
Lanthanum loaded CuO nanoparticles: synthesis and characterization of a recyclable catalyst for the synthesis of 1,4-disubstituted 1,2,3-triazoles and propargylamines†
R. Sasikala,
S. Kutti Rani*,
D. Easwaramoorthy and
K. Karthikeyan*
Department of Chemistry, B. S. Abdur Rahman University, Vandalur, Chennai-600 048, India. E-mail: karthiclri@gmail.com; skrani@bsauniv.ac.in; Fax: +91-44-22750520; Tel: +91-44-22750520
First published on 9th June 2015
Abstract
Lanthanum loaded CuO (LCO) nanoparticles (NPs) were successfully synthesized by a precipitation–thermal decomposition method and characterized by X-ray diffraction (XRD), field emission scanning electron microscopy (FESEM), energy dispersive spectra (EDS), diffuse reflectance spectra (DRS), photoluminescence (PL), X-ray photoelectron spectroscopy (XPS) and BET surface area measurements. The synthesized LCO NPs were used as a nanocatalyst for the synthesis of 1,4-disubstituted 1,2,3-triazoles by a click reaction of substituted benzyl azides and alkynes and the synthesis of propargylamines via three-component coupling reactions of aldehydes, alkynes and amines under ultrasonication. The one-pot operation, atom-economical nature, regioselectivity and good yields are the noteworthy features of this protocol. The reusability of the prepared nanocatalyst was successfully examined six times without any appreciable loss in catalytic activity.
Introduction
During the last few decades, great attention has been given to click1 and green chemistry2 for the development of efficient and environmentally benign protocols. Recently nanoparticles and nanoparticles supported on metal oxides have emerged as a sustainable and competitive alternative to conventional catalysts, and have been extensively studied in the field of medicine for drug delivery systems.3,4 Mesoporous nanomaterials have also gained increasing importance for their use as catalysts in various organic reactions,5 as sensors for the detection of hydrazine,6 in optoelectronics7,8 and as electron transfer mediators in bio-electrochemical systems.91,2,3-Triazoles are important sub-units in heterocyclic chemistry because of their unique structure, chemical and biological properties.10 Huisgen 1,3-dipolar azide–alkyne cycloaddition is traditionally applied for accessing 1,2,3-triazoles.11 However, the major limitations of this non-catalyzed process are the requirement of high temperature and the poor regioselectivity giving a mixture of 1,4- and 1,5-disubstituted triazoles. Up to now, several efforts to control the 1,4- versus 1,5-regioselectivity have been reported.12 As a prototype for click chemistry,13 the recent advance in Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reported by Sharpless14 and Meldal15 has enabled access to 1,2,3-triazoles in short reaction times under mild conditions and as only one 1,4-regioisomer.16–19 The major limitation of the existing CuAAC protocols realized as homogeneous catalysts is the problem of separation of the catalyst and product(s), and another is the requirement of adding reducing agents and stabilizing ligands which limits their utilization for practical processes. Recent research in this area has concentrated on heterogeneous catalytic systems, which have several advantages, such as faster and simpler isolation of the reaction products by filtration, as well as recovery and recycling of the catalyst systems. Thus, particles of copper20 or its oxide derivatives,21 copper salts supported on charcoal22 or on organic materials,23 as well as on inorganic supports24 have been tested for this transformation.
Furthermore, propargylamines are key intermediates for the synthesis of natural products and nitrogen containing biologically active compounds.25 Recently, propargylamines have been prepared by transition metal-catalyzed A3 coupling reactions of aldehydes, alkynes and amines. A3 coupling reactions, which employ amines as nitrogen sources, have received much attention due to their simple operation and high efficiency. Several homogenous catalysts like nickel26a and copper salts,26b CuI/ligand,26c and heterogeneous systems such as Ag(I),27a and Cu(I),27b in ionic liquids, silica-immobilized CuI,27c Cu(0)-nanoparticles on montmorillonite,27d copper ferrite nanoparticles,27e CuNPs/MagSilica,27f Au–SBA27g and Au–CeO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27h have been developed for propargylamine synthesis.
27h have been developed for propargylamine synthesis.
On the other hand, ultrasound irradiation has been well established as an energy source to promote chemical reactions.28 Many homogeneous and heterogeneous reactions can be conducted smoothly by sonication under milder conditions and shorter reaction times to afford improved yields and increased selectivity.29 Ultrasound irradiation has some potential effects on a heterogeneous catalytic system, like increasing the active catalyst surface area, promotion of cavitation bubble formation, and removal of impurities deposited on the catalyst.30,31
Modern research has focused on finding new catalysts to improve the efficiency of these azide–alkyne cycloaddition and A3 coupling reactions. As the above mentioned methods are not compatible with heat sensitive substrates there is a need to develop an effective synthesis of triazoles and propargylamines employing a more eco-friendly catalyst. With this view, we wish to describe a heterogeneous lanthanum loaded CuO-catalyst for the synthesis of 1,4-disubstituted 1,2,3-triazole and propargylamine derivatives under ultrasonication.
Results and discussion
Catalyst preparation and characterization
The LCO NPs were prepared using a precipitation thermal decomposition method. Two aqueous solutions of copper nitrate and oxalic acid were separately brought to their boiling points. The oxalic acid solution was added to the copper nitrate solution, copper oxalate was precipitated, then the La(NO3)3 solution was mixed with the copper oxalate suspension and stirred for 1 h at 60–70 °C. After the mixture cooled to room temperature, the La-containing copper oxalate was washed with distilled water, air dried overnight and dried at 100 °C for 5 h. Calcination of the mixed precipitate at 450 °C for 5 h resulted in the decomposition of copper oxalate to CuO NPs. La-loaded CuO NPs were obtained after the sample was cooled to room temperature. The bare CuO NPs were prepared without addition of La(NO3)3 by the above procedure.The prepared LCO NPs were characterized by XRD, FE-SEM, EDS, DRS, PL, XPS and BET surface area measurements. The crystalline nature of the bare CuO NPs was identified from their powder XRD patterns (Fig. 1a). All the diffractions were well matched with the monoclinic phase of CuO NPs (standard JCPDS File no. 048-1548): the 2θ diffraction peaks were 35.45°, 38.73°, 42.50°, 48.92°, 61.99° and 66.49°, corresponding to the (002), (111), (200), (202), (220) and (113) planes of the monoclinic phase of CuO NPs.32 In the XRD spectrum of the LCO NPs (Fig. 1b), no additional peaks appeared but the peaks were slightly broadened due to the reduction in the particle size compared to bare CuO NPs. The inset of Fig. 1 shows that the peak corresponding to LCO NPs was slightly shifted to a lower angle, which revealed that the La was loaded on the surface of CuO material.33 The crystalline sizes of the LCO and CuO NPs were determined using the Debye–Scherrer equation:
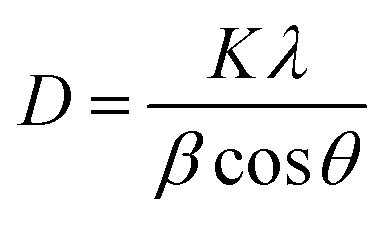
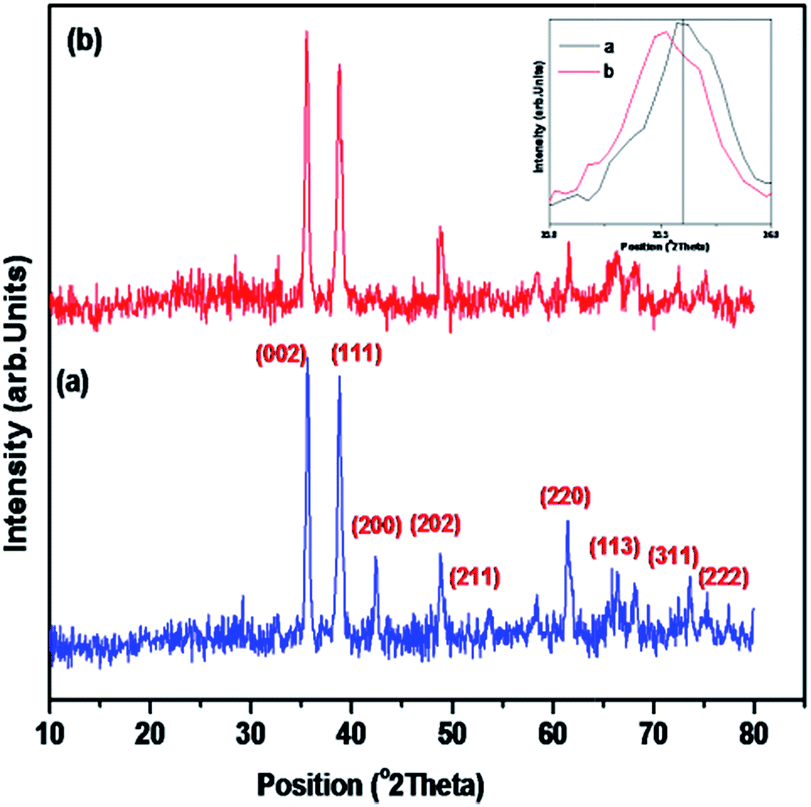 | ||
| Fig. 1 XRD patterns of (a) bare CuO and (b) LCO NPs (the inset figure shows a magnification of the XRD patterns of (a) bare CuO and (b) LCO NPs). | ||
The FE-SEM images of CuO and LCO NPs (Fig. 2) show that the particle sizes are in the nanometer region, which is in agreement with the grain size based on Scherrer’s formula. Fig. 2(a) shows the almost uniform formation of spherical shaped CuO (3 μm) particles. In addition, it shows that there are more gaps between the spherical shaped CuO NPs. The LCO NPs (3 μm) exhibit agglomerate formations; Fig. 2(b) reveals non-uniform formation of spherical shaped nanoparticles. It shows that the LCO NPs morphology is much better than the CuO NPs morphology.
