
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-temperature redox activity and alcohol ammoxidation performance on Cu- and Ru-incorporated ceria catalysts†
Chaoqi
Chen
a,
Satoru
Ikemoto
a,
Gen-ichi
Yokota
a,
Kimitaka
Higuchi
b,
Satoshi
Muratsugu
 *ac and
Mizuki
Tada
*acd
*ac and
Mizuki
Tada
*acd
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8602, Japan. E-mail: muratsugu.satoshi.a5@f.mail.nagoya-u.ac.jp
bInstitute of Materials and Systems for Sustainability, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8602, Japan
cIntegrated Research Consortium on Chemical Sciences (IRCCS), Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8602, Japan
dResearch Center for Materials Science (RCMS), Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8602, Japan. E-mail: tada.mizuki.u6@f.mail.nagoya-u.ac.jp
First published on 10th May 2024
Abstract
Transition-metal-incorporated cerium oxides with Cu and a small amount of Ru (Cu0.18Ru0.05CeOz) were prepared, and their low-temperature redox performance (<423 K) and catalytic alcohol ammoxidation performance were investigated. Temperature-programmed reduction/oxidation under H2/O2 and in situ X-ray absorption fine structure revealed the reversible redox behavior of the three metals, Cu, Ru, and Ce, in the low-temperature redox processes. The initially reduced Ru species decreased the reduction temperature of Cu oxides and promoted the activation of Ce species. Cu0.18Ru0.05CeOz selectively catalyzed the production of benzonitrile in the ammoxidation of benzyl alcohol. H2-treated Cu0.18Ru0.05CeOz showed a slightly larger initial conversion of benzyl alcohol than O2-treated Cu0.18Ru0.05CeOz, suggesting that the reduced structure of Cu0.18Ru0.05CeOz was active for the ammoxidation. The integration of both Cu and Ru resulted in the efficient promotion of ammoxidation, in which the Ru species were involved in the conversion of benzyl alcohol and Cu species were required for selective production of benzonitrile.
Introduction
Transition metal-incorporated cerium oxides (cerias) are promising candidates for catalysts that contain cooperating multiple metal species.1–3 The metal ion species create oxygen species that are more labile and oxygen vacancy sites on the surface that enhance the redox performance of ceria and are the active sites that generate the unique catalytic performance.4–10 For instance, it was reported that the deposition of Au species to form Au/CexZr1−xO2 improved the physicochemical properties of CexZr1−xOz. The Au species assisted the distribution of metal particles and also affected the Lewis/Brønsted acidity, which enhanced catalyst selectivity in the oxidation of 5-hydroxymethylfurfural.11 The catalyst 1%Pt–1%Co/CeO2 was prepared by combining a noble metal (Pt) and a 3d transition metal (Co), and the addition of Co altered the chemical states of Pt. This alternation resulted in catalytic activity in hydrogen-borrowing amination by facilitating dehydrogenation and resisting overhydrogenation.12 The singly dispersed Ni and Ru cations on CeO2 were developed to enhance the reforming of CH4 with H2O. This combination promoted the synergistic effect of Ni activated C–H and Ru activated H2O to form CO and H2.13 We reported that Cr- and Rh-incorporated ceria, Cr0.19Rh0.06CeOz, showed superior redox performance at low temperatures (<373 K) compared with pure ceria.14–16 Cr0.19Rh0.06CeOz exhibited the redox performance of all three metal species and had remarkable NO reduction performance toward N2 because Rh activated the CO and NO species and Cr cleaved NO species.14,15 Thus, incorporating multiple metal species into ceria surface leads to efficient oxidation catalysis.Nitriles, one of the most important organic chemical groups, are used in many industrial applications, such as building blocks for high-performance polymers, useful organic intermediates in pharmaceuticals, and fine chemicals in agriculture.17–21 Traditionally, nitriles have been produced by the Sandmeyer reaction, Wittig reaction, and cyanation of aryl halides with metal cyanides, all of which share major disadvantages, including the use of toxic reagents, harsh reaction conditions, and formation of harmful waste.22–25 Ammoxidation is a reaction that shows promise to address recent shortages of nitriles by forming nitriles directly from the corresponding alcohols and hydrocarbons.17,26 Ammoxidation uses O2 and NH3 as the oxidant and nitrogen source, respectively, and produces H2O as the only by-product.17,26 Compared with other functional groups, alcohols have attracted attention as reactants because they can be obtained easily as derivatives from biomass with high stability.18–20 Pioneering research identified Cu-based and Fe-based homogeneous catalysts with catalytic activity in the ammoxidation of alcohol, although they usually suffer from a low substrate-to-catalyst ratio, difficult separation, and the need for additives, such as 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO).27–31 Heterogeneous catalysts, such as noble metal-based oxides (Ru,32,33 Pt,34 and Ag35) and transition metal-based carbon (Co,36,37 Fe,37,38 La,39 and Mn40), also produce benzonitrile. However, the ammoxidation performance of multiple metal species and the role of metal species in each reaction step are still unrevealed.
In this paper, we report new ceria catalysts containing Cu and a trace amount of Ru (denote as Cu0.18Ru0.05CeOz) for efficient ammoxidation catalysis. Cu0.18Ru0.05CeOz showed superior redox performance at low temperatures near 373 K and efficient catalytic performance for the ammoxidation of benzyl alcohol. Temperature-programmed reduction/oxidation (TPR/TPO) under H2/O2 and in situ X-ray absorption fine structure (XAFS) showed that the incorporation of the Ru species shifted the activation of Cu to a lower temperature range, which also contributed to the activation of Ce species. The reduced Cu0.18Ru0.05CeOz exhibited activity for the selective conversion of benzyl alcohol to benzonitrile, and the reversible redox reaction of Cu0.18Ru0.05CeOz was suggested to be related to the ammoxidation catalysis with ceria containing the two cooperating transition metal species.
