
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplications of all-inorganic perovskites for energy storage
Ziyang
Jia
ab,
Caipeng
Cheng
a,
Xi
Chen
a,
Lili
Liu
 a,
Rui
Ding
*b,
Jilei
Ye
a,
Jing
Wang
a,
Lijun
Fu
a,
Yuhui
Cheng
a and
Yuping
Wu
*ac
a,
Rui
Ding
*b,
Jilei
Ye
a,
Jing
Wang
a,
Lijun
Fu
a,
Yuhui
Cheng
a and
Yuping
Wu
*ac
aSchool of Energy Science and Engineering, Nanjing Tech University, Nanjing 211816, China. E-mail: wuyp@fudan.edu.cn
bSchool of Chemistry, Xiangtan University, Xiangtan 4100XX, China. E-mail: drm8112@xtu.edu.cn
cSchool of Energy and Environment, Southeast University, Nanjing 210096, China
First published on 22nd October 2022
Abstract
In recent years, electrode materials of perovskite structure with controllable properties and structural advantages have been widely studied in the field of electrochemical energy storage. In this review, the research progress and application potential of a series of novel all-inorganic perovskite electrode materials in the fields of batteries and supercapacitors are reviewed. Strategies to modulate perovskite materials are discussed, including tailoring chemical composition and synthesis methods, controlling crystallinity and morphology, surface/interface defect engineering, and elemental doping. In addition, the correlation between the microscopic properties of the materials and the macroscopic electrode performance is discussed. Furthermore, some directions are pointed out.
 Yuping Wu | Dr Yuping Wu FRSC, is a Distinguished Professor of Nanjing Tech University and Southeast University. He got his PhD degree from the Institute of Chemistry, CAS, in 1997. In 2003, he was promoted to full professor at Fudan University, China. In 2015, he moved to Nanjing Tech University and shifted to Southeast University in 2021. He has published over 390 papers in peer-reviewed journals with H-index >89, was awarded the World's Most Influential Minds (2015) by Thomson Reuters, and Albert Nelson Marquis Lifetime Achievement Award (2019), and is one of the Highly Cited Researchers in the world. He has made pioneering research work on ARLBs, gel-type and pore-free separators for lithium batteries with high safety, and hybrid capacitors. |
1. Introduction
As the need to address environmental pollution and the energy crisis becomes more pressing, it is critical to identify new renewable energy materials/resources and to create superior clean energy utilization/conversion/storage technologies. Solar energy and wind energy will be significant pollution-free energy sources in the upcoming years, but due to their intermittent and uneven distribution, they should be combined with efficient energy storage technology for practical use. Electrochemical energy storage systems (EES) have attracted significant attention and research interest as they can harvest sustainable and renewable energy for important applications such as electric vehicles, electronic communication devices, and backup power sources for home use. They store and release energy by turning chemical energy into electrical energy and reversing the process, which is associated with electron and ion transport in electrode materials.1–3 The ideal EES device should feature high energy density, high power density, long cycling life, environmental friendliness, etc. To achieve this goal, a series of new rechargeable energy storage devices and new electrode materials have emerged and been widely studied.4–6Every significant advancement in electrochemical energy storage technology has been linked to the discovery and enhancement of electrode materials. The discovery of LiCoO2 and graphite has led to the marketization of Li-ion batteries;7 the discovery of LiFePO4 marked the arrival of a new era of Li-ion batteries;8 activated carbon (AC) has facilitated the application of higher power density devices;9 and the concept of pseudocapacitance was first introduced by the investigation of the electrochemical energy storage capacity of RuO2 materials.10 Consequently, finding and creatively designing new electrode materials and tuning their physical, chemical, and electrochemical properties are essential to achieve more efficient electrochemical energy storage.
At the same time, the physical and chemical properties of electrode materials mainly affect the energy storage capacity of electrochemical energy storage devices. In practical applications, the key issues that need to be addressed for electrode materials are specific capacity, kinetic behavior affected by ion and electron transfer, and structural stability in electrochemical reactions.11 The preparation and optimization of the materials usually start with the selection of elements, structures, and morphologies. In terms of element selection, higher energy storage generally benefits from transition metal elements that can undergo multiple electron transfers.12 For structure, high stability usually is related to certain structural features, such as dense rows of anionic skeletons occupied by cations in tetrahedral and octahedral sites.13 With respect to the morphological composition, different morphologies change their electron and ion transport ways/lengths, interfaces between electrolytes and electrodes, etc.14 Electrode materials such as carbon and carbon derivatives, metal oxides/hydroxides/halides, conducting polymers and MXene are now widely studied as key components in electrochemical energy storage devices.
Unlike the common electrode materials with crystal structures such as layered, spinel, olivine, and calcite,15–17 perovskites have been recognized as a leader among inexpensive and highly photochemically and electrochemically active materials for electrochemical energy applications with their robust open frame, intersecting tetragonal cavity chains and three-dimensional diffusion channels.18 In terms of the physical properties of the material itself, perovskite materials have high crystalline quality, facile and controllable preparation technology as well as excellent electron transfer capability, thus exhibiting excellent electrical conductivity and ion mobility rate, high charge storage capacity, and notable catalytic performance in the application of electrochemical energy storage devices.19–22
CaTiO3, which features a special structure and properties, was first discovered in 1839 and was named perovskite in honor of the mineralogist Lev Perovski. With the discovery of more perovskite materials, perovskite refers to all substances that have the general molecular formula of ABX3 and have a crystal arrangement similar to CaTiO3.23,24 The structure of ABX3 perovskites is usually based on a cubic array of corner-sharing BX6 octahedra, where the A-site cations are located within the cubic octahedral cavities. Another way to represent the structure is a cubic dense array of AX3, where the B-site cations are located within the octahedral holes. The A-site is a cation or an organic group, usually coordinated to 12 X-site anions, and the B-site cation coordinated to 6 X-site anions has a smaller radius. Most of the existing elements can be doped into the perovskite structure, resulting in an increasing number of compounds with perovskite structures.21
The tolerance factor (t) is an important parameter to measure the characteristics of perovskites at the structural level.25,26 Currently, the most accepted one is the Goldschmidt tolerance factor t, t = (rA + rX)/√2(rB + rX), where rA, rB and rX are the cation/anion radii of different sites in the perovskite structure, respectively. The calculated value of the tolerance factor can be used to determine the inter-cavity match between the A-site cation and the cavity in BX3. When the value of t is in the range of 0.8 to 1, a perovskite is usually formed. Likewise, tolerance factors have been very successful in describing and predicting the stability of oxide and fluoride perovskites. The higher the electronegativity of the X-site is, the stronger the bonds in the structure are, making the predictions of the hard-sphere model more successful.27 The ideal perovskite-type ABX3 structure has cubic symmetry with the space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. In addition to the primary perovskite structure ABX3, there are also some structures related to the A2BB′O6 double perovskite, in which the angles in the crystal structure share BO6 and B′O6 units in an ordered rock salt-like arrangement, and the B–O–B angles and B–O bond lengths in the structure determine the structural properties. In contrast, anti-perovskites, whose cation and anion components are in opposite positions in the cell structure to normal calcite, have superconductivity.28,29 A range of different impressive material characteristics, including ferroelectric, pyroelectric, magnetic, catalytic, photovoltaic, electronic conduction, ionic conduction, mixed conduction, and superconducting properties, can be generated by tuning the composition and structure of perovskite compounds.30–34 Therefore, perovskite materials have a variety of potential applicability as a result of these features, which is encouraging their developments for energy storage devices.
m. In addition to the primary perovskite structure ABX3, there are also some structures related to the A2BB′O6 double perovskite, in which the angles in the crystal structure share BO6 and B′O6 units in an ordered rock salt-like arrangement, and the B–O–B angles and B–O bond lengths in the structure determine the structural properties. In contrast, anti-perovskites, whose cation and anion components are in opposite positions in the cell structure to normal calcite, have superconductivity.28,29 A range of different impressive material characteristics, including ferroelectric, pyroelectric, magnetic, catalytic, photovoltaic, electronic conduction, ionic conduction, mixed conduction, and superconducting properties, can be generated by tuning the composition and structure of perovskite compounds.30–34 Therefore, perovskite materials have a variety of potential applicability as a result of these features, which is encouraging their developments for energy storage devices.
According to the different types of constituent ions, perovskite materials are mainly divided into organic–inorganic hybrid perovskites and all-inorganic perovskites. Organic–inorganic hybrid perovskites have shown promising applications in solar cells with the highest certified efficiency, exceeding 25%, but that of all-inorganic perovskites exceeds only 18%.35 Nevertheless, as one of the most important material families in condensed matter physics and materials science, all-inorganic perovskites have also attracted extensive research interest in the field of electrochemical energy storage in recent years. Based on the improved structural and integrated properties of perovskite materials, here recent advances in energy storage devices based on all-inorganic perovskite materials (organic groups are not included in the composition of perovskite compounds) are reviewed (Fig. 1), e.g., within the areas of lithium/sodium/potassium ion batteries, Li–O2 batteries, zinc–air batteries, zinc ion batteries, and supercapacitors. Tailoring tools commonly used in the application of perovskites in a variety of energy storage devices are also discussed, including tuning chemical makeup and processing techniques, controlling crystallinity and morphology, and engineering surface/interface defects, as well as the exploration of the correlation between microscopic material properties and macroscopic device performance.
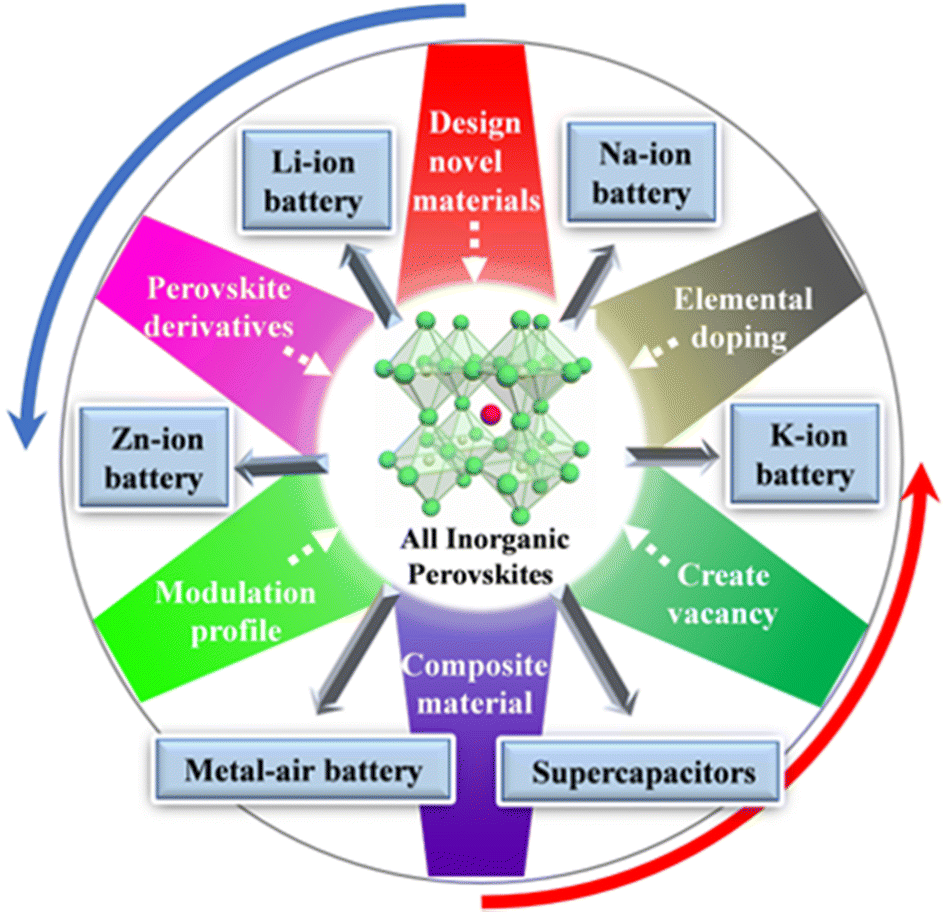 | ||
| Fig. 1 Modulation methods of all-inorganic perovskite electrode materials and the applied energy storage fields. | ||
2. All-inorganic perovskites for lithium-based energy storage devices
Li-ion-based energy storage devices typically consist of cathodes and anodes for ion storage and electrolytes for ion transport. Designing materials suitable for ion storage/transfer is critical for Li-ion batteries.36 Electrode materials are responsible for reversibly storing certain forms of lithium, either as ions that fit into interstitial sites or by intercalation into the crystal structure, or undergo conversion reactions with lithium to form a series of compounds.37 The electrode material should ensure efficient ion and electron transport. During discharge, the anode releases lithium ions through the electrolyte to the cathode. This causes electrons to flow from the anode to the cathode through an external circuit, and the current powers external loads such as cell phones and laptops. During charging, lithium ions move from the cathode to the anode through the electrolyte. The voltage is determined by the chemical potential difference between the anode and the cathode.38Common anodes for lithium-ion batteries (LIBs) typically rely on graphite, which possesses a theoretical capacity of 372 mA h g−1. To obtain higher capacities, silicon and its surface-treated derivatives are used to replace graphite electrodes because silicon has ten times the capacity of graphite. Nevertheless, silicon electrodes dramatically swell after a few cycles, leading to severe degradation of performance. In addition, graphene can also be used as an anode material, but has a higher cost and high initial irreversible capacity. Cathodes are typically made from lithium-containing materials such as LiCoO2, LiFePO4, and LiMn2O4, which typically provide storage capacities of 150–170 mA h g−1. Nickel- and manganese-based electrodes can be used to improve stability and reduce costs. Li-ion cathode materials should meet the requirements of being compatible with other components in the cell, providing high voltages associated with the Fermi energy difference between the two electrodes, and avoiding crystal structure collapse when large amounts of lithium ions enter and exit the cathode.39 Therefore, it is important to find new cathode and anode electrode materials.
