. H. Cho and Y. Kim, Desalination, 2020, 495, 114666 CrossRef CAS.
- M. E. Suss and V. Presser, Joule, 2018, 2, 10–15 CrossRef.
- J. Lee, P. Srimuk, R. L. Zornitta, M. Aslan, B. L. Mehdi and V. Presser, ACS Sustainable Chem. Eng., 2019, 7, 10132–10142 CrossRef CAS.
- S. Z. Liang, Y. J. Cheng, J. Zhu, Y. G. Xia and P. Müller-Buschbaum, Small Methods, 2020, 4, 2000218 CrossRef CAS.
- K. Pfeifer, S. Arnold, Ö. Budak, X. Luo, V. Presser, H. Ehrenberg and S. Dsoke, J. Mater. Chem. A, 2020, 8, 6092–6104 RSC.
- H. Tan, D. Chen, X. Rui and Y. Yu, Adv. Funct. Mater., 2019, 29, 1808745 CrossRef.
- H. Hou, Y. Yang, Y. Zhu, M. Jing, C. Pan, L. Fang, W. Song, X. Yang and X. Ji, Electrochim. Acta, 2014, 146, 328–334 CrossRef CAS.
- H. Park, K. Jung, M. Nezafati, C.-S. Kim and B. Kang, ACS Appl. Mater. Interfaces, 2016, 8, 27814–27824 CrossRef CAS PubMed.
- M. Zeiger, N. Jäckel, M. Aslan, D. Weingarth and V. Presser, Carbon, 2015, 84, 584–598 CrossRef CAS.
- D. Weingarth, M. Zeiger, N. Jäckel, M. Aslan, G. Feng and V. Presser, Adv. Energy Mater., 2014, 4, 1400316 CrossRef.
- J. Qian, Y. Chen, L. Wu, Y. Cao, X. Ai and H. Yang, Chem. Commun., 2012, 48, 7070–7072 RSC.
- Y.-X. Wang, Y.-G. Lim, M.-S. Park, S.-L. Chou, J. H. Kim, H.-K. Liu, S.-X. Dou and Y.-J. Kim, J. Mater. Chem. A, 2014, 2, 529–534 RSC.
- A. Ponrouch, D. Monti, A. Boschin, B. Steen, P. Johansson and M. R. Palacin, J. Mater. Chem. A, 2015, 3, 22–42 RSC.
- A. Ponrouch, A. R. Goñi and M. R. Palacín, Electrochem. Commun., 2013, 27, 85–88 CrossRef CAS.
- V. Simone, L. Lecarme, L. Simonin and S. Martinet, J. Electrochem. Soc., 2016, 164, A145–A150 CrossRef.
- K. Xu, S. P. Ding and T. R. Jow, J. Electrochem. Soc., 1999, 146, 4172–4178 CrossRef CAS.
- J. Lee, P. Srimuk, S. Carpier, J. Choi, R. L. Zornitta, C. Kim, M. Aslan and V. Presser, ChemSusChem, 2018, 11, 3460–3472 CrossRef CAS PubMed.
- S. Liu, J. Feng, X. Bian, J. Liu and H. Xu, Energy Environ. Sci., 2016, 9, 1229–1236 RSC.
- L. Wu, X. Hu, J. Qian, F. Pei, F. Wu, R. Mao, X. Ai, H. Yang and Y. Cao, Energy Environ. Sci., 2014, 7, 323–328 RSC.
- X. Zhou, Z. Dai, J. Bao and Y.-G. Guo, J. Mater. Chem. A, 2013, 1, 13727–13731 RSC.
- A. Darwiche, C. Marino, M. T. Sougrati, B. Fraisse, L. Stievano and L. Monconduit, J. Am. Chem. Soc., 2012, 134, 20805–20811 CrossRef CAS PubMed.
- P. R. Abel, Y.-M. Lin, T. de Souza, C.-Y. Chou, A. Gupta, J. B. Goodenough, G. S. Hwang, A. Heller and C. B. Mullins, J. Phys. Chem. C, 2013, 117, 18885–18890 CrossRef CAS.