Results and discussion
Structure of Cu0.18Ru0.05CeOz
Cu0.18Ru0.05CeOz and reference compounds (Cu0.18CeOz, Ru0.04CeOz, CeOz) were prepared by a hydrothermal method using three metal precursors, (Cu(NO3)2·3H2O, RuCl3, and Ce(NO3)3·6H2O) simultaneously, similar to our previous work on Cr0.19Rh0.06CeOz.14,15 The XRD patterns of as-prepared Cu0.18Ru0.05CeOz and CeOz prepared without Ru and Cu showed a similar peak position for the (111) CeO2 fluorite structure at 28.6° (Cu0.18Ru0.05CeOz) and 28.5° (CeOz), and there were no major peaks attributed to Ru and Cu compounds (Ru, RuO2, Cu, and CuO) in the XRD pattern of Cu0.18Ru0.05CeOz (Fig. 1(a)). The negligible shift of the CeO2 (111) peak of the prepared Cu0.18Ru0.05CeOz sample suggested that doping of Cu and Ru into the CeO2 fluorite structure was negligible.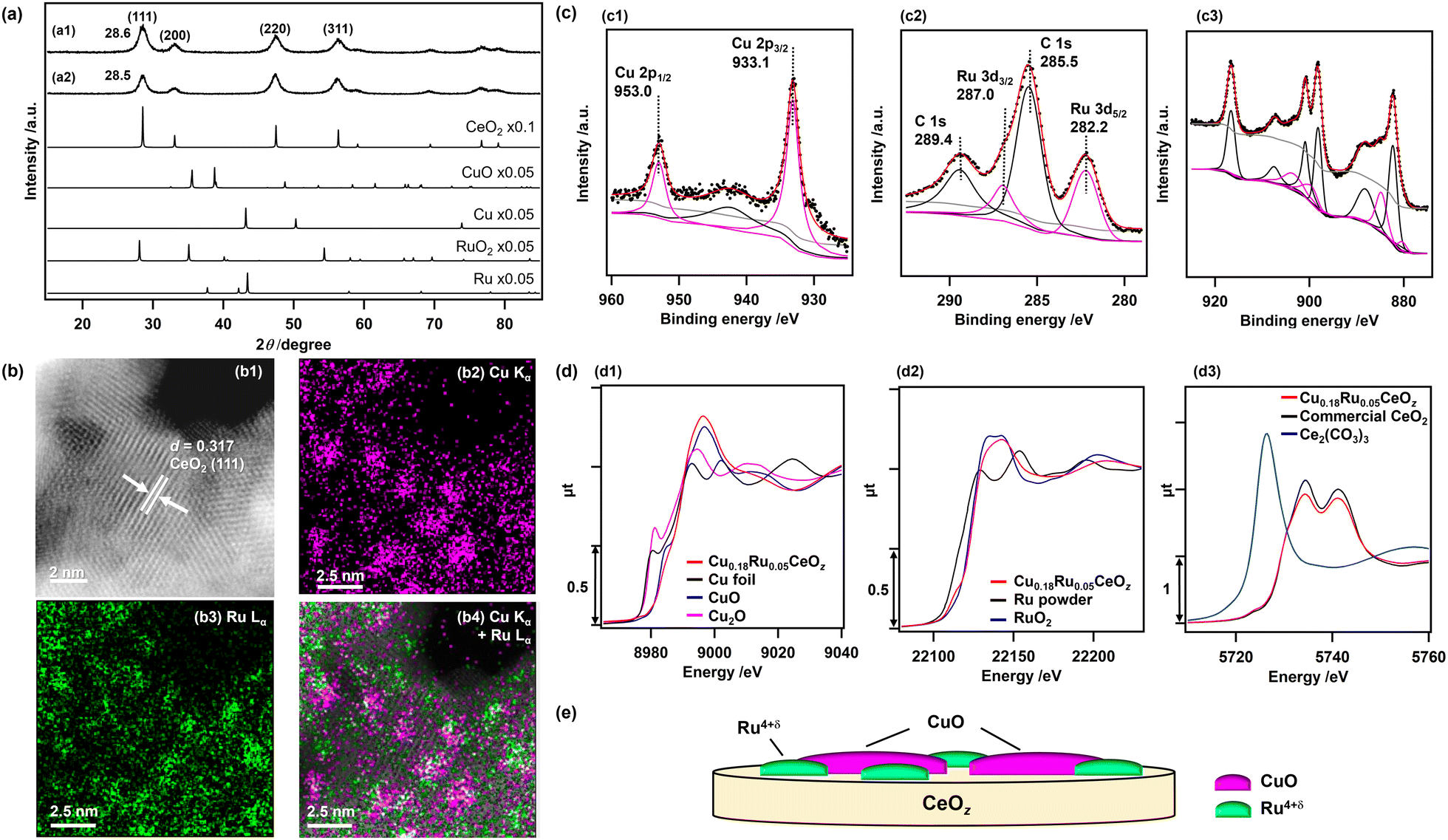 | ||
| Fig. 1 (a) XRD patterns of (a1) Cu0.18Ru0.05CeOz and (a2) CeOz. (b) HAADF-STEM-EDS images of Cu0.18Ru0.05CeOz. (b1) HAADF-STEM image of Cu0.18Ru0.05CeOz, (b2) EDS mapping of Cu, (b3) EDS mapping of Ru, and (b4) overlapped EDS mapping of Cu and Ru. (c) (c1) Cu 2p, (c2) Ru 3d, and (c3) Ce 3d XPS spectra of Cu0.18Ru0.05CeOz. Black dots: observed spectra; red lines: total fitted curves; gray lines: Shirley backgrounds. (c1) Pink line: fitted curves attributed to CuO species; black line: fitted curves attributed to satellite peaks. (c2) Pink line: fitted curves attributed to Ru4+δ species; black line: fitted curves attributed to C 1s overlapped in the Ru 3d spectrum. (c3) Black line: fitted curves attributed to Ce4+ species; pink line: fitted curves attributed to Ce3+ species. (d) (d1) Cu K-edge, (d2) Ru K-edge, and (d3) Ce LIII-edge XANES spectra of Cu0.18Ru0.05CeOz with standard reference samples. (e) Schematic of the surface structure of as-prepared Cu0.18Ru0.05CeOz. | ||
The Cu, Ru, and Ce contents in Cu0.18Ru0.05CeOz were determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES) (Table S1, ESI†). The BET surface area of Cu0.18Ru0.05CeOz was estimated to be 1.24 × 102 m2 g−1, which was similar to that of CeOz (1.20 × 102 m2 g−1) (Table S1, ESI†). Transmission electron microscopy (TEM) (Fig. S1, ESI†) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Fig. 1(b1)) of Cu0.18Ru0.05CeOz showed a lattice image (d = 0.317 nm) that matched the (111) facet of fluorite CeO2 (d = 0.312 nm).41 HAADF-STEM images with energy dispersive X-ray spectroscopy (EDS) showed the spatial distribution of Cu and Ru on Cu0.18Ru0.05CeOz and that Cu and Ru were highly dispersed (Fig. 1(b2–b4)). The domain sizes of Cu and Ru species were found to be of nm size as shown in Fig. 1(b).
The oxidation state of Cu in the as-prepared Cu0.18Ru0.05CeOz sample was characterized by X-ray photoelectron spectroscopy (XPS). The Cu 2p3/2 XPS peak was observed at 933.1 eV (Fig. 1(c1)) and it agreed with bivalent Cu (933.6 ± 0.4 eV).42 The slight negative shift of the observed Cu 2p3/2 peak suggested interaction with the CeOz surface, such as Cu2+–[O]–Ce (on CeO2) (reported at 933.0 eV).43 In addition, a broad satellite peak near 940.0–950.0 eV was observed, which is also characteristic of CuO44 or Cu2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 45 species (Fig. 1(c1)).
45 species (Fig. 1(c1)).
The Ru valence state was estimated by analyzing the energy at the 50% level of the absorption edge of Ru K-edge XANES (Fig. 1(d2)),46,47 and metallic Ru powder, Ru(acac)3, RuO2, and KRuO4 with different Ru valence were used as references. The average valence state of Ru in Cu0.18Ru0.05CeOz was calculated to be +4.5 (Fig. S2 and Table S2, ESI†). The observed Ru 3d5/2 XPS peak at 282.2 eV (Fig. 1(c2)) agreed with the Ru valence judging from the binding energies of reference compounds (279.9 (Ru0), 281.4 (RuO2), and 282.4 eV (RuO3)).48
The oxidation state of Ce was characterized by Ce 3d and O 1s XPS spectra. The Ce 3d XPS spectrum of as-prepared Cu0.18Ru0.05CeOz (Fig. 1(c3)) were deconvoluted to Ce 3d peaks of Ce3+ and Ce4+ (see Experimental section and the ESI†). The ratio of the summed peak areas of Ce3+ and Ce4+ was 0.27/0.73, suggesting that there was a significant amount of reduced Ce3+ species at the oxide surface (Table S3, ESI†).41 The Ce LIII-edge XANES spectrum of as-prepared Cu0.18Ru0.05CeOz showed slight differences in the XANES region from that of commercial CeO2 (Fig. 1(d3)), and the average oxidation state of Ce was estimated to be +3.85 from the curve-fitting analysis of the Ce LIII-edge XANES spectra of commercial CeO2 and Ce2(CO3)3 (Fig. S3, ESI†). The O 1s spectra contained two peaks, which were attributed to the lattice oxygen of CeO2 (529.6 eV),14 and either hydroxyl groups or oxygen vacancies that were generated from Ce3+ of CeO2 (531.7 eV)49,50 (Fig. S4, ESI†).
The curve-fitting analysis of extended X-ray absorption fine structure (EXAFS) Fourier transforms suggested the local coordination structures of Cu and Ru in as-prepared Cu0.18Ru0.05CeOz. Cu–O bonds at 0.196 ± 0.002 nm (coordination number [CN] of Cu–O: 4.6 ± 1.2) and Ru–O bonds at 0.194 ± 0.002 nm (CN of Ru–O: 3.2 ± 1.0) were found (Fig. S5, S6 and Tables S4–S7, ESI†). These structural characterization studies indicated that Cu and Ru were not incorporated into the CeO2 fluorite framework and bivalent Cu oxide and Ru4+δ oxide were highly dispersed on the surface of CeOz as nm-sized domain structures illustrated in Fig. 1(e).