In addition, the differences in the chemical activity of electrode materials and electrolytes lead to the formation of the SEI in liquid electrolyte batteries.40 The SEI is composed of two groups of components, one organic and the other inorganic. The organic components originate from the decomposition of solvents in the electrolyte such as esters and ethers.41 Inorganic substances are derived from the decomposition of salts and usually contain fluorides, oxides, and carbonates.42 Inorganic substances with thermodynamic stability, such as LiF, Li2CO3, and Li2O, are preferentially formed because they are compatible with lithium metal or graphite anodes in lithium/lithium-ion batteries.43 The chemical compatibility between the organic material and the native electrolyte improves the wettability, and in turn, the composition of the SEI will affect the electron transfer and ion transfer resistance in the cell. The anion in the lithium salt has a significant effect on the impedance of the SEI. Organic species are not considered to have a significant effect on SEI impedance, but their reversibility affects the specific capacity of the converted anode. In addition to the solute and solvent, the nature of the electrode material also affects the distribution and composition of the SEI.
2.1 ABX3 (X = Cl, Br, I) type perovskite halide
Due to the crystal structure characteristics and soft lattice properties of perovskite materials, they have good defect tolerance properties that allow more ions to be doped into the lattice without causing destructive damage.44,45 Likewise, perovskite halides have been used in the anode of lithium batteries because of their superiority in storing multiple lithium ions, which can migrate to the inside of the perovskite crystal to form LixABX3.46,47 Hence, they can be used as excellent materials for storing Li+. Simultaneously, the doping of Li+ shifts the Fermi energy level of the metal halide perovskite CsPbX3 (CsPbBr3) to the conduction band, which suggests that Li+ can act as a sender-type impurity in CsPbBr3 materials (Fig. 2a and b). CsPbX3 for Li-ion batteries has an unchanged crystal structure of the main body during cycling. This means that the perovskite halide can accept Li+ with topological intercalation without structural distortion (Fig. 2c and d). Of note, perovskite halides with different halogen species have widely diverse energy storage capacities, e.g., the halogen species affects the Li+ storage capacity of CsPbX3. Tufan et al. applied the all-inorganic CsPbBr3 perovskite as an active anode material for lithium-ion batteries, showing first charge/discharge capacities of ∼403 mA h g−1 and 376 mA h g−1, respectively. Voltage plateaus at around 0.6–0.72 V for charging and at around 0.72–0.98 and 1.59 V for discharging were observed at the current density of 30 mA g−1. These plateau voltages for charging–discharging are also consistent with the CV profile of the half-cells (Fig. 2e and f). The good rate performance suggested that CsPbBr3 does not present much capacity decay at different charging and discharging rates, indicating good structural stability of the CsPbBr3 perovskite as the anode for LIB applications (Fig. 2g). Likewise, the electrode of CsPbBr3 retained 79% of the original specific capacity of the button cell after 100 cycles at 60 mA g−1, and the Li+ absorption/adsorption process did not significantly affect the crystal structure of the perovskite (Fig. 2e–h).48 Additionally, a lithium-ion half-cell assembled with a gel polymer electrolyte and a CsPbCl3 anode by Pulak et al. had a specific capacity of 612.3 mA h g−1 at a current density of 50 mA g−1, with a Coulomb efficiency of 88%.49 Similarly, Nahid et al. prepared CsPbI3 as an anode material for lithium-ion batteries using a simple solution approach. The capacity of CsPbI3 decreased from 151 mA h g−1 to 149.8 mA h g−1 in the first six charge/discharge cycles and subsequently increased to 235 mA h g−1 in the 100th cycle at 40 mA g−1.50 The CsPbCl3 compound features a greater Li+ storage capacity.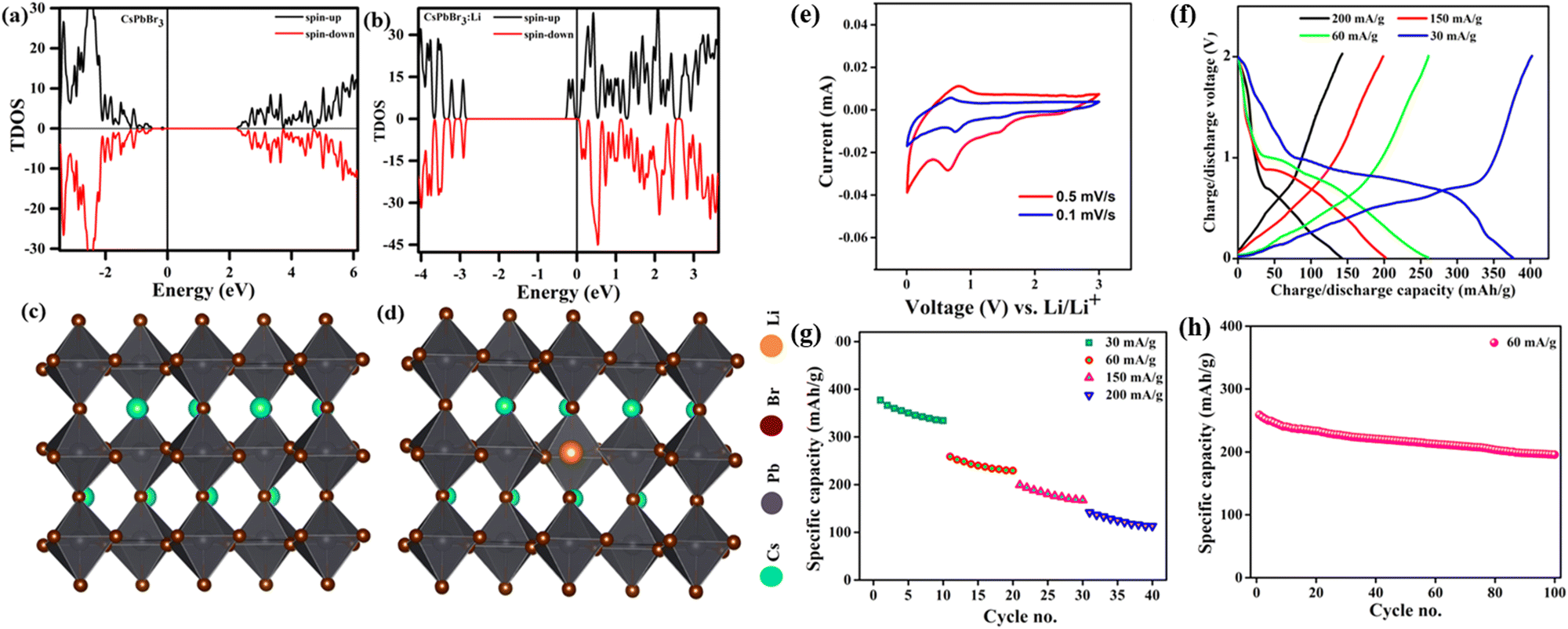 | ||
| Fig. 2 TDOS of CsPbBr3 without (a) and with Li+ (b). Side views (c and d). CV curves (e), GCD curves (f), specific discharge capacity (g), and cycling diagram (h) of CsPbBr3.48 Copyright © 2021 American Chemical Society. | ||
Generally speaking, hydrophobic carbon nanotubes (CNTs) can protect water-sensitive perovskite materials from the effects of moisture, thus improving the overall material stability under environmental conditions.51 More importantly, CNTs are regarded as an excellent conductive material and have been shown to decrease the charge transfer resistance of perovskites through doping.52 Liu et al. synthesized CsPbBr3@CNTs composites for lithium-ion batteries using a simple co-precipitation method. The electroactive materials with pseudocapacitance-controlled behavior exhibited high reversible capacity (644.6 mA h g−1 at 100 mA g−1), long cycle stability (470.2 mA h g−1 after 200 cycles at 100 mA g−1), and excellent rate capacity in LIBs.53
Nevertheless, the toxicity of lead-based peroxides may have a negative effect on further expansion of applications in energy storage. Materials with a double-perovskite structure, as a derivative based on the 113-type perovskite, have been widely used in recent years as an effective means to develop lead-free perovskite materials based on the replacement of Pb with monovalent and trivalent cations.54,55 The double-perovskite structure offers higher tunability and a wider range of elemental incorporation possibilities than the “113” perovskite structure. Therefore, lead-free double-perovskite materials doped with different elements at the B-site show a promising application prospect. Wu et al. showed the possibility of double-perovskite materials in lithium storage by synthesizing all-inorganic double perovskite Cs2NaBiCl6 as a new electrode material with a specific capacity of about 300 mA h g−1 and a coulombic efficiency of more than 99%. Nonetheless, the poor performance of Cs2NaBiCl6-based batteries, especially the poor cycling stability, limits their practical applications.56 Yang et al., for the first time, synthesized a rare-earth-based double perovskite Cs2NaErCl6:Li+ doped with a high concentration of lithium ions for LIBs by adjusting the elements doped at the B-site. Embedding of different concentrations of Li+ did not affect the crystal structure of the material (Fig. 3a and b). Benefiting from its excellent structural stability, Cs2NaErCl6 possessed relatively good cycling stability with a specific capacity of 120 mA h g−1 at 300 mA g−1 after 500 cycles, with a coulombic efficiency close to 100% (Fig. 3c–f). However, it can be seen that the decay of the specific capacity was severe in the first 20 cycles (Fig. 3c and e). The irreversible capacity of the first few cycles was mainly caused by irreversible side reactions and the formation of a solid electrolyte interface (SEI) film. Furthermore, the too high doping concentration of Li+ also changed the crystal structure, thereby limiting the Li+ migration and affecting the electrochemical performance. The addition of rare earth elements into the lead-free double perovskite opens up a way for the exploration of new promising anode materials for lithium storage applications.57
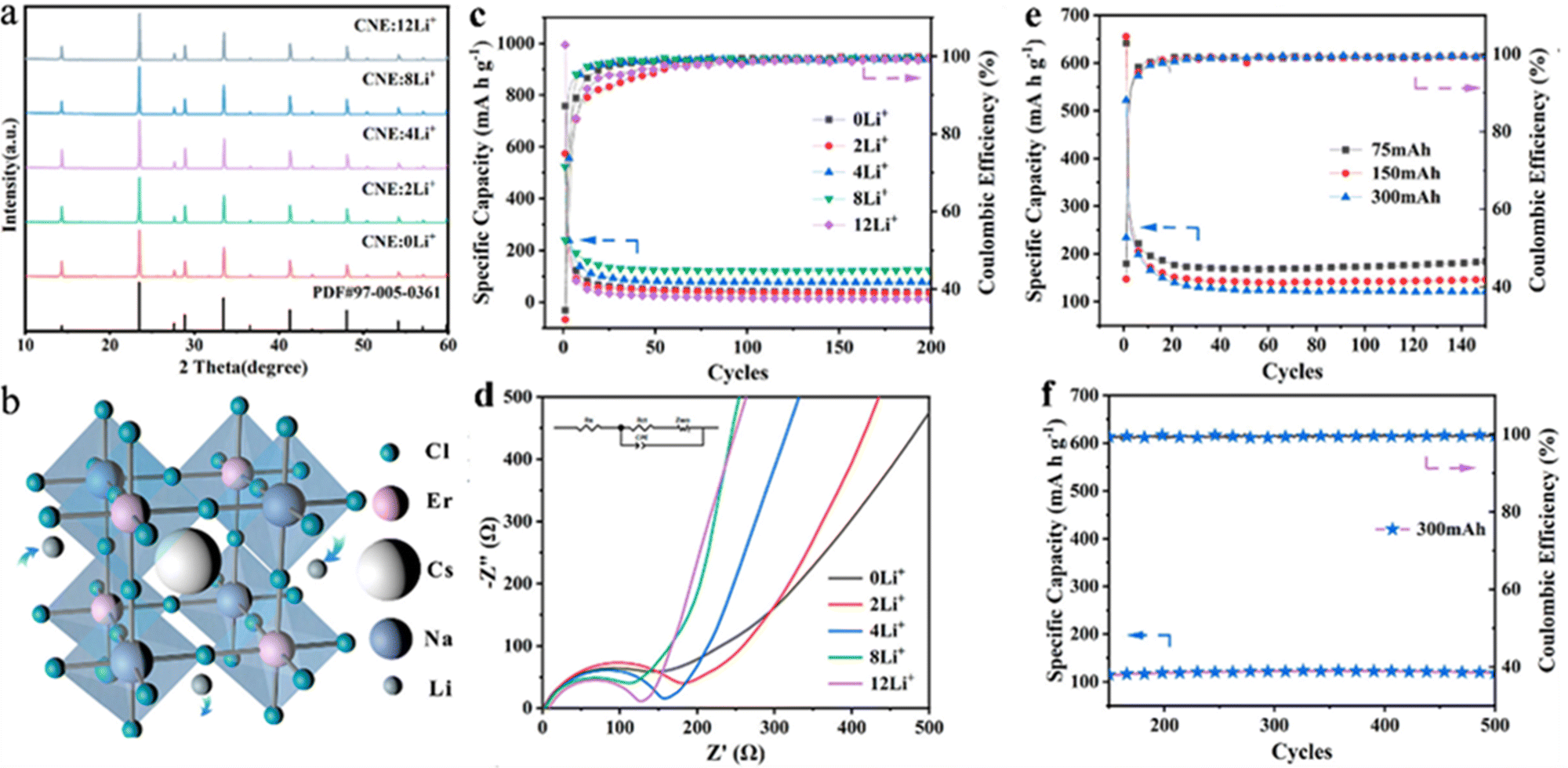 | ||
| Fig. 3 XRD patterns of Cs2NaErCl6 with various Li+ compositions (a). 3D structure patterns of Cs2NaErCl6 (b). Specific capacity (c) and EIS (d) of Cs2NaErCl6 anodes with different Li+ concentrations. Cycling performance of Cs2NaErCl6:8Li+ anodes in the initial 150 cycles at different charging current densities of 75, 150, and 300 mA g−1 (e) and the following 150–500 cycles at a charging current density of 300 mA g−1 (f).57 Copyright © 2022 American Chemical Society. | ||
2.2 ABF3 type perovskite fluoride
Unlike the above-mentioned perovskite halides, perovskite fluorides (ABF3) showed high redox potential, high energy density and good cycling stability due to the highly ionic nature of the M–F bond and the presence of the strongest electronegative F element. In recent years, ABF3 (A = K, Na, NH4+, etc.; B = Fe, Co, Ni, Mn, Zn, Cu, etc.) has drawn extensive research interest as an electrode material in the area of electrochemical energy storage.58–60 The structural advantages of perovskites can address some of the disadvantages of traditional transition metal-based materials in energy storage. For example, the channel size of the FeF3 backbone hindered ion diffusion, thus making long term storage of larger ions (e.g., sodium and potassium) difficult or impossible.61 AFeF3 (A = K, Na, NH4+, etc.) allows the direct insertion of lithium in the structure (after the first delocalization of the alkali metal and/or the NH4+ cation) and enables its circulation in larger alkali metal ions (e.g. sodium and potassium). For LIBs, perovskite LiFeF3 would be a suitable candidate, but it has not been synthesized due to its thermodynamic instability, decomposing into LiF and FeF2.62,63 Different elemental substitution at the A-site directly affects the tolerance factor of the perovskite structure, and in fact, NaFeF3 was shown to distort to an orthogonal symmetric structure.62,64 The major difference was also reflected in the application of Li+ storage.The NaFeF3//Li half-cell constructed by Andréa et al. retained a capacity higher than 200 mA h g−1 after 50 cycles at C/3 and higher than 180 mA h g−1 after 100 cycles at 1 C.59 The 3D stabilized channels are favorable to increase the migration rate of alkali metal ions in the framework of the perovskite structure. In addition, Martin et al. used a fluorinated sol–gel process to synthesize perovskite materials with different cations at the A-site (Fig. 4a). For example, KFeF3 is in the Pnma conformation after the insertion of Na+ and in the cubic conformation after the insertion of Li+, and the volume of its device remained nearly constant during a long charge/discharge period (Fig. 4b). This change in structure can explain the excellent rate performance (maintaining 70% of the theoretical capacity at 5 C), still with a capacity of 132 mA h g−1. The GCD curves of KFeF3, when cycled toward lithium, are shown in Fig. 3e. After deinsertion of the potassium ions characterized by a first slopped curve, a single clear plateau can be observed between 3.5 and 2.8 V. Unfortunately, the cycling performance of KFeF3 was far from ideal (Fig. 4g). NH4FeF3, as the cathode material, was observed to have a significant decrease in crystallinity during the initial decomposition of NH4+ ions (Fig. 4c). Fig. 4d displays galvanostatic charge and discharge cycles of NH4FeF3 toward Li. The first step was the deinsertion of the ammonium cations from the crystalline framework represented by a slopped plateau between 3.3 and 4.5 V versus Li/Li+. The theoretical capacity (Cth (NH4FeF3) = 206 mA h g−1)) was achieved at a current rate of C/2 with a relatively well-defined plateau around 3 V and a capacity retention of 94% after 100 cycles (Fig. 4f). However, the cubic structure was retained in the next cycles. The low crystallinity of NH4FeF3 cannot store ions like KFeF3 (only 50% of the theoretical capacity was retained at 5 C (Fig. 4g)). After the first insertion of Li+, its crystallinity was essentially unchanged and had spectacular capacity retention during cycling.65
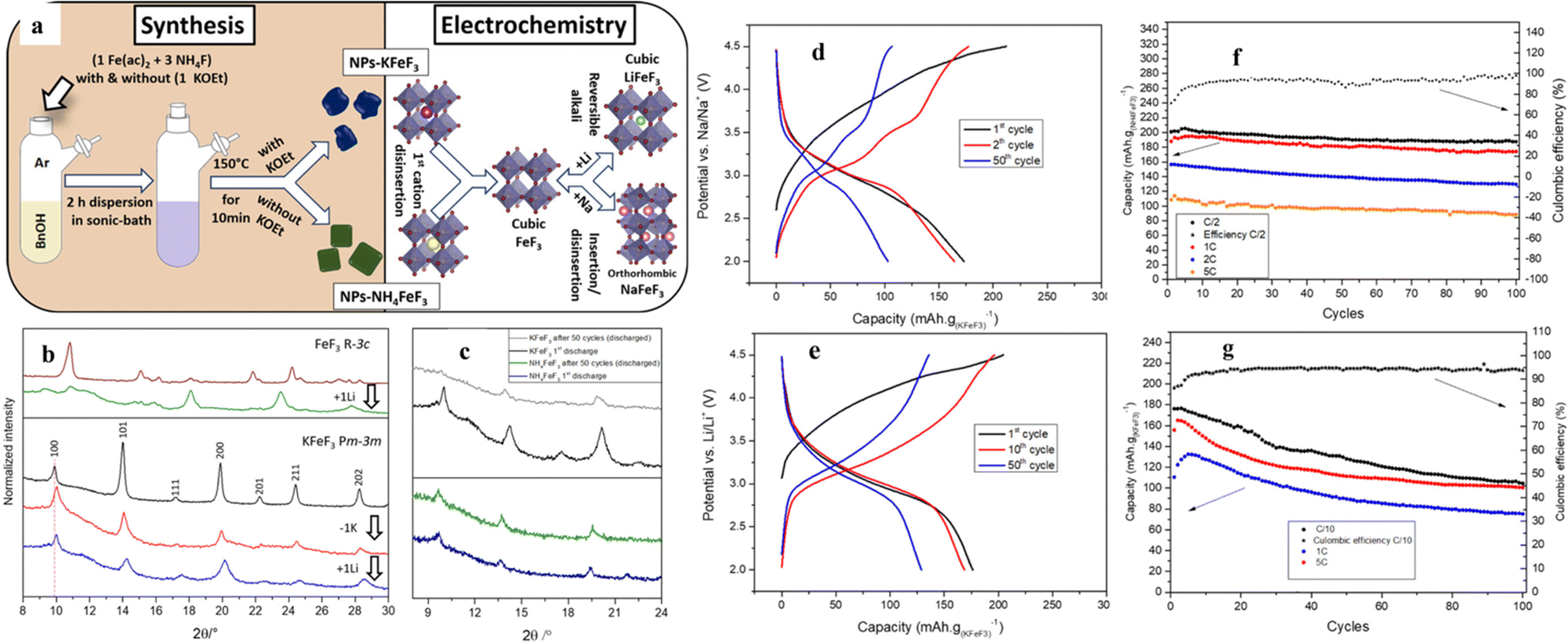 | ||
| Fig. 4 Synthesis roadmap and model diagram of K/NH4+FeF3 (a). Ex situ XRD of KFeF3 during charging and discharging (b). Comparison of ex situ XRD of NH4FeF3 and KFeF3 (c). GCD of NH4FeF3 at C/10 (d) and KFeF3 at C/2 (e) toward Li. Cycling performance of NH4+FeF3 (f) and KFeF3 (g).65 Copyright © 2019 American Chemical Society. | ||
A-site elements play a supporting role in the crystal structure, whereas B-site elements affect the reactivity and catalytic activity of perovskite-type compounds. Therefore, the modulation of B-site elements is necessary for electrochemical performance improvement for application in Li-ion batteries. Ying et al. synthesized a new cost-competitive KZnF3 (KZF) material and constructed an advanced Li-DIB. KZF displayed a conversion/alloying hybrid mechanism with pseudocapacitance-dominated kinetics for Li-ion storage, which differed from the previously described conversion/insertion hybrid process with pseudocapacitive characterization for Na0.85Ni0.45Co0.55F3.56.60 Due to the high natural reserves of Zn, KZF not only had the advantage of being inexpensive but also exhibited fast pseudocapacitive kinetic behavior and good cycling stability. Specifically, the KZF electrode can achieve a high capacity of 134 mA h g−1 at a current density of 100 mA g−1 and also maintain 136% of the initial capacity after 1000 cycles.66 Similarly, given the finite and high cost of nickel/cobalt resources, Huang et al. synthesized a tetragonal KCuF3 (KCF) material with lower cost as an advanced anode for Li+ storage devices, demonstrating the conversion mechanism and pseudo-capacitance kinetic properties. The KCF electrode had a specific capacity of 65.6 mA h g−1 at a current density of 100 mA g−1 and an excellent stability of 191% of the initial capacity after 1000 cycles.67 It can be seen that the different B-site elements in the perovskite structure had a significant effect on cost control, the lithium storage mechanism and management of electrochemical properties.
Generally, polymetallic candidates exhibited better behavior than the corresponding monometallic candidates, mainly due to the synergetic effects of polymetallic redox substances. For redox substances containing Ni, Co, Zn and Mn, Ni-containing substances can enhance the electrochemical activity, whereas Co-/Mn-based materials can contribute to electrical conductivity and structural stability, respectively. Accordingly, depending on the characteristics of the various elements, a compound composed of polymetallic elements would contribute to the overall performance of the material. Xu et al. optimized the performance as LIC and DIB anode for dual-ion batteries via tuning the Ni/Co ratio in the perovskite K–Ni–Co–F (denoted as KNCF). The optimal material, KNi0.1Co0.9F3, had a bulk nanocrystalline morphology with a hierarchical porous structure, thus showing the surface conversion mechanism for Li+ energy storage, in which Ni and Co electroactive substances had a synergistic effect on Li-ion storage.68 In addition, Yan et al. synthesized ABF3 materials (K0.97Ni0.31C0.34Mn0.35F2.97, KNCMF-111) doped by three elements of metal Ni, Co and Mn at the B-site and explored their electrochemical properties and energy storage characteristics in a Li+ storage device. During cycling, KNCMF-111 underwent conversion reactions and showed a pseudocapacitive kinetic behavior, exhibiting good kinetics and specific capacity, reaching a maximum capacity of 343 mA h g−1 at 100 mA g−1.69 Ying et al. synthesized KZMF(1-3)(K1.1Zn0.17Mn0.83F3.03) demonstrating pseudocapacitance-diffusion hybrid control kinetics with the conversion–alloying hybrid mechanism (Fig. 5a and b), which contributed to significantly improved rate performance and cycling stability, especially at high current densities (Fig. 5c and d). The KZMF(1-3) electrode exhibited a higher reversible capacity of 111–47 mA h−1 at 0.1–3.2 A g−1 and an excellent retention of 334% at 2 A g−1 for 1000 cycles. Therefore, KZMF(1-3) electrodes are expected to be promising low-cost candidates for lithium-ion and dual-ion energy storage because Zn and Mn elements are much more abundant than Ni and Co species.70