 | ||
| Fig. 2 FE-SEM images of (a) CuO (3 μm) and (b) LCO NPs (3 μm). | ||
The EDS spectra of the bare CuO and LCO NPs are depicted in Fig. 3, showing the presence of Cu and O in the bare CuO and La, Cu, O in the LCO NPs. This indicates clearly that the La ions were successfully loaded on the surface of CuO NPs.
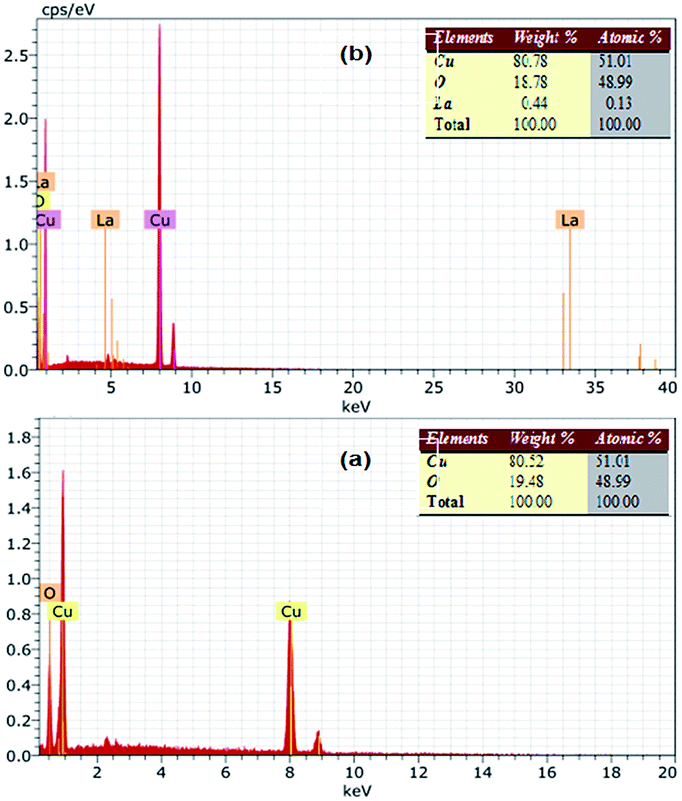 | ||
| Fig. 3 EDS of (a) bare CuO and (b) LCO NPs. | ||
The diffuse reflectance spectra (DRS) of pure CuO and LCO NPs are depicted in Fig. 4. La3+ might covalently interact with CuO and slightly reduce the band gap of LCO NPs. There is no significant change of absorption in the UV region but LCO NPs have a higher absorption than bare CuO NPs in the visible region. Photoluminescence (PL) occurs due to the recombination of electron–hole pairs in the semiconductor. PL (Fig. 5) spectra reveal that the PL intensity of the LCO NPs is less than that for the CuO NPs. This is due to the suppression of recombination of the photo generated electron–hole pairs by the La loaded on to the CuO. Inhibition of the electron–hole recombination makes the catalyst more active.
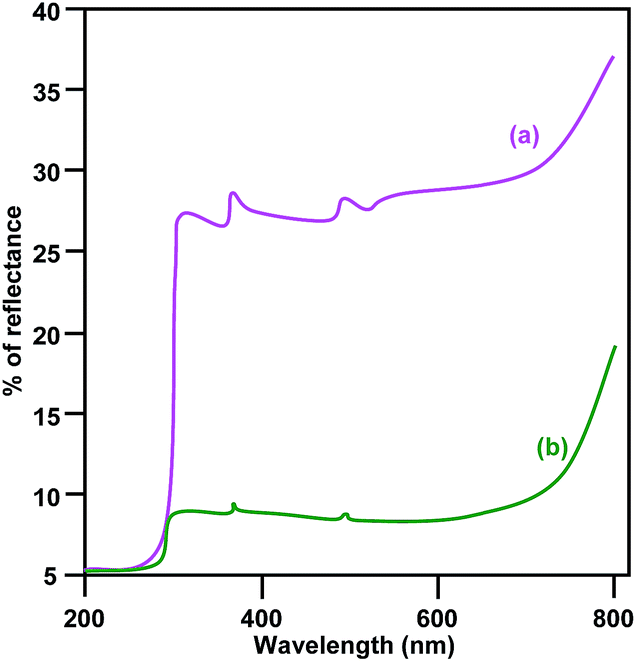 | ||
| Fig. 4 DRS of (a) bare CuO and (b) LCO NPs. | ||
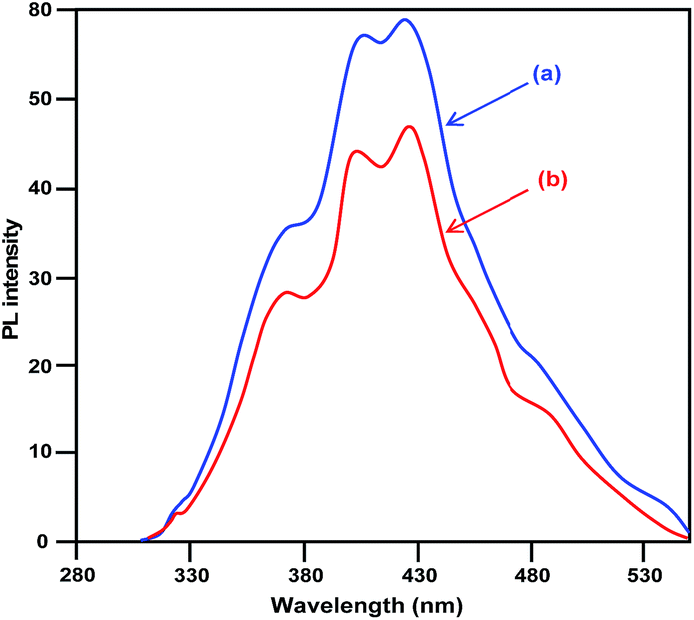 | ||
| Fig. 5 Photoluminescence spectra of (a) bare CuO and (b) LCO NPs. | ||
The elemental composition and oxidation states of the CuO NPs and LCO NPs were analyzed using XPS. The binding energies obtained in the XPS analysis were corrected for each specimen by referencing the C 1s to 284.0 eV (Fig. 6a). A selected area scan for the individual elements in the LCO NPs (O 1s, Cu 2p, and La 3d) are shown in Fig. 6b–d, respectively. The Cu 2p core level spectrum (Fig. 6c) shows two peaks located at 932.0 and 953.7 eV which corresponds to the Cu 2p3/2 and Cu 2p1/2 levels, respectively. These values match well with the data reported for Cu 2p in CuO.34 In addition, the shake-up satellite peaks located at 940.8 eV and 960.5 eV are characteristic of materials with a d9 configuration.35 The strong shake-up satellites recorded in the LCO NP spectra confirm the Cu2+ oxidation state and rule out the possibility of the existence of a Cu2O phase.34 The O 1s core level XPS signal is presented in Fig. 6b. The peak at the binding energy value of 529.0 eV is due to the oxygen in the CuO (the O 1s signal for Cu2O is generally found at 530.5 ± 0.2 eV in the literature).36 Fig. 6d shows that the binding energy of the La 3d5/2 and La 3d3/2 levels are 835.0 and 851.5 eV, respectively.37 The XPS data revealed that La exists in the LCO NPs as La2O3 only and not as the hydroxide or carbonate from the following evidence: the binding energy of O 1s at 529.0 eV is characteristic of O2− (the O 1s signal for hydroxide and carbonate appeared at 531.6 eV in the literature).41 The XPS analysis of bare CuO NPs revealed that the oxidation state of Cu was +2.