Redox properties of Cu0.18Ru0.05CeOz
The redox properties of Cu0.18Ru0.05CeOz were investigated by using TPR with H2 and the amounts of H2 consumed were estimated (Fig. 2 and Table S8, ESI†). The first reduction of as-prepared Cu0.18Ru0.05CeOz began at 333 K and contained two TPR peaks near 353 and 363 K (“First” in Fig. 2(b)). The H2 consumption in the first reduction of Cu0.18Ru0.05CeOz was calculated to be 2.39 mmol g_cat−1 (Fig. 2(a) and Table S8, ESI†). In contrast, the first TPR of Cu0.18CeOz showed different features from that of Cu0.18Ru0.05CeOz; the reduction started at around 423 K and the H2 consumption was smaller than that of Cu0.18Ru0.05CeOz (1.44 mmol g_cat−1) up to 523 K. For Ru0.04CeOz, the reduction occurred in the low-temperature range, as for Cu0.18Ru0.05CeOz at 339 K, but H2 consumption was only 1.60 mmol g_cat−1, which was only 67% of that of Cu0.18Ru0.05CeOz. CeOz prepared via a similar method was not reduced at less than 673 K,14 and CuO and RuO2 were reduced at 477 and 429 K, respectively. These results indicated that incorporating Cu and Ru into Cu0.18Ru0.05CeOz contributed to decreasing the reduction temperature and enhancing its redox properties in the low-temperature range (303–523 K).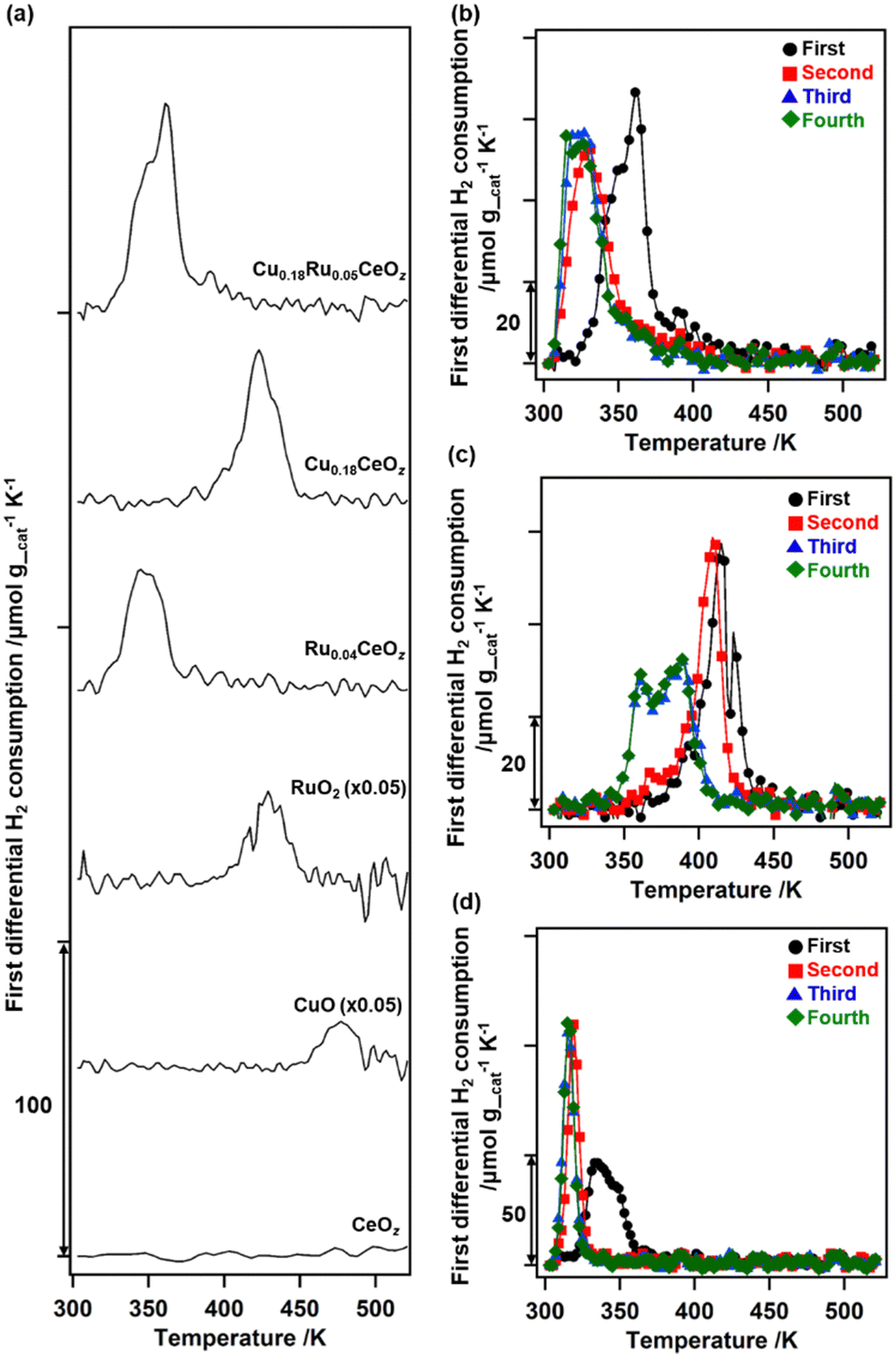 | ||
| Fig. 2 (a) The first differential plots of the TPR measurements with H2 of Cu0.18Ru0.05CeOz, Cu0.18CeOz, Ru0.04CeOz, RuO2, CuO, and CeOz. First differential plots of continuous TPR with H2 on (b) Cu0.18Ru0.05CeOz, (c) Cu0.18CeOz, and (d) Ru0.04CeOz (black: first cycle; red: second cycle; blue: third cycle; green: fourth cycle). TPR was conducted from 303 to 523 K, and TPO was conducted from 303 to 573 K after TPR up to 523 K. | ||
After the first TPR, the reduced sample was re-oxidized with O2 and the amount of O2 consumption was estimated. The oxidation of Cu0.18Ru0.05CeOz started at 303 K and 0.92 mmol g_cat−1 of O2 was consumed on Cu0.18Ru0.05CeOz below 573 K (Table S8, ESI†). The O2 consumption was comparable to the H2 consumption in the first reduction.
We repeated the reduction and oxidation cycles on Cu0.18Ru0.05CeOz (Fig. 2(b)). In the second TPR cycle, a large shift in the reduction temperature was observed at 333 K and the trend was reversible for the third and fourth reduction cycles (Fig. 2(b) and Table S8, ESI†). For Cu0.18CeOz without Ru, the second cycle was similar to the first cycle but third and fourth cycles were different (Fig. 2(c) and Table S8, ESI†). For Ru0.04CeOz, the peak shift was observed in the second cycle and the H2 consumption was also decreased after the second TPR cycle (Fig. 2(d) and Table S8. ESI†). These results suggested that the incorporation of Cu and Ru into CeOz not only affected the reversible redox properties of Cu0.18Ru0.05CeOz but also contributed to the increase in the redox capacity of Cu0.18Ru0.05CeOz.
Changes in the oxidation states of Ru, Cu, and Ce in Cu0.18Ru0.05CeOz during the TPR/TPO cycles
Changes in the oxidation state of each metal species on as-prepared Cu0.18Ru0.05CeOz during TPR and TPO were investigated by in situ XANES analyses. In the first H2 reduction, all the in situ Ru K-edge, Cu K-edge, and Ce LIII-edge XANES spectra of as-prepared Cu0.18Ru0.05CeOz changed between 303 and 473 K (Fig. 3). The Ru K-edge XANES spectra initially changed at 333 K; the lowest reduction temperature among the metal species. Followed by the reduction of Ru, changes in the Cu K-edge XANES spectra were observed at 353 K and the Ce LIII-edge XANES spectra changed at 373 K (Fig. 3(a–c)). The quantitative analyses of the oxidation states of Ru, Cu, and Ce from the XANES spectra revealed that the average oxidation state of Ru changed from +4.37 to +1.06, that of Cu changed from +2.00 to +0.30, and that of Ce changed from +3.85 to +3.62, respectively (Fig. 3(d) and Fig. S7, Tables S9–S11, ESI†). The sum of the oxidation state changes in the three metal species was converted to the H2 consumption required for reduction (1.81 H2 mmol g_cat−1) (Table S12, ESI†), which was comparable to that observed by TPR measurements (1.9–2.4 H2 mmol g_cat−1). These results strongly suggested that Cu, Ru, and surface Ce species cooperated in the reduction of Cu0.18Ru0.05CeOz, similar to Cr0.19Rh0.06CeOz.14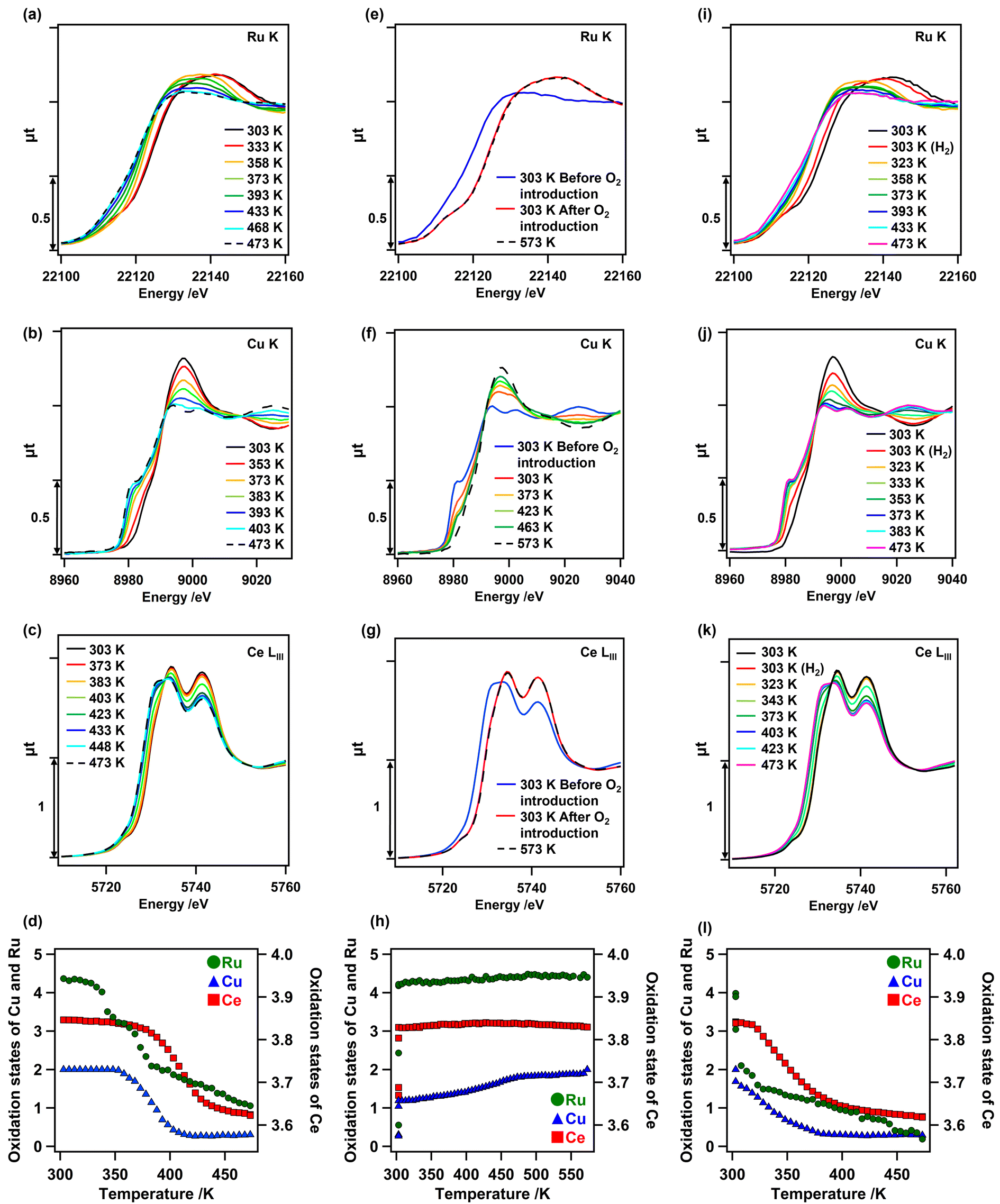 | ||