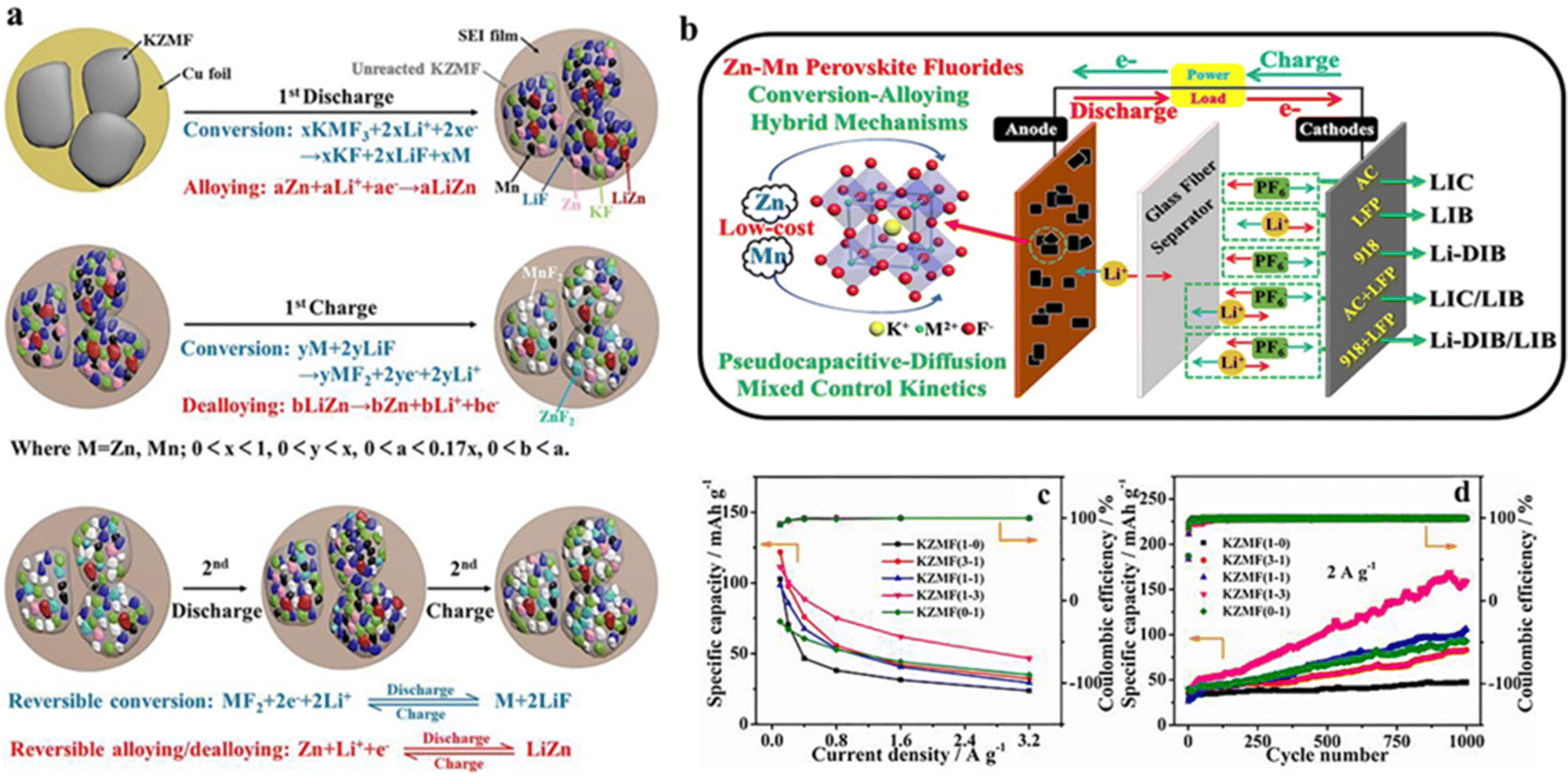 | ||
| Fig. 5 Reaction mechanism of the KZMF electrode (a). Schematic diagram of potential applications of KZMF for Li+ storage (b). Specific capacity of KZMF (c) and cycling performance (d).70 Copyright © 2020 Elsevier. | ||
To further promote the application of perovskite fluoride electrode materials in energy storage fields, the construction of composite materials in combination with other materials with complementary properties is also an effective means. For example, graphene has been widely used for electrochemical energy storage due to its excellent electrical conductivity, stability and flexibility. Ying et al. constructed electrode materials (KCMF(3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2)/rGO) consisting of Co and Mn element-doped perovskite fluoride anchored on graphene with robust heterogeneous nanostructures that exhibited fast kinetics of pseudocapacitive behavior in the storage of Li+.71 The electrochemical performance of the KCMF(3:2)/rGO electrode was better compared to that of KCMF(3:2) electrodes. And the KCMF(3:2)/rGO electrode had a reversible capacity of 288–106 mA h g−1 at a current density of 0.1–3.2 A g−1, which was twice that of bare KCMF(3:2) electrodes. With the introduction of rGO, the particle size and specific surface area of KCMF(3:2) become smaller and larger, respectively, contributing to the increase in electrode/electrolyte contact area. Second, rGO in KCMF(3:2)/rGO is also an active material that can store Li+. The introduction of particles at the nanoscale level can prevent aggregation between graphene sheets, which can expose more active surface area, which is beneficial to increase the overall capacity of KCMF(3:2)/rGO. The presence of rGO builds a conductive network in the composite, ensuring higher electron conductivity,72 reducing the internal resistance of the LIBs, thus resulting in higher reversible capacity and better rate capability.73
:2)/rGO) consisting of Co and Mn element-doped perovskite fluoride anchored on graphene with robust heterogeneous nanostructures that exhibited fast kinetics of pseudocapacitive behavior in the storage of Li+.71 The electrochemical performance of the KCMF(3:2)/rGO electrode was better compared to that of KCMF(3:2) electrodes. And the KCMF(3:2)/rGO electrode had a reversible capacity of 288–106 mA h g−1 at a current density of 0.1–3.2 A g−1, which was twice that of bare KCMF(3:2) electrodes. With the introduction of rGO, the particle size and specific surface area of KCMF(3:2) become smaller and larger, respectively, contributing to the increase in electrode/electrolyte contact area. Second, rGO in KCMF(3:2)/rGO is also an active material that can store Li+. The introduction of particles at the nanoscale level can prevent aggregation between graphene sheets, which can expose more active surface area, which is beneficial to increase the overall capacity of KCMF(3:2)/rGO. The presence of rGO builds a conductive network in the composite, ensuring higher electron conductivity,72 reducing the internal resistance of the LIBs, thus resulting in higher reversible capacity and better rate capability.73
The introduction of defects in the material is another strategy. The resulting increases in active sites, specific surface area and conductivity all contribute to the improved specific capacity.74 Cation vacancies have unique advantages for electrode materials in that they allow reversible embedding/de-embedding of ions in the crystal structure with little effect on the crystal structure, thus maintaining excellent performance at high rate and long cycling life.75 The vacancies of F− are new active sites that favor strong adsorption of Li ions and enhanced electrical conductivity.76 Shi et al. successfully synthesized Na0.85Ni0.45Co0.55F3.56 (NNCF) anode materials with sodium ion vacancies, and due to the presence of Na vacancies, lithium ions can be inserted into the NNCF electrodes dominated by the Ni sites, thus exhibiting excellent lithium ion storage behavior.60 Huang et al. successfully synthesized the first ternary Ni/Co/Mn perovskite containing K+ vacancies with excellent lithium ion storage performance (K0.89Ni0.02Co0.03Mn0.95F3.0, KNCMF-3#), exhibiting excellent overall performance and conversion/insertion mechanism dominated by pseudocapacitive behavior, which lead to the excellent rate and cycling performance. By forming composites with rGO, the specific capacity of the KNCMF-3# material was increased again, and KNCMF-3#@rGO exhibited excellent electrochemical performance (217 mA h g−1@100 mA g−1, 150%/1000 cycles@2 A g−1).58 Huang et al. also explored a perovskite anode for LIBs (K1.00Ni0.06Co0.14Mn0.80F2.92, KNCMF-3#) with an F− vacancy and a different Ni/Co/Mn doping ratio than before. The presence of the F− vacancy led to an increase in activity and conductivity. The change of the elemental doping ratio altered the synergistic effect and electrode cost. KNCMF-3# showed a conversion-type charge storage mechanism with excellent overall electrochemical performance.77
2.3 ABO3 type perovskite fluoride
As a metal oxide, perovskite oxides have been studied by many researchers due to their high electrical conductivity and accessible oxygen vacancies.78 In the case of ABO3-type perovskite oxides, their characteristics are that the B-site transition element can change its valence and improve the electrical conductivity. Mahata et al. found a significant increase in conductivity during the reduction of Mn4+ to Mn3+ in La(1−x)CaxMnO3, which is suitable as an electrode material.79 The materials with ordered structures in the A-site generally have outstanding thermal and chemical stability, high electrical conductivity, and oxygen dynamics.28 The ability to easily tune the structure of A+ and B+ cations brings tremendous improvements in the performance of perovskite oxides in various applications. On the other hand, perovskite structural versatility depends on the ability to accommodate a large number of oxygen vacancies in the ABO3−δ stoichiometry of the oxide. This is crucial for energy conversion materials because the emergence of oxygen vacancies enables oxygen to diffuse through the solid.80 In addition, the size and morphology of the nanocrystals are two elements that affect the electrochemical performances of perovskites. The quality of the structured nanocrystals, the presence of defects in the lattice, the A- and/or B-site doping of the perovskite lattice, the porosity of the nanocrystals and the presence of synergistic effects in the bifunctional morphology all play vital roles in the ultimate electrochemical behavior.It is an interesting work to explore perovskite oxides with multiple morphological features. Yan et al. synthesized NaNbO3 as the anode for lithium-ion batteries with bulk morphology (Fig. 6a), which can reach a reversible specific capacity of 130–55 mA h g−1 at the current densities of 0.1–3.2 A g−1.81 In particular, NaNbO3 can achieve a cycle retention rate of 185% after 1000 cycles (Fig. 6b). Unlike the bulk material, the flower-like nanostructured KTaO3 prepared by Sumedha et al. had a high interfacial area and a short charge transport length, which were the main reasons for exhibiting favorable electrochemical properties (Fig. 6c–f). It still had a high specific capacity of 256 mA h g−1 after 200 cycles at a current rate of 0.1 C and a high coulomb efficiency (∼99%). The material exhibited an excellent specific capacity of 74 mA h g−1 at high current rates (3 C).82
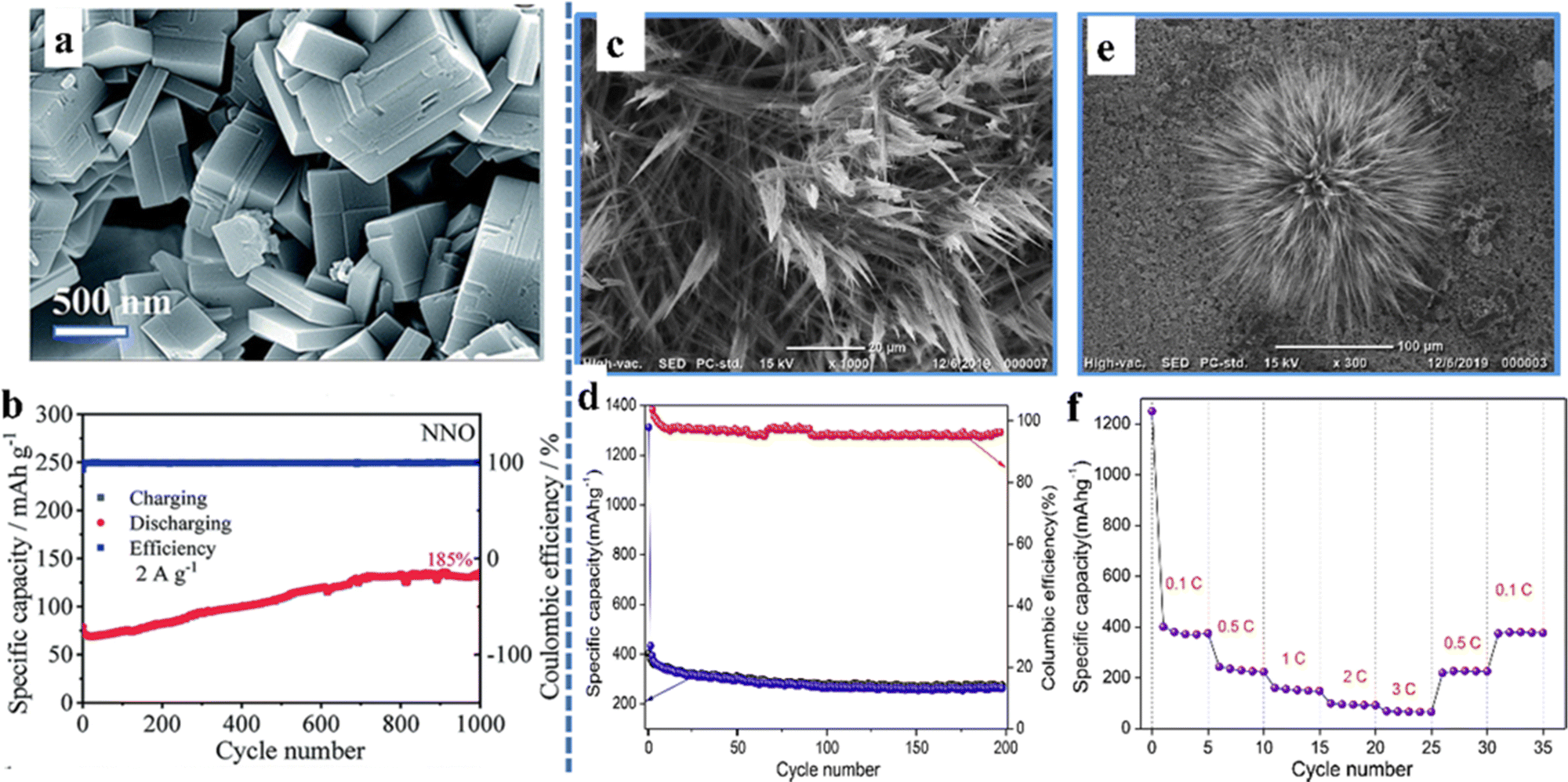 | ||
| Fig. 6 SEM micrograph (a) and cycling performance (b) of NaNbO3.81 Copyright © 2019 Royal Society of Chemistry. SEM micrographs (c and e), cycling performance (d) and rate performance (f) of nanostructured flowerlike KTaO3.82 Copyright © 2019 Elsevier. | ||
The design of new perovskite materials with special combinatorial arrangements by A- or B-site doping to boost their optical and electronic properties is also a means to promote the practical implementation of perovskite oxides in energy storage. Ko et al. first explored the Mn-doped Nd0.9Mn0.1FeO3 perovskite as the anode material with fascinating specific capacity in place of the common graphite anodes. The perovskite was prepared by a simple hydrothermal strategy and was structurally confirmed for the occurrence of manganese in its distorted octahedral structure. The introduction of manganese significantly accelerated the electron conduction of the material, thus shortening the ion/electron transport length and improving the electrochemical properties. The material had a reversible specific capacity of 763 mA h g−1 after 100 cycles at 0.5 A g−1.83 Similarly, Liu et al. synthesized SmFeO3 and Sm0.92Bi0.08FeO3 as electrode materials in lithium-ion batteries. The first discharge capacities of SmFeO3 and Sm0.92Bi0.08FeO3 electrodes were 450 and 550 mA h g−1, respectively, and their corresponding reversible capacities were 300 and 400 mA h g−1, respectively. It is very interesting to note that SmFeO3 still retained 98% of the discharge capacity of the previous cycle, while Sm0.92Bi0.08FeO3 seemed to lose capacity extremely quickly after the 20th cycle. Discharging resulted in partial loss of crystallinity, whereas charging slightly restored the crystallinity of SmFeO3 but not that of the Bi-doped SmFeO3. The lack of crystallinity restoration may be associated with the poorer rate properties observed in this doping solution.84
The defect engineering in perovskite oxides (ABO3) has been extensively investigated as a key factor affecting ion and charge transfer properties.85 The study of defect chemistry in these oxides has allowed us to tailor novel materials with suitable oxygen vacancy densities, mixed ionic/electrical conductivity, and other features. The oxygen defect concentration in perovskites can be substantially adjusted through doping with low and/or high valence cations at the B/A-site vacancies. As a consequence, perovskites containing transition metals facilitate the subsequent tuning of the structure to promote electrocatalytic and electronic processes.86 Yang et al. synthesized cationic defective perovskite CeNb3O9 (CNO) with particle size in the order of microns, which can store adequate amounts of lithium at high charge/discharge rates. At 60 C (15 A g−1), the CNO anode provided more than 52.8% of the theoretical capacity. Additionally, the CNO anode material had excellent long-term cycling stability at an ultrahigh rate of 50 C (12.5 A g−1). The superior cycling performance was ascribed to the formation of atomic short-range ordering, which significantly prevented the local and long-range structural rearrangements and stabilized the bulk structure responsible for small volume variation. The insertion sites, migration paths and the corresponding energy distribution of Li+ within the CNO structure were calculated by DFT (Fig. 7). The A-site cation vacancies in the Ce atomic layer provided more pathways for Li+ migration, increasing the dimensionality of long-distance diffusion to three dimensions. Therefore, the infinite three-dimensional Li+ migration paths within CNO promoted the diffusion of Li+ and produced excellent rate performance of the CNO.87
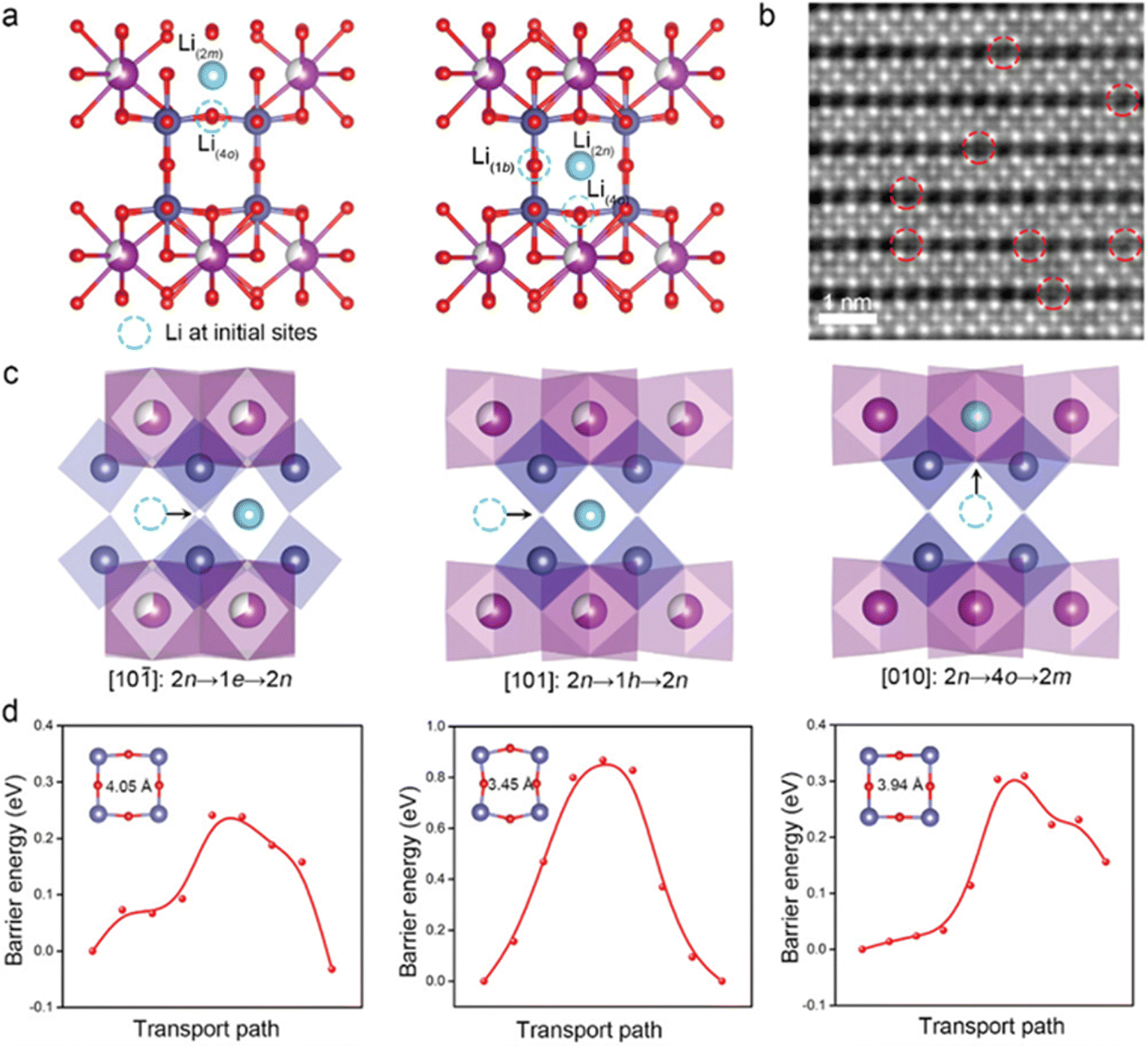 | ||
| Fig. 7 Li+ insertion point of CNO calculated by DFT (a). The iDPC image along the [101] direction after charging (b). The Li+ migration pathway (c) and the energy profile (d).87 Copyright © 2022 Wiley VCH. | ||
Gao et al. elucidated the characteristics of cation ordering in A-site defective perovskite single crystals of La(1−x)/3LixNbO3 (x = 0 and x = 0.04). Both materials were found to have a complex modulated crystal structure, which possessed two kinds of A-type cation orderings, i.e., long-range layer ordering in the alternating (001)p plane and short-range (in-domain) columnar ordering in the La-rich (001)p layer, respectively. It was also observed that even a small amount of Li substitution for La significantly affected the columnar arrangement, resulting in a suite of structural and even microstructural changes that may have detrimental effects on the lithium ion conductivity of the material.88
The construction of composites can also improve the conductivity and stability of perovskite oxides during lithium storage. Ganapathy et al. reported the excellent specific capacity and cycling behavior of perovskite BaSnO3 nanoparticles (NPs) decorated on two-dimensional graphene flakes. The prepared BaSnO3/rGO20 (20 wt% rGO) composite provided a maximum capacity of 1200 mA h g−1 at 0.5 C (650 mA g−1), which was far higher than that of the single BaSnO3 and graphene anodes.89 Qin et al. used few-layer Nb2CTx MXene (f-Nb2CTx) as a precursor and synthesized S-P-NNO/f-Nb2CTx hybrids on the basis of single crystal perovskite NaNbO3 nanocubes (S-P-NNO NCs) by a freeze-drying process. Profiting from the optimized composition and synergistic effect between S-P-NNO NCs and f-Nb2CTx, the optimal S-P-NNO/f-Nb2CTx nanohybrids exhibited a large reversible capacity of 157 mA h g−1 at 2.0 A g−1.90 The FeMnO3-CNTCN (carbon nanotube conductive network) was prepared using an electrostatic self-assembly strategy by Li et al., which showed splendid cyclability and rate performance.91 FeMnO3 microspheres with multi-step redox reactions can provide abundant active sites, while the highly conductive and flexible CNT substrate ensures fast lithium-ion transport and electron transfer. Table 1 summarizes the electrochemical properties of all-inorganic perovskite materials for lithium-based energy storage devices.