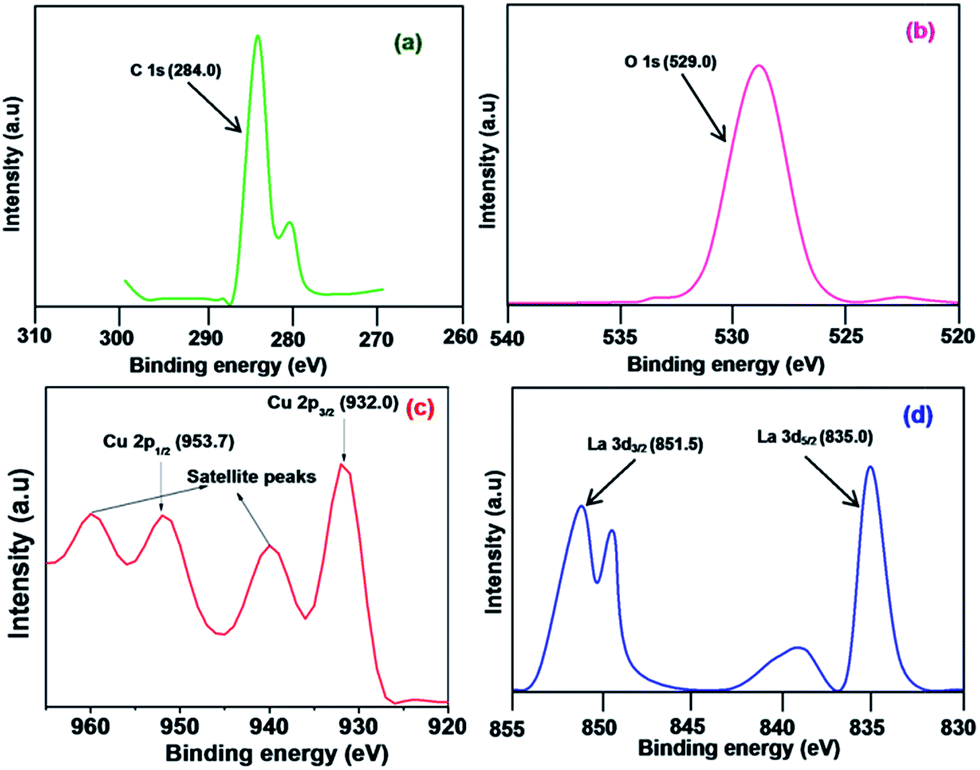 | ||
| Fig. 6 XPS spectrum of LCO NPs: (a) C 1s peak, (b) O 1s peak, (c) Cu 2p peak, and (d) La 3d peak. | ||
In general, the surface area of the catalyst is the most important factor influencing the catalytic activity. The BET surface area analysis of bare CuO and LCO NPs was performed using the N2 gas adsorption method. The BET surface area of LCO NPs (4.26 m2 g−1) was higher than that of the bare CuO NPs (1.82 m2 g−1). This is in accordance with the IUPAC classifications of type IV isotherms with type H1 hysteresis. Type H1 hysteresis indicates that, for spherical pores, the pore opening is smaller than the diameter of the main cavity. As the desorption portion of the isotherms moves from a higher partial pressure to a lower partial pressure, a gradual decrease in pore volume was observed. The BET surface and pore volume of the bare CuO and LCO NPs are given in Table 1.
| Properties | Bare CuO | LCO |
|---|---|---|
| BET surface area | 1.82 (m2 g−1) | 4.26 (m2 g−1) |
| Total pore volume (single point) | 0.01 (cm3 g−1) | 0.04 (cm3 g−1) |
The catalytic activity of the synthesized LCO NPs was tested for two different terminal-alkyne transformations: the 1,3-dipolar cycloaddition of terminal alkynes with azide and the three component synthesis of propargylamines from aldehydes, amines and terminal alkynes (A3 coupling). The optimal reaction conditions were determined independently for both reactions studied.