- M.-S. Balogun, Y. Luo, W. Qiu, P. Liu and Y. Tong, Carbon, 2016, 98, 162–178 CrossRef CAS.
- Y. Kim, Y. Park, A. Choi, N.-S. Choi, J. Kim, J. Lee, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2013, 25, 3045–3049 CrossRef CAS PubMed.
- M. Bahdanchyk, M. Hashempour and A. Vicenzo, Electrochim. Acta, 2020, 332, 135503 CrossRef CAS.
- D. Weingarth, H. Noh, A. Foelske-Schmitz, A. Wokaun and R. Kötz, Electrochim. Acta, 2013, 103, 119–124 CrossRef CAS.
- S. Narayanan, S. Reid, S. Butler and V. Thangadurai, Solid State Ionics, 2019, 331, 22–29 CrossRef CAS.
- A. Jalalian-Khakshour, C. O. Phillips, L. Jackson, T. O. Dunlop, S. Margadonna and D. Deganello, J. Mater. Sci., 2020, 55, 2291–2302 CrossRef.
- S. Naqash, D. Sebold, F. Tietz and O. Guillon, J. Am. Ceram. Soc., 2019, 102, 1057–1070 CrossRef CAS.
- S.-M. Lee, S.-T. Lee, D.-H. Lee, S.-H. Lee, S.-S. Han and S.-K. Lim, Journal of Ceramic Processing Research, 2015, 16, 49–53 Search PubMed.
- H. Shao, Y. C. Wu, Z. Lin, P. L. Taberna and P. Simon, Chem. Soc. Rev., 2020, 49, 3005–3039 RSC.
- Y. Liu, C. Y. Nie, X. J. Liu, X. T. Xu, Z. Sun and L. K. Pan, RSC Adv., 2015, 5, 15205–15225 RSC.
- C. Kim, J. Lee, P. Srimuk, M. Aslan and V. Presser, ChemSusChem, 2017, 10, 4914–4920 CrossRef CAS PubMed.
- A. Shrivastava, S. Rosenberg and M. Peery, Desalination, 2015, 368, 181–192 CrossRef CAS.
- R. Semiat, Environ. Sci. Technol., 2008, 42, 8193–8201 CrossRef CAS PubMed.
- A. Al-Karaghouli and L. L. Kazmerski, Renewable Sustainable Energy Rev., 2013, 24, 343–356 CrossRef CAS.
- T. Mtombeni, J. Maree, C. Zvinowanda, J. Asante, F. Oosthuizen and W. Louw, Int. J. Environ. Sci. Technol., 2013, 10, 545–550 CrossRef CAS.
- S. Porada, L. Weinstein, R. Dash, A. van der Wal, M. Bryjak, Y. Gogotsi and P. M. Biesheuvel, ACS Appl. Mater. Interfaces, 2012, 4, 1194–1199 CrossRef CAS PubMed.
- M. Aslan, M. Zeiger, N. Jäckel, I. Grobelsek, D. Weingarth and V. Presser, J. Phys.: Condens. Matter, 2016, 28, 114003 CrossRef CAS PubMed.
- P. Srimuk, F. Kaasik, B. Krüner, A. Tolosa, S. Fleischmann, N. Jäckel, M. C. Tekeli, M. Aslan, M. E. Suss and V. Presser, J. Mater. Chem. A, 2016, 4, 18265–18271 RSC.
- P. Srimuk, J. Lee, S. Fleischmann, S. Choudhury, N. Jäckel, M. Zeiger, C. Kim, M. Aslan and V. Presser, J. Mater. Chem. A, 2017, 5, 15640–15649 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: chemical analysis data, supporting electrochemical characterization, schematic illustration of the cell operation, post mortem material characterization (SEM, XRD). See DOI: 10.1039/d0ta09806j |
| This journal is © The Royal Society of Chemistry 2021 |