| Fig. 3 In situ (a) Ru K-edge, (b) Cu K-edge, and (c) Ce LIII-edge XANES spectra of Cu0.18Ru0.05CeOz under H2 reduction conditions (H2 + N2, 50 + 50 sccm) from 303 to 473 K (black solid lines: 303 K before reduction; black dashed lines: 473 K under H2). (d) Changes in the oxidation states of Ru (green), Cu (blue), and Ce (red) calculated from the XANES spectra (a–c) for the first reduction. In situ (e) Ru K-edge, (f) Cu K-edge, and (g) Ce LIII-edge XANES spectra of Cu0.18Ru0.05CeOz under O2 oxidation conditions (O2 + N2, 50 + 50 sccm) from 303 to 573 K (blue solid lines: 303 K before O2 introduction; red solid lines: 303 K after O2 introduction; black dashed lines: 573 K after holding for 1 h). (h) Changes in the oxidation states of Ru (green), Cu (blue), and Ce (red) calculated from the XANES spectra during the first oxidation. In situ (i) Ru K-edge, (j) Cu K-edge, and (k) Ce LIII-edge XANES spectral changes of Cu0.18Ru0.05CeOz under H2 reduction conditions (H2 + N2 50 + 50 sccm) from 303 to 473 K (black solid lines: 303 K before reduction; pink solid lines: 473 K under H2). (l) Changes in the oxidation states of Ru (green), Cu (blue), and Ce (red) calculated from XANES spectra during the second reduction. | ||
The in situ Ru K-edge, Cu K-edge, and Ce LIII-edge XANES spectra in the first reoxidation after the TPR measurement were similarly evaluated (Fig. 3(e–g)). Instantaneous oxidation occurred on Ru and Ce at 303 K, whereas the oxidation of Cu proceeded steadily from 303 K to 573 K. We also plotted the changes in the oxidation states of Cu, Ru, and Ce (Fig. 3(h)), showing the reversible changes on Cu0.18Ru0.05CeOz. The average oxidation states of Ru and Ce changed immediately to +4.40 (Ru) and to +3.83 (Ce) (Fig. 3(h) and Tables S9, S11, ESI†), respectively. The average oxidation state of Cu gradually reverted to +2.00 (Fig. 3(h) and Fig. S7, Table S10, ESI†). Thus, the structural transformation occurred rapidly on Ru and the surface layer of CeOz, followed by the gradual oxidation of Cu. This behaviour was also similar to Cr0.19Rh0.06CeOz.14
We investigated the in situ Ru K-edge, Cu K-edge, and Ce LIII-edge XANES measurements for the second reduction of Cu0.18Ru0.05CeOz under H2 flow conditions (Fig. 3(i–k)). The oxidation states of Ru, Cu, and Ce were again changed to +0.19, +0.29, and +3.62, respectively (Fig. 3(l) and Fig. S7, Tables S9–S11, ESI†). The sum of the H2 consumption for the second reduction was calculated to be 1.85 H2 mmol g_cat−1 (Table S12, ESI†), which was in agreement with the result from the second TPR cycle (1.98 mmol g_cat−1). The in situ XANES analysis suggested that Ru promoted the subsequent reduction of Cu and Ce species in the reduction.
Changes in the local coordination structures of Ru and Cu in Cu0.18Ru0.05CeOz during the TPR/TPO cycles
The changes in the local coordination structures of both Cu and Ru species on Cu0.18Ru0.05CeOz during the TPR/TPO cycles were investigated by the Cu K-edge and Ru K-edge EXAFS curve-fitting analyses (Fig. 4(a), (b) and Fig. S8, S9, Table S13, ESI†). After the H2 reduction, the Cu K-edge EXAFS changed completely from that of the as-prepared sample; the Cu–O bonds disappeared, and a new peak attributed to Cu–Cu bonds was observed at 0.253 ± 0.001 nm (CN = 6.1 ± 0.9). The Cu–Cu bond peak was similar to that of bulk Cu (0.256 nm) and the EXAFS results suggested the transformation to metallic Cu nanoparticles after the reduction with H2 (Fig. 4(a2) and Fig. S8, Table S13, ESI†). The Ru K-edge EXAFS after the H2 reduction showed the appearance of the peak of Ru–Ru bonds (0.2–0.3 nm in the Fourier transform spectra) (Fig. 4(b2), (b6) and Fig. S9, ESI†), suggesting the formation of small metallic Ru nanoclusters.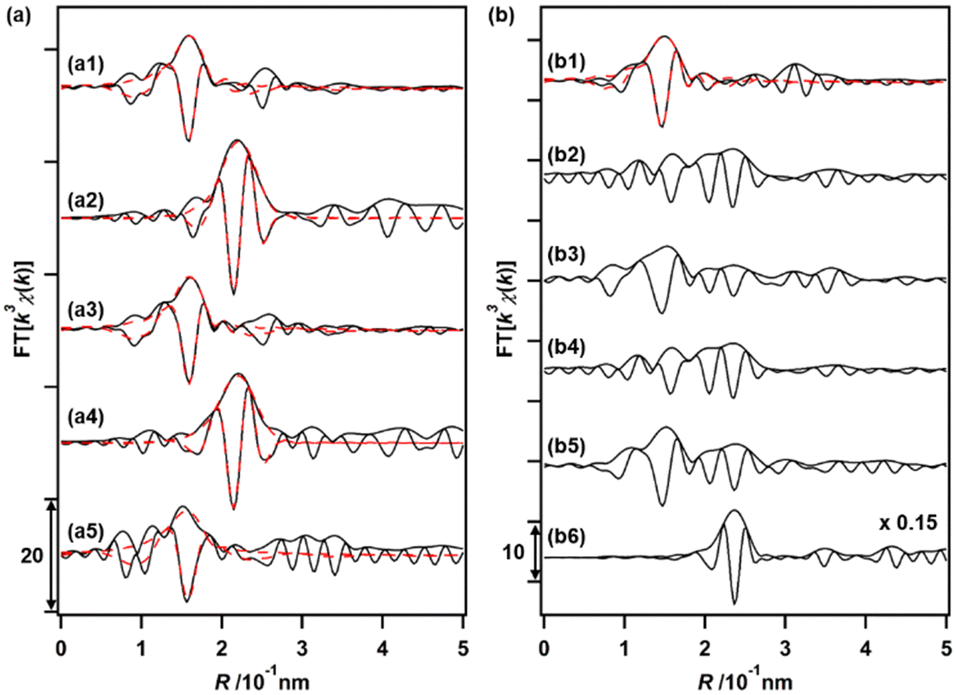 | ||
| Fig. 4 (a) Cu K-edge EXAFS Fourier transforms of Cu0.18Ru0.05CeOz (k = 30–140 nm−1); (a1) as-prepared, (a2) after first H2 reduction, (a3) after first O2 oxidation, (a4) after third H2 reduction, and (a5) after third O2 oxidation. (b) Ru K-edge EXAFS Fourier transforms for Cu0.18Ru0.05CeOz (k = 30–130 nm−1); (b1) as-prepared, (b2) after first H2 reduction, (b3) after first O2 oxidation, (b4) after third H2 reduction, (b5) after third O2 oxidation, and (b6) reference Ru powder (×0.15 in magnitude, k = 30–180 nm−1). H2 reduction conditions: (27 kPa H2) from 303 to 523 K, O2 oxidation conditions: (27 kPa O2) from 303 to 573 K and held for 2 h after H2 reduction up to 523 K. Black solid lines showed observed data and red dashed lines with fitted data. | ||
After the reoxidation, the Cu species reverted to the local coordination structure before the introduction of H2, with a Cu–O bond at 0.196 ± 0.002 nm (CN = 4.7 ± 0.8) (Fig. 4(a3) and Fig. S8, Table S13, ESI†). The peak of Ru–O bonds (0.1–0.18 nm) became also dominant (Fig. 4(b3) and Fig. S9, ESI†). A similar EXAFS analysis was also conducted after repeating the reduction/oxidation cycles (Fig. 4(a4), (a5), (b4), (b5) and Fig. S8, S9, Table S13, ESI†). After the reduction, Cu–Cu bonds were observed and the Cu–O bonds were consumed after the third oxidation cycle. A similar reversible feature on the structural changes on Ru was observed in the third reduction and oxidation cycles, suggesting drastic structural changes in both Cu and Ru during the reversible redox cycles of Cu0.18Ru0.05CeOz.
Based on the TPR/TPO results and the in situ XAFS characterization, the redox structural transformation of Cu0.18Ru0.05CeOz was proposed as follows (Fig. 5).