Click reaction based synthesis of 1,4-disubstituted 1,2,3-triazoles
We started our study by examining the model reaction of benzyl azides and phenyl acetylene affording 1-benzyl-4-phenyl-1H-1,2,3-triazole (3a). Initially, the reaction was investigated using various catalysts and solvents (Table 2) with a view to finding optimal conditions to maximize the yield of the product. The reaction in the absence of catalyst in water at room temperature and with sonication failed to afford the product (Table 2, entries 1 and 2). Then we used LCO NPs as a heterogeneous catalyst to carry out azide cycloaddition. Under grinding, the triazole was obtained as a sole product with a 30% yield (Table 2, entry 3) and the product was confirmed by NMR spectrum analysis. When the same reaction was performed using ultrasonic irradiation without solvent, the yield increased to 40% (Table 2, entry 4). Further optimization was carried out in different organic solvents such as toluene and DMF, which gave yields of 30% and 15% (Table 2, entry 5 and 6) respectively. While the reaction proceeded moderately in acetonitrile (Table 2, entry 7), a huge improvement was observed in polar protic solvents (82–85%), (Table 2, entries 8–10) and combinations of polar protic solvents with water such as methanol:water, ethanol:water, t-butanol:water and acetonitrile:water, which gave 93–95% yields (Table 2, entries 11–14). Inspired by the observed determinant effect of water and polar protic solvents on the reaction, we further investigated the feasibility of using water as the solvent for this reaction. As we expected, the reaction progressed very well and 99% yield was obtained under identical conditions (Table 2, entry 15). It should be noted that the model reaction in CuO NPs gave only a 65% yield (Table 2, entry 20). This revealed that the La in the La loaded CuO increases the yield of the reaction. Further investigation revealed that the result is affected by the amount of catalyst. The catalyst loading varied from 3 to 30 mg (Table 2, entries 16–19), the maximum yield was obtained when 10 mg catalyst was used. Above 10 mg, no significant change in the conversion occurred (Fig. 7). Performing the model reaction in H2O not only offers a high reaction yield, but also avoids: (i) generation of toxic waste; (ii) the use of large amount of organic solvent; and (iii) tedious post treatment. Therefore, our methodology is an attractive strategy for the synthesis of the target compound from the view point of green synthesis.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Condition | Catalyst | Weight % of catalyst (mg) | Solvent | Time (min) | Yielda (%) |
| a Isolated yield. | ||||||
| 1 | RT stirring | — | — | Water | 17 h | — |
| 2 | Sonication | — | — | Water | 60 | — |
| 3 | Grinding | LCO | 10 | Without solvent | 60 | 30 |
| 4 | Sonication | LCO | 10 | Solvent free | 60 | 40 |
| 5 | Sonication | LCO | 10 | Toluene | 30 | 30 |
| 6 | Sonication | LCO | 10 | Dimethylformamide | 30 | 15 |
| 7 | Sonication | LCO | 10 | Acetonitrile | 30 | 65 |
| 8 | Sonication | LCO | 10 | Methanol | 30 | 85 |
| 9 | Sonication | LCO | 10 | Ethanol | 30 | 85 |
| 10 | Sonication | LCO | 10 | t-Butanol | 30 | 82 |
| 11 | Sonication | LCO | 10 | Methanol:water |
15 | 95 |
| 12 | Sonication | LCO | 10 | Ethanol:water |
15 | 95 |
| 13 | Sonication | LCO | 10 | t-Butanol:water |
15 | 93 |
| 14 | Sonication | LCO | 10 | Acetonitrile:water |
30 | 95 |
| 15 | Sonication | LCO | 10 | Water | 15 | 99 |
| 16 | Sonication | LCO | 3 | Water | 15 | 94 |
| 17 | Sonication | LCO | 7.5 | Water | 15 | 97 |
| 18 | Sonication | LCO | 15 | Water | 15 | 98 |
| 19 | Sonication | LCO | 30 | Water | 15 | 98 |
| 20 | Sonication | CuO | 10 | Water | 30 | 65 |
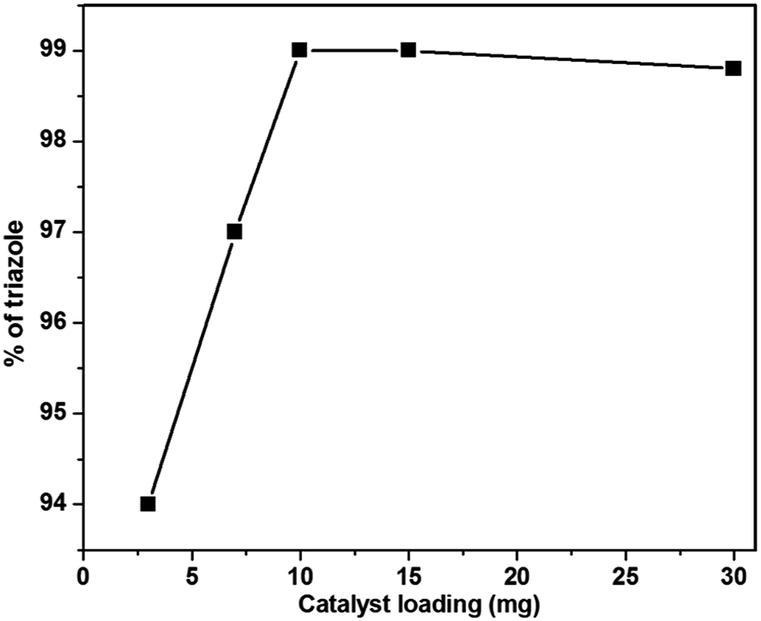 | ||
| Fig. 7 Effect of catalyst loading for the synthesis of 1,4-disubstituted 1,2,3-triazole under the ultrasonication method. | ||