| Material | Electrolyte | Operating potential (V) | Specific capacity (mA h g−1/mA g−1) | Cycling behavior (%/cycles/mA g−1) | Ref. |
|---|---|---|---|---|---|
| CsPbBr3 | 0.5 M LiTFSI in BMIMTFSI | 0–2 | 376–142/30–200 | 79/100/60 | 48 |
| CsPbCl3 | 1 M LiBF4 in BMIMBF4 | 0.01–3.6 | 612.3–275.2/50–250 | — | 49 |
| CsPbI3 | 1 M LiPF6 in EC:DMC = 1:1 |
0.1–3 | — | 156/100/40 | 50 |
| CsPbBr3@CNTs | 1 M LiPF6 inEC:DMC = 1:1 |
0.01–3 | 644.6/100 | 73/200/100 | 53 |
| Cs2NaBiCl6 | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 |
0.01–2.5 | 775/75 | 38/25/75 | 56 |
| Cs2NaErCl6 | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 |
0–2.5 | 522/300 | 71/500/300 | 57 |
| K0.89Ni0.02Co0.03Mn0.95F3.0@rGO | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 |
0.01–3 | 217–97/100–3200 | 150/1000/2000 | 58 |
| Na0.85Ni0.45Co0.55F3.56 | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 |
0.01–3 | 286–115/100–3200 | 187/600/1000 | 60 |
| KFeF3; NH4FeF3 | 1 M LiPF6 in EC:PC:DMC = 1:1:1 |
2–4.5; 2–4.5 | 60/40 C (1 C = 176); 206/1/2 C | 63/100/C/10; 74/300/5 C | 65 |
| KZnF3 | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 |
0.01–3 | 134–33/100–3200 | 136/1000/1000 | 66 |
| KCuF3 | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 |
0.01–3 | 66–48/100–3200 | 191/1000/1000 | 67 |
| KNi0.1Co0.9F3 | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 |
0.01–3 | 166–73/100–3200 | 197/1000/1000 | 68 |
| K0.97Ni0.31Co0.34Mn0.35F2.98 | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 |
0.01–3 | 343–63/100–3200 | 80/400/1000 | 69 |
| K1.1Zn0.17Mn0.83F3.03 | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 |
0.01–3 | 111–47/100–3200 | 334/1000/2000 | 70 |
| KCo0.54Mn0.46F3/rGO | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 |
0.01–3 | 288–106/100–3200 | 169/1000/2000 | 71 |
| K1.00Ni0.06Co0.14Mn0.80F2.92 | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 |
0.01–3 | 240–110/100–3200 | 72/1000/2000 | 77 |
| NaNbO3 | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 with 1% VC |
0.01–3 | 130–55/100–3200 | 185/1000/2000 | 81 |
| KTaO3 | 1 M LiPF6 in EC:DEC:DMC = 2:1:2 |
0.01–3 | 401–102/0.1–2 C | 59/200/0.1 C | 82 |
| Nd0.9Mn0.1FeO3 | 1 M LiPF6 in EC:DEC:DMC = 1:1:1 |
0.02–3 | — | 86/50/500 | 83 |
| CeNb3O9(CNO) | 1 M LiPF6 in EC:DEC:DMC = 1:1:1 |
0.8–3 | 195–103/0.2–60 C (1 C = 250) | 96.6/200/1 C | 87 |
| BaSnO3/rGO | 1 M LiPF6 in EC:DEC = 1:1 |
0.01–3 | 1059/10 C | 1200 mA h g−1/100th/650 | 89 |
| S-P-NaNbO3/f-Nb2CTx | 1 M LiPF6 in EC:DEC = 1:1 |
0.01–3 | 398–157/0.05–2 | 170/1500/1000 | 90 |
| FeMnO3-CNTCN | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 |
0.01–3.0 | 1074–383/100–5000 | 83/1000/1000 | 91 |
3. All-inorganic perovskite for other energy storage batteries
3.1 Sodium ion battery
Considering the high price, safety issues, and poor lifespan of LIBs, researchers have started to explore other alternative energy storage systems that can achieve sustainable energy storage.92,93 Because of the low cost and abundant natural reserves of sodium metal, SIBs are regarded as a potential substitution for lithium-ion battery technology. Sodium is chemically comparable to lithium, because it is the second lightest alkali metal after lithium. In addition to these, sodium is available in the earth crust in various mineral and ore forms, which makes sodium metal cost-competitive compared to lithium metal. Since sodium–aluminum alloys cannot be formed in the operating voltage range of SIBs, aluminum foil can also be used as a collector for cathode and anode instead of the more expensive and heavier copper foil, further reducing the cost of SIBs. In addition, owing to the larger ionic radius of Na+ (1.02 Å for Na+ and 0.76 Å for Li +), the desolvation energy of Na+ in various organic solvents is lower, which can facilitate diffusion kinetics.94 Nevertheless, the lack of more suitable electrode materials hinders the practical application of SIBs, and it is necessary to explore new materials.Wang et al. synthesized polycrystalline ZnSnO3 as an anode material for SIBs, followed by a simple etching reaction to obtain hollow structures, which achieved a reversible capacity of 315 mA h g−1 at 30 mA g−1.95 Ammu et al. synthesized BiFeO3 (BFO) electrode materials with conversion and alloying mechanisms for Na+ storage, exhibiting intriguing specific capacity (770 mA h g−1).96 The use of perovskite based insertion-type hosts is a promising method to conquer the huge volume swelling due to alloying/de-alloying in the SIB anode. Bharathi et al. synthesized perovskite Na0.5Bi0.5TiO3 (NBTO) which exhibited high sodium storage capacity, fast sodium storage diffusion kinetics and splendid recyclability through alloying/de-alloying reactions. It also provided spectacular capacity (470–230 mA h g−1@100–230 mA g−1).97 NaFeF3 had the potential to be used as a cathode material in sodium ion batteries.98 Unfortunately, the electrochemical property of NaFeF3 was restricted even after the synthesis of nanostructured chalcogenides by complex chemical or mechanochemical methods, because the Na-filled channels are still not enough to provide fast (de)embedding.99 Equimolarly larger K can be used instead of filled Na to expand the lattice channels without causing degradation of the perovskite structure. Cao et al. proposed a pre-expanded calixarene iron fluoride (KFeF3) skeleton by filling it with the large size K+ cation as a channel filler. The FeF6 octahedra were not deformed during the electrochemical exchange between Na and K and Na insertion cycles, and the K+ filling can maintain a more “regular” cubic phase, where rapid diffusion occurred. The electrode achieved high rates of sodium storage with a reversible capacity of 110 mA h g−1 at 0.1 C.100 Furthermore, K+ vacancies can be viewed as another host site to accommodate the insertion of alkali metal cations (Na-ions), thus promoting the diffusion of Na-ions during electrochemical cycling. Tan et al. reported a new perovskite fluoride K0.82Co0.43Mn0.57F2.66@reduced graphene oxide (defined as KCMF(1-1)@rGO) nanoparticle with vacancy defects as an anode material, which showed a pseudocapacitance-dominated transition–insertion hybrid mechanism (Fig. 8a and b). The presence of defects in the structure can be seen by TEM and ICP-OES (Fig. 8c and d). The KCMF(1-1)@rGO anode showcased excellent specific capacity, rate performance and cycling stability (176–84 mA h g−1 at 0.05–1 A g−1, 67% retention@500 cycles and 0.3 A g−1) due to the synergistic effects of Co and Mn active species, fast pseudocapacitive kinetics, enhanced ion storage capacity of K ion vacancies and unique nano-heterogeneous structure (Fig. 8e and f).101
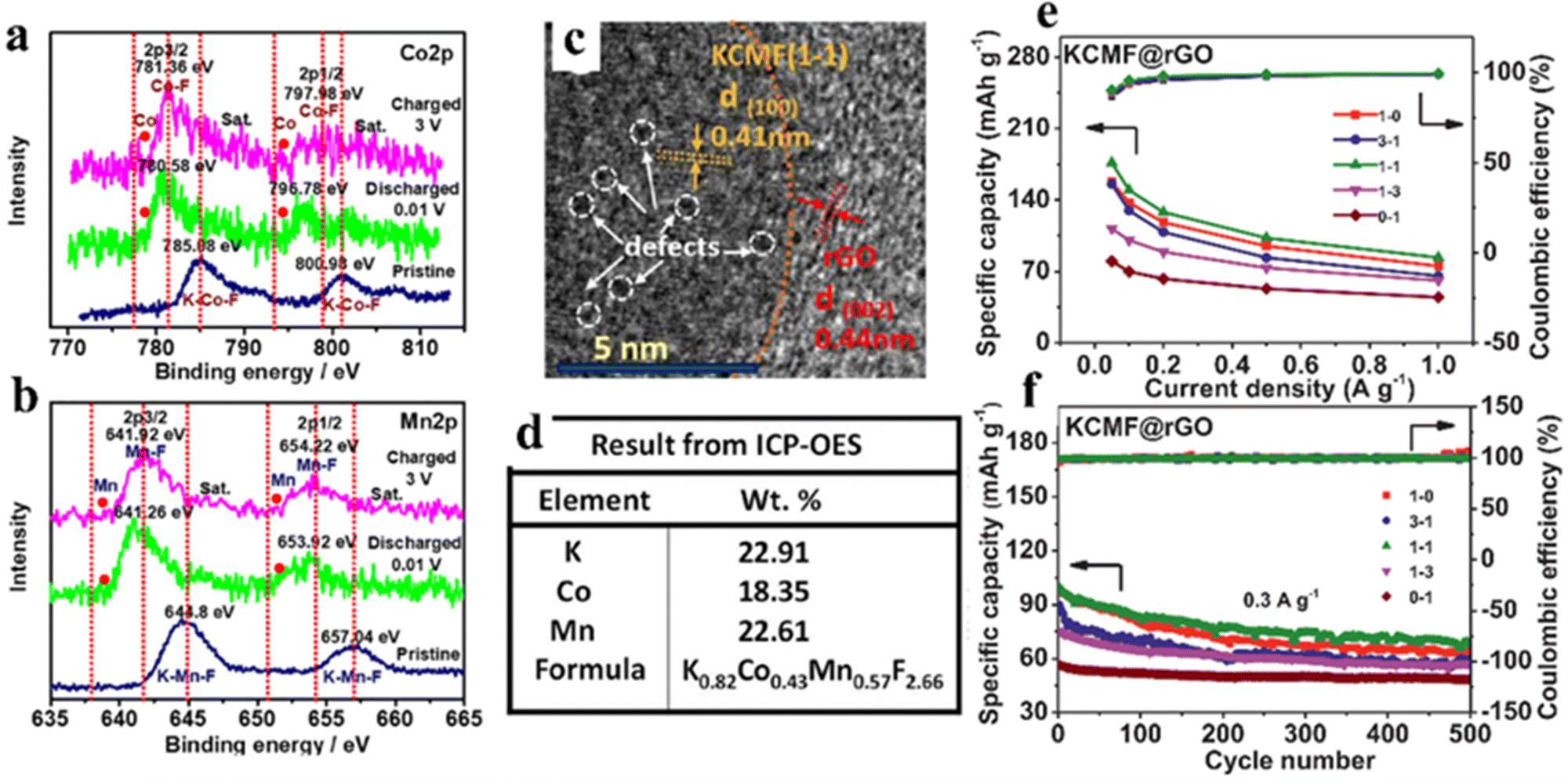 | ||
| Fig. 8 Co2p (a) and Mn2p (b) of KCMF(1-1)@rGO at different voltage points (ex situ XPS). TEM (c) and ICP-OES (d) of KCMF(1-1)@rGO samples. The capacity and coulombic efficiency of KCMF@rGO with different Co/Mn ratios were obtained in the fifth cycle (e). Cycling stability (f).101 Copyright © 2021 Elsevier. | ||
3.2 Potassium–ion battery
The scarcity, high price and the uneven geographical distribution of lithium resources seriously confine the sustainability of lithium-based batteries for large-scale energy storage purposes. Therefore, there is an urgent need to develop EES that can replace Li-ion batteries based on low budget and sufficient natural resources (e.g., Na and K).102 In recent years, potassium ion batteries (PIBs) have drawn much attention owing to the higher abundance, lower cost as well as lower standard redox potential of K+ than Li among the available energy storage technologies. The key to developing PIBs with excellent performance is to design cathode materials with reasonable microstructures to achieve ideal K+ embedding/detachment. Manganese-based materials can be employed as anode and cathode materials for various types of batteries because of their abundant reserves and environmental friendliness.103,104 As the cathode of PIBs, manganese-based fluoride electrode materials can overcome the drawback that manganese-based oxide materials (K0.3MnO2 and K0.5MnO2) cannot reach charging voltages greater than 4 V or higher.105 Manganese-based perovskite fluoride KMnF3 has attracted extensive research interest from scholars as a cathode material for PIBs.Poor electrical conductivity of manganese-based fluorides caused by high band gap is a common issue when employed in electrode materials.106,107 Improving the conductivity of fluoride by a range of approaches, such as doping and nanosizing, is an essential realm of research to enhance the electrochemical property of electrode materials. Oxide has a narrow forbidden band gap in comparison to fluoride, so doping a small amount of oxygen in fluoride can boost its conductivity to a larger degree.108,109 Meanwhile, the Bowling radius of O2− similar to that of F− (1.40 Å vs. 1.36 Å) makes O doping easier. Wang et al. synthesized O-doped KMnF3 as a cathode for PIBs by a carbon coating cladding and etching method (Fig. 9a). The doping of O atoms increased the conductivity and slightly reduced the migration barrier of K (Fig. 9b–e). The carbon coating served to prevent the direct contact of Mn-ions with the electrolyte, thus possibly hindering the dissolution of Mn-ions in the electrolyte. The carbon-coated KMnO0.125F2.875 electrode retained a specific charge capacity of 113.8 mA h g−1 and a specific discharge capacity of 103.6 mA h g−1 after 40 cycles (Fig. 9f and g).110
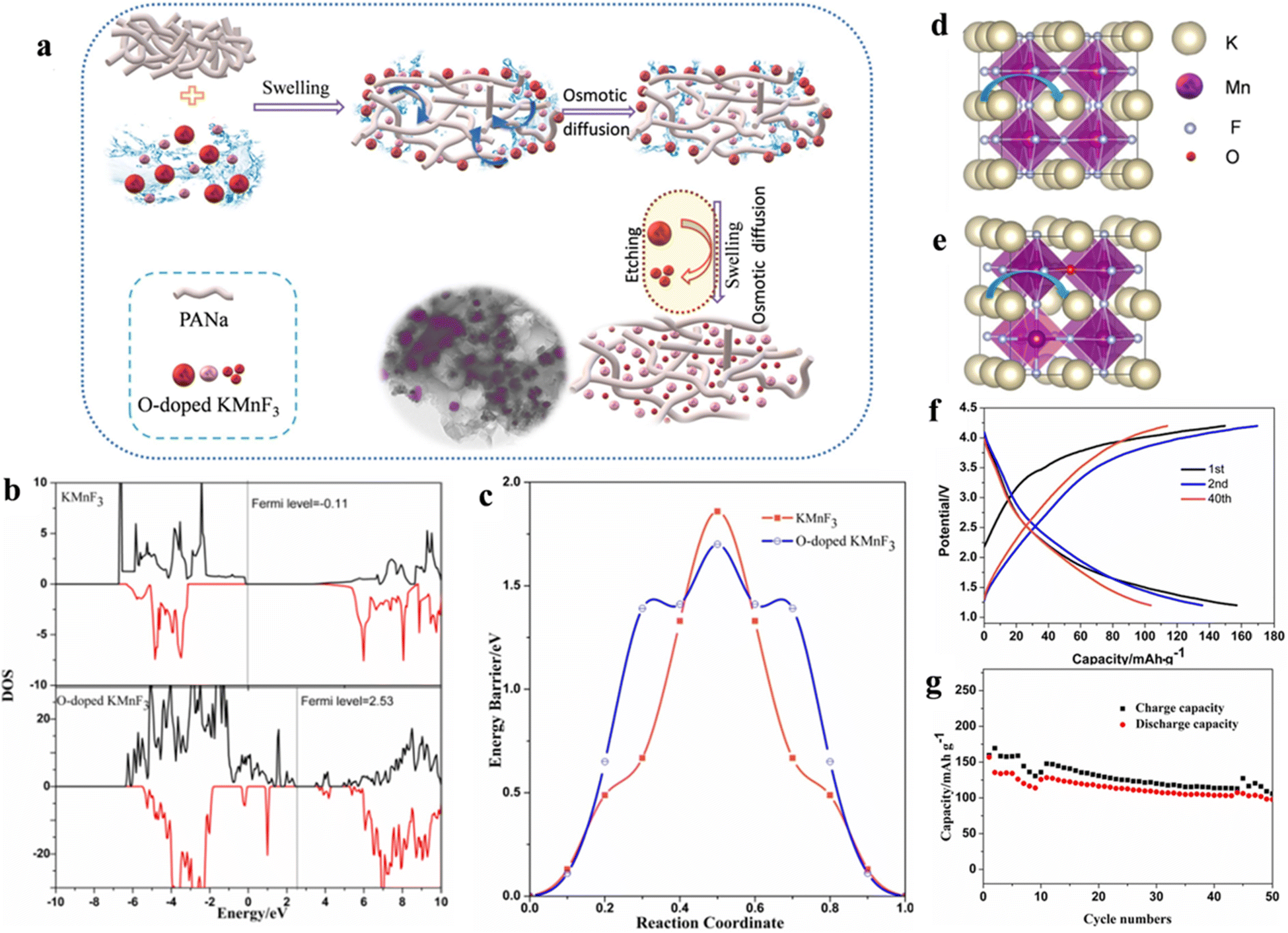 | ||
| Fig. 9 Schematic diagram of the synthesis process. (a) Charge/discharge curves (b) and cycling performance (c) of KMnO0.125F2.875. Spin polarization of DOS (d) and energy profile (e). Schematic diagram of K migration (f and g).110 Copyright © 2020 Elsevier. | ||
Wang et al. prepared Co-doped KMnF3 as a cathode material for potassium ion batteries by a co-precipitation method under mild conditions. By controlling the amount of cobalt doping, the dissolution of manganese could be effectively mitigated and the cycling behavior of the material could be boosted, so that the material could still reach 118.2 and 103.4 mA h g−1 of charging and discharging capacities after 60 cycles at 40 mA g−1.111 Wang et al. fabricated δ-MnO2/KMnF3-30 (KMnF3 mass%: 30%) with the multivalence manganese via a homogeneous precipitation method and used it as the cathode for PIBs wherein δ-MnO2 nanowires with different lengths can fully take advantage of short-range filling of short nanowires and cross-linking of long nanowires to produce more stable network connectivity.
Moreover, the homogeneous insertion of KMnF3 nanograins in δ-MnO2 nanowires can effectively mitigate the volume swing pertaining to ion embedding during the repeated cycling. The composite combines the advantages of Mn-based fluoride and oxide, and was used as an anode material, which displayed high capacity, good cycling and rate performances. Interestingly, the capacity can still be as high as 90 mA h g−1 after 200 cycles at 100 mA g−1.112
The low capacity and poor stability of PIBs, especially the much huge volume change in comparison to traditional Li-ion batteries, greatly hinder their rapid development. Liu et al. demonstrated a new etching method to construct a new zero-strain manganese-containing perovskite fluoride (KMnF-LE) with a hollow nanocube structure to improve the performance of the nanocube KMnF-L with respect to capacity, rate performance, and cycling behavior (Fig. 10a–e). The prepared KMNF-LE, rich in K/F vacancies, exhibited high capacity and ultrahigh stability, maintained its structure well during K+ embedding and delocalization (Fig. 10f), and showed a specific capacity of 110 mA h g−1 even after 10000 cycles (Fig. 10g and h).113
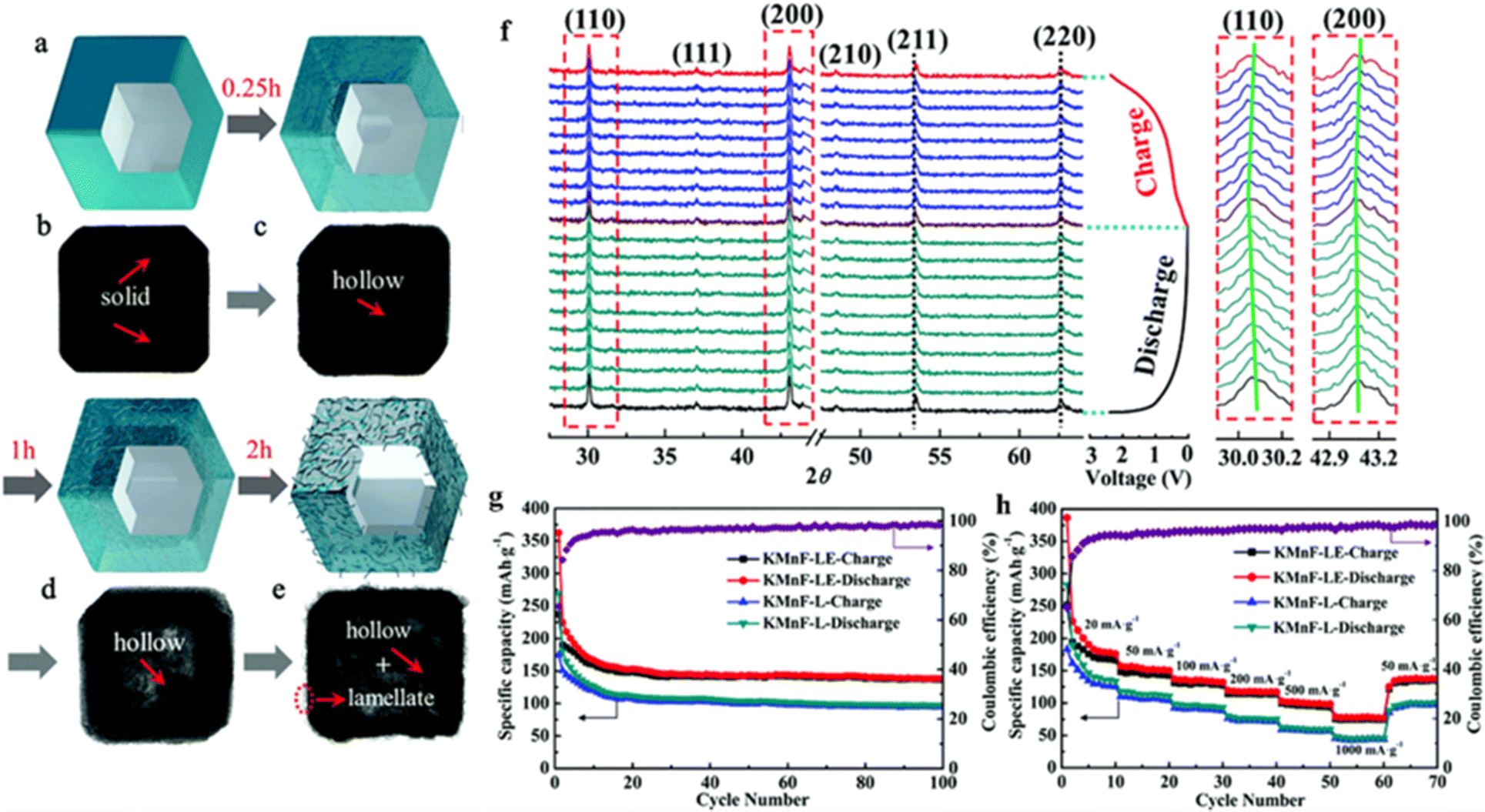 | ||
| Fig. 10 Synthesis and characterization of hollow KMnF-LE (a–e). In situ XRD patterns of KMnF-LE (f). Cyclic stability (g) and rate performance (h) of KMnF-L and KMnF-LE.113 Copyright © 2018 Royal Society of Chemistry. | ||
3.3 Zinc-ion battery
As a novel energy storage device with cost-effectiveness and environmental friendliness, aqueous zinc-ion batteries (ZIBs) have received broad interest from researchers.114,115 The cathode is an important component of ZIBs, and significantly affects the electrochemical performance of the batteries. Currently, the cathode materials used for ZIBs mainly include vanadium/manganese-based oxides, transition metal sulfides and Prussian blue analogs. Vanadium-based oxides are good in their capacity and cycling performance, but have a low voltage window and high toxicity. Transition metal sulfides are used as electrode materials in ZIBs owing to their layered structure and large layer spacing. However, the strong electrostatic interactions between Zn2+ ions and the primary structure lead to still unsatisfactory capacity and rate performance.116 Prussian blue materials are of interest because of their non-toxicity and low price. However, their capacity is low and rate capability is poor.117 Manganese-based oxides have high discharge voltage and specific capacity, but their conductivity is poor and active sites are insufficient.118 As such, exploring innovative cathode materials for ZIBs is an effective way to further enhance their application prospects.Zhang et al. synthesized a cathode material with perovskite structure of bimetallic oxides (NiMnO3) for ZIBs, which exhibited a high capacity of 300 mA g−1 at a current density of 280 mA h g−1 and an excellent cycling capability of 120 mA h g−1 even after 10000 cycles at high current density.119
Manganese-based cathodes are usually affected by manganese leaching and structural deformation, resulting in a continuous capacity decay during cycling. The by-products generated during the long cycling period will be deposited on the surface of the cathode materials, such as ZnO, Mn3O4, Zn(OH)2, and Mn(OH)2, which seriously affect the cycling life. Zhu et al. synthesized porous filamentous Ni-doped LaMnO3 as a new cathode material for weakly acidic aqueous ZIBs (Fig. 11a and b), which has a morphology similar to that of porous loofah-like particles of several micrometers in diameter. The synergistic effect of nickel doping and La3+ cation vacancies can significantly improve the electrochemical properties. The vacancy of La (rLa3+ = 1.15 Å) promoted the insertion/exaction of Zn2+ with a small ionic radius (rZn2+ = 0.74 Å), while the structure does not undergo deformation (Fig. 11c and d). The small amount of the Zn4SO4(OH)6·xH2O phase is associated with the side reaction of the electrolyte. The doping of Ni improves the electrical conductivity and hinders the dissolution of the active material. The LaNixMn1−xO3 perovskite exhibited better electrochemical properties than LaMnO3 with quick diffusion kinetics of Zn2+ (Fig. 11e and f). The prepared LaNixMn1−xO3 (x = 0.2) still showed a specific discharge capacity of 243 mA h g−1 after the 100th cycle at 0.1 A g−1.120Table 2 summarizes the electrochemical properties of the above-mentioned all-inorganic perovskite materials for K/Na/Zn-ion batteries.