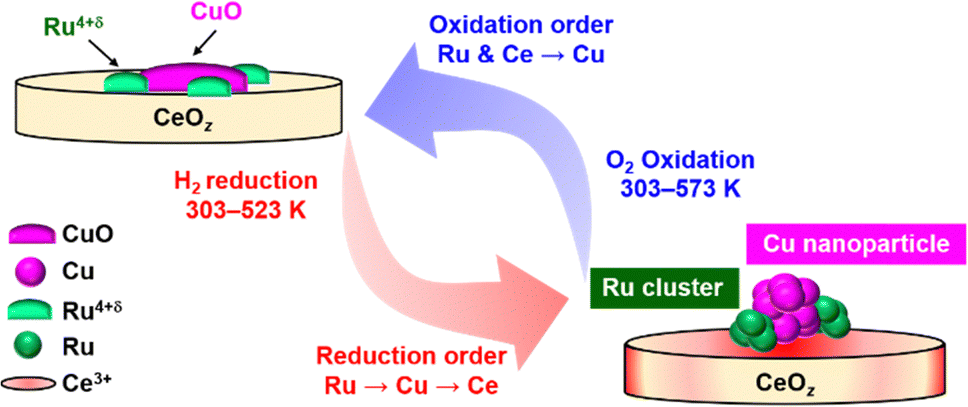 | ||
| Fig. 5 Suggested structural transformation of the reversible low-temperature redox cycle of Cu0.18Ru0.05CeOz. | ||
(1) As-prepared Cu0.18Ru0.05CeOz: bivalent Cu oxide and Ru4+δ oxide were dispersed on the CeO2 fluorite surface.
(2) After the first reduction in the low-temperature range: Ru oxide was first reduced and converted to metallic Ru nanoclusters at around 333 K. Then, the reduction of Cu and Ce proceeded around 373 K with the formation of Cu nanoparticles and Ce3+ species.
(3) After the subsequent oxidation the Ru nanoclusters and surface/subsurface Ce3+ species were oxidized quickly at 303 K. Then, the Cu nanoparticles smoothly reverted to Cu oxide at 303–573 K.
(4) Reversible redox transformation after the second cycle: Cu0.18Ru0.05CeOz was reduced at a lower temperature range than in the first cycle, and the reversible redox transformation between the reduced structure and the oxidized structure was observed.
Ammoxidation performance of benzylic alcohol on Cu0.18Ru0.05CeOz
The catalytic performance of Cu0.18Ru0.05CeOz for the ammoxidation of benzyl alcohol (1) was investigated compared with reference catalysts, such as Cu0.18CeOz, Ru0.04CeOz, and CeOz (Table 1). Cu0.18Ru0.05CeOz showed high benzyl alcohol conversion (95%) with high benzonitrile selectivity (2a, 95%) (Table 1, entry 1). Previously reported Cr0.19Rh0.06CeOz14 showed the conversion of benzyl alcohol (82%), with low benzonitrile selectivity (11%) (Table 1, entry 2). Cu0.18CeOz without Ru showed lower conversion (70%) and benzonitrile selectivity (78%) than Cu0.18Ru0.05CeOz (Table 1, entry 3). Ru0.04CeOz without Cu showed low benzonitrile selectivity (29%), despite the high conversion of benzyl alcohol (91%) (Table 1, entry 4). CeOz without both Cu and Ru showed low benzonitrile selectivity (10%) (Table 1, entry 5). The physical mixture of Cu0.18CeOz and Ru0.04CeOz effectively promoted the reaction (99%) but benzonitrile selectivity was 75% and benzamide was formed (Table 1, entry 6). These results demonstrated that the combination of Cu and Ru on the same CeOz surface is key for the ammoxidation of benzyl alcohol to benzonitrile, and Cu0.18Ru0.05CeOz acted as an efficient catalyst for the ammoxidation.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Reaction atmosphere | Conversion of 1% | Select. of 2a % | Select. of 2b % | Select. of 2c % | Select. of 2d % | Select. of 2e % |
| Reaction conditions: 1 (1.93 mmol), toluene (1.0 mL), dodecane (internal standard; 0.07 mL), 423 K, 24 h. The selectivity of 2a–2e was calculated by product amount/consumed benzyl alcohol amount. Mass balance was over 0.9 except for entries 2, 4, and 5.a Cu0.18Ru0.05CeOz (74 mg), Ru/Cu/1/NH3/dodecane = 1/3.5/100/191/100.b Cr0.19Rh0.06CeOz (61 mg), Rh/Cr/1/NH3/dodecane = 1/3.5/100/191/100.c Cu0.18CeOz (77 mg), Cu/1/NH3/dodecane = 1/29/58/29.d Ru0.04CeOz (72 mg), Ru/1/NH3/dodecane = 1/100/191/100.e CeOz (74 mg), 1/NH3/dodecane = 1/1.91/1.f Cu0.18CeOz (77 mg), Ru0.04CeOz (72 mg), Ru/Cu/1/NH3/dodecane = 1/3.5/100/191/100. | ||||||||
| 1a | Cu0.18Ru0.05CeOz | 0.32 MPa NH3, 1.0 MPa O2 | 95 | 95 | 5 | 0 | 0 | 3 |
| 2b | Cr0.19Rh0.06CeOz | 0.32 MPa NH3, 1.0 MPa O2 | 82 | 11 | 21 | 1 | 6 | 3 |
| 3c | Cu0.18CeOz | 0.32 MPa NH3, 1.0 MPa O2 | 70 | 78 | 11 | 0 | 0 | 4 |
| 4d | Ru0.04CeOz | 0.32 MPa NH3, 1.0 MPa O2 | 91 | 29 | 12 | 1 | 4 | 12 |
| 5e | CeOz | 0.32 MPa NH3, 1.0 MPa O2 | 76 | 10 | 13 | 1 | 6 | 11 |
| 6f | Cu0.18CeOz + Ru0.04CeOz | 0.32 MPa NH3, 1.0 MPa O2 | 99 | 75 | 17 | 0 | 0 | 7 |
| 7a | Cu0.18Ru0.05CeOz (after first TPR) | 0.32 MPa NH3, 1.0 MPa O2 | >99 | 93 | 4 | 0 | 0 | 1 |
| 8a | Cu0.18Ru0.05CeOz (after first TPO) | 0.32 MPa NH3, 1.0 MPa O2 | 90 | 95 | 6 | 0 | 0 | 2 |
| 9 | None | 0.32 MPa NH3, 1.0 MPa O2 | 0 | 0 | 0 | 0 | 0 | 0 |
| 10a | Cu0.18Ru0.05CeOz | 1.0 MPa N2 | 10 | 0 | 0 | 59 | 0 | 46 |
| 11a | Cu0.18Ru0.05CeOz | 0.32 MPa NH3, 1.0 MPa N2 | 20 | 0 | 0 | 0 | >99 | 0 |
| 12c | Cu0.18CeOz | 0.32 MPa NH3, 1.0 MPa N2 | 9 | 0 | 0 | 0 | >99 | 0 |
| 13d | Ru0.04CeOz | 0.32 MPa NH3, 1.0 MPa N2 | 20 | 4 | 0 | 0 | 66 | 0 |
| 14e | CeOz | 0.32 MPa NH3, 1.0 MPa N2 | <1 | 0 | 0 | 0 | 0 | 0 |
| 15 | None | 0.32 MPa NH3, 1.0 MPa N2 | 0 | 0 | 0 | 0 | 0 | 0 |
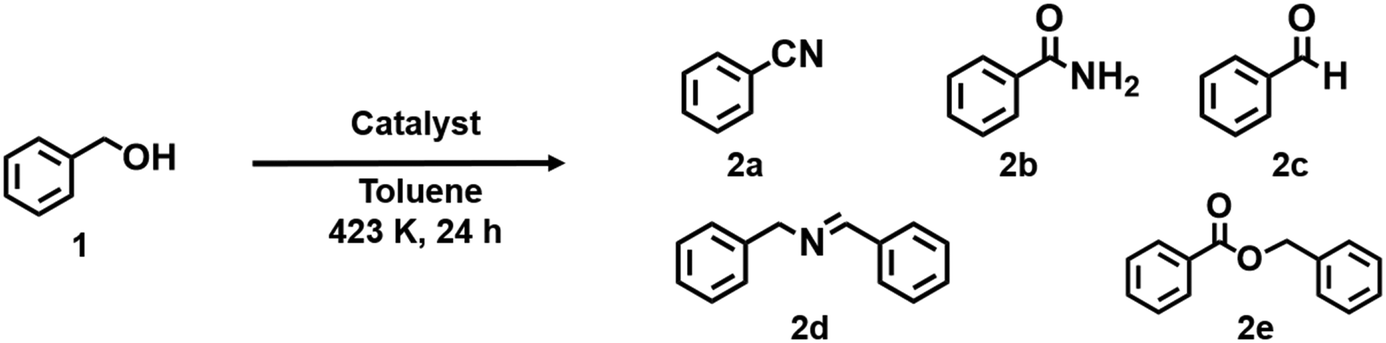
Fig. 6(a) shows the time profiles of benzyl alcohol conversion and selectivity for the products (2a–2e) on Cu0.18Ru0.05CeOz. At the beginning of the reaction, a small amount of benzaldehyde was observed, indicating that it was an intermediate in the ammoxidation. Benzonitrile production rate was 2.37 × 10−4 mol g_cat−1 min−1 on Cu0.18Ru0.05CeOz, which was higher than those on Cu0.18CeOz, Ru0.04CeOz, and the physical mixture of Cu0.18CeOz and Ru0.04CeOz (Fig. S10(a), ESI)†. The initial reaction rate of benzyl alcohol conversion on Cu0.18Ru0.05CeOz was also higher than those on Cu0.18CeOz and Ru0.04CeOz (Fig. S10(b), ESI†).