With these results in hand, we further investigated the substrate scope and the results are presented in Table 3. Many azides including 4-nitro benzyl, 4-methoxy benzyl, 4-chloro benzyl, 2-thiophenyl methyl, n-octyl and n-hexyl react with various terminal alkynes and afford the corresponding products in good to excellent yields (Table 3, entries 1–9 and 11–14). The reaction condition is also applicable to internal alkynes to afford the corresponding product with moderate yield (Table 3, entry 10). The reusability of the catalyst was checked six times without any treatment (Table 4) and no appreciable loss in the yield was found (Fig. 8).
|
||||
|---|---|---|---|---|
| Entry | Alkyne | Azide (R) | Product | Yielda (%) |
| a Isolated yield. | ||||
| 1 | 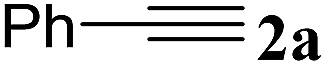 |
C6H5, 1a | 3a | 99 |
| 2 | 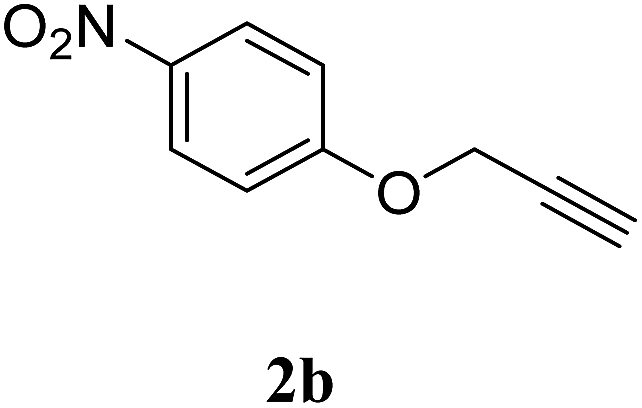 |
1a | 3b | 96 |
| 3 | 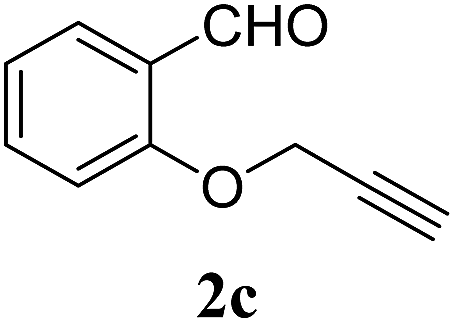 |
1a | 3c | 97 |
| 4 | 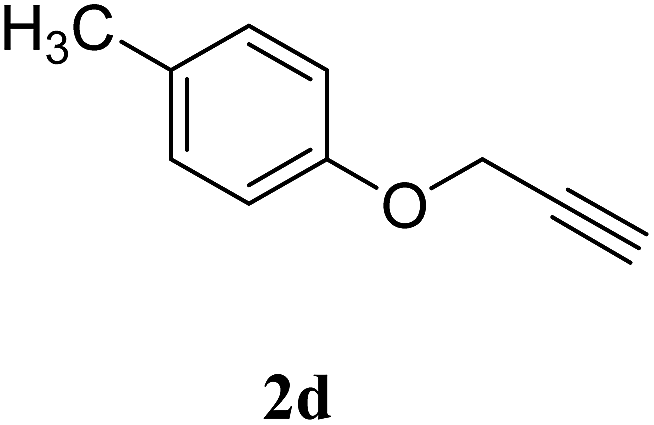 |
1a | 3d | 90 |
| 5 | 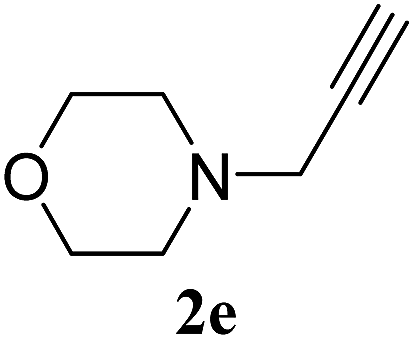 |
1a | 3e | 87 |
| 6 | 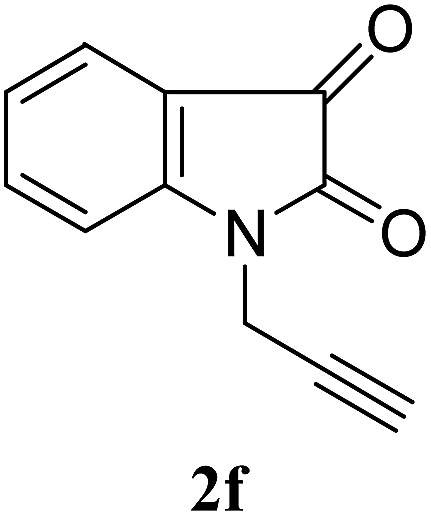 |
1a | 3f | 89 |
| 7 | 2a | 4-NO2C6H4, 1b | 3g | 93 |
| 8 | 2a | 4-OCH3C6H4, 1c | 3h | 97 |
| 9 | 2a | 4-ClC6H4, 1d | 3i | 95 |
| 10 | 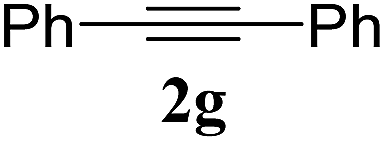 |
1a | 3j | 65 |
| 11 | 2a | 2-Thiophenyl, 1e | 3k | 82 |
| 12 | 2a | CH3(CH2)6, 1f | 3l | 87 |
| 13 | 2a | CH3(CH2)4, 1g | 3m | 91 |
| 14 | 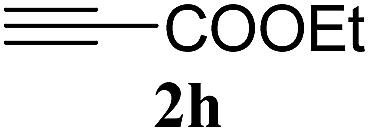 |
1a | 3n | 88 |

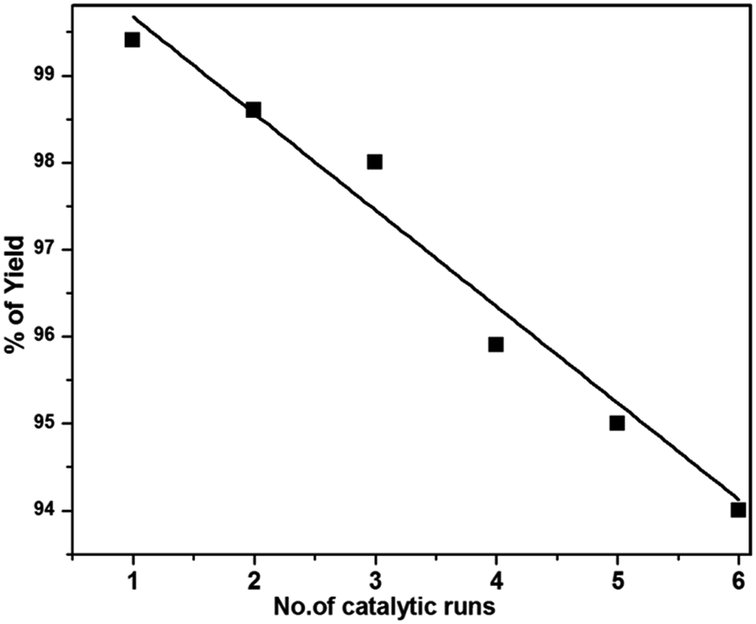 | ||
| Fig. 8 Reusability of the catalyst for the synthesis of 1,4-disubstituted 1,2,3-triazole under the ultrasonication method. | ||
By analogy with previous reports, the synthesis involves a stepwise mechanism (Scheme 1). In the first step, the LCO NPs coordinate with the alkyne 2 to form copper acetylide (2′) followed by coordination of the organic azide 1 in the second step. The coordination event is synergistic for both reaction partners. Coordination of the azide reveals the β-nucleophilic, vinylidene-like properties of the acetylide, whereas the azide’s terminus becomes more electrophilic, and a strained copper triazolide (3′) is formed. Protonation of the triazole–copper derivative followed by dissociation of the product 3 completes the reaction and regenerates the catalyst.
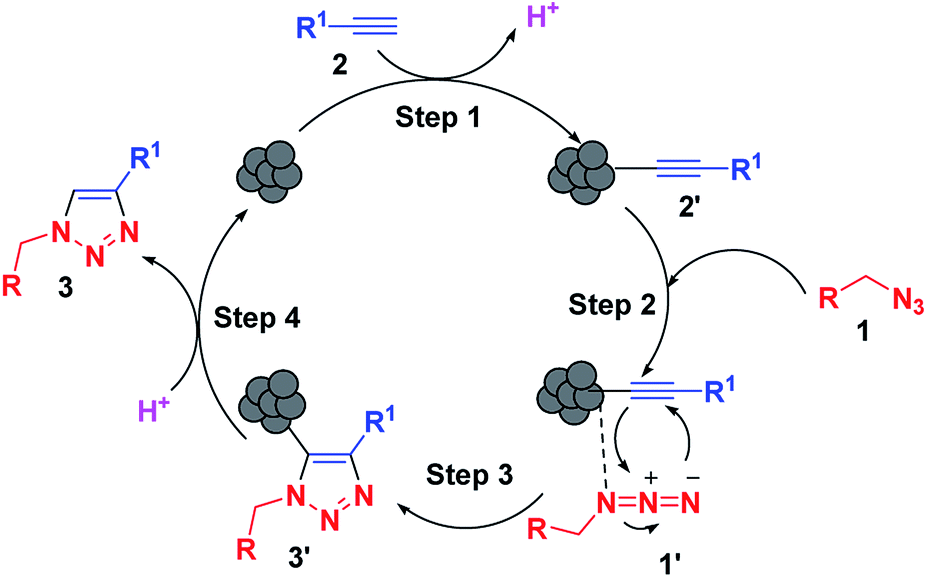 | ||
| Scheme 1 Tentative mechanism for the click reaction using the LCO NP catalyst. | ||
A3 coupling reaction based one-pot synthesis of propargylamines
The catalytic activity of the LCO NPs was also examined in the synthesis of propargylamines, the reactions of benzaldehyde, morpholine and phenylacetylene were examined and the results are presented in Table 5. Among the various solvents and solvent/co-solvent mixtures, toluene was found to be the most effective medium for this transformation (Table 5, entry 7). After finding the best solvent, the amount of catalyst (Table 5, entries 8–11) was optimized. The highest yield of propargylamine was achieved when 10 mg of catalyst in toluene was used (Table 5, entry 7). The lowest yield was obtained when the bare CuO NPs were used as a catalyst (Table 5, entry 12), which clearly demonstrates the effect of LCO NPs in this reaction.
|
|||
|---|---|---|---|
| Entry | Catalyst (mg) | Solvent | Yielda (%) |
| a Isolated yield.b Reaction time 1 h. | |||
| 1 | LCO (10) | Water | 40b |
| 2 | LCO (10) | Ethanol | 30 |
| 3 | LCO (10) | Methanol | 45 |
| 4 | LCO (10) | DCM | 40 |
| 5 | LCO (10) | DMF | 15 |
| 6 | LCO (10) | CH3CN | 58 |
| 7 | LCO (10) | Toluene | 98 |
| 8 | LCO (3) | Toluene | 93 |
| 9 | LCO (7.5) | Toluene | 95 |
| 10 | LCO (15) | Toluene | 98 |
| 11 | LCO (30) | Toluene | 98 |
| 12 | CuO (10) | Toluene | 60 |
The scope of this LCO NP catalyzed A3 coupling was further expanded with a variety of aldehydes and amines and these results are summarized in Table 6. The reactions proceeded well to obtain products in encouraging yields. Electron donating substituents like methyl, as well as a methoxy group at the para position (Table 6, entries 2, 3 and 7, 8) and an electron withdrawing chlorine and bromine atom at the para position of the aldehyde (Table 6, entries 4, 5 and 9, 10) did not induce appreciable changes in the reaction efficiency. In the presence of an NO2 group at the para position of the aldehyde no product was isolated (Table 6, entry 6).