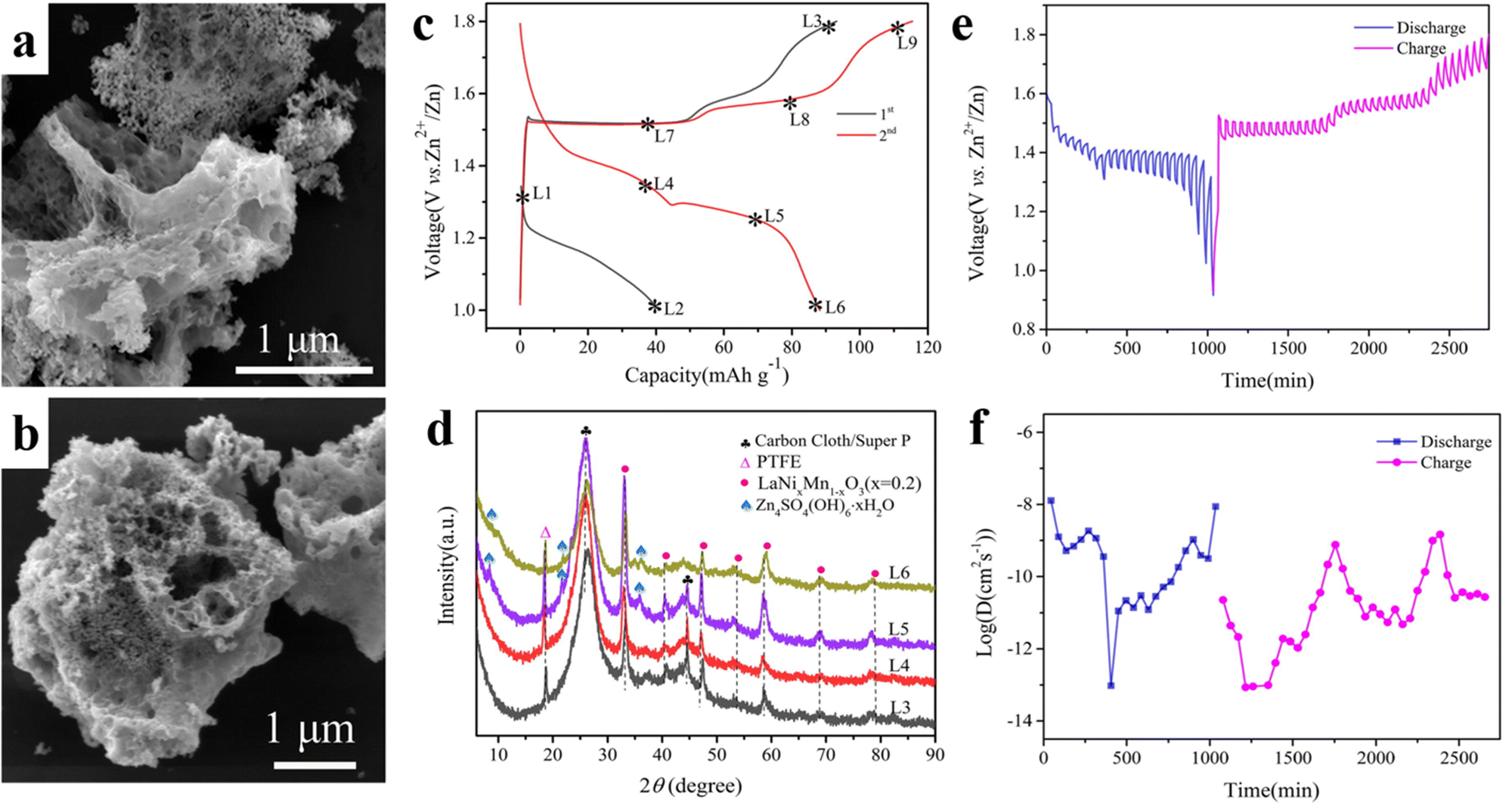 | ||
| Fig. 11 SEM of LaNixMn1−xO3 (x = 0.2) (a) and LaMnO3 (b). Charge/discharge curves (c), XRD at different voltages (d), GITT curves (e) and diffusion coefficients (f) of LaNixMn1−xO3 (x = 0.2).120 Copyright © 2022 Elsevier. | ||
| Material | Electrolyte | Operating potential (V) | Specific capacity | Cycling behavior (%/cycles/mA g−1) | Ref. |
|---|---|---|---|---|---|
| ZnSnO3 | 1 M NaClO4 in EC:PC = 1:1 |
0.01–3.00 | 315 mA h g−1 at 30 mA g−1 | 92/100/250 | 95 |
| Na0.5Bi0.5TiO3 | 1 M NaClO4 in PC with 2% FEC | 0.001–3 | 470–230 mA h g−1 at 100–250 A g−1 | 45.7/50/100 | 97 |
| NaFeF3 | 1 M NaClO4 in PC | — | 130 mA h g−1 at 0.2 mA cm−2 | — | 98 |
| KFeF3 | 1 M NaClO4 in EC:PC = 1:1 |
1.5–4.5 | 110–40 mA h g−1 at 0.1–10 C | — | 100 |
| K0.82Co0.43Mn0.57F2.66@rGO | 1 M NaPF6 in EC:EMC:DMC = 1:1:1 |
0.01–3 | 176–84 mA h g−1 at 0.5–1 A g−1 | 67/500/300 | 101 |
| Carbon-coated KMnO0.125F2.875 | 0.8 M KPF6 in EC:DEC = 1:1 |
1.2–4.2 | 113.8 mA h g−1 at 35 mA g−1 | 61 mA h g−1/200th/100 | 110 |
| K(Mn,Co)F3 (Mn:Co = 10:1) |
0.85 M KPF6 in EC:DEC = 1:1 |
1.2–4.2 | 170.5 mA h g−1 at 40 mA g−1 | 60.6/60/40 | 111 |
| δ-MnO2/KMnF3-30 | 0.85 M KPF6 in EC:EMC = 1:1 |
1.0–4.3 | — | 90 mA h g−1/200th/100 | 112 |
| K0.6Mn1F2.7-NC frame | 0.8 M KPF6 in EC:DEC = 1:1 |
0.01–2.6 | 182–78 mA h g−1 at 20–1000 mA g−1 | 110 mA h g−1/10000th/400 |
113 |
| NiMnO3 | 2 M ZnSO4 + 0.1 M MnSO4 | 0.8–1.8 | 291–152 mA h g−1 at 0.1–1 A g−1 | 236 mA h g−1/1000th/300 | 119 |
| LaNi0.2Mn0.8O3 | 2 M ZnSO4 + 0.2 M MnSO4 | 1.0–1.8 | 208–93 mA h g−1 at 0.1–3.2 A g−1 | 46.5/1000/500 | 120 |
3.4 Zinc–air battery
Rechargeable lithium-ion batteries are the most abundantly used rechargeable batteries on the market today, but their energy density (200–250 W h kg−1) is not high enough, thereby hindering their practical application in more fields. In recent years, the metal–air battery has emerged into the limelight with a theoretical energy density several times higher than that of LIBs, while the low cost, environmental friendliness and safety features further promote the metal–air battery to be one of the most promising devices among a variety of future energy storage devices.121 The high energy of the metal–air battery originates from the redox reaction between the metal anode and the oxygen in the cathode air. In general, the cathode has an open pore structure that facilitates the continuous supply/giving-off of oxygen.As an important type of metal–air battery, zinc–air batteries (ZABs) have received a high degree of attention. The zinc anode features not only low cost and abundant natural reserves, but also spectacular energy density by virtue of the participation of two electrons in the redox process.122 The energy storage process of ZABs is a reversible reaction: during the discharge process, the zinc anode undergoes an oxidation reaction to form Zn(OH)2, while the oxygen of the air participates in the reduction reaction to form OH−. During charging, zinc hydroxide is reduced to metallic zinc, while OH− is oxidized to form oxygen. As a result, the oxygen evolution reaction (OER) and the oxygen reduction reaction (OER) play critical roles in the energy storage process of ZABs, determining their overall performance and cost. However, the sluggish kinetic process has a detrimental effect on the power density and energy utilization efficiency of ZABs, thus making them fall short of their theoretical capacity. Therefore, the design and construction of high-efficiency electrocatalysts with excellent stability for OER and ORR is one of the most significant strategies in current research on ZABs.123
Although noble metals have high catalytic activity compared to other researched catalysts, their high cost and inability to simultaneously satisfy ORR and OER requirements make them a less desirable option. Carbon-based nanomaterials, which are among the non-precious metal catalysts, have a large specific surface area and strong electrical conductivity/mass transfer capabilities, yet they are gradually oxidized and corroded during OER. Other metal compounds enjoy bifunctional catalytic activity, but suffer from the shortcomings of low active sites as well as inferior electron and mass transport features.124 An ideal electrocatalyst should possess the advantages of a high density of active sites, a robust structure, good catalytic activity and high electrical conductivity.
In ZABs, perovskite materials like LaCoO3 display excellent catalytic activity. A number of studies have been carried out in the latest years to boost the overall performance of perovskite catalysts. The larger the specific surface area of the material, the greater the number of active sites that can participate in catalysis, which facilitates the enhancement of catalytic performance. Shim et al. prepared nanofibrous LaCoO3, which had a bigger surface area and more active sites than bulk LaCoO3, showing better catalytic activity.125 Ishihara et al. created La0.6Ca0.4CoO3 with a substantial percentage of mesopores as well as a high specific surface area. The addition of mesopores enhanced the catalytic stability of La0.6Ca0.4CoO3, and similarly, the potential of OER and ORR of La0.6Ca0.4CoO3 remained stable after 90 cycles at a current density of 20 mA cm−2.126
The electrical conductivity and electronic configuration of perovskite materials have an impact on the ability of electron conduction to the active site; hence the electrical conductivity of the material cannot be overlooked. Furthermore, defects in the structure of catalysts not only allow the fabrication of more edge structures to increase the active sites but also reduce the potential barriers of the rate-determining step to strengthen the catalytic activity. Ran et al. synthesized sulfur-doped LaCoO3 (S-LCO) catalysts for ZABs, where sulfur doping and the introduction of oxygen defects promoted the electrical conductivity and simultaneously optimized the filling state of the eg, thus enhancing the electrocatalytic performance of LaCoO3. When used as the cathode of ZABs, S-LCO had a power density of 92 mW cm−2, a high specific capacity of 747 mA h g−1 at 5 mA cm−2, and a long cycling life (100 h).127 Gui et al. demonstrated the oxygen vacancy-rich Ce0.9Gd0.1O2−δ (GDC) modified Pr0.5Ba0.5CoO3−δ (PBC) as a bifunctional catalyst for ZABs (Fig. 12a). The surface modification of GDC not only introduced abundant electrochemically active oxygen vacancies for ORR and OER (Fig. 12b and c), but also enhanced the structural stability of the perovskite. In contrast to the pristine PBC catalyst, the 20 wt% GDC decorated PBC catalyst delivered a higher signal intensity, denoting more surface oxygen vacancies after GDC decoration (Fig. 12b). The concentration of O22−/O− representing surface active oxygen vacancies increased from 24.86% on the pristine PBC catalyst to 35.96% for the 20 wt% GDC decorated PBC catalyst, fairly agreeing with the EPR result (Fig. 12c). From the calculation results, it can be seen that the presence of oxygen vacancies increased the adsorption energy of O2 and H2O on the surface of GDC, which was beneficial to the OER and ORR reactions (Fig. 12d–g). The ZABs assembled with this composite exhibited a maximum peak power density of 207 mW cm−2 and a significant durability of 200 h.128
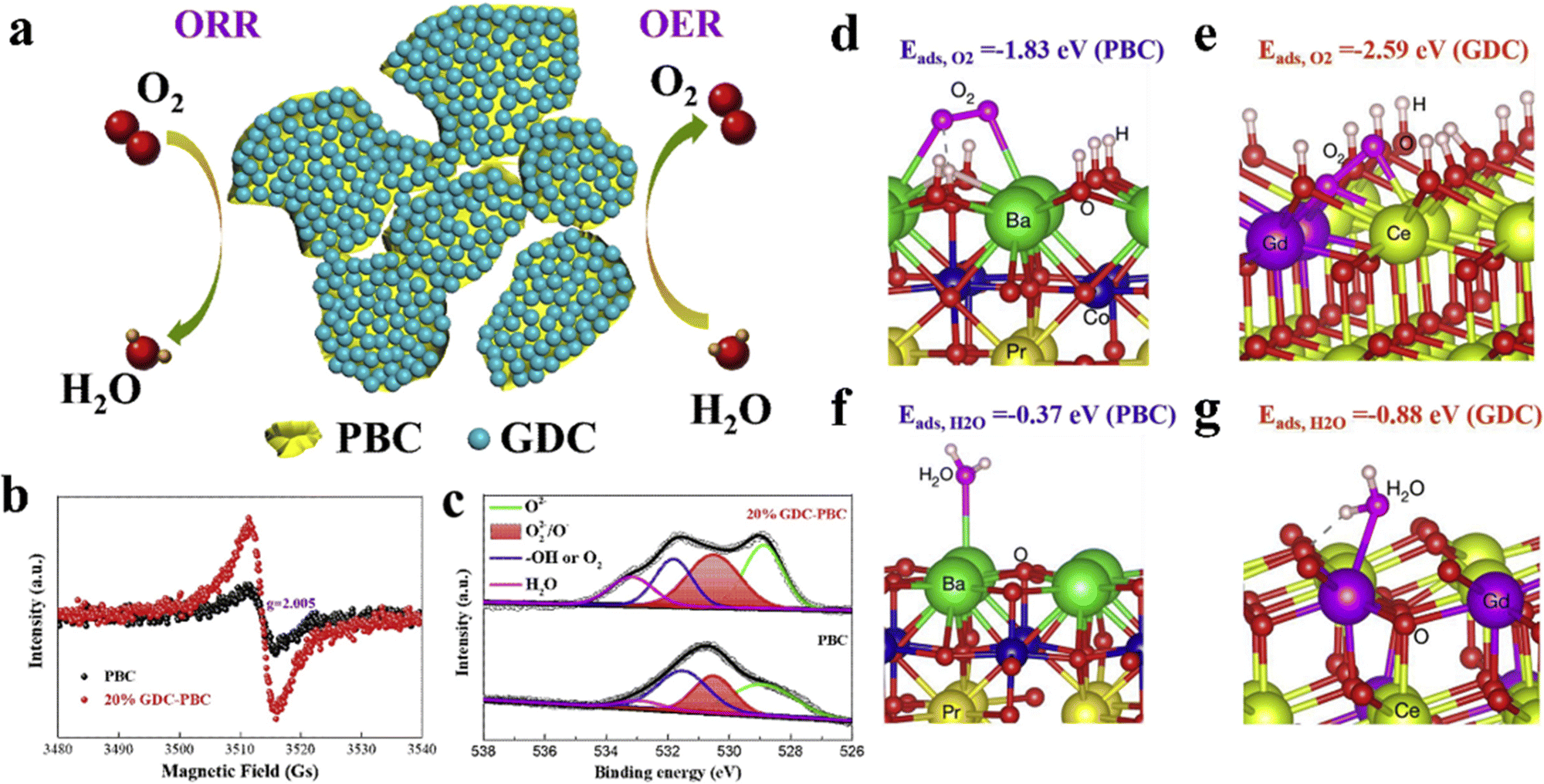 | ||
| Fig. 12 Schematic diagram of the GDC-modified PBC bi-functional catalyst (a). Comparison of the EPR spectra (b) and O 1s spectra (c) of PBC before and after GDC modification. Schematic diagram of adsorption of O2 and H2O molecules on PBC (d and f) and GDC (e and g).128 Copyright © 2021 Wiley VCH. | ||
The preparation of composites to overcome some of the drawbacks of a single material is one of the typical methods to improve the catalytic activity of perovskite catalysts. Especially in ZABs, catalysts are required to meet both ORR and OER high performance requirements. Generally, the design of suitable composites is a simple and efficient method. Considering that CoO can provide more suitable active sites for oxygen adsorption and boost the electrical conductivity, Zheng et al. prepared the composite electrocatalyst by introducing CoO onto LaMnO3. In particular, CoO solved the problem of poor OER performance of LaMnO3. The assembled ZABs based on the composite possessed a power density of 101.48 mW cm−2 and intriguing cycling stability.129 Wang et al. synthesized bifunctional composites (Pt-SCFP) with excellent electrochemical activity and durability based on Pt and perovskite Sr(Co0.8Fe0.2)0.95P0.05O3−δ (SCFP). Pt–O–Co bonds existed in the Pt-SCFP/C-12 samples from the ball milling method, and a large number of electrons were transferred from Pt to SCFP through these Pt–O–Co bonds, thus changing the electronic structure of Pt and SCFP and forming a strong electronic interaction between Pt and SCFP in the Pt-SCFP/C-12 composites. Meanwhile, the spillover effect also favored the enhancement of catalytic activity. The best sample, Pt-SCFP/C-12, exhibited impressive bifunctional activity for the oxygen reduction and oxygen evolution reactions with a potential difference of 0.73 V. Remarkably, the initial discharge and charging potentials of the ZABs based on this catalyst were 1.25 and 2.02 V at 5 mA cm−2, accompanied by excellent cycling stability.130
Furthermore, A2B′B′′O6 compounds can contain elements that are not possible in individual perovskites because they can allow the expansion of their composition space. Bhardwaj et al. investigated a new-type perovskite material Sr2TiMnO6 (STMO) for rechargeable ZABs. The constructed ZABs exhibited a maximum peak power density of 97 mW cm−2 and a cycling stability of 6.6 h cycles.131Table 3 summarizes the electrochemical properties of the above-mentioned all-inorganic perovskite materials for ZABs.
| Catalysts | Electrolyte | E 1/2 (vs. RHE) (V) | E OER (vs. RHE) | ΔE (V, EOER − EORR) | Maximum power density (mW cm−2) | Ref. |
|---|---|---|---|---|---|---|
| S-LaCoO3 | 6 M KOH and 0.2 M Zn(Ac)2 | 0.704 | 1.594 V/10 mA cm−2 | 0.89 | 92 | 127 |
| 20 wt% GDC-Pr0.5Ba0.5CoO3-δ | 6 M KOH and 0.2 M Zn(Ac)2 | 0.71 | 1.65 V/10 mA cm−2 | 0.94 | 207 | 128 |
| LaMnO3–CoO | 6 M KOH and 0.2 M Zn(Ac)2 | 0.56 | 1.78 V/10 mA cm−2 | 1.22 | 101.48 | 129 |
| Pt-Sr(Co0.8Fe0.2)0.95P0.05O3−δ/C-12 | 6 M KOH and 0.2 M ZnCl2 | 0.87 | 1.6 V/10 mA cm−2 | 0.73 | 122 | 130 |
| Sr2TiMnO6 | 6 M KOH and 0.2 M Zn(Ac)2 | 0.67 | 1.91 V/5 mA cm−2 | 1.24 | 97 | 131 |
3.5 Lithium–air battery
As another type of metal–air battery, the lithium–air (Li–O2) battery has also attracted research interest. Among the various types of Li–O2 batteries, the nonaqueous type is the most promising one. Its energy density is ultra-high, but all other electrochemical performance aspects are unsatisfactory, such as coulombic efficiency, rate performance, and cycling life.132 During the discharge process, O2 is reduced and reacts with Li+ (ORR-LO) and forms soluble Li2O2, which later decomposes into Li+ and O2 (OER-LO) during charging. Considering the difference from the Zn–air batteries, the oxygen reduction reaction and oxygen evolution reaction in the Li–air batteries in this review are denoted as ORR-LO and OER-LO, respectively. Slow charge transfer and insufficient catalyst activity lead to sluggish kinetics of ORR-LO and OER-LO, which in turn affect the electrochemical performance of Li–O2 batteries.133 Air cathodes with high surface area, excellent electrical conductivity, good electrochemical stability, and efficient bifunctional catalytic activity for ORR-LO and OER-LO are extremely important for high-performance Li–O2 batteries.ABO3-type perovskite oxide is a cost-effective and stable bifunctional cathode catalyst for Li–O2 batteries with tunable catalytic activity, good electrochemical stability, etc. Yang et al. designed a new graphene/LaSrMnO3 composite with a layered porous structure to address the porosity and conductivity of ABO3. The macropores between the nanosheets promoted the diffusion of oxygen molecules and lithium ions. In parallel, the mesopores facilitated the impregnation of the electrolyte and the deposition of products. LaSrMnO3 reduced the reaction overpotential, and graphene accelerated the electron transport capability. When used as the cathode of Li–O2 batteries, it exhibited high specific capacity, excellent rate performance and good cycling stability.134 Moreover, there is no doubt that the loaded metal nanoparticles favor the improvement of catalyst activity. Cong et al. proposed a new solution to obtain a perovskite co-decorated with CoFe alloy and Co metal on the basis of La0.8Fe0.9Co0.1O3−δ by varying the annealing temperature. Benefiting from the change of cation exchange behavior at different temperatures, the Co cations in the perovskite gradually exsolved onto the surface of nanoparticles, and the Fe atoms in the CoFe alloy gradually migrated back to the perovskite. The obtained composites demonstrated an excellent specific discharge capacity of 6549.7 mA h g−1 and a cycle stability of 215 cycles when used in Li–O2 batteries.135
In the structure of perovskite oxides, doping at the A-site may form O defects, which indirectly affects the ORR/OER activity of perovskite oxides. The doping of different ions in the B-site directly can influence the B–O–B angle, thereby increasing the intrinsic activity. Sung et al. synthesized Sr- and V-doped perovskite La0.8Sr0.2VO3 with a nanofibrous shape and investigated the kinetic behavior of the composed Li–O2 battery (Fig. 13). Doping Sr at the A-site may be accompanied by an increase in the number of oxygen vacancies. The variable valence states of vanadium ions (V3+, V 4+ and V5+) contributed to provide donor–acceptor chemisorption sites to facilitate ORR-LO/OER-LO. The doped electrode can maintain 253 reversible cycles at a current of 2000 mA g−1.136 The Li0.34La0.55MnO3-δ perovskites designed by Sang et al. exhibited high electronic (2.04 × 10−3 S cm−1) and lithium ion (8.53 × 10−5 S cm−1) conductivity in experiments, and when applied in the carbon-free cathode of lithium–air batteries, they showcased excellent reversibility at 0.21 mA h cm−2 over 100 cycles while avoiding the degradation related to carbon-containing materials.137 Li et al. introduced surface permeable nanolayers and oxygen vacancies on mesoporous hollow LaCoxMn1−xO3−σ perovskite catalysts by in situ cation replacement using surface structure and defect engineering strategies. The O2-electrode using the LaCo0.75Mn0.25O3−σ catalyst manifested an extremely impressive discharge capacity of 10301 mA h g−1 in the initial cycle at 200 mA g−1. The excellent electrochemical performance of LaCo0.75Mn0.25O3−σ may be owing to the synergistic effect of the graded hollow structure and the large amount of oxygen vacancies covered over the catalyst surface. The introduced oxygen vacancy can act as an effective substrate for the combination of intermediate products and decomposition of Li2O2 during discharge and charge protocols.138 Hou et al. demonstrated a surface reconstruction strategy on perovskite La0.8Fe0.9Co0.1O3-δ (LFCO) to prepare a LaF3/LFCO composite, which induces a modified surface and also manipulates the electronic structure.139 The optimized surface and electronic structure adjusted the discharge reaction pathway and formed thin petal-like F-doped Li2O2, which displays better conductivity and benefits Li–O2 battery performance. Table 4 summarizes the electrochemical properties of the above-mentioned all-inorganic perovskite materials for Li–O2 batteries.