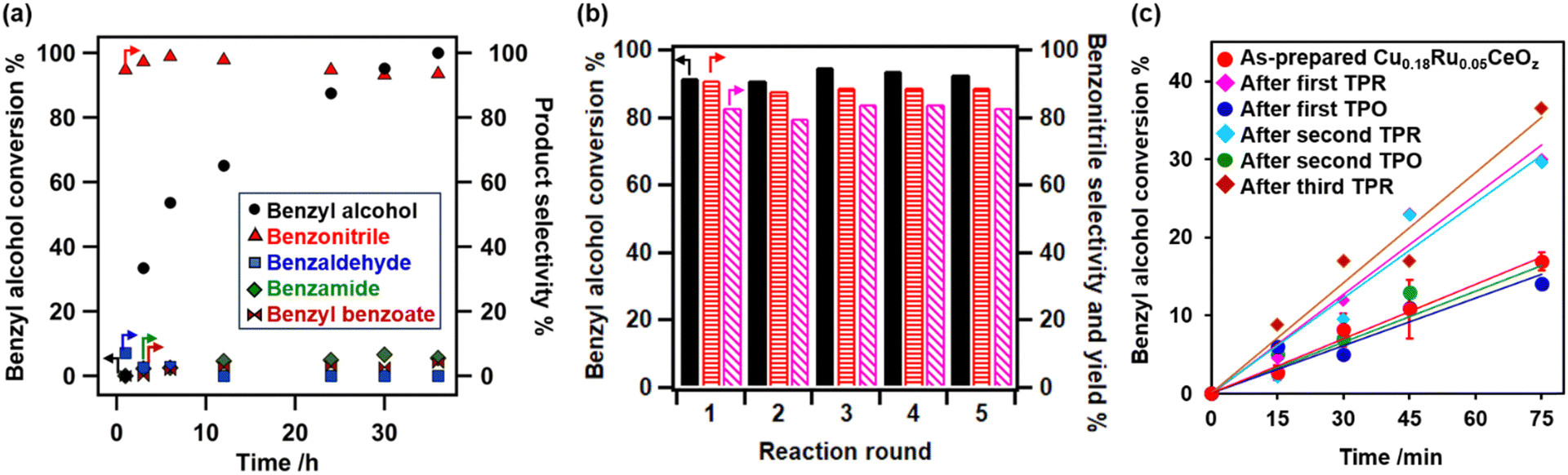 | ||
| Fig. 6 (a) Time-conversion plots of the ammoxidation of benzyl alcohol on Cu0.18Ru0.05CeOz at 423 K. (b) Recycling tests for the ammoxidation of benzyl alcohol on Cu0.18Ru0.05CeOz at 423 K, 24 h (black bars: benzyl alcohol conversion; red horizontal line pattern bars: benzonitrile selectivity; pink diagonal line pattern bars: benzonitrile yield). (c) Initial reaction rates of Cu0.18Ru0.05CeOz with different pretreatments. | ||
The heterogeneity test of Cu0.18Ru0.05CeOz suggested that the ammoxidation proceeded on the solid catalyst (Fig. S11, ESI†). The recycled Cu0.18Ru0.05CeOz catalyst (collected by centrifugation and reactivated through calcination at 573 K under air) showed steady performance in the fifth ammoxidation cycle (Fig. 6(b)). ICP-OES showed that negligible amounts of Ru and Ce leaching were observed after each recycling round. The amount of leached Cu after fourth recycling round was approximately 1% of the total amount of Cu in Cu0.18Ru0.05CeOz. Cu0.18Ru0.05CeOz after the reaction showed a similar XRD pattern to that before (Fig. S12, ESI†). HAADF-STEM-EDS images (Fig. S13, ESI†) and Cu K-edge, Ru K-edge, and Ce LIII-edge XANES spectra (Fig. S14, ESI†) were similar to those of the as-prepared catalyst. Cu K-edge and Ru K-edge EXAFS analyses (Fig. S15 and Tables S14, S15, ESI†) also showed a similar local coordination structure to the as-prepared one. These results indicated that Cu0.18Ru0.05CeOz was stable under the ammoxidation conditions.
We also investigated differences in the ammoxidation activity on reduced and oxidized Cu0.18Ru0.05CeOz. The initial reaction rates of Cu0.18Ru0.05CeOz after TPR and TPO were measured. Cu0.18Ru0.05CeOz after treatment with H2 (after the first TPR) showed higher benzyl alcohol conversion, and Cu0.18Ru0.05CeOz after treatment with O2 (after the first TPO) showed similar activity to as-prepared Cu0.18Ru0.05CeOz (Fig. 6(c)). The differences in the activity between them were clear, and similar differences were also observed for the second TPR and TPO (Fig. 6(c)). The reversibility strongly indicated that the reduced state was more active than the oxidized state for Cu0.18Ru0.05CeOz and that Ru nanoclusters and Cu nanoparticles on reduced Cu0.18Ru0.05CeOz may promote the activation of benzyl alcohol. However, both reduced and oxidized (as-prepared) catalysts showed similar performance for the ammoxidation after 24 h (Table 1, entries 7 and 8); therefore, the ammoxidation proceeded on Cu0.18Ru0.05CeOz independent of the pretreatment conditions.
Role of Cu and Ru in Cu0.18Ru0.05CeOz in the ammoxidation of benzyl alcohol
We investigated the activity of lattice oxygen for benzyl alcohol conversion in the absence of O2. The conversion of benzyl alcohol on Cu0.18Ru0.05CeOz in the absence of O2 and NH3 was only 10% (Table 1, entry 10), and this value, which was equivalent to 1.04 mmol g_cat−1 of O2, was comparable to the amount of active lattice oxygen (0.92 mmol g_cat−1 O2, from TPR/TPO) (Table S8, ESI†). The reaction of benzyl alcohol with NH3 in the absence of O2 also occurred (20% conversion), although the selectivity switched to N-benzylidenebenzamine (2d, >99%) (Table 1, entry 11). In contrast, CeOz showed no conversion of benzyl alcohol (Table 1, entry 14). The Ru K-edge, Cu K-edge, and Ce LIII-edge XANES spectra of Cu0.18Ru0.05CeOz after the reaction suggested that all the metal species changed oxidation states (Fig. S16, ESI†). These results demonstrated that the lattice oxygen on Cu0.18Ru0.05CeOz, which was responsible under the redox conditions, involved in the conversion of the alcohol.The steps in the ammoxidation of benzyl alcohol are the aerobic oxidation of alcohol to form benzaldehyde, self-condensation of the imine through the aldehyde-ammonia interaction, and aerobic oxidation of the imine to form the nitrile.19 We tested the oxidation of benzyl alcohol, which was the initial step, without the addition of NH3 (Table S16, ESI†). Cu0.18Ru0.05CeOz (>99%) and Ru0.04CeOz (>99%) showed high conversions of benzyl alcohol in the absence of NH3 with the formation of benzoic acid (Table S16, entries 1 and 3, ESI†). In contrast, Cu0.18CeOz (30%) and CeOz (26%) showed moderate conversions of benzyl alcohol with benzaldehyde as the major product, demonstrating that Cu was not mainly responsible for the first benzyl alcohol oxidation (Table S16, entries 2 and 4, ESI†). Thus, Ru was the key element for the first benzyl alcohol oxidation to benzaldehyde.
The second ammoxidation of benzaldehyde was evaluated by the reaction of benzaldehyde with NH3. The consumption of benzaldehyde occurred without catalysts via a spontaneous reaction between benzaldehyde and ammonia,51 but the selectivity for benzonitrile varied widely among the catalysts (Table S17, ESI†). The benzonitrile selectivity was high for Cu0.18Ru0.05CeOz (91%) and Cu0.18CeOz (81%) (Table S17, entries 1 and 2, ESI†), whereas those for Ru0.04CeOz (5%) and CeOz (2%) were low (Table S17, entries 3 and 4, ESI†). These results demonstrated that Cu was the active site for benzonitrile formation (Scheme S1, ESI†). Ru species selectively oxidized benzyl alcohol using the lattice oxygen of the catalyst, and then Cu converted the intermediate benzaldehyde to benzonitrile, showing that the incorporation of both Cu and Ru on the same CeOz surface of Cu0.18Ru0.05CeOz was required for the selective ammoxidation catalysis.
Conclusions
A Cu- and Ru-incorporated ceria-based catalyst (Cu0.18Ru0.05CeOz) was prepared, and it exhibited low-temperature, reversible redox performance (< 423 K) and high catalytic activity for the ammoxidation of benzyl alcohol to form benzonitrile selectively. In situ analysis suggested that the redox behaviors of the mixed oxides were as follows: (i) the reduction of Ru occurred first and promoted the reduction of Cu and Ce below 423 K; and (ii) the oxidation proceeded immediately on Ru and Ce, followed by Cu. The catalytic activity of the benzyl alcohol oxidation depended on the structure of the Cu0.18Ru0.05CeOz catalyst and the reduced structure was more active than the oxidized structure. The small Ru nanoclusters in the reduced form converted the benzyl alcohol to benzaldehyde, and then the Cu species promoted the formation of benzonitrile with high selectivity. The incorporation of highly dispersed Cu and Ru on the same CeOz surface of Cu0.18Ru0.05CeOz (they might be located in close proximity to each other) enhanced the redox properties and the catalytic ammoxidation of benzyl alcohol to benzonitrile.Experimental
Catalyst preparation
All chemicals were purchased from commercial companies and used without further purification. Cu(NO3)2·3H2O (0.67 mmol; Wako), RuCl3 (0.22 mmol; Sigma-Aldrich), and Ce(NO3)3·6H2O (3.58 mmol; Sigma-Aldrich) were dissolved in ultrapure water (Milli-Q; 18 mL) and continuously stirred at 293 K for 1 h. Aqueous NaOH solution (7 M, 53 mL) was added to the mixture with continuous stirring for 30 min. The slurry was transferred to a 100 mL Teflon-lined stainless steel vessel autoclave that was sealed and placed in an electric oven and kept at 358 K for 24 h. After the autoclave cooled to room temperature, the precipitate was separated by centrifugation and washed with ultrapure water and ethanol several times followed by drying under air at 353 K for 12 h. The as-dried solid was ground and calcined at 573 K for 2 h to obtain Cu0.18Ru0.05CeOz.Cu0.18CeOz and Ru0.04CeOz were prepared using similar procedures without the addition of the Ru or Cu precursor. CeOz without Ru and Cu was also prepared using a similar procedure without the Cu and Ru precursors. The hydrothermal temperature of the CeOz preparation was 373 K, as in our previous paper.14
Characterization
XAFS spectra at the Ru K-edge were measured at the NW10A station of the Photon-Factory Advanced Ring (PF-AR) at KEK-IMSS and at the BL11S2 station of the Aichi Synchrotron Radiation Center. The energy and current of the electrons in the storage ring were 6.5 GeV and 60 mA at PF-AR, and 1.2 GeV and 300 mA at the Aichi Synchrotron Center, respectively. X-rays from the storage ring were monochromatized with a Si(311) double-crystal monochromator. At the NW10A station of the Photon Factory, two ionization chambers filled with pure Ar were used to monitor the incident (I0) and transmitted (I) X-rays. At the BL11S2 station of the Aichi Synchrotron Center, two ionization chambers filled with pure Ar and a mixture of pure Ar and Kr (9/1, v/v) were used to monitor both the incident (I0) and transmitted (I) X-rays, respectively. Ru(acac)3 (diluted with boron nitride), RuO2 (Sigma-Aldrich; diluted with boron nitride), KRuO4 (Sigma-Aldrich; diluted with boron nitride), and Ru powder (diluted with boron nitride) were used as references. As-prepared Cu0.18Ru0.05CeOz and Cu0.18Ru0.05CeOz after the ammoxidation (25 mg) were ground and packed into a cell (4 mmϕ), and the cell was sealed with a Kapton film under air. Cu0.18Ru0.05CeOz after the first and third TPR/TPO, and after the ammoxidation under N2 was ground and packed into a cell (4 mmϕ) in the glove box filled with Ar, and the cell was sealed with a Kapton film under an Ar atmosphere. All samples were measured in transmission mode at 298 K.
XAFS spectra were analysed using ATHENA and ARTEMIS with IFEFFIT (version 1.2.11).52,53 Threshold energies were tentatively set at 8980.3 eV (Cu K-edge; the inflection point of Cu foil54), 22119.3 eV (Ru K-edge; the inflection point of Ru0 powder54), and 5734.4 eV (Ce LIII-edge; the first peak top of the white line for CeO2), and background was subtracted using the Autobk program and the spline smoothing algorithm in ATHENA.55,56 The average oxidation states of Ce were determined from the changes at the second peak top of the CeO2 white line (5741.3 eV). The average oxidation states of Cu were determined by linear combination fitting using the XANES spectra of Cu foil, Cu2O, and CuO (8960.0–9010.0 eV). The average oxidation states of Ru were determined from the Ru K-edge energy (at the 50% level of the edge jump in the spectrum) versus the calibration graph, which was constructed by using Ru powder (+0), Ru(acac)3 (+3), RuO2 (+4), and KRuO4 (+7) as references for different oxidation states.46,47
The k3-weighted EXAFS oscillation was Fourier transformed into R-space and curve-fitting analysis was performed. For the Cu K-edge, S02 was fixed as 1 for metallic Cu based on the fitting of Cu foil and 0.70 for Cu2+ based on the fitting of CuO (Fig. S5 and Table S4, ESI†). For the Ru K-edge, S02 was fixed as 0.86 based on the fitting of RuO2 (Fig. S5 and Table S5, ESI†). Phase shifts and backscattering amplitudes for Cu–O, Cu–Cu, Ru–O, and Ru–Ru were calculated with FEFF857 code by using structural parameters obtained from the crystal structures of CuO (ICSD-16025), Cu (ICSD-53246), RuO2 (ICSD-23961), and Ru (ICSD-40354). Coordination number (CN), interatomic distance (R), Debye–Waller factor (σ2, mean-square displacement), and correction-of-edge energy (ΔE) were calculated as fitting parameters for each shell.
Catalytic reactions
| Benzyl alcohol conversion (%) = ((mole of benzyl alcohol)initial − (mole benzyl alcohol)final)/(mol of benzyl alcohol)initial × 100% | (1) |
| Benzonitrile selectivity (%) = (mole of benzonitrile)final/((mole of benzyl alcohol)initial − (mol of benzyl alcohol)final) × 100% | (2) |
| By-product selectivity (%) = (mole of by-product)final/((mole benzyl alcohol)initial − (mol benzyl alcohol)final) × 100% | (3) |
| Mass balance = ((mole of benzyl alcohol)final + (mole of all products)final)/(mol of benzyl alcohol)initial | (4) |
To evaluate the ammoxidation performance of different redox phases, Cu0.18Ru0.05CeOz treated with 27 kPa of H2 (99.99999%) (denoted as Cu0.18Ru0.05CeOz after first TPR), and Cu0.18Ru0.05CeOz treated with 27 kPa of H2 followed by 27 kPa of O2 (denoted as Cu0.18Ru0.05CeOz after first TPO) were prepared (The detailed procedure is described in the section “TPR with H2 and TPO with O2”.). The sample was transferred into a Teflon tube of the autoclave in an Ar-filled glove box, and toluene and benzyl alcohol (both degassed) were added in the glove box. Then the autoclave was tightly sealed and taken out of the glove box, and the inner atmosphere was changed accordingly.
Benzyl alcohol oxidation was performed using a similar procedure without the addition of 0.32 MPa of NH3. The ammoxidation of benzaldehyde was performed by changing the starting substrate to benzaldehyde (0.2 mL; Sigma-Aldrich; 1.96 mmol, 1.63 mol L−1). The ammoxidation of benzyl alcohol under a N2 atmosphere was performed with the addition of all the starting substrates in the Ar-filled glove box. The autoclave was placed under vacuum before the addition of 0.32 MPa of NH3 and 1.0 MPa of N2 (99.99995%) to avoid O2 appearing in the autoclave.
Author contributions
C. C. prepared catalysts, carried out the redox performance measurements, investigated the ammoxidation reactions, and wrote the manuscript. S. I. contributed to XPS measurements and in situ XAFS measurements. G. Y. designed the setup of the redox measurement and evaluation of the ammoxidation reaction. K. H. carried out TEM and HAADF-STEM measurements. S. M. conceived the project and wrote the manuscript. M. T. directed this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. C. acknowledges a fellowship from the Japan Society for the Promotion of Science (JSPS) (No. JP23KJ1103). This work was financially supported in part by MEXT/JSPS KAKENHI (Grant No. JP20H04808 (hybrid catalysis), JP22H02031, JP23H01973, and JP23K20034 (International Leading Research)), the Japan Science and Technology Agency (JST) PRESTO program (No. JPMJPR15S7), the JST CREST (No. JPMJCR2235), the Ministry of Education, Culture, Sports, Science and Technology (MEXT) project “Integrated Research on Chemical Synthesis, the Integrated Research Consortium on Chemical Sciences”, the World Research Unit (B-1) at Nagoya University “Reaction Infography (R-ing)”, the Iketani Science and Technology Foundation, the Hori Science and Arts Foundation, and the Chubei Itoh Foundation. XAFS measurements were performed with the approval of PF-PAC (No. 2019G105, 2021G113, and 2023G162) and the Aichi Synchrotron Radiation Center (No. 202102001, 202102055, 202203022, 202303024, and 202304025). TEM/HAADF-STEM-EDS measurements were conducted at the High-Voltage Electron Microscope Laboratory, Institute of Materials and Systems for Sustainability, Nagoya University, supported by the “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of MEXT, Japan (Grant Number JPMXP1223NU0017). Part of this work was also conducted at the Institute for Molecular Science, supported by the Nanotechnology Platform Program <Molecule and Material Synthesis> (JPMXP09S20MS1064 and JPMXP09S21MS1061) and “Advanced Research Infrastructure for Materials Nanotechnology” (JPMXP1222MS1011 and JPMXP1223MS1023) of MEXT, Japan.Notes and references
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- P. Li, X. Chen, Y. Li and J. W. Schwank, Catal. Today, 2019, 327, 90–115 CrossRef CAS.
- E. W. McFarland and H. Metiu, Chem. Rev., 2013, 113, 4391–4427 CrossRef CAS PubMed.
- M. Ozawa, M. Kimura and A. Isogai, J. Alloys Compd., 1993, 193, 73–75 CrossRef CAS.
- P. Zhang, H. Lu, Y. Zhou, L. Zhang, Z. Wu, S. Yang, H. Shi, Q. Zhu, Y. Chen and S. Dai, Nat. Commun., 2015, 6, 8446 CrossRef CAS PubMed.
- J. Beckers and G. Rothenberg, Dalton Trans., 2008, 6573–6578 RSC.
- Y. Xiong, L. Li, L. Zhang, Y. Cao, S. Yu, C. Tang and L. Dong, Phys. Chem. Chem. Phys., 2017, 19, 21840–21847 RSC.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, ACS Catal., 2018, 8, 6203–6215 CrossRef CAS.
- N. C. Nelson, J. S. Manzano, A. D. Sadow, S. H. Overbury and I. I. Slowing, ACS Catal., 2015, 5, 2051–2061 CrossRef CAS.
- T. Rajkumar, A. Sápi, M. Ábel, J. Kiss, I. Szenti, K. Baán, J. F. Gómez-Pérez, Á. Kukovecz and Z. Kónya, Catal. Lett., 2021, 151, 3477–3491 CrossRef CAS.
- C. Megías-Sayago, K. Chakarova, A. Penkova, A. Lolli, S. Ivanova, S. Albonetti, F. Cavani and J. A. Odriozola, ACS Catal., 2018, 8, 11154–11164 CrossRef.
- T. Tong, W. Guo, X. Liu, Y. Guo, C.-W. Pao, J.-L. Chen, Y. Hu and Y. Wang, J. Catal., 2019, 378, 392–401 CrossRef CAS.
- Y. Chen, B. deGlee, Y. Tang, Z. Wang, B. Zhao, Y. Wei, L. Zhang, S. Yoo, K. Pei, J. H. Kim, Y. Ding, P. Hu, F. F. Tao and M. Liu, Nat. Energy, 2018, 3, 1042–1050 CrossRef CAS.
- S. Ikemoto, X. Huang, S. Muratsugu, S. Nagase, T. Koitaya, H. Matsui, G. Yokota, T. Sudoh, A. Hashimoto, Y. Tan, S. Yamamoto, J. Tang, I. Matsuda, J. Yoshinobu, T. Yokoyama, S. Kusaka, R. Matsuda and M. Tada, Phys. Chem. Chem. Phys., 2019, 21, 20868–20877 RSC.
- S. Ikemoto, S. Muratsugu, T. Koitaya and M. Tada, ACS Catal., 2022, 12, 431–441 CrossRef CAS.
- S. Ikemoto, S. Muratsugu, T. Koitaya, Y. Tsuji, M. Das, K. Yoshizawa, F. Glorius and M. Tada, J. Am. Chem. Soc., 2023, 145, 1497–1504 CrossRef CAS PubMed.
- A. Martin and V. N. Kalevaru, ChemCatChem, 2010, 2, 1504–1522 CrossRef CAS.
- G. Yan, Y. Zhang and J. Wang, Adv. Synth. Catal., 2017, 359, 4068–4105 CrossRef CAS.
- Z. Qi, C. Hu, Y. Zhong, C. Cai and G.-P. Lu, Org. Chem. Front., 2021, 8, 3137–3149 RSC.
- Y. Wang, S. Furukawa, X. Fu and N. Yan, ACS Catal., 2020, 10, 311–335 CrossRef CAS.
- J. Kim, H. J. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 11948–11959 CrossRef CAS PubMed.
- H. H. Hodgson, Chem. Rev., 1947, 40, 251–277 CrossRef CAS PubMed.
- V. V. Grushin and H. Alper, Chem. Rev., 1994, 94, 1047–1062 CrossRef CAS.
- M. Ezawa and H. Togo, Eur. J. Org. Chem., 2017, 2379–2384 CrossRef CAS.
- P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049–5067 RSC.
- J. F. Brazdil, Catal. Today, 2021, 363, 55–59 CrossRef CAS.
- J.-B. Xie, J.-J. Bao, H.-X. Li, D.-W. Tan, H.-Y. Li and J.-P. Lang, RSC Adv., 2014, 4, 54007–54017 RSC.
- D.-W. Tan, J.-B. Xie, Q. Li, H.-X. Li, J.-C. Li, H.-Y. Li and J.-P. Lang, Dalton Trans., 2014, 43, 14061–14071 RSC.
- W. Yin, C. Wang and Y. Huang, Org. Lett., 2013, 15, 1850–1853 CrossRef CAS PubMed.
- G. Zhang, D. Ma, Y. Zhao, G. Zhang, G. Mei, J. Lyu, C. Ding and S. Shan, ChemistryOpen, 2018, 7, 885–889 CrossRef CAS PubMed.
- S. U. Dighe, D. Chowdhury and S. Batra, Adv. Synth. Catal., 2014, 356, 3892–3896 CrossRef CAS.
- T. Oishi, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2009, 48, 6286–6288 CrossRef CAS PubMed.
- H. Wang, D. Xu, E. Guan, L. Wang, J. Zhang, C. Wang, S. Wang, H. Xu, X. Meng, B. Yang, B. C. Gates and F.-S. Xiao, ACS Catal., 2020, 10, 6299–6308 CrossRef CAS.
- Y. Preger, T. W. Root and S. S. Stahl, ACS Omega, 2018, 3, 6091–6096 CrossRef CAS PubMed.
- A. N. Hashemi, H. Eshghi and K. Lamei, Appl. Organomet. Chem., 2019, 33, e4835 CrossRef.
- F. Mao, Z. Qi, H. Fan, D. Sui, R. Chen and J. Huang, RSC Adv., 2017, 7, 1498–1503 RSC.
- R. V. Jagadeesh, H. Junge and M. Beller, Nat. Commun., 2014, 5, 4123 CrossRef CAS PubMed.
- T. Yasukawa, X. Yang and S. Kobayashi, J. Org. Chem., 2020, 85, 7543–7548 CrossRef CAS PubMed.
- F. Ziaee, M. Gholizadeh and S. M. Seyedi, Appl. Organomet. Chem., 2018, 32, e4253 CrossRef.
- T. Ishida, H. Watanabe, T. Takei, A. Hamasaki, M. Tokunaga and M. Haruta, Appl. Catal., A, 2012, 425–426, 85–90 CrossRef CAS.
- D. R. Mullins, Surf. Sci. Rep., 2015, 70, 42–85 CrossRef CAS.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- F. Chen, Y. Xia, J. Lao, D. Cheng and X. Zhan, Ind. Eng. Chem. Res., 2021, 60, 9068–9079 CrossRef CAS.
- S. Cao, B. Zou, J. Yang, J. Wang and H. Feng, ACS Appl. Nano Mater., 2022, 5, 11689–11698 CrossRef CAS.
- Z. Liu, Q. Wang, J. Wu, H. Zhang, Y. Liu, T. Zhang, H. Tian and S. Zeng, ACS Appl. Mater. Interfaces, 2021, 13, 35804–35817 CrossRef CAS PubMed.
- N. Planas, L. Vigara, C. Cady, P. Miró, P. Huang, L. Hammarström, S. Styring, N. Leidel, H. Dau, M. Haumann, L. Gagliardi, C. J. Cramer and A. Llobet, Inorg. Chem., 2011, 50, 11134–11142 CrossRef CAS PubMed.
- K. Sardar, E. Petrucco, C. I. Hiley, J. D. B. Sharman, P. P. Wells, A. E. Russell, R. J. Kashtiban, J. Sloan and R. I. Walton, Angew. Chem., Int. Ed., 2014, 53, 10960–10964 CrossRef CAS PubMed.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- Z. Li, K. Werner, K. Qian, R. You, A. Płucienik, A. Jia, L. Wu, L. Zhang, H. Pan, H. Kuhlenbeck, S. Shaikhutdinov, W. Huang and H.-J. Freund, Angew. Chem., Int. Ed., 2019, 58, 14686–14693 CrossRef CAS PubMed.
- C. Yang, X. Yu, S. Heißler, A. Nefedov, S. Colussi, J. Llorca, A. Trovarelli, Y. Wang and C. Wöll, Angew. Chem., Int. Ed., 2017, 56, 375–379 CrossRef CAS PubMed.
- J.-M. Huang, J.-F. Zhang, Y. Dong and W. Gong, J. Org. Chem., 2011, 76, 3511–3514 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- M. Newville, B. Ravel, D. Haskel, J. J. Rehr, E. A. Stern and Y. Yacoby, Phys. B, 1995, 208–209, 154–156 CrossRef.
- J. A. Bearden and A. F. Burr, Rev. Mod. Phys., 1967, 39, 125–142 CrossRef CAS.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS PubMed.
- M. Newville, P. Līviņŝ, Y. Yacoby, J. J. Rehr and E. A. Stern, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 14126–14131 CrossRef CAS PubMed.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- M. Repoux, Surf. Interface Anal., 1992, 18, 567–570 CrossRef CAS.
- G. Yan, Y. Tang, Y. Li, Y. Li, L. Nguyen, T. Sakata, K. Higashi, F. F. Tao and P. Sautet, Nat. Catal., 2022, 5, 119–127 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Structural characterization data (TEM, BET, XPS, and XAFS), TPR/TPO data, in situ XANES data, EXAFS data after TPR/TPO, and catalytic reaction results. See DOI: https://doi.org/10.1039/d4cp01432d |
| This journal is © the Owner Societies 2024 |