A tentative mechanism (Scheme 2) is proposed for the probable sequence of events involving the activation of the C–H bond of alkynes (6) by LCO NPs. The Cu–acetylide intermediate (A) generated by the reaction of acetylene and LCO reacts with the iminium ion (B), formed in situ by condensation of the aldehyde with secondary amine to afford the desired propargylamines (7), and the catalyst is regenerated.
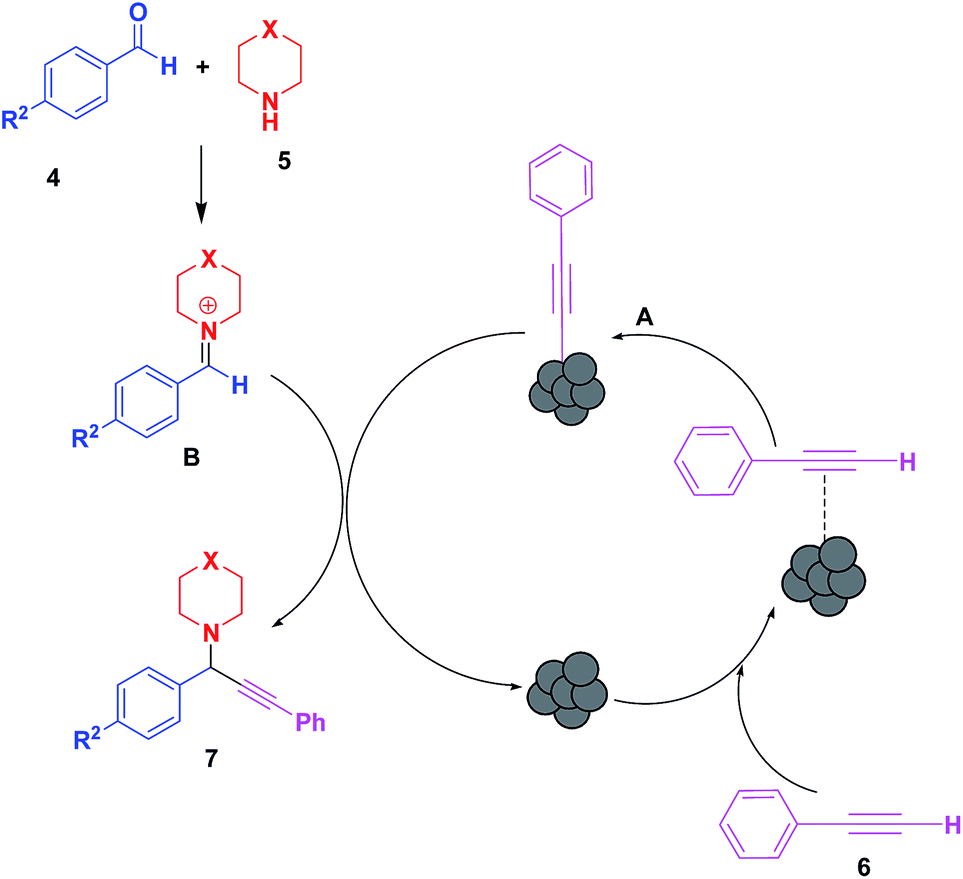 | ||
| Scheme 2 The tentative mechanism of the A3 coupling reaction using the LCO NP catalyst. | ||
Conclusions
We developed efficient La loaded CuO nanoparticles as a heterogeneous catalyst for the synthesis of 1,4-disubstituted 1,2,3-triazoles by a click reaction of azide and alkynes, and propargyl amines by A3 coupling reaction of aldehyde, secondary amine and terminal alkynes under ultrasonic irradiation. In this method, the catalyst was collected easily by filtration and the reusability of the prepared nanocatalyst was successfully examined over six cycles and was found to be effective with only a very slight loss of catalytic activity. This protocol is a clean and safe process, and can be used to generate a diverse range of products in good to excellent yields.Experimental section
All chemicals were obtained from the Sigma-Aldrich Company and used as received. All azides were prepared as per the procedure reported earlier.38 All the solvents and aldehydes were used after distillation. Melting points were measured on Guna melting point apparatus and are uncorrected. NMR spectra were obtained on a Bruker Avance 400 NMR Spectrometer (1H NMR: 400 MHz; 13C NMR: 100 MHz). Analytical TLC was carried out with Merck plates precoated with silica gel 60 F254 (0.25 mm thick). Mass spectra were recorded on a JEOL GC Mate using electron impact ionization (EI) techniques. The masses were analyzed by using a electrospray ionisation method with a Thermo Finnigan Mass spectrometer. Elemental analyses were recorded using a Thermo Finnigan FLASH EA 1112 CHN analyzer. Powder X-ray diffraction patterns were obtained using an X’Per PRO diffractometer equipped with Cu Kα radiation (wavelength 1.5406 Å) at 2.2 kW max. Peak positions were compared with the standard files to identity the crystalline phase. The morphology of the catalyst was examined using a JEOL JSM-6701F cold field emission scanning electron microscope (FE-SEM). The specific surface areas of the samples were determined through nitrogen adsorption at 77 K on the basis of the BET equation using a micrometrics ASAP 2020 V3.00 H. Diffuse reflectance spectra were recorded using a Shimadzu UV-2450. Photoluminescence (PL) spectra at room temperature were recorded using a Perkin Elmer LS 55 fluorescence spectrometer. The nanoparticles were dispersed in carbon tetrachloride and excited using light of wavelength 300 nm. X-ray photoelectron spectra of the catalysts were recorded in an ESCA-3 Mark II spectrometer (VG Scientific Ltd., England) using Al Kα (1486.6 eV) radiation as the source. Spectra were referenced to the binding energy of C 1s (284 eV).Preparation of LCO nanoparticles
The La-loaded CuO NPs were prepared by a precipitation thermal decomposition method. Aqueous solutions consisting of copper nitrate and oxalic acid in deionized water were separately brought to their boiling points. When the oxalic acid solution was added to solution of copper nitrate, copper oxalate was precipitated. Then, La(NO3)3 solution was mixed with the copper oxalate suspension and stirred for 1 h at 60–70 °C. After the mixture had cooled to room temperature, the La containing copper oxalate was washed with distilled water, air dried overnight, and dried at 100 °C for 5 h. Calcination of the mixed precipitate at 450 °C for 5 h decomposed copper oxalate to CuO. The La loaded CuO was obtained after the sample had cooled to room temperature. The bare CuO was prepared without the addition of La(NO3)3 by the above procedure.General procedure for the synthesis of 1,4-disubstituted 1,2,3-triazoles (3a–j)
The heterogeneous LCO NPs (10 mg) were added to a mixture of azide (1 mmol) and alkyne (1.5 mmol) in 5 mL of water. The solution was subjected to ultrasonic irradiation for a particular time duration. The progress of the reaction was monitored by TLC (20% ethyl acetate/hexane). When the reaction was complete, the catalyst was removed by centrifugation. The filtrate was extracted with ethyl acetate (3 × 10 mL). The combined organic solutions were dried with Na2SO4. The pure products were isolated without column purification only by the process of crystallization.:n-hexane); 1H NMR (400 MHz, CDCl3): δH 7.80 (d, J = 7.6 Hz, 2H), 7.66 (s, 1H), 7.42–7.38 (m, 5H), 7.32–7.30 (m, 3H), 5.58 (s, 2H); 13C NMR (100 MHz, CDCl3): δc 149.0, 134.7, 130.5, 129.2, 128.8, 128.2, 128.1, 125.7, 119.5, 54.3.:n-hexane); 1H NMR (400 MHz, CDCl3): δH 8.19 (d, J = 8.8 Hz, 2H), 7.56 (s, 1H), 7.40–7.38 (m, 3H), 7.30–7.26 (m, 2H), 7.06 (d, J = 8.8 Hz, 2H), 5.55 (s, 2H), 5.27 (s, 2H); 13C NMR (100 MHz, CDCl3): δc 163.1, 141.9, 134.2, 129.2, 128.9, 128.2, 125.9, 122.9, 114.9, 62.5, 54.4; HRMS: calculated for C16H14N4O3 Mw: 310.1066, found: 310.1062.:n-hexane); 1H NMR (400 MHz, CDCl3): δH 10.42 (s, 1H), 7.83 (d, J = 7.6 Hz, 1H), 7.58 (s, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.39–7.37 (m, 3H), 7.29–7.27 (m, 2H), 7.16 (d, J = 8.4 Hz, 1H), 7.06 (t, J = 7.6 Hz, 1H), 5.56 (s, 2H), 5.32 (s, 2H); 13C NMR (100 MHz, CDCl3): δc 189.6, 160.5, 135.9, 134.3, 129.2, 128.9, 128.7, 128.1, 125.1, 121.4, 113.1, 62.6, 54.4; HRMS: calculated for C17H15N3O2 Mw: 293.1164, found: 293.1162.:n-hexane); 1H NMR (400 MHz, CDCl3): δH 7.52 (s, 1H), 7.37–7.35 (m, 3H), 7.27–7.25 (m, 2H), 7.06 (d, J = 8.0 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 5.51 (s, 2H), 5.15 (s, 2H), 2.27 (s, 3H); 13C NMR (100 MHz, CDCl3): δc 156.1, 134.5, 130.5, 129.9, 129.2, 128.8, 128.1, 122.7, 114.7, 62.3, 54.3, 20.5; MS m/z = 280 M+ + 1; anal. calcd for C17H17N3O (279.14): C, 73.10; H, 6.13; N, 15.04. Found: C, 73.18; H, 6.06; N, 15.14.:n-hexane); 1H NMR (400 MHz, CDCl3): δH 7.39–7.36 (m, 4H), 7.28–7.26 (m, 2H), 5.51 (s, 2H), 3.70–3.68 (m, 4H), 3.64 (s, 2H), 2.51–2.48 (m, 4H); 13C NMR (100 MHz, CDCl3): δc 144.5, 134.6, 129.1, 128.8, 128.1, 122.5, 66.8, 54.2, 53.7, 53.4; MS m/z = 259 M+ + 1; anal. calcd for C14H18N4O (258.15): C, 65.09; H, 7.02; N, 21.69. Found: C, 65.02; H, 7.05; N, 21.75.:n-hexane); 1H NMR (400 MHz, CDCl3): δH 7.60–7.56 (m, 2H), 7.52 (s, 1H), 7.37–7.35 (m, 2H), 7.32 (d, J = 8.0 Hz, 1H), 7.25–7.23 (m, 2H), 7.11 (t, J = 7.2 Hz, 1H), 5.48 (s, 2H), 4.99 (s, 2H); 13C NMR (100 MHz, CDCl3): δc 183.1, 157.9, 150.2, 142.1, 138.6, 134.0, 129.2, 129.0, 128.3, 125.3, 124.0, 122.8, 117.5, 111.6, 54.4, 35.4. HRMS: calculated for C18H14N4O2 Mw: 318.1117, found: 318.1115.:n-hexane); 1H NMR (400 MHz, CDCl3): δH 8.26–8.23 (m, 2H), 7.82 (d, J = 6.8, 2H), 7.75 (s, 1H), 7.46–7.40 (m. 4H), 7.36–7.32 (m, 1H), 5.70 (s, 2H). 13C NMR (100 MHz, CDCl3): δc 148.1, 141.7, 130.1, 128.9, 128.6, 128.5, 125.8, 124.4, 119.7, 53.2.:n-hexane); 1H NMR (400 MHz, CDCl3): δH 7.78 (d, J = 7.2 Hz, 2H), 7.62 (s, 1H), 7.39 (t, J = 7.6 Hz, 2H), 7.32–7.25 (m, 3H), 6.90 (d, J = 8.8, 2H), 5.49 (s, 2H), 3.80 (s, 3H); 13C NMR (100 MHz, CDCl3): δc 159.9, 148.1, 130.6, 129.7, 128.8, 128.1, 126.7, 125.7, 119.3, 114.5, 55.4, 53.8.:n-hexane); 1H NMR (400 MHz, CDCl3): δH 7.80 (d, J = 7.2 Hz, 2H), 7.67 (s, 1H), 7.42–7.30 (m, 5H), 7.24 (d, J = 8.4 Hz, 2H), 5.54 (s, 2H); 13C NMR (100 MHz, CDCl3): δc 134.9, 133.2, 130.5, 129.5, 129.4, 129.0, 128.9, 128.3, 125.7, 119.5, 53.5.:n-hexane); 1H NMR (400 MHz, CDCl3): δH 7.55 (d, J = 6.8 Hz, 2H), 7.46 (d, J = 6.8 Hz, 1H), 7.41 (t, J = 7.6 Hz, 2H), 7.25–7.22 (m, 6H), 7.14 (d, J = 7.2 Hz, 2H), 7.03–7.01 (m, 2H), 5.40 (s, 2H); 13C NMR (100 MHz, CDCl3): δc 144.6, 139.4, 135.5, 133.9, 131.0, 130.2, 129.8, 129.3, 128.8, 128.5, 128.2, 127.9, 127.6, 126.8, 52.1.:n-hexane); 1H NMR (400 MHz, CDCl3): δH 7.71 (d, J = 7.2 Hz, 2H), 7.64 (s, 1H), 7.33–7.29 (m, 2H), 7.25–7.22 (m, 2H), 7.05–7.04 (m, 1H), 6.93–6.91 (m, 1H), 5.64 (s, 2H); 13C NMR (100 MHz, CDCl3): δc 147.2, 135.1, 129.5, 127.8, 127.2, 127.1, 126.3, 126.1, 124.7, 118.2, 47.5.:n-hexane); 1H NMR (400 MHz, CDCl3): δH 7.75 (d, J = 7.6 Hz, 2H), 7.67 (s, 1H), 7.34 (t, J = 7.2 Hz, 2H), 7.26–7.22 (m, 1H), 4.29 (t, J = 7.6 Hz, 2H), 1.86–1.83 (m, 2H), 1.26–1.18 (m, 10H), 0.79 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δc 147.7, 130.8, 128.8, 128.1, 125.7, 119.4, 50.4, 31.7, 30.4, 29.1, 28.9, 26.5, 22.6, 14.1.:n-hexane); 1H NMR (400 MHz, CDCl3): δH 7.75 (d, J = 6.8 Hz, 2H), 7.67 (s, 1H), 7.34 (t, J = 7.6 Hz, 2H), 7.26–7.22 (m, 1H), 4.30 (t, J = 7.2 Hz, 2H), 1.89–1.82 (m, 2H), 1.29–1.21 (m, 6H), 0.81 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δc 146.7, 129.7, 127.8, 127.0, 124.7, 118.3, 49.4, 30.1, 29.3, 25.1, 21.4, 12.9.:n-hexane); 1H NMR (400 MHz, CDCl3): δH 8.05 (s, 1H), 7.26–7.19 (m, 5H), 5.83 (s, 2H), 4.26 (q, J = 7.2 Hz, 2H), 1.26 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δc 157.3, 137.1, 134.0, 127.7, 127.4, 126.9, 126.8, 60.8, 52.3, 13.1.General procedure for the synthesis of propargylamine derivatives (7a–j)
In a 25 mL glass vial, a mixture of benzaldehydes 4a–j (1 mmol), amine 5a and 5b (1.2 mmol), phenylacetylene 6 (1.5 mmol) and LCO nanoparticles (10 mg) in 5 mL toluene, were sonicated for 30 min. The progress of the reaction was monitored by TLC (20% ethyl acetate/hexane). After completion of the reaction, the catalyst was removed by centrifugation. The reaction mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic phases were dried with Na2SO4, filtered, concentrated, and the residue was purified by flash chromatography on silica gel (elutant: hexane/ethyl acetate, 3:1, v/v).
Acknowledgements
The authors are grateful to the DST-SERB (Project no. SB/FT/CS-033/2013) for financial support and author R.S. thanks B S Abdur Rahman University for providing a fellowship.References
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS
.
-
(a) R. A. Sheldon, I. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007 Search PubMed
- S. Tan, X. Li, Y. Guo and Z. Zhang, Nanoscale, 2013, 5, 860 RSC
- K. T. Yong, I. Roy, M. T. Swihart and P. N. Prasad, J. Mater. Chem., 2009, 19, 4655 RSC
-
(a) P. Song, D. Wen, Z. X. Guo and T. Korakianitis, Phys. Chem. Chem. Phys., 2008, 10, 5057 RSC
-
(a) Y. Ding, Y. Wang, L. Zhang, H. Zhang, C. M. Li and Y. Lei, Nanoscale, 2011, 3, 1149 RSC
- D. Jariwala, V. K. Sangwan, L. J. Lauhon, T. J. Marks and M. C. Hersam, Chem. Soc. Rev., 2013, 42, 2824 RSC
- J. Liao, Z. Yang, H. Wu, D. Yan, J. Qiu, Z. Song, Y. Yang, D. Zhou and Z. Yin, J. Mater. Chem. C, 2013, 1, 6541 RSC
- H. Kang, Y. Zhu, X. Yang, J. Shen, C. Chen and C. Li, New J. Chem., 2010, 34, 2166 RSC
-
(a) H. Wamhoff, Comprehensive Heterocyclic Chemistry 1, ed. A. R. Katritzky and C. W. Rees, Pergamon, Oxford, vol. 5, 1984, p. 669 Search PubMed
-
(a) R. Huisgen, In 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, John Wiley, New York, 1984, pp. 1–176 Search PubMed
-
(a) F. Palacios, A. M. Ochoa de Retana, J. Pagalday and J. M. Sanchez, Org. Prep. Proced. Int., 1995, 27, 603–612 CrossRef CAS PubMed
-
(a) K. Wang, X. Bi, S. Xing, P. Liao, Z. Fang, X. Meng, Q. Zhang, Q. Liu and Y. Ji, Green Chem., 2011, 13, 562–565 RSC
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed
-
(a) V. D. Bock, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2006, 51–68 CrossRef CAS PubMed
- Q. Wang, S. Chittaboina and H. N. Barnhill, Lett. Org. Chem., 2005, 2, 293–301 CrossRef CAS
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed
- S. Quader, S. E. Boyd, I. D. Jenkins and T. A. Houston, J. Org. Chem., 2007, 72, 1962–1979 CrossRef CAS PubMed
-
(a) T. Jin, M. Yan, Menggernbateer, T. Minato, M. Bao and Y. Yamamoto, Adv. Synth. Catal., 2011, 353, 3095–3100 CrossRef CAS PubMed
-
(a) J. Y. Kim, J. C. Park, H. Kang, H. Song and K. H. Park, Chem. Commun., 2010, 46, 439–441 RSC
-
(a) B. H. Lipshutz and B. R. Taft, Angew. Chem., Int. Ed., 2006, 45, 8235–8238 CrossRef CAS PubMed
-
(a) Y. Wang, J. Liu and C. Xia, Adv. Synth. Catal., 2011, 353, 1534–1542 CrossRef CAS PubMed
-
(a) K. Namitharan, M. Kumarraja and K. Pitchumani, Chem.–Eur. J., 2009, 15, 2755–2758 CrossRef CAS PubMed
-
(a) E.-S. Lee, H.-S. Yeom, J.-H. Hwang and S. Shin, Eur. J. Org. Chem., 2007, 3503–3507 CrossRef CAS PubMed
-
(a) S. Samai, G. C. Nandi and M. S. Singh, Tetrahedron Lett., 2010, 51, 5555–5558 CrossRef CAS PubMed
-
(a) Z. Li, C. Wei, L. Chen, R. S. Varmab and C.-J. Li, Tetrahedron Lett., 2004, 45, 2443–2446 CrossRef CAS PubMed
-
(a) R. Cella and H. A. Stefani, Tetrahedron, 2009, 65, 2619 CrossRef CAS PubMed
- L. Q. Ding, W. Wang and A. Q. Zhang, Ultrason. Sonochem., 2007, 14, 563 CrossRef CAS PubMed
- J. K. Kim, F. Martinez and I. S. Metcalfe, Catal. Today, 2007, 124, 224 CrossRef CAS PubMed
-
(a) P. Cintas, A. Barge, S. Tagliapietra, L. Boffa and G. Cravotto, Nat. Protoc., 2010, 5, 607 CrossRef CAS PubMed
-
(a) R. Srivastava, M. U. A. Prathap and R. Kore, Colloids Surf., A, 2011, 392, 271–282 CrossRef CAS PubMed
- A. S. Priya, I. B. S. Banu and M. Chavali, Arabian J. Sci. Eng., 2015, 40, 2079–2084, DOI:10.1007/s13369-015-1668-z
-
(a) J. Moulder, W. Sticke, P. Sobol and K. Bomben, Standard ESCA spectra of the elements and line energy information. in Handbook of X-ray Photoelectron Spectroscopy, ed. J. Chastain, Perkin Elmer Coorporation, Physical Electronics Division, USA, 1992 Search PubMed
-
(a) J. Ghijsen, L. H. Tjeng, J. V. Elp, H. Eskes, J. Westerink, G. A. Sawatzky and M. T. Czyzyk, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 11322 CrossRef CAS
-
(a) S. Poulston, P. M. Parlett, P. Stone and M. Bowker, Surf. Interface Anal., 1996, 24, 811–820 CrossRef CAS
-
(a) X. Ma, H. Zhang, Y. Ji, J. Xu and D. Yang, Mater. Lett., 2004, 58, 1180–1182 CrossRef CAS PubMed
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC
-
(a) K. Kamata, Y. Nakagawa, K. Yamaguchi and N. Mizuno, J. Am. Chem. Soc., 2008, 130, 15304 CrossRef CAS PubMed
-
(a) L. L. Chang, J. Yang, Y. Wei and J. Y. Ying, Adv. Synth. Catal., 2009, 351, 2887–2896 CrossRef PubMed
-
(a) S. N. Shkerin, M. V. Kuznetsov and N. A. Kalashnikova, Russ. J. Electrochem., 2003, 39, 591–599 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all compounds. See DOI: 10.1039/c5ra05468k |
| This journal is © The Royal Society of Chemistry 2015 |