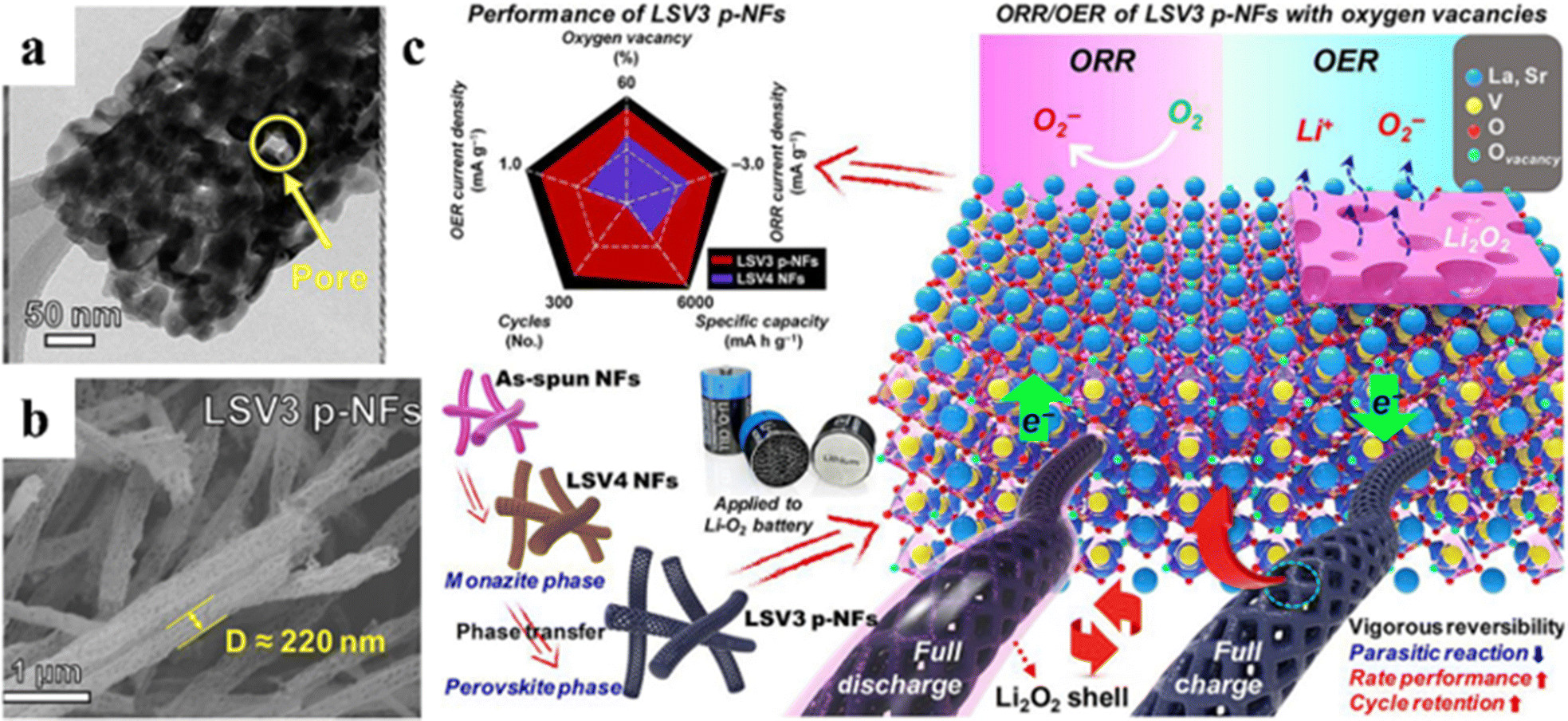 | ||
| Fig. 13 TEM (a) and SEM (b) of La0.8Sr0.2VO3 (LSV3 p-NFs). Schematic illustration of LSV3 p-NFs used in rechargeable Li–O2 batteries (c).136 Copyright © 2020 Elsevier. | ||
| Material | Electrolyte | Charge potential (VLi) | A restricted capacity (mA h g−1) | Current density (mA g−1) | Cycling life | Ref. |
|---|---|---|---|---|---|---|
| G/meso-LaSrMnO3 | 1 M LiTFSI in TEGDME | 4.21 | 500 | 500 | 50 | 134 |
| CoFe@La0.8Fe0.9Co0.1O3−δ | 1 M LiTFSI in TEGDME | 4.4 | 500 | 200 | 215 | 135 |
| La0.8Sr0.2VO3 | 1 M LiNO3 in DMAc | 4.8 | 1000 | 2000 | 200 | 136 |
| LixLayMO3 | 1 M LiTFSI in PC | 4.2 | 0.21 mA h cm−2 | 100 | 100 | 137 |
| LaCoxMn1−xO3−σ | 1 M LiTFSI in TEGDME | 4.5 | 500 | 200 | 60 | 138 |
| LaF3/La0.8Fe0.9Co0.1O3−δ | 1 M LiTFSI in TEGDME | 4.4 | 500 | 200 | 157 | 139 |
4. All-inorganic perovskite for supercapacitors
Both rechargeable batteries and supercapacitors (SCs) as energy storage devices are expected to meet the characteristics of fast storage mechanism, high energy density, high power density, longer life cycle and environmental protection. The power density of batteries is inferior to that of SCs. SCs have high power density and long cycling life but poor energy density.140 The functions and properties of SCs originate from the interaction of their electrodes and electrolyte materials. According to the energy storage mechanism, materials used for SCs are generally classified into two categories, including capacitive materials and pseudocapacitive materials.141,142 Electrode materials for double-layer capacitors rely on purely physical electron accumulation occurring on the surface of the material, such as activated carbon, carbon fiber cloth, graphene, carbon nanotubes and so on.143 Materials with pseudocapacitive properties have completely different electrochemical properties, where the current response is neither purely capacitive nor bulk Faraday (battery-like).144 The redox reaction occurs during the energy storage process which endows pseudocapacitive materials with higher capacity. The energy storage mechanisms of pseudocapacitive materials can generally be divided into three categories. The first is through the adsorption/desorption of ions from/into the electrolyte, which usually occurs as a monolayer on the electrode surface. The second is the redox reaction that occurs on the surface of the material. The third mechanism is that ions in solution intercalate/deintercalate into/out of pores or layers of the material, then react with the surrounding atoms and transport electrons through redox reactions. From the kinetic analysis, most of the first two mechanisms are dominated by surface control, while for the third one, diffusion control accounts for a large part. The energy density and comprehensive performance of SCs are significantly affected by many factors, such as the electrochemical properties of the electrode material, the choice of the electrolyte, and the potential window of the electrode. Therefore, a lot of research efforts have been focused on developing advanced materials for SC electrodes with appropriate structural design to facilitate efficient electron transport and ion diffusion.145–147 Recently, perovskite materials have shown excellent potential for supercapacitor applications.4.1 ABO3 type perovskite fluoride
Recently, perovskite oxide (ABO3) materials have attracted considerable attention due to their valence change at the “B” transition metal, oxygen ion mobility and electron conduction, and three-dimensional BO6 octahedral network. Transition metal-containing perovskite oxides, with their stable structure and wide potential window, are suitable for the construction of supercapacitor electrodes.148 Mefford et al. revealed the mechanism of oxygen vacancy-mediated redox pseudocapacitance of nanostructured lanthanide-based perovskite LaMnO3. This was the first example of anion-based intercalation pseudocapacitance and the first use of oxygen intercalation for fast energy storage. The oxygen embedding mechanism also occurred in other lanthanide-based perovskites such as LaNiO3 with similar charge storage behavior.31 But the main problems are its lower conductivity and shorter cycling life. The multiple possibilities of altering the electronic and physical properties of perovskites and perovskite-derived structures may lead to materials with high energy density. Agglomeration easily occurs during the synthesis of perovskite materials, resulting in a large number of active sites being encapsulated inside the particles. Morphology changes could improve the overall utility of the active site.Zhang et al. prepared a honeycomb LaMnO3 material using a carbon sphere material as a template to control the morphology.149 It has an excellent specific capacitance (535 F g−1), higher than that of the LaMnO3 perovskite (247 F g−1) without the addition of carbon spheres as a template. The crystallinity, microstructure, morphology, elemental composition and chemical bonding state of LaMO3 perovskites were studied in detail by Hu et al. to elucidate the electrochemical properties of LaMO3 perovskite nanofibers as supercapacitor electrodes. The specific capacitances of LaFeO3, LaCoO3 and LaNiO3 nanofibers at a current density of 1 A g−1 were 183.6, 95.8 and 116.3 F g−1, respectively.150 By replacing the metal cations at the A- or B-site with other cations, non-chemo-metric perovskites can be developed to increase the oxygen vacancies. A common approach to increase the conductivity of the LaBO3 perovskite is to partially replace the trivalent lanthanides at the A-site with divalent alkaline earth metals (Ca2+, Sr2+, etc.), which can introduce more oxygen vacancies. La0.75Sr0.25Cr0.5Mn0.5O3 (LSCM) electrode materials were synthesized by Rehman et al.151 The charge storage capacity of LSCM was enhanced by the oxygen vacancies created by doping with Sr. The specific capacitance of LSCM can be as high as 752 F g−1 at 1 mV s−1.
Tomar et al. demonstrated cation and anion (de)embedded hybrid electrodes that were nanocomposites (MLMO) containing MXene (Ti3C2Tx) and perovskite oxide (LaMnO3−δ, LMO) (Fig. 14a). Multilayer MXene and anoxic LMO showed embedded pseudocapacitance based on cations (K+) and anions (OH−), respectively, contributing to total charge storage. Thus, MLMO exhibits high capacitance retention (70.82%) even at a high current density of 30 A g−1 (313.6 F g−1) (Fig. 14b and c).152 However, the increase in oxygen vacancies cannot solve the problems of high internal resistance and slow charge transfer rate of the material. Sun et al. combined Ca0.5Ca0.5MnO3 (LCM) with Ag by a two-step process based on sol–gel and silver mirror reaction. The prepared LCM@Ag composite had an excellent specific capacitance of 287 C g−1 (179 F g−1) at 1.5 A g−1, whereas pure LCM provided only 187 C g−1 (117 F g−1). The key to the improved performance can be attributed to the introduction of silver nanoparticles that enhanced the electron transport and ion diffusion capabilities of the composites.153 Tian et al. constructed LaMnO3@NiCo2O4 materials with layered nanosheet morphology on nickel foam for SCs. The encapsulation of LaMnO3 nanoparticles in NiCo2O4 nanosheets effectively suppressed the leaching of Mn species during reversible intercalation and significantly improved the electrical conductivity. Materials with flower-like nanosheets with open surfaces facilitate mass transfer and can provide abundant electroactive sites for surface redox reactions.154
 | ||
| Fig. 14 Schematic illustration of reversible cation and anion (de)intercalation in MLMO (a). Comparison of CV curves (b) and specific capacitances (c) of LMO, MXene and MLMO.152 Copyright © 2022 Elsevier. | ||
The double perovskite structure has a more ordered structural arrangement than element-doped perovskites, which can prevent lattice distortion and improve cyclic stability. Tomar et al. revealed that the B-site cation ordering in the double perovskite Sr2CoMoO6−δ (DP-SCM) tends to favor the rock salt structure (0D arrangement). The synergistic effect of Co/Mo with excellent redox ability further promotes high oxygen mobility. The high content of oxygen vacancies helps to increase the oxygen anion diffusion rate. Additionally, the fast kinetics of charge storage prevents any phase transition, reflecting excellent cycling life (125% retention up to 5000 cycles). DP-SCM achieved a specific capacitance of 747 F g−1 at 1 A g−1, and the rate can still be maintained at 56% at currents up to 10 A g−1.155 Li et al. fabricated 2D perovskite LaNiO3 nanosheet assemblies with abundant pores by a facile sol–gel method and subsequent thermal treatment. The crystal structure, morphology, and electrochemical performance of LaNiO3 can be simply tuned by adjusting the heating temperature and time. The optimized sample achieved a high specific capacitance and cycling stability.156
4.2 ABF3 type perovskite fluoride
In recent years, perovskite fluorides containing transition metals have shown good potential for application in the field of SCs. They have a similar structure to ABO3-type perovskites, but the strong electronegativity of F makes it possible to lower the Fermi energy level and increase the operating voltage window as anode materials.157 The perovskite fluoride ABF3 features a robust open framework, intersecting tetragonal cavity chains and three-dimensional diffusion channels that favor ion mobility, and thus shows excellent rate performance in SCs. In SCs with alkaline electrolytes, unlike the oxygen embedding mechanism of ABO3, ABF3 usually undergoes mainly conversion reactions for charge storage. In addition, monometallic perovskite fluoride was found to have low conductivity and poor stability when applied in SCs.158–160 Therefore, it is necessary to design perovskite fluorides with improved properties in terms of crystal properties, morphology, and electrical conductivity for better electrochemical performance.Modulation by adjusting the morphology and surface area of the material is a technique that has been extensively studied to improve the specific capacity and cycling stability in energy storage processes.161 Micro/nanostructures with hollow spheres have the unique structural advantage of adsorbing more ions, significantly shortening the diffusion path of ions and electrons, and significantly reducing the volume expansion during cycling. Hussain et al. synthesized a fluorinated perovskite (NaNiF3) with hollow spheres and explained the mechanism of hollow sphere formation in depth, and the method helped to construct other ABF3 materials with hollow structures. As a cathode material for SCs, it had a high specific capacitance (1342 F g−1), excellent rate performance and long cycle stability (90%/8000 cycles).162
Materials with redox properties containing transition metal components are commonly used in SCs and have a higher specific capacity than carbon materials. Compared to their corresponding monometallic counterparts, the binary/polymetallic candidates typically have higher activity and conductivity and exhibit excellent integrated electrochemical behavior, which is mainly caused by the synergistic effect between multiple transition metal redox species. Previous studies have found Ni species to show enhanced electrochemical activity, while Co and Mn species exhibit enhanced electrical conductivity and electrochemical stability.163 Li et al. synthesized perovskite KNi0.8Co0.2F3 nanocrystals by a one-pot solvothermal strategy for use as an electrode material for SCs. Due to the synergistic effect of Ni and Co, it exhibited excellent specific capacity and rate performance (673–619 C g−1, 1–32 A g−1), and its comprehensive performance was superior to that of monometallic perovskites KNiF3 and KCoF3.164 Shi et al. prepared a Co- and Mn-doped (K–Co–Mn–F, Co/Mn = 6:1) KMF3 electrode material doped at the B-site, and the anode showed a specific capacity of 113 C g−1 at a current density of 1 A g−1 due to the synergistic effect of Co and Mn redox substances. Its large particle size and many active sites could not be fully utilized resulting in a low capacity, but after activating the material during cycling, the capacity reached 118% of the initial capacity at the 5000th cycle.165 Doping of zinc in electrode materials or electrocatalysts usually contributes to special features such as increased reactivity, electrical conductivity and surface roughness, which can improve electrochemical performance.166,167 Jia et al. synthesized a novel Zn-doped ABF3 electrode material K0.95Ni0.30Co0.38Zn0.32F3.05 (KNCZF) and applied it to SCs. The monometallic perovskite fluoride KZnF3 has no capacity in alkaline electrolytes, but the introduction of Zn in KNCZF enhanced the activity of Ni and Co species. It was confirmed by multiple characterization means that KNCZF has more electroactive sites, larger charge transfer ability, and stronger hydroxyl adsorption capacity compared to K1.03Ni0.39Co0.61F3.35 (KNCF) (Fig. 15). The KNCZF electrode demonstrates excellent specific capacity (368 C g−1/1 A g−1) and cycle retention (75%/10000 cycles/15 A g−1).168
 | ||
| Fig. 15 Top view of the structures of KNCZF and KNCF (a and b). Eads (OH−) (c). The distance between OH− and the metal atom (dM–O) (d). Top view (e, h, k and n) and side view (f, i, l and o) of different sites after OH− adsorption. Differential charge densities of selected Ni/Co sites with OH− and Bader charge analysis (g, j, m and p). Peak positions of Ni2p3/2 (q) and Co2p3/2 (r) and the percentage of corresponding species for KNCF and KNCZF at the first complete charge (C), complete discharge (D) and pristine state (P).168 Copyright © 2021 Wiley VCH. | ||
The construction of composites is also one of the effective means to enhance the performance of fluorinated perovskites in SCs. Jing et al. prepared composites (NaCo0.2Ni0.8F3/rGO) consisting of perovskite and rGO, considering the superiority of rGO materials. More redox reactions can be promoted by the adjustment of the Co/Ni ratio of the variable metal ions, which helps to increase the specific capacity of the material. In addition, the addition of rGO improves the electrical conductivity of the material and reduces the degree of agglomeration of the perovskite nanoparticles, resulting in improved cycling stability and rate performance. As an anode the material shows a specific capacity of 805.7 C g−1 at a current density of 50 mA g−1, and can maintain 92% of the initial capacity after 5000 cycles.169 Ying et al. prepared a polymetallic Ni–Co–Mn perovskite (K1.0Ni0.4Co0.2Mn0.4F3.2/Ag) composite electrode promoted by nanosilver, showing the synergistic effect of Ni, Co and Mn redox substances and a substantial increase in the comparative capacity of Ag active substances. It exhibited a multi-electron phase transition mechanism and a kinetic behavior of pseudocapacitance and diffusion mixing. The best composite electrode has a specific capacity of 489 C g−1 at a current density of 1 A g−1.170 This new nanoscale composite (MnO2@NH4MnF3) has less Mn2+ and more Mn4+, and more O defects, exhibiting better electrical conductivity and moderate capacitive contribution. What's more, the graded porous morphology facilitates the diffusion of ions in the electrolyte, which can add more interfaces. The specific capacitance of the nanorods reached 240 F g−1 at a current density of 1 A g−1 and maintained 98.0% of the initial value after 1000 cycles.171Table 5 summarizes the electrochemical properties of all-inorganic perovskite materials for SCs.
| Material | Electrolyte | Operating potential (V) | Specific capacity | Cycle (%/cycles/A g−1) | Ref. |
|---|---|---|---|---|---|
| NaNiF3 | 3 M KOH and 0.5 M LiOH | 0–0.5 | 1342–1080 F g−1 at 5–50 A g−1 | >90%/8000/20 | 162 |
| KNi0.8Co0.2F3 | 3 M KOH and 0.5 M LiOH | 0.1–0.55 | 1530–1407 F g−1 at 1–16 A g−1 | — | 164 |
| K–Co–Mn–F3(Co/Mn = 6:1) |
3 M KOH and 0.5 M LiOH | 0–0.55 | 113–100 C g−1 at 1–16 A g−1 | 118%/5000/8 | 165 |
| K0.95Ni0.30Co0.38Zn0.32F3.05 | 3 M KOH and 0.5 M LiOH | 0–0.55 | 368–294 C g−1 at 1–32 A g−1 | 75%/10000/15 | 168 |
| NaCo0.2Ni0.8F3/rGO | 3 M KOH and 0.5 M LiOH | 0–0.5 | 805.7–661.4 C g−1 at 0.5–16 A g−1 | 92%/5000/8 | 169 |
| KNi0.4Co0.2Mn0.4F3/Ag(37%) | 3 M KOH and 0.5 M LiOH | 0–0.6 | 489–278 C g−1 at 1–128 Ag−1 | 73%/10000/30 | 170 |
| MnO2@NH4MnF3 | 1 M Na2SO4 | 0–1.0 | 240–38.9 F g−1 at 1–30 A g−1 | 98%/1000/1 | 171 |
5. Summary and outlook
As one of the most prominent material classes, all-inorganic perovskite-type compounds have recently received significant attention as the functional materials in the field of energy storage, such as lithium-ion batteries, potassium–ion batteries, zinc-ion batteries, sodium-ion batteries, zinc–air batteries, Li–O2 batteries and supercapacitors. Table 6 summarizes the charge storage mechanism of various all-inorganic perovskites in different types of devices. However, there are no practical commercial energy storage devices based on perovskite materials. Although perovskite materials have shown diverse merits in different energy-related applications, some key performance factors must be optimized before they can be widely used in real devices. The modulation of materials in terms of microstructure, morphology and surface area can enhance the capacity, cycling stability, etc., such as designing nanoscale electrode materials with hollow, porous, flower-like, and core–shell shapes. In addition, the strategy of A-site, B-site or a combination of A-site and B-site doping can tune the crystal structure, reactivity, catalytic activity, etc., and significantly increase the overall electrochemical performance of the material. The double-perovskite structure possesses a more ordered structural arrangement than the element-doped perovskite, which prevents lattice distortion and improves cycle stability. The defect engineering of perovskite oxides and fluorides has been extensively studied, which is a key factor affecting the quality and charge transfer performance. The ion size and the valence change of B-site ions, as well as oxygen vacancies in perovskite ABO3, determine the occupancy site, the electrical conductivity and the ionic conductivity, respectively, while the ionic conductivity in ABF3 is related to the vacancies of A-site and F−. Loading certain amounts of metals or making composites with good electron conductors such as Ag, carbon nanotubes, graphene or MXene can effectively improve the electron conductivity and cyclic stability. All the above means of material modification can promote the application of all-inorganic perovskite-type materials in energy storage.| Material | Types of devices | Electrolyte | Reaction mechanism | Ref. |
|---|---|---|---|---|
| CsPbBr3 | Li-ion battery | 0.5 M LiTFSI in BMIMTFSI | CsPbBr3 + xLi+ + xe− ↔ CsPbBr3Lix | 48 |
| CsPbI3 | Li-ion battery | 1 M LiPF6 in EC:DMC = 1:1 |
Discharge: CsPbI3 + Li+ + e− → LixCsPbI3 | 50 |
| CsPbBr3@CNTs | Li-ion battery | 1 M LiPF6 in EC:DMC = 1:1 |
Discharge: CsPbBr3 + Li+ + e− → Li-CsPbBr3 | 53 |
| Li-CsPbBr3 + Li+ + e− → CsBr + LiBr + Pb | ||||
| Pb + Li+ + e− → LixPb (1≤ x ≤ 4.5) | ||||
| K0.89Ni0.02Co0.03Mn0.95F3.0@rGO | Li-ion battery | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 with 1% VC |
xK0.89M(II)0.89 M(III)0.11F3 + 2.11xLi+ + 2.11xe− → xM + 0.89xKF +2.11xLiF | 58 |
| yM +2yLiF ↔ yMF2 + 2yLi+ + 2ye− | ||||
| K0.89M(II)0.89 M(III)0.11F3 + zLi+ + ze− ↔ LizK0.89M(II)0.89+z M(III)0.11−zF3 | ||||
| (M = Ni, Co, Mn; 0 < x < 1; 0 < z < 0.11, 0 < y < x) | ||||
| KCuF3 | Li-ion battery | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 with 1% VC |
xKCuF3 + 2xLi+ + 2xe− → xCu + xKF + 2xLiF | 67 |
| yCu + 2yLiF ↔ yCuF2 + 2yLi+ + 2ye− | ||||
| (0 < x < 1; 0 < y < x) | ||||
| KNi0.1Co0.9F3 | Li-ion battery | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 with 1% VC |
yKMF3 + 2yLi+ + 2ye− → yKF + 2yLiF + yM | 68 |
| yM + 2yLiF ↔ yMF2 + 2yLi+ | ||||
| (M = Ni, Co; 0 < y < 1) | ||||
| K0.97Ni0.31Co0.34Mn0.35F2.98 | Li-ion battery | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 with 1% VC |
xABF3 + 2xLi+ + 2xe− → xB + xAF + 2xLiF | 69 |
| yB + 2yLiF ↔ yBF2 + 2yLi+ + 2ye− | ||||
| (A = K; B = Ni, Co, Mn; 0 < x < 1; 0 < y < x) | ||||
| K1.1Zn0.17Mn0.83F3.03 | Li-ion battery | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 with 1% VC |
xKMF3 + 2xLi+ + 2xe− → xKF + 2xLiF + xM | 70 |
| yM + 2yLiF ↔ yMF2 + 2yLi+ + 2ye− | ||||
| aZn + aLi+ + ae− ↔ aLiZn | ||||
| (M = Zn, Mn; 0 < x < 1; 0 < y < x; 0 < a < 0.17x) | ||||
| NaNbO3 | Li-ion battery | 1 M LiPF6 in EC:EMC:DMC = 1:1:1 with 1% VC |
xNaNb5+O3 + xLi+ + xe− ↔ xLiNaNb4+O3, 0 < x < 1 | 81 |
| yLiNaNb4+O3 + yLi+ + ye− ↔ yLi2NaNb3+O3, 0 < y < x | ||||
| KTaO3 | Li-ion battery | 1 M LiPF6 in EC:DEC:DMC = 2:1:2 |
2KTaO3 + 10Li + + 10e− → 2Ta + K2O + 5Li2O | 82 |
| Ta + 3Li2 ↔ LiTaO3 + 5Li+ + 5e− | ||||
| BaSnO3/rGO | Li-ion battery | 1 M LiPF6 in EC:DEC = 1:1 |
BaSnO3 + 4Li+ + 4e− → BaO + 2Li2O + Sn | 89 |
| BaO + 2Li + + 2e− ↔ Li2O + Ba | ||||
| Sn + 4.4Li + + 4.4e− ↔ Li4.4Sn | ||||
| Ba + 4Li + + e− ↔ Li4Ba | ||||
| ZnSnO3 | Na-ion battery | 1 M NaClO4 in EC:PC = 1:1 |
ZnSnO3 + 6Na+ + 6e− → 3Na2O + Zn + Sn | 95 |
| 4Sn + 15Na+ + 15e− ↔ Na15Sn4 | ||||
| 13Zn + Na+ + e− ↔ NaZn13 | ||||
| Na0.5Bi0.5TiO3 | Na-ion battery | 1 M NaClO4 in PC with 2%FEC | Na0.5Bi0.5TiO3 + 12Na+ + 12e− → TiO2 + Na2O + Bi | 97 |
| Bi + 4Na+ + 4e− → NaxBi | ||||
| Bi + Na+ + e− ↔ BiO + Na | ||||
| K0.82Co0.43Mn0.57F2.66@rGO | Na-ion battery | 0.85 M NaPF6 in EC:EMC:DMC = 1:1:1 with 1% FEC |
xK0.82MF2.66 + 1.84xNa+ + 1.84xe− → xM + 0.82xKF + 1.84xNaF | 101 |
| yM + 2yNaF ↔ yMF2 + 2yNa+ + 2ye− | ||||
| K0.82MF2.66 + zNa+ + ze− ↔ NazK0.82MF2.66 | ||||
| (M = Co; Mn. 0 < x < 1; 0 < z < 1; 0 < y <x) | ||||
| LaNi0.2Mn0.8O3 | Zn-ion battery | 2 M ZnSO4 + 0.2 M MnSO4 | LaNixMn1−xO3 + yZn2+ + 2ye− ↔ LaZnyNixMn1−xO3 | 120 |
| NaNiF3 | SCs | 3 M KOH and 0.5 M LiOH | Ni2+ + 3OH− → NiOOH + H2O + e− | 162 |
| KNi0.8Co0.2F3 | SCs | 3 M KOH and 0.5 M LiOH | Ni2+ + 3OH− ↔ NiOOH + H2O + e− | 164 |
| Co2+ + 3OH− ↔ CoOOH + H2O + e− | ||||
| K–Co–Mn–F3(Co/Mn =6:1) |
SCs | 3 M KOH and 0.5 M LiOH | M2+ + 3OH− ↔ MOOH + H2O + e− | 165 |
| MOOH + OH− ↔ MO2 + H2O + e− (M = Mn, Co) | ||||
| K0.95Ni0.30Co0.38Zn0.32F3.05 | SCs | 3 M KOH and 0.5 M LiOH | K0.95[M2+0.58M3+0.10]Zn0.32F3.05 + 2.68OH− → 0.95K+ + 3.05F− + 0.68MOOH+ 0.32ZnO + H2O +0.58e− | 168 |
| CoOOH + OH− ↔ CoO2 + H2O + e− | ||||
| NiOOH + H2O + e− ↔ Ni(OH)2 + OH− | ||||
| CoOOH + H2O + e− ↔ Co(OH)2 + OH− | ||||
| NiOOH + e− ↔ NiO + OH− | ||||
| 3CoOOH + e− ↔ Co3O4 + H2O + OH− | ||||
| Co3O4 + 4H2O + 2e− ↔ 3Co(OH)2 + 2OH− (M = Ni, Co) | ||||
| NaCo0.2Ni0.8F3/rGO | SCs | 3 M KOH and 0.5 M LiOH·H2O | Ni2+ + 3OH− ↔ NiOOH + H2O + e− | 169 |
| Co2+ + 3OH− ↔ CoOOH + H2O + e− | ||||
| K1.0Ni0.4Co0.2Mn0.4F3.2/Ag(37%) | SCs | 3 M KOH and 0.5 M LiOH | K1.0[M2+0.8M3+0.2]F3.2 + 3OH− → K+ + 3.2F− + MOOH + H2O + 0.8 e− | 170 |
| CoOOH + OH− ↔ CoO2 + H2O + e− | ||||
| MnOOH + OH− ↔ MnO2 + H2O + e− | ||||
| NiOOH + H2O + e− ↔ Ni(OH)2 + OH− NiOOH + e− ↔ NiO + OH− | ||||
| CoOOH + H2O + e− ↔ Co(OH)2 + OH− | ||||
| 3CoOOH + e− ↔ Co3O4 + H2O + OH− | ||||
| 3MnOOH + e− ↔ Mn3O4 + H2O + OH− | ||||
| 2Ag + 2OH− ↔ AgO2 + H2O + 2e− | ||||
| Ag2O + 2OH− ↔ 2AgO + H2O + 2e− (M = Ni/Co/Mn) |
It should also be emphasized that there are still some challenges when perovskite materials are used in the field of energy storage. (1) The exploration of new all-inorganic perovskite-type materials is still necessary, and the material discovery process to find more perovskite materials can be accelerated through data mining, machine learning, and high-throughput computing techniques.172 (2) Previous studies have demonstrated the essential role of electrode material morphology in boosting electrochemical behavior. Nanoscale materials can optimize kinetic behavior and increase active sites, but with the corresponding particle aggregation and more marked irreversible interfacial reactions. (3) Numerous fresh perovskites have been designed and applied for different energy storage devices, but the basic energy storage/catalysis mechanism and structure–performance relationship have not been fully revealed. More advanced in situ characterization techniques and DFT calculations can be used to obtain further structural information and interaction rules. (4) At present, there are several synthetic methods for perovskites, but they are still at the experimental level, and it is difficult to repeat the preparation on a large scale. And hence, it is particularly crucial to formulate simpler, lower-cost, and more repeatable process routes. (5) Too much research has concentrated solely on enhancing electrochemical performance, ignoring the relevance to real-world applications, such as the investigation of adaptability to load variations and toughness under adverse conditions. (6) When perovskites are used as electrode materials for energy storage devices, the entire electrode material is usually composed of active materials, conductive agents, and binders. Therefore, the design of self-supporting electrodes is an attractive research area, which can enhance active material utilization, charge transfer efficiency, and structural stability. (7) Perovskites still need to be further enhanced in terms of catalytic activity, stability and conductivity when applied as catalysts in metal–air batteries. Doping, defect, facet, and morphology engineering all could strengthen OER/ORR catalytic performance, but composites with internal synergies are far more promising catalysts for future metal–air batteries.
In conclusion, all-inorganic perovskites have made great progress in the field of electrochemical energy storage in the past few decades, and we believe that a deep understanding of the fundamental principles, optimization methods, and application requirements will further advance the development of energy storage devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Key R & D Program of China (2021YFB2400400), the Key Project of National Natural Science Foundation of China (52131306), Project on Carbon Emission Peak and Neutrality of Jiangsu Province (BE2022031-4) and the Research Foundation of State Key Lab (ZK201805, ZK201717) is greatly appreciated.Notes and references
- Y.-M. Chiang, Science, 2010, 330, 1485–1486 CrossRef CAS PubMed.
- D. Larcher and J.-M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- Y. C. Wang, F. L. Chu, J. Zeng, Q. J. Wang, T. Naren, Y. Y. Li, Y. Cheng, Y. P. Lei and F. X. Wu, ACS Nano, 2021, 15, 210–239 CrossRef CAS.
- M. K. Aslam, Y. B. Niu and M. W. Xu, Adv. Energy Mater., 2021, 11, 2000681 CrossRef CAS.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 1–16 Search PubMed.
- R. Hou, G. S. Gund, K. Qi, P. Nakhanivej, H. Liu, F. Li, B. Y. Xia and H. S. Park, Energy Storage Mater., 2019, 19, 212–241 CrossRef.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188 CrossRef CAS.
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 CrossRef.
- S. Trasatti and G. Buzzanca, J. Electroanal. Chem. Interfacial Electrochem., 1971, 29, A1–A5 CrossRef.
- K. F. Chen and D. F. Xue, J. Mater. Chem. A, 2016, 4, 7522–7537 RSC.
- F. Wu and C. Wu, Chin. Sci. Bull., 2014, 59, 3369–3376 CrossRef CAS.
- G. M. Liang, Z. B. Wu, C. Didier, W. C. Zhang, J. Cuan, B. H. Li, K. Y. Ko, P. Y. Hung, C. Z. Lu and Y. Z. Chen, Angew. Chem., 2020, 132, 10681–10689 CrossRef.
- J. Cheng, X. Chen, J. S. Wu, F. Liu, X. B. Zhang and V. P. Dravid, CrystEngComm, 2012, 14, 6702–6709 RSC.
- X. R. Gao, X. M. Liu, D. J. Wu, B. Qian, Z. K. Kou, Z. H. Pan, Y. J. Pang, L. Q. Miao and J. Wang, Adv. Funct. Mater., 2019, 29, 1903879 CrossRef.
- J. M. Gonçalves, M. N. Silva, K. K. Naik, P. R. Martins, D. P. Rocha, E. Nossol, R. A. Munoz, L. Angnes and C. S. Rout, J. Mater. Chem. A, 2021, 9, 3095–3124 RSC.
- N. Tolganbek, Y. Yerkinbekova, S. Kalybekkyzy, Z. Bakenov and A. Mentbayeva, J. Alloys Compd., 2021, 882, 160774 CrossRef CAS.
- P. Goel, S. Sundriyal, V. Shrivastav, S. Mishra, D. P. Dubal, K.-H. Kim and A. Deep, Nano Energy, 2021, 80, 105552 CrossRef CAS.
- J. P. Liu, J. Wang, A. Belotti and F. Ciucci, ACS Appl. Energy Mater., 2019, 2, 5472–5480 CrossRef CAS.
- S. Narayanan, N. Parikh, M. M. Tavakoli, M. Pandey, M. Kumar, A. Kalam, S. Trivedi, D. Prochowicz and P. Yadav, Eur. J. Inorg. Chem., 2021, 1201–1212 CrossRef CAS.
- L. Zhu, R. Ran, M. Tade, W. Wang and Z. P. Shao, Asia-Pac. J. Chem. Eng., 2016, 11, 338–369 CrossRef CAS.
- W. S. Hou, Y. X. Yang, L. Fang, Y. Q. Mao, W. Sun, Y. Bai, K. N. Sun and Z. H. Wang, Chem. Eng. J., 2021, 409, 128079 CrossRef CAS.
- S. Q. Hu, J. Wang, J. Zhang, J. Lim, Y. Gao and S. G. Zhang, Appl. Catal., B, 2021, 282, 119593 CrossRef CAS.
- M. L. Medarde, J. Phys.: Condens. Matter, 1997, 9, 1679 CrossRef CAS.
- Y. Jiang, C. Q. Zhu, P. Y. Zhao, K. Bi, J. M. Liu, L. M. Guo, X. H. Wang and L. T. Li, J. Eur. Ceram. Soc., 2021, 41, 6465–6473 CrossRef CAS.
- Y. R. Yu, Z. Wang and G. S. Shao, J. Mater. Chem. A, 2018, 6, 19843–19852 RSC.
- W. Travis, E. Glover, H. Bronstein, D. Scanlon and R. Palgrave, Chem. Sci., 2016, 7, 4548–4556 RSC.
- S. Sengodan, S. Choi, A. Jun, T. H. Shin, Y.-W. Ju, H. Y. Jeong, J. Shin, J. T. Irvine and G. Kim, Nat. Mater., 2015, 14, 205–209 CrossRef CAS PubMed.
- X. J. Lü, J. W. Howard, A. P. Chen, J. L. Zhu, S. Li, G. Wu, P. Dowden, H. W. Xu, Y. S. Zhao and Q. X. Jia, Adv. Sci., 2016, 3, 1500359 CrossRef.
- T. Zheng, H. M. Deng, W. L. Zhou, X. Z. Zhai, H. Y. Cao, L. Yu, P. X. Yang and J. H. Chu, Ceram. Int., 2016, 42, 6033–6038 CrossRef CAS.
- J. T. Mefford, W. G. Hardin, S. Dai, K. P. Johnston and K. J. Stevenson, Nat. Mater., 2014, 13, 726–732 CrossRef CAS PubMed.
- Z. Y. Wu, C. M. Ji, L. N. Li, J. Kong, Z. H. Sun, S. G. Zhao, S. S. Wang, M. C. Hong and J. H. Luo, Angew. Chem., Int. Ed., 2018, 57, 8140–8143 CrossRef CAS PubMed.
- H. Kozuka, K. Ohbayashi and K. Koumoto, Sci. Technol. Adv. Mater., 2015, 16, 026001 CrossRef PubMed.
- S. Fop, K. S. McCombie, E. J. Wildman, J. M. Skakle and A. C. Mclaughlin, Chem. Commun., 2019, 55, 2127–2137 RSC.
- J. Wang, J. Zhang, Y. Z. Zhou, H. B. Liu, Q. F. Xue, X. S. Li, C.-C. Chueh, H.-L. Yip, Z. L. Zhu and A. K. Jen, Nat. Commun., 2020, 11, 1–9 CrossRef.
- C. K. Sun, X. Zhang, C. Li, K. Wang, X. Z. Sun and Y. W. Ma, Energy Storage Mater., 2020, 24, 160–166 CrossRef.
- J. Galos, K. Pattarakunnan, A. S. Best, I. L. Kyratzis, C. H. Wang and A. P. Mouritz, Adv. Mater. Technol., 2021, 6, 2001059 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS.
- P. Patel, ACS Cent. Sci., 2015, 1, 161–162 CrossRef CAS.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- D. Bar-Tow, E. Peled and L. Burstein, J. Electrochem. Soc., 1999, 146, 824 CrossRef CAS.
- J. Christensen and J. Newman, J. Electrochem. Soc., 2004, 151, A1977 CrossRef CAS.
- X. W. Yu and A. Manthiram, Acc. Chem. Res., 2017, 50, 2653–2660 CrossRef CAS PubMed.
- J. Kang and L.-W. Wang, J. Phys. Chem. Lett., 2017, 8, 489–493 CrossRef CAS PubMed.
- W. B. Chu, W. A. Saidi, J. Zhao and O. V. Prezhdo, Angew. Chem., Int. Ed., 2020, 59, 6435–6441 CrossRef CAS PubMed.
- N. Vicente and G. Garcia-Belmonte, J. Phys. Chem. Lett., 2017, 8, 1371–1374 CrossRef CAS PubMed.
- Q. L. Jiang, M. M. Chen, J. Q. Li, M. C. Wang, X. Q. Zeng, T. Besara, J. Lu, Y. Xin, X. Shan and B. C. Pan, ACS Nano, 2017, 11, 1073–1079 CrossRef CAS.
- T. Paul, S. Maiti, B. K. Chatterjee, P. Bairi, B. K. Das, S. Thakur and K. K. Chattopadhyay, J. Phys. Chem. C, 2021, 125, 16892–16902 CrossRef CAS.
- P. Pal and A. Ghosh, Phys. Rev. Appl., 2020, 14, 064010 CrossRef CAS.
- N. Kaisar, T. Paul, P.-W. Chi, Y.-H. Su, A. Singh, C.-W. Chu, M.-K. Wu and P. M. Wu, Materials, 2021, 14, 5718 CrossRef CAS PubMed.
- X. M. Li, D. J. Yu, J. Chen, Y. Wang, F. Cao, Y. Wei, Y. Wu, L. Wang, Y. Zhu and Z. G. Sun, ACS Nano, 2017, 11, 2015–2023 CrossRef CAS PubMed.
- S. Ko, J. I. Lee, H. S. Yang, S. Park and U. Jeong, Adv. Mater., 2012, 24, 4451–4456 CrossRef CAS PubMed.
- S. Liu, K. Zhang, L. Tan, S. P. Qi, G. N. Liu, J. X. Chen and Y. B. Lou, Electrochim. Acta, 2021, 367, 137352 CrossRef CAS.
- F. Giustino and H. J. Snaith, ACS Energy Lett., 2016, 1, 1233–1240 CrossRef CAS.
- G. Volonakis, A. A. Haghighirad, R. L. Milot, W. H. Sio, M. R. Filip, B. Wenger, M. B. Johnston, L. M. Herz, H. J. Snaith and F. Giustino, J. Phys. Chem. Lett., 2017, 8, 772–778 CrossRef CAS PubMed.
- H. Wu, J. C. Pi, Q. Liu, Q. M. Liang, J. B. Qiu, J. M. Guo, Z. W. Long, D. C. Zhou and Q. Wang, J. Phys. Chem. Lett., 2021, 12, 4125–4129 CrossRef CAS PubMed.
- S. Yang, Q. M. Liang, H. Wu, J. C. Pi, Z. L. Wang, Y. X. Luo, Y. Liu, Z. W. Long, D. C. Zhou and Y. G. Wen, J. Phys. Chem. Lett., 2022, 13, 4981–4987 CrossRef PubMed.
- Y. F. Huang, R. Ding, D. F. Ying, Y. X. Huang, T. Yan, C. N. Tan, X. J. Sun and E. H. Liu, Rare Met., 2022, 1–14 Search PubMed.
- A. Martin, M. L. Doublet, E. Kemnitz and N. Pinna, Adv. Funct. Mater., 2018, 28, 1802057 CrossRef.
- W. Shi, R. Ding, Q. L. Xu, T. Yan, Y. X. Huang, C. N. Tan, X. J. Sun, P. Gao and E. H. Liu, Chem. Commun., 2019, 55, 6739–6742 RSC.
- R. E. Doe, K. A. Persson, Y. S. Meng and G. Ceder, Chem. Mater., 2008, 20, 5274–5283 CrossRef CAS.
- N. Dimov, A. Nishimura, K. Chihara, A. Kitajou, I. D. Gocheva and S. Okada, Electrochim. Acta, 2013, 110, 214–220 CrossRef CAS.
- R. E. Doe, K. A. Persson, Y. S. Meng and G. Ceder, Chem. Mater., 2008, 20, 5274–5283 CrossRef CAS.
- Y. Yamada, T. Doi, I. Tanaka, S. Okada and J.-I. Yamaki, J. Power Sources, 2011, 196, 4837–4841 CrossRef CAS.
- A. Martin, E. S. Santiago, E. Kemnitz and N. Pinna, ACS Appl. Mater. Interfaces, 2019, 11, 33132–33139 CrossRef CAS PubMed.
- D. F. Ying, R. Ding, Y. F. Huang, T. Yan, Y. X. Huang, C. N. Tan, X. J. Sun, P. Gao and E. H. Liu, Chem. – Eur. J., 2020, 26, 2798–2802 CrossRef CAS PubMed.
- Y. X. Huang, R. Ding, Q. L. Xu, W. Shi, D. F. Ying, Y. F. Huang, T. Yan, C. N. Tan, X. J. Sun and E. H. Liu, Dalton Trans., 2021, 50, 8671–8675 RSC.
- Q. L. Xu, R. Ding, W. Shi, D. F. Ying, Y. F. Huang, T. Yan, P. Gao, X. J. Sun and E. H. Liu, J. Mater. Chem. A, 2019, 7, 8315–8326 RSC.
- T. Yan, Y. F. Huang, R. Ding, W. Shi, D. F. Ying, Z. Y. Jia, C. N. Tan, Y. X. Huang, X. J. Sun and E. H. Liu, Nanoscale Adv., 2021, 3, 5703–5710 RSC.
- D. F. Ying, Q. L. Xu, R. Ding, Y. F. Huang, T. Yan, Y. X. Huang, C. N. Tan, X. J. Sun, P. Gao and E. H. Liu, Chem. Eng. J., 2020, 388, 124154 CrossRef CAS.
- D. F. Ying, R. Ding, Y. F. Huang, W. Shi, Q. L. Xu, C. N. Tan, X. J. Sun, P. Gao and E. H. Liu, J. Mater. Chem. A, 2019, 7, 18257–18266 RSC.
- Z. S. Wu, W. C. Ren, L. B. Gao, B. L. Liu, C. B. Jiang and H. M. Cheng, Carbon, 2009, 47, 493–499 CrossRef CAS.
- Z. S. Wu, W. C. Ren, L. Wen, L. B. Gao, J. P. Zhao, Z. P. Chen, G. M. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS PubMed.
- J. F. Xie, H. Zhang, S. Li, R. X. Wang, X. Sun, M. Zhou, J. F. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- P. Gao, Z. Chen, Y. X. Gong, R. Zhang, H. Liu, P. Tang, X. H. Chen, S. Passerini and J. L. Liu, Adv. Energy Mater., 2020, 10, 1903780 CrossRef CAS.
- M. A. Nowroozi, I. Mohammad, P. Molaiyan, K. Wissel, A. R. Munnangi and O. Clemens, J. Mater. Chem. A, 2021, 9, 5980–6012 RSC.
- Y. Huang, R. Ding, D. Ying, Y. Huang, C. Tan, T. Yan, X. Sun and E. Liu, Chem. Commun., 2021, 57, 7705–7708 RSC.
- Z. A. Elsiddig, H. Xu, D. Wang, W. Zhang, X. L. Guo, Y. Zhang, Z. M. Sun and J. Chen, Electrochim. Acta, 2017, 253, 422–429 CrossRef CAS.
- A. Mahata, P. Datta and R. N. Basu, Ceram. Int., 2017, 43, 433–438 CrossRef CAS.
- C. W. Sun, J. A. Alonso and J. J. Bian, Adv. Energy Mater., 2021, 11, 2000459 CrossRef CAS.
- T. Yan, R. Ding, D. F. Ying, Y. F. Huang, Y. X. Huang, C. N. Tan, X. J. Sun, P. Gao and E. H. Liu, J. Mater. Chem. A, 2019, 7, 22884–22888 RSC.
- H. Sumedha, M. Shashank, F. A. Alharthi, M. S. Santosh, B. Praveen and G. Nagaraju, Int. J. Hydrogen Energy, 2021, 46, 28214–28220 CrossRef CAS.
- K. Ogunniran, G. Murugadoss, R. Thangamuthu and P. Periasamy, J. Alloys Compd., 2018, 766, 1014–1023 CrossRef CAS.
- J. Liu, E. Sheha, S. I. El-Dek, D. Goonetilleke, M. Harguindeguy and N. Sharma, CrystEngComm, 2018, 20, 6165–6172 RSC.
- C. C. Sorrell, J. Nowotny and S. Sugihara, Materials for energy conversion devices, Elsevier, 2005 Search PubMed.
- C. W. Sun, J. A. Alonso and J. J. Bian, Adv. Energy Mater., 2021, 11, 2000459 CrossRef CAS.
- L. T. Yang, X. H. Xiong, G. S. Liang, X. Li, C. Wang, W. B. You, X. B. Zhao, X. H. Liu and R. C. Che, Adv. Mater., 2022, 34, 2200914 CrossRef CAS PubMed.
- X. Gao, C. A. Fisher, Y. H. Ikuhara, Y. Fujiwara, S. Kobayashi, H. Moriwake, A. Kuwabara, K. Hoshikawa, K. Kohama and H. Iba, J. Mater. Chem. A, 2015, 3, 3351–3359 RSC.
- G. Veerappan, K. Zhang, M. Ma, B. Kang and J. H. Park, Electrochim. Acta, 2016, 214, 31–37 CrossRef CAS.
- L. Qin, Y. Liu, S. H. Zhu, D. X. Wu, G. Y. Wang, J. Y. Zhang, Y. Y. Wang, L. R. Hou and C. Z. Yuan, J. Mater. Chem. A, 2021, 9, 20405–20416 RSC.
- S. L. Li, M. D. Zhang, Z. X. Feng, Y. C. Huang, T. Qian, H. Hu, X. Zheng, P. F. Liu, H. Y. Liu and T. Xing, Chem. Eng. J., 2021, 424, 130315 CrossRef CAS.
- M. Walter, M. V. Kovalenko and K. V. Kravchyk, New J. Chem., 2020, 44, 1677–1683 RSC.
- S. W. Kim, D. H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS.
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed., 2018, 57, 102–120 CrossRef CAS PubMed.
- L. P. Wang, Y. Zhao, C. Wei, C. L. Wong, M. Srinivasan and Z. J. Xu, J. Mater. Chem. A, 2015, 3, 14033–14038 RSC.
- A. Surendran, H. Enale, A. Thottungal, A. Sarapulova, M. Knapp, S. Nishanthi, D. Dixon and A. Bhaskar, ACS Appl. Mater. Interfaces, 2022, 14, 7856–7868 CrossRef CAS PubMed.
- K. K. Bharathi, B. Moorthy, H. K. Dara, L. Durai and D. K. Kim, J. Mater. Sci., 2019, 54, 13236–13246 CrossRef CAS.
- I. D. Gocheva, M. Nishijima, T. Doi, S. Okada, J.-I. Yamaki and T. Nishida, J. Power Sources, 2009, 187, 247–252 CrossRef CAS.
- A. Kitajou, H. Komatsu, K. Chihara, I. D. Gocheva, S. Okada and J.-I. Yamaki, J. Power Sources, 2012, 198, 389–392 CrossRef CAS.
- D. P. Cao, C. L. Yin, D. R. Shi, Z. W. Fu, J. C. Zhang and C. L. Li, Adv. Funct. Mater., 2017, 27, 1701130 CrossRef.
- C. Tan, R. Ding, Y. Huang, T. Yan, Y. Huang, F. Yang, X. Sun, P. Gao and E. Liu, Electrochim. Acta, 2021, 389, 138713 CrossRef CAS.
- W. X. Yang, J. H. Zhou, S. Wang, Z. C. Wang, F. Lv, W. S. Zhang, W. Y. Zhang, Q. Sun and S. J. Guo, ACS Energy Lett., 2020, 5, 1653–1661 CrossRef CAS.
- Q. Ma, Q. Deng, H. Sheng, W. Ling, H. R. Wang, H. W. Jiao, X. W. Wu, W. X. Zhou, X. X. Zeng and Y. X. Yin, Sci. China: Chem., 2018, 61, 732–738 CrossRef CAS.
- D. H. Liu, W. H. Li, Y. P. Zheng, Z. Cui, X. Yan, D. S. Liu, J. W. Wang, Y. Zhang, H. Y. Lü and F. Y. Bai, Adv. Mater., 2018, 30, 1706317 CrossRef PubMed.
- C. Vaalma, G. A. Giffin, D. Buchholz and S. Passerini, J. Electrochem. Soc., 2016, 163, A1295 CrossRef CAS.
- S. W. Kim, D. H. Seo, H. Gwon, J. Kim and K. Kang, Adv. Mater., 2010, 22, 5260–5264 CrossRef CAS PubMed.
- K. Rui, Z. Y. Wen, Y. Lu, J. Jin and C. Shen, Adv. Energy Mater., 2015, 5, 1401716 CrossRef.
- Z. H. Yang, X. Y. Wang, L. Liu, S. Y. Yang and X. P. Su, Comput. Mater. Sci., 2011, 50, 3131–3135 CrossRef CAS.
- A. Kitajou, H. Komatsu, R. Nagano and S. Okada, J. Power Sources, 2013, 243, 494–498 CrossRef CAS.
- S. Y. Wang, Y. X. Chen, B. Cui, B. Li, S. X. Wang, Y. H. Cui, Z. C. Ju and Q. C. Zhuang, Appl. Surf. Sci., 2020, 514, 145954 CrossRef CAS.
- S. Y. Wang, B. Cui, Q. C. Zhuang, Y. L. Shi and H. Zheng, J. Electrochem. Soc., 2019, 166, A1819 CrossRef CAS.
- S. Y. Wang, Y. L. Cui, B. Cui, Q. C. Zhuang, B. Li, H. Zheng and Z. C. Ju, Adv. Mater. Interfaces, 2019, 6, 1901362 CrossRef CAS.
- Z. W. Liu, P. Li, G. Q. Suo, S. Gong, W. A. Wang, C. Y. Lao, Y. J. Xie, H. Guo, Q. Y. Yu and W. Zhao, Energy Environ. Sci., 2018, 11, 3033–3042 RSC.
- J. W. Gao, X. S. Xie, S. Q. Liang, B. A. Lu and J. Zhou, Nano-Micro Lett., 2021, 13, 1–12 CrossRef CAS PubMed.
- P. H. Yang, P. Sun and W. J. Mai, Mater. Today, 2016, 19, 394–402 CrossRef CAS.
- T. Y. Wei, Q. Li, G. Z. Yang and C. X. Wang, J. Mater. Chem. A, 2018, 6, 8006–8012 RSC.
- R. Trócoli and F. La Mantia, ChemSusChem, 2015, 8, 481–485 CrossRef PubMed.
- H. L. Pan, Y. Y. Shao, P. F. Yan, Y. W. Cheng, K. S. Han, Z. M. Nie, C. M. Wang, J. H. Yang, X. L. Li and P. Bhattacharya, Nat. Energy, 2016, 1, 1–7 Search PubMed.
- Q. Q. Zhuang, B. Z. Wu, H. L. Wang, X. K. Wu, P. Xu, H. M. Yi, Z. N. Xiong, G. J. Shi, Q. G. Wang and B. F. Wang, Ionics, 2021, 27, 4811–4818 CrossRef CAS.
- T. T. Zhu, K. Zheng, P. P. Wang, X. Cai, X. Wang, D. M. Gao, D. M. Yu, C. G. Chen and Y. P. Liu, J. Colloid Interface Sci., 2022, 610, 796–804 CrossRef CAS PubMed.
- Q. F. Liu, Z. F. Pan, E. D. Wang, L. An and G. Q. Sun, Energy Storage Mater., 2020, 27, 478–505 CrossRef.
- J. Ming, J. Guo, C. Xia, W. X. Wang and H. N. Alshareef, Mater. Sci. Eng. R Rep., 2019, 135, 58–84 CrossRef.
- K. W. Leong, Y. F. Wang, M. Ni, W. D. Pan, S. J. Luo and D. Y. Leung, Renewable Sustainable Energy Rev., 2022, 154, 111771 CrossRef CAS.
- J. Pan, Y. Y. Xu, H. Yang, Z. H. Dong, H. F. Liu and B. Y. Xia, Adv. Sci., 2018, 5, 1700691 CrossRef PubMed.
- J. Shim, K. J. Lopez, H.-J. Sun, G. Park, J.-C. An, S. Eom, S. Shimpalee and J. W. Weidner, J. Appl. Electrochem., 2015, 45, 1005–1012 CrossRef CAS.
- T. Ishihara, L. Guo, T. Miyano, Y. Inoishi, K. Kaneko and S. Ida, J. Mater. Chem. A, 2018, 6, 7686–7692 RSC.
- J. Ran, T. Wang, J. Zhang, Y. Liu, C. Xu, S. Xi and D. Gao, Chem. Mater., 2020, 32, 3439–3446 CrossRef CAS.
- L. Q. Gui, Z. B. Wang, K. Zhang, B. B. He, Y. Z. Liu, W. Zhou, J. M. Xu, Q. Wang and L. Zhao, Appl. Catal., B, 2020, 266, 118656 CrossRef CAS.
- Q. L. Zheng, Y. D. Zhang, C. C. Wang, C. H. Zhang and Y. M. Guo, Energy Fuels, 2021, 36, 1091–1099 CrossRef.
- X. X. Wang, J. Sunarso, Q. Lu, Z. L. Zhou, J. Dai, D. Q. Guan, W. Zhou and Z. P. Shao, Adv. Energy Mater., 2020, 10, 1903271 CrossRef CAS.
- U. Bhardwaj, A. Sharma, A. Mathur, A. Halder and H. S. Kushwaha, Energy Storage, 2022, 4, e293 CrossRef CAS.
- M. Balaish, J. W. Jung, I. D. Kim and Y. Ein-Eli, Adv. Funct. Mater., 2020, 30, 1808303 CrossRef CAS.
- Z. W. Chang, J. J. Xu and X. B. Zhang, Adv. Energy Mater., 2017, 7, 1700875 CrossRef.
- Y. B. Yang, W. Yin, S. T. Wu, X. D. Yang, W. Xia, Y. Shen, Y. H. Huang, A. Y. Cao and Q. Yuan, ACS Nano, 2016, 10, 1240–1248 CrossRef CAS PubMed.
- Y. G. Cong, Z. B. Geng, Q. Zhu, H. W. Hou, X. F. Wu, X. Y. Wang, K. K. Huang and S. H. Feng, Angew. Chem., 2021, 133, 23568–23575 CrossRef.
- M. C. Sung, G. H. Lee and D. W. Kim, InfoMat, 2021, 3, 1295–1310 CrossRef CAS.
- S. B. Ma, H. J. Kwon, M. Kim, S. M. Bak, H. Lee, S. N. Ehrlich, J. J. Cho, D. Im and D. H. Seo, Adv. Energy Mater., 2020, 10, 2001767 CrossRef CAS.
- X. D. Li, Z. Y. Qian, G. K. Han, B. Y. Sun, P. J. Zuo, C. Y. Du, Y. L. Ma, H. Huo, S. F. Lou and G. P. Yin, ACS Appl. Mater. Interfaces, 2020, 12, 10452–10460 CrossRef CAS PubMed.
- H. W. Hou, Y. G. Cong, Q. Zhu, Z. B. Geng, X. Y. Wang, Z. Y. Shao, X. F. Wu, K. K. Huang and S. H. Feng, Chem. Eng. J., 2022, 448, 137684 CrossRef CAS.
- W. Raza, F. Ali, N. Raza, Y. W. Luo, K.-H. Kim, J. H. Yang, S. Kumar, A. Mehmood and E. E. Kwon, Nano Energy, 2018, 52, 441–473 CrossRef CAS.
- Z. S. Iro, C. Subramani and S. Dash, Int. J. Electrochem. Sci., 2016, 11, 10628–10643 CrossRef CAS.
- S. Najib and E. Erdem, Nanoscale Adv., 2019, 1, 2817–2827 RSC.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- S. Kumar, G. Saeed, L. Zhu, K. N. Hui, N. H. Kim and J. H. Lee, Chem. Eng. J., 2021, 403, 126352 CrossRef CAS.
- A. González, E. Goikolea, J. A. Barrena and R. Mysyk, Renewable Sustainable Energy Rev., 2016, 58, 1189–1206 CrossRef.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P.-L. Taberna, C. P. Grey, B. Dunn and P. Simon, Nat. Energy, 2016, 1, 1–10 Search PubMed.
- R. Liu, A. Zhou, X. R. Zhang, J. B. Mu, H. W. Che, Y. M. Wang, T. T. Wang, Z. X. Zhang and Z. K. Kou, Chem. Eng. J., 2021, 412, 128611 CrossRef CAS.
- P. M. Shafi, D. Mohapatra, V. P. Reddy, G. Dhakal, D. R. Kumar, D. Tuma, T. Brousse and J.-J. Shim, Energy Storage Mater., 2022, 45, 119–129 CrossRef.
- C. Zhang, W. Q. Zhang, X. Li, Z. R. Zhu, Q. Wang, S. P. Luo and A. J. Xie, Energy Fuels, 2021, 35, 13457–13465 CrossRef CAS.
- Q. L. Hu, B. Yue, H. Y. Shao, F. Yang, J. H. Wang, Y. Wang and J. H. Liu, J. Alloys Compd., 2021, 852, 157002 CrossRef CAS.
- Z. U. Rehman, M. A. Raza, A. Tariq, U. N. Chishti, M. F. Maqsood, N. Lee, M. H. Awais, S. M. Z. Mehdi and A. Inam, J. Energy Storage, 2020, 32, 101951 CrossRef.
- A. K. Tomar, T. Kshetri, N. H. Kim and J. H. Lee, Energy Storage Mater., 2022, 50, 86–95 CrossRef.
- X. C. Sun, Z. Y. Hao, H. S. Nan, J. Xu and H. W. Tian, Mater. Res. Express, 2021, 8, 075502 CrossRef CAS.
- H. W. Tian, X. Q. Lang, H. S. Nan, P. An, W. Zhang, X. Y. Hu and J. S. Zhang, Electrochim. Acta, 2019, 318, 651–659 CrossRef CAS.
- A. K. Tomar, A. Joshi, S. Atri, G. Singh and R. K. Sharma, ACS Appl. Mater. Interfaces, 2020, 12, 15128–15137 CrossRef CAS PubMed.
- Z. J. Li, W. Y. Zhang, H. Y. Wang and B. C. Yang, Electrochim. Acta, 2017, 258, 561–570 CrossRef CAS.
- H. G. Fan, X. Zhang, Y. C. Wang, R. J. Gao and J. W. Lang, J. Colloid Interface Sci., 2019, 557, 546–555 CrossRef CAS PubMed.
- H. G. Fan, X. Zhang, Y. C. Wang, J. W. Lang and R. J. Gao, J. Power Sources, 2020, 474, 228603 CrossRef CAS.
- W. Mi, C. J. Dai, S. Zhou, J. Yang, Q. Li and Q. Y. Xu, Mater. Lett., 2018, 227, 66–69 CrossRef CAS.
- K.-P. Cheng, R.-J. Gu and L.-X. Wen, RSC Adv., 2020, 10, 11681–11693 RSC.
- W. Jin and G. Maduraiveeran, Mater. Today Energy, 2019, 13, 64–84 CrossRef.
- N. Hussain, F. F. Wu, W. Younas and L. Q. Xu, New J. Chem., 2019, 43, 11959–11967 RSC.
- X. Zhou, H. Dai, X. Huang, Y. Ren, Q. Wang, W. Wang, W. Huang and X. Dong, Mater. Today Energy, 2020, 17, 100429 CrossRef.
- R. Ding, X. D. Li, W. Shi, Q. L. Xu, X. L. Han, Y. Zhou, W. F. Hong and E. H. Liu, J. Mater. Chem. A, 2017, 5, 17822–17827 RSC.
- W. Shi, R. Ding, X. D. Li, Q. L. Xu, D. F. Ying, Y. F. Huang and E. H. Liu, Chem. – Eur. J., 2017, 23, 15305–15311 CrossRef CAS PubMed.
- J. H. Li, Z. C. Liu, Q. B. Zhang, Y. Cheng, B. T. Zhao, S. G. Dai, H. H. Wu, K. L. Zhang, D. Ding and Y. P. Wu, Nano Energy, 2019, 57, 22–33 CrossRef CAS.
- X. J. Liu, Z. Chang, L. Luo, T. H. Xu, X. D. Lei, J. F. Liu and X. M. Sun, Chem. Mater., 2014, 26, 1889–1895 CrossRef CAS.
- Z. Y. Jia, R. Ding, W. J. Yu, Y. Li, A. L. Wang, M. Liu, F. Yang, X. J. Sun and E. H. Liu, Adv. Funct. Mater., 2022, 32, 2107674 CrossRef CAS.
- Y. W. Jing, W. L. Li, D. Wang, X. W. Chang, M. He and Z. Y. Ren, J. Alloys Compd., 2022, 905, 164188 CrossRef CAS.
- D. F. Ying, Y. Li, R. Ding, W. Shi, Q. L. Xu, Y. F. Huang, Z. Y. Jia, W. J. Yu, X. J. Sun and P. Gao, Adv. Funct. Mater., 2021, 31, 2101353 CrossRef CAS.
- B. Li, X. H. Zhang, J. H. Dou and P. X. Zhang, Electrochim. Acta, 2020, 347, 136257 CrossRef CAS.
- R. M. Geilhufe, S. S. Borysov, D. Kalpakchi and A. V. Balatsky, Phys. Rev. Mater., 2018, 2, 024802 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |