
Evolution of adsorption heights in the on-surface synthesis and decoupling of covalent organic networks on Ag(111) by normal-incidence X-ray standing wave†
Lukas
Grossmann
 ab,
David A.
Duncan
c,
Samuel P.
Jarvis
d,
Robert G.
Jones
e,
Soumen
De
f,
Johanna
Rosen
g,
Michael
Schmittel
f,
Wolfgang M.
Heckl
ab,
Jonas
Björk
g and
Markus
Lackinger
*ab
ab,
David A.
Duncan
c,
Samuel P.
Jarvis
d,
Robert G.
Jones
e,
Soumen
De
f,
Johanna
Rosen
g,
Michael
Schmittel
f,
Wolfgang M.
Heckl
ab,
Jonas
Björk
g and
Markus
Lackinger
*ab
aDeutsches Museum, Museumsinsel 1, 80538 München, Germany. E-mail: markus@lackinger.org
bTechnische Universität München, Physics Department, James-Franck-Strasse 1, 85748 Garching, Germany
cDiamond Light Source, Harwell Science and Innovation Campus, Didcot OX11 0QX, UK
dLancaster University, Physics Department, Lancaster LA1 4YB, UK
eUniversity of Nottingham, Department of Physical Chemistry, School of Chemistry, Nottingham NG7 2RD, UK
fCenter of Micro and Nanochemistry and Engineering, Organische Chemie I, Universität Siegen, Adolf-Reichwein-Str. 2, 57068 Siegen, Germany
gLinköping University, Department of Physics, Chemistry and Biology, IFM, 581 83 Linköping, Sweden
First published on 2nd December 2021
Abstract
Structural characterization in on-surface synthesis is primarily carried out by Scanning Probe Microscopy (SPM) which provides high lateral resolution. Yet, important fresh perspectives on surface interactions and molecular conformations are gained from adsorption heights that remain largely inaccessible to SPM, but can be precisely measured with both elemental and chemical sensitivity by Normal-Incidence X-ray Standing Wave (NIXSW) analysis. Here, we study the evolution of adsorption heights in the on-surface synthesis and post-synthetic decoupling of porous covalent triazine–phenylene networks obtained from 2,4,6-tris(4-bromophenyl)-1,3,5-triazine (TBPT) precursors on Ag(111). Room temperature deposition of TBPT and mild annealing to ∼150 °C result in full debromination and formation of organometallic intermediates, where the monomers are linked into reticulated networks by C–Ag–C bonds. Topologically identical covalent networks comprised of triazine vertices that are interconnected by biphenyl units are obtained by a thermally activated chemical transformation of the organometallic intermediates. Exposure to iodine vapor facilitates decoupling by intercalation of an iodine monolayer between the covalent networks and the Ag(111) surface. Accordingly, Scanning Tunneling Microscopy (STM), X-ray Photoelectron Spectroscopy (XPS) and NIXSW experiments are carried out for three successive sample stages: organometallic intermediates, covalent networks directly on Ag(111) and after decoupling. NIXSW analysis facilitates the determination of adsorption heights of chemically distinct carbon species, i.e. in the phenyl and triazine rings, and also for the organometallic carbon atoms. Thereby, molecular conformations are assessed for each sample stage. The interpretation of experimental results is informed by Density Functional Theory (DFT) calculations, providing a consistent picture of adsorption heights and molecular deformations in the networks that result from the interplay between steric hindrance and surface interactions. Quantitative adsorption heights, i.e. vertical distances between adsorbates and surface, provide detailed insight into surface interactions, but are underexplored in on-surface synthesis. In particular, the direct comparison with an in situ prepared decoupled state unveils the surface influence on the network structure, and shows that iodine intercalation is a powerful decoupling strategy.
New conceptsOn-surface synthesis is a thriving research field that has produced unique covalent nanostructures that remain largely inaccessible by any other synthetic means. Structure resolution almost exclusively relies on Scanning Probe Microscopy. The bird's eye view offers high lateral resolution but lacks quantitative vertical resolution. Yet, prevalent height variations are highly relevant as conformations cannot only influence the network's physicochemical properties, but may also have a bearing on the reaction pathway. Currently, only one study has deployed Normal-Incidence X-ray Standing Wave (NIXSW) for analyzing heights of on-surface synthesized covalent nanostructures (Saywell et al., Commun. Chem., 2020, 3, 166). Even though this pioneering work demonstrates the utility of NIXSW, our work reaches significantly beyond by (1) exploiting the chemical sensitivity of NIXSW to separately resolve adsorption heights of the vertices (triazines) and spokes (phenyls) in hexagonal covalent networks; (2) also studying relaxations upon in situ decoupling of the networks from the metal growth substrate by intercalation of an iodine monolayer; and (3) full comparison of measured adsorption heights to elaborate structure simulations by Density Functional Theory. Especially the comparison of accurate experimental data with state-of-the-art calculations allows for important benchmarking: Albeit average adsorption heights are well reproduced by theory, phenyl tilt angles as a gauge of molecular conformation are seriously underestimated by DFT. |
Introduction
Robust covalent nanostructures with unique properties have been synthesized on solid surfaces by covalent coupling of functional monomers.1–6 However, most reactions, in particular those involving C–C coupling, require metal surfaces that are impractical for electronic applications. Consequently, synthesis on inert and eventually insulating surfaces is an important long-term goal.7–10 Meanwhile, decoupling by introducing an inert spacer layer between the nanostructures and the metal represents a viable means to reduce the strong influence of the underlying surface on the covalent nanostructures for fundamental studies of their intrinsic properties. The most obvious and severe impact of underlying metal surfaces is hybridization with the molecular electronic states which results in their broadening and band gap renormalization. Yet, metal surfaces can similarly affect atomic structures, for instance by enforcing more planar geometries of networks that are intrinsically corrugated,11 with concomitant effects on their electronic structure. Accordingly, various approaches have been proposed for in situ decoupling, comprising chemical transformation of the upper metal layers into an inert compound (e.g. silicide or oxide),12,13 lateral manipulation with a scanning probe on top of a posteriori deposited thin insulating films14,15 and intercalation.11,16 For assessing the efficacy of decoupling and also for an atomistic understanding of surface influences, the precise experimental determination of adsorption heights that reflect the strength and specificity of adsorbate-surface bonds is crucial. This was previously demonstrated for individual molecules by comparing adsorption heights on pristine and hexagonal boron-nitride (hBN) passivated Cu(111).17,18Structural characterization in on-surface synthesis heavily relies on Scanning Probe Microscopy (SPM), whose unprecedented real space resolving power provides unique insights into submolecular details of the often less regular structures. However, these techniques allow at best a semi-quantitative determination of adsorption heights as typically inferred with respect to lattice planes of the substrate.19 Yet, the length and strength of vertical bonds are crucial, because these can also affect the reactivity of adsorbed molecules with far reaching implications for the reaction outcome. For instance, surface proximity can activate relatively inert C–H bonds through reduced adsorption heights.20 Consequently, the interpretation of experimental results often relies on adsorption heights from Density Functional Theory (DFT) calculations.21–23 Even though the accuracy of this prevalent quantum chemical method has been proven in many instances, outcomes still depend on the choice of the exchange–correlation functional, possibly resulting in non-unique and misleading solutions.24,25 Thus, benchmarking with accurate experimental results remains essential.26–30 Moreover, the complementary chemical characterization by X-ray photoelectron spectroscopy (XPS), also for real time monitoring of chemical changes that often accompany structural changes in reactive systems, has become state-of-the-art in on-surface synthesis.31–33
Here, we determine adsorption heights for all stages of the on-surface synthesis and decoupling of porous covalent triazine–phenylene networks obtained from 2,4,6-tris(4-bromophenyl)-1,3,5-triazine (TBPT, Fig. 1a) precursors on Ag(111) by NIXSW. A relevant asset of this model system is the possibility to individually measure triazine and phenyl moieties by virtue of the large chemical shift of their respective C 1s core levels (vide infra). Experiments were conducted for the commonly observed organometallic intermediates on Ag(111) and for the resulting covalent networks, both directly on Ag(111) and after decoupling by intercalation of an iodine monolayer. Each sample stage was additionally characterized by Scanning Tunneling Microscopy (STM) and XPS to obtain complementary information on the network's structure and chemical state. The experimental data were compared to DFT calculations, using the version of the van der Waals density functional34 denoted as rev-vdWDF235 that was previously shown to provide accurate descriptions of molecule-metal systems.28 Thereby, we obtain a fully consistent picture of adsorption heights on the bare metal and their substantial increase upon intercalation of an iodine monolayer. By separately addressing the chemically distinct carbon atoms, we also unveil deformations in the molecular networks that arise from the interplay between steric hindrance and surface interactions.
Results and discussion
On-surface synthesis and decoupling by STM
We intentionally used a low coverage of ∼15% with respect to a regular organometallic monolayer to avoid influences from molecular crowding that may play a role at higher coverages. Room temperature deposition of TBPT onto Ag(111) and subsequent mild annealing at 150 °C resulted in full debromination and formation of extended two-dimensional organometallic networks, where the molecules were linked by C–Ag–C bonds (cf.Fig. 1b and ESI† for overview images). The initial occurrence of organometallic, rather than covalent, linkages between the molecules is corroborated by three experimental signatures: (1) triazine-to-triazine distances between adjacent molecules were measured to be (1.50 ± 0.04) nm. For comparison, we calculated the lattice parameters of a regular fully covalent hexagonal network (ESI†). These calculations yielded an optimized triazine-to-triazine distance of 1.27 nm, i.e. ∼0.23 nm shorter than the experimental value. (2) STM images showed bright protrusions at the center of intermolecular linkages that can be consistently assigned to organometallic Ag atoms (cf.Fig. 1b for STM data and Fig. 2a for corresponding image simulation);36–38 (3) C 1s XP spectra exhibited a low binding energy (BE) shoulder due to a chemical shift in the organometallic carbon atoms that directly bind to Ag (Fig. 3a).33,37–39 The dissociated bromine atoms remained adsorbed on the surface. They were frequently observed in the vicinity of the organometallic networks or in its pores, presumably due to attractive electrostatic interactions (Fig. 1b).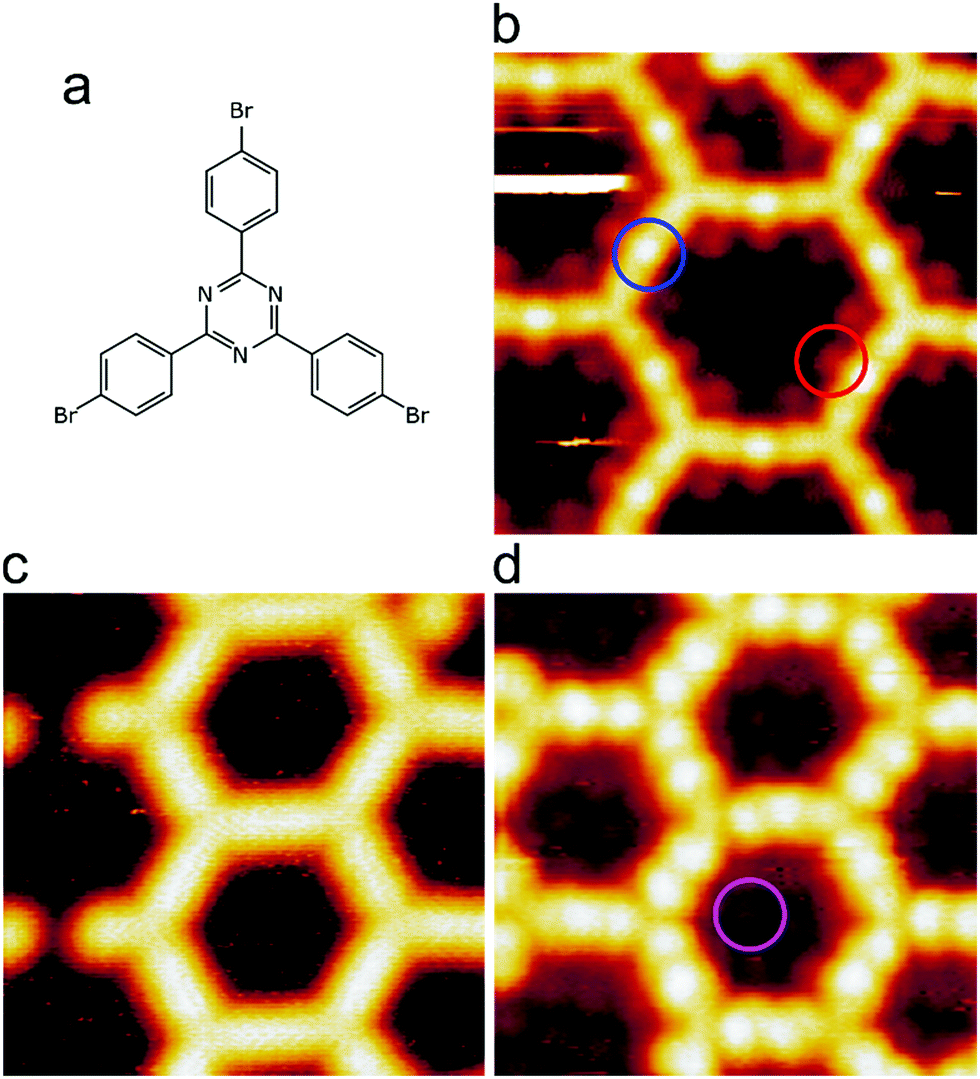 | ||
| Fig. 1 STM imaging of the network formation and decoupling. (a) Molecular structure of the TBPT precursor. Images of (b) organometallic network obtained after annealing to 150 °C. The bright dots at the center of the intermolecular bonds arise from the organometallic Ag atoms (one example is highlighted by the blue circle). The dissociated bromine atoms adsorb at the rim inside the pores (one example is highlighted by the red circle). (c) Covalent network directly on Ag(111) obtained after annealing to 350 °C. The network does not show pronounced internal contrast, only the triazine vertices appear as slight depressions. Owing to the relatively high annealing temperature in STM experiments, the dissociated bromine atoms were already desorbed; (d) decoupled covalent network obtained by iodine intercalation. The internal contrast has changed markedly. The interconnecting biphenyl units exhibit internal contrast and the triazine rings appear as pronounced depression due to electronic effects. Individual iodine atoms of the decoupling layer can be discerned inside the pores (one example is highlighted by the pink circle). (Size of all STM images: 5 × 5 nm2. Tunneling parameters: b −1.0 V, 50 pA; c −1.5 V, 50 pA; d −0.8 V, 80 pA). | ||
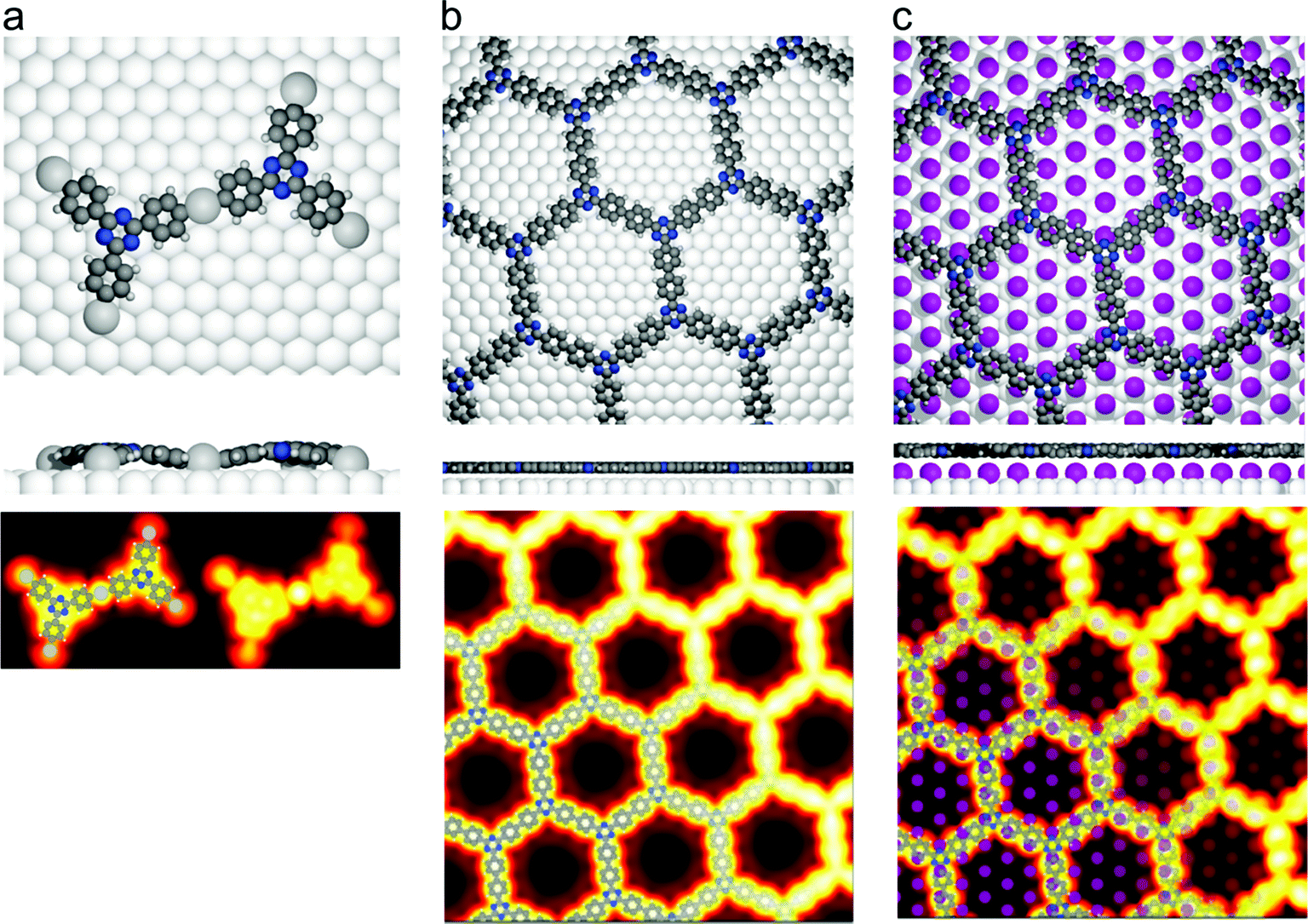 | ||
Fig. 2 DFT calculations. Top views (upper row) and side views (middle row) of the three DFT-optimized structures and corresponding STM image simulations (lower row): (a) organometallic intermediates on Ag(111) modelled by an Ag-terminated C–Ag–C linked dimer; covalent networks (b) directly on Ag(111) and (c) on top of a commensurate 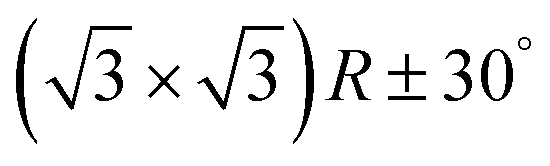 iodine monolayer. For (b) and (c) the covalent networks were simulated as commensurate (2 × 2) superstructure on a iodine monolayer. For (b) and (c) the covalent networks were simulated as commensurate (2 × 2) superstructure on a 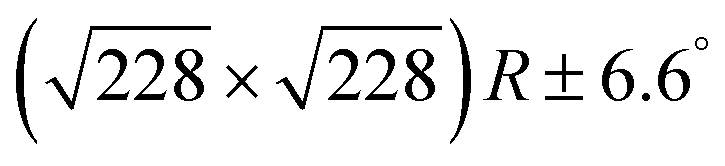 unit cell with respect to Ag(111) or unit cell with respect to Ag(111) or 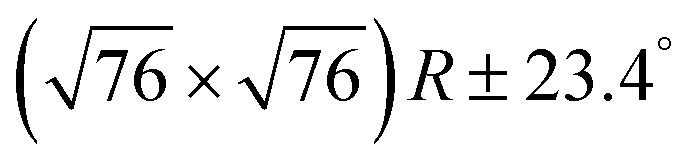 with respect to the iodine monolayer. The matrix notation of these superstructures is given in the ESI.† This yields a lattice parameter of 2.18 nm for the adsorbed covalent networks, which conforms to the optimized lattice parameter of 2.20 nm for free-standing networks with less than 1% compressive strain. with respect to the iodine monolayer. The matrix notation of these superstructures is given in the ESI.† This yields a lattice parameter of 2.18 nm for the adsorbed covalent networks, which conforms to the optimized lattice parameter of 2.20 nm for free-standing networks with less than 1% compressive strain. | ||
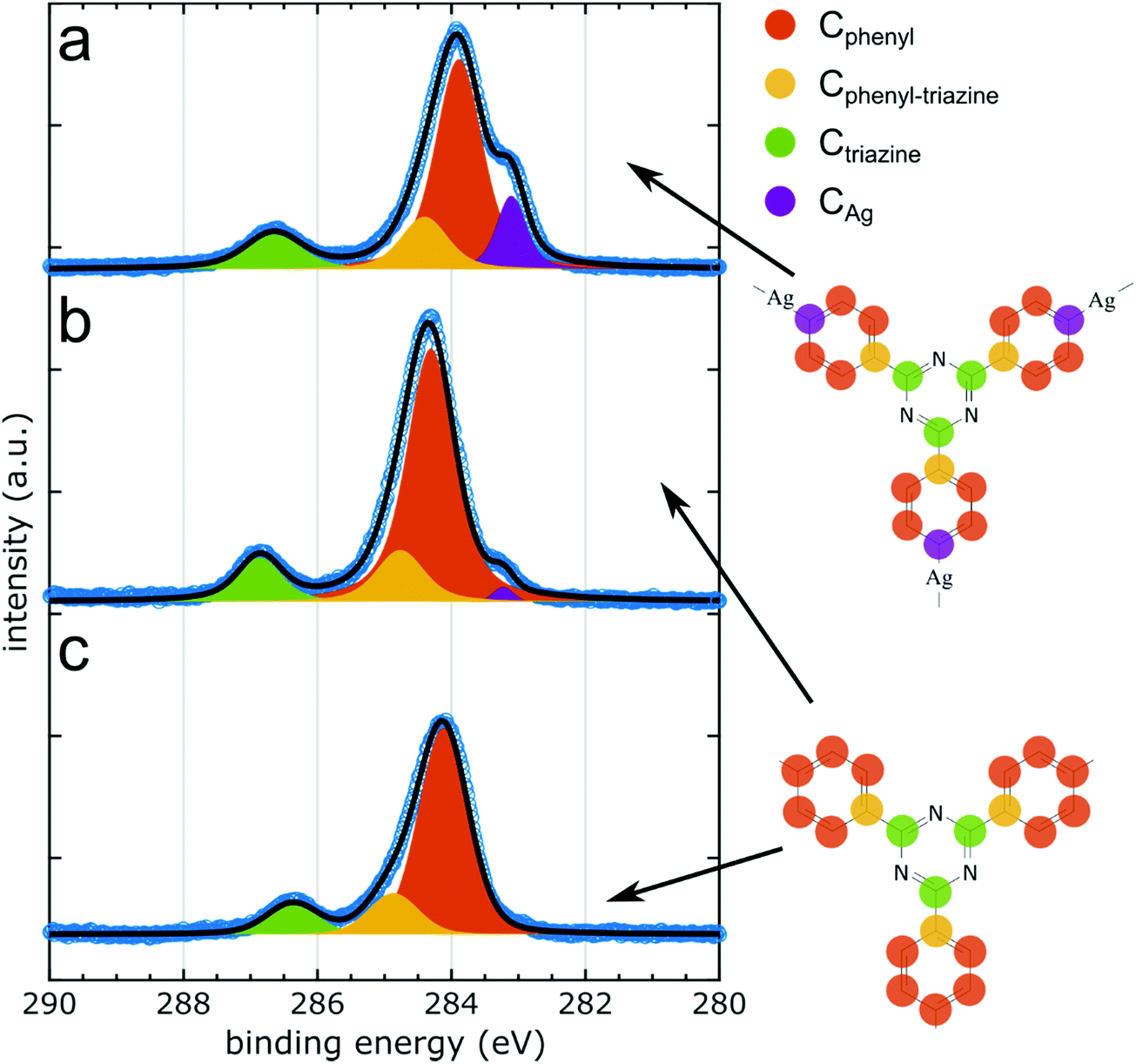 | ||
| Fig. 3 C 1s XP spectra of all successive sample stages. (a) Organometallic networks. (b) Covalent networks directly on Ag(111). (c) Decoupled covalent networks obtained upon iodine intercalation. The color code is explained by the respective sketches on the right-hand side. The asymmetry of Cphenyl is accounted for by fitting with an additional component that arises from the carbon atoms in the phenyl rings that are bound to the triazine rings (Cphenyl–triazine, depicted in yellow). | ||
Further annealing above 200 °C activated the progressive transformation of organometallic C–Ag–C bonds into covalent C–C bonds by reductive elimination of the Ag atoms in accordance with previous studies on Ag(111).32,36,40 The triazine-to-triazine distance between bonded molecules as measured by STM shrunk to (1.29 ± 0.03) nm, i.e. in excellent agreement with DFT results for covalent networks (1.27 nm, ESI†). Moreover, the characteristic bright protrusions at the center of the linkages were no longer observed (Fig. 1c). DFT also indicated that the mixed triazine–phenylene network is intrinsically more planar compared to an isostructural network that exclusively consists of phenyl rings (ESI,† Fig. S10 and ref. 11), because the bonds between the central triazine and the peripheral phenyl moieties are not sterically hindered.
In the next step, the covalent networks prepared on bare Ag(111) were lifted by intercalation of an iodine monolayer between them and the metal surface as a facile means for the post-synthetic in situ decoupling.11,16 To this end, the sample was exposed to iodine vapor (5 × 10−7 mbar, ∼5 min.), whereby intercalation was driven by the high adsorption strength of iodine on Ag(111). Iodination is self-limiting to one monolayer, because under the applied conditions I2 dissociation and subsequent chemisorption of atomic iodine is only feasible on the bare metal. Interestingly, iodine exposure at room temperature that readily induced decoupling of polyphenylene networks from Ag(111),11 only resulted in iodine co-adsorption in the present work, indicating stronger adsorption of the triazine–phenylene networks. However, the networks could be detached by carrying out the iodination at elevated sample temperatures (ESI†). Iodine intercalation could be directly monitored by STM. First of all, the triazine-to-triazine distance remained unaltered (1.28 ± 0.03) nm, confirming integrity of the covalent networks. STM images showed a densely packed hexagonal monolayer of round entities, which coherently extended into the pores of the network (Fig. 1d). The measured lattice parameter of (0.48 ± 0.03) nm is in accord with the known commensurate (√3 × √3) R ± 30° iodine superstructure on Ag(111).41 More importantly, iodine intercalation profoundly altered the STM contrast of the covalent networks. While these appeared mostly featureless without pronounced internal structure when still adsorbed directly on Ag(111) (only the triazine moieties appeared as slight depressions), appreciable submolecular features emerged after decoupling. This observation is in accord with polyphenylene networks, where the STM contrast of decoupled networks resembled the frontier molecular orbitals of free-standing networks,11 as expected for a vastly diminished surface influence. Even though the change of STM contrast by itself does not unambiguously prove successful decoupling, the complementary data obtained by XPS/NEXAFS (ref. 11) and XPS/NIXSW (this work, vide infra) provides entirely consistent, strong evidence for iodine intercalation. Here, the triazine rings appeared as clearly developed depressions with respect to the phenyl rings, while the interconnecting biphenyls exhibited internal structure. To explain these pronounced changes, we performed STM image simulations within the framework of the Tersoff–Hamann approach, based on the atomic and electronic structure obtained from DFT calculations (vide infra). An excellent agreement between experiment and simulation can be seen for all sample stages by the comparison of Fig. 1 and 2. Accordingly, the clearly diminished apparent height in STM of the triazine rings in the decoupled state originates in an electronic effect, i.e. a reduced local density of electronic states in comparison to the phenyl rings, hence does not reflect differences in adsorption height.
Tracking chemical changes by XPS
The structural changes observed by STM are driven and accompanied by chemical changes that can be tracked by XPS. The samples studied by STM and NIXSW are necessarily prepared in two different UHV systems. Normally, preparation temperatures are not directly comparable due to limited measurement accuracy, but also temperature profiles are subject to different dynamics of the sample heating. Hence, verifying the sample's chemical state prior to NIXSW experiments by XPS is imperative. Moreover, XPS provides an overview over the chemically distinct species that can be individually assessed by NIXSW.XPS data collected in the C 1s region for each sample stage are summarized in Fig. 3 (cf.Table 1 for a summary of binding energies). Deposition of TBPT onto Ag(111) held at room temperature yielded partly debrominated species in accord with previous studies.36,38 To form the organometallic networks, the sample was annealed until debromination was completed as monitored in XPS by the disappearance of the molecule-bound species in Br 3p. In the C 1s XP spectrum in Fig. 3a three chemically distinct carbon species can readily be discerned: the two main components correspond to carbon atoms in the triazine rings (Ctriazine) at a BE of 286.7 eV and in the phenyl rings (Cphenyl) at a BE of 283.9 eV. This assignment is based on the stoichiometrically anticipated intensity ratio of Cphenyl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ctriazine = 6:1 and is consistent with reported chemical shifts.42 Moreover, the carbon atoms in the phenyl rings that directly bind to Ag (CAg) give rise to a shoulder at a lower BE of 283.1 eV as also previously reported.33,38,39 Fitting resulted in peak area ratios of Cphenyl:Ctriazine:CAg = 5.0:0.8:0.8, which agree with the stoichiometric ratio of 5:1:1 within an error margin of 20%.
:Ctriazine = 6:1 and is consistent with reported chemical shifts.42 Moreover, the carbon atoms in the phenyl rings that directly bind to Ag (CAg) give rise to a shoulder at a lower BE of 283.1 eV as also previously reported.33,38,39 Fitting resulted in peak area ratios of Cphenyl:Ctriazine:CAg = 5.0:0.8:0.8, which agree with the stoichiometric ratio of 5:1:1 within an error margin of 20%.
| Sample stage | Core level | Species | Binding energy (eV) | f 111 | P 111 | h adexp (Å) | h adDFT (Å) |
|---|---|---|---|---|---|---|---|
| Organometallic | C 1s | Phenyl | 283.9 | 0.56 ± 0.03 | 0.19 ± 0.02 | 2.81 ± 0.04 | 2.88 |
| Organometallic | 283.1 | 1.0 ± 0.2 | 0.96 ± 0.07 | 2.3 ± 0.2 | 2.53 | ||
| Triazine | 286.7 | 0.9 ± 0.2 | 0.34 ± 0.06 | 3.1 ± 0.2 | 3.23 | ||
| N 1s | Triazine | 398.4 | 0.8 ± 0.2 | 0.30 ± 0.07 | 3.1 ± 0.2 | 3.23 | |
| Br 2p | Surface-bound | 188.5 (3p1/2), 181.8 (3p3/2) | 0.90 ± 0.03 | 0.93 ± 0.01 | 2.19 ± 0.03 | 2.13 | |
| Covalent | C 1s | Phenyl | 284.3 | 0.56 ± 0.04 | 0.31 ± 0.02 | 3.08 ± 0.05 | 3.16 |
| Triazine | 286.9 | 0.8 ± 0.2 | 0.35 ± 0.09 | 3.2 ± 0.2 | 3.19 | ||
| N 1s | Triazine | 398.6 | 0.8 ± 0.2 | 0.32 ± 0.06 | 3.1 ± 0.2 | 3.19 | |
| Br 2p | Surface-bound | 188.2 (3p1/2), 181.5 (3p3/2) | 0.93 ± 0.04 | 0.92 ± 0.02 | 2.18 ± 0.04 | 2.13 | |
| Decoupled | C 1s | Phenyl | 284.1 | 0.37 ± 0.03 | 0.51 ± 0.02 | 5.92 ± 0.04 | 5.70 |
| Triazine | 286.4 | 0.7 ± 0.2 | 0.49 ± 0.07 | 5.9 ± 0.2 | 5.69 | ||
| N 1s | Triazine | 398.1 | 0.5 ± 0.1 | 0.46 ± 0.06 | 5.8 ± 0.2 | 5.69 | |
| Br 2p | Surface-bound | 188.2 (3p1/2), 181.5 (3p3/2) | 0.9 ± 0.2 | 0.90 ± 0.06 | 2.1 ± 0.2 | 2.13 | |
| I 3d | Surface-bound | 630.3 (3d3/2), 618.8 (3d5/2) | 0.96 ± 0.03 | 0.99 ± 0.02 | 2.32 ± 0.03 | 2.31 | |
The main phenyl peak is asymmetric, and the quality of the fit can be improved by representing Cphenyl with a second component centered at 284.6 eV BE. We presume that the second component arises from the carbon atoms in the phenyl rings that are bound to the triazine rings (Cphenyl–triazine). Satisfying fits were obtained by constraining the intensity ratio of the two distinct phenyl components to the anticipated intensity ratio, whereby the remaining CAg was also considered. Accordingly, the total amount of phenyl carbons corresponds to the sum of Cphenyl and Cphenyl–triazine. Noteworthy, Ctriazine, Cphenyl and Cphenyl–triazine peaks exhibit a similar Full Width at Half Maximum (FWHM) of ∼1.0 eV, whereas CAg shows a markedly smaller FWHM of ∼0.5 eV in accord with previous studies.43 We presume the FWHM is largely determined by inhomogeneous broadening. Thereby, the lower FWHM of CAg is readily explained by the higher degree of uniformity in its binding environment, imprinted by the preferred adsorption of the organometallic Ag atoms in the three-fold hollow or two-fold bridge sites.19,44 By contrast, the phenyl and triazine rings do not exhibit preferred adsorption sites owing to the incommensurability of the less regular networks. XP spectra of N 1s and Br 3p core levels consistently show single components (cf. ESI† for XP spectra and Table 1 for binding energies), corresponding to the nitrogen atoms in the triazine rings and surface-bound bromine, respectively.
In the next step, the sample was further annealed to obtain the covalent networks. Thereby, the intensity of the CAg peak decreased, but did not disappear completely. The annealing was stopped when the organometallic shoulder did not change further, i.e. reached its minimum of ∼12% of the original amount, whereby the total intensity of C 1s remained constant. We attribute the non-negligible intensity of CAg to dangling phenyl rings at the periphery of the covalent networks (cf. ESI† for overview STM images), whose debrominated carbon atoms are presumed to likewise bind to Ag. The STM image simulations shown in Fig. 2a suggest that the peripheral organometallic Ag atoms can appear less bright as compared to those in intermolecular C–Ag–C bonds, which is attributed to their reduced adsorption height.
Upon formation of covalent networks, Cphenyl exhibits a shift of 0.39 eV to higher BE, whereas the related shift of both Ctriazine and Ntriazine only amounts to 0.20 eV. This shift is well documented for the conversion from organometallic to covalent,31,33,39 and the appreciable difference between Cphenyl and Ctriazine is in accord with the expected stronger influence of covalent bond formation on the phenyl rings. By contrast, Br 3p associated with the chemisorbed bromine atoms shifts by 0.26 eV to lower BE (ESI†) despite the absence of chemical changes. This points to an additional influence of work function changes, possibly related to the ∼9% intensity loss of Br 3p which we observed upon heating, with presumably further influences from changes in the charge transfer to the organic networks. Furthermore, the FWHM of Ctriazine decreases by 0.2 eV, indicating a more uniform chemical environment of the triazine rings with respect to Ag(111). Fitting results in a peak area ratio of Cphenyl:Ctriazine = 6.0:0.8, consistently showing a ∼20% too low amount of Ctriazine that might originate in photoelectron diffraction.45
In the last step, the covalent networks were decoupled from Ag(111) by intercalation of an iodine monolayer. Iodine adsorption was directly observed in XPS by the emergence of a strong I 3d doublet at 618.8 eV BE (I 3d5/2) (ESI†). Although iodine intercalation should not chemically alter the covalent networks, we nevertheless observed characteristic changes in C 1s. The most obvious is a non-uniform shift of Cphenyl and Ctriazine to lower BE by 0.2 eV and 0.5 eV, respectively, (Fig. 3b and c). An analogous, but significantly larger, shift of 0.92 eV was previously observed for polyphenylene networks.11 Interestingly, the peak shape of Cphenyl did not become more symmetric as previously observed for the polyphenylene networks.11 This corroborates our assignment to a further chemically distinct carbon species in the phenyl rings (Cphenyl–triazine), and largely rules out surface influences as core hole screening. We hypothesize that the larger shift of Ctriazine is related to stronger and more site-specific interactions of its nitrogen atoms with the Ag(111) surface that are eliminated by the decoupling. Moreover, upon iodination the total C 1s intensity was reduced by ∼27%. This may be because iodination for the XPS/NIXSW experiments was carried out at a relatively high temperature, where fragmentation of the covalent networks and desorption of smaller fragments cannot be completely ruled out. The FWHM of the Ctriazine peak increased slightly by 0.14 eV. Fitting results in a peak area ratio Cphenyl:Ctriazine = 6.0:0.75, where again Ctriazine remains 25% below the stoichiometric ratio of 6:1.
It is noteworthy that the residual organometallic shoulder that remained for the covalent networks directly on Ag(111) vanished completely after iodination. This observation is in accord with the previous assignment to dangling phenyl groups at the network's periphery. Directly on Ag(111), the carbon radicals bind to Ag atoms and experience the observed core level shift. After iodination, these carbon atoms do not bind to Ag atoms anymore but to iodine atoms of the monolayer.7,46 The associated chemical shift goes in opposite direction to higher BE and is also significantly smaller,37 implying that the respective carbon species merges in the main phenyl peak. In summary, the characteristic changes observed by XPS corroborate detachment of the networks by iodine intercalation.
Adsorption heights by NIXSW
We extracted adsorption heights for all adsorbed elements for all sample stages from NIXSW analysis.47 This synchrotron-based technique utilizes a standing wave of X-rays generated by the interference of the incident and reflected beams that arise from Bragg diffraction at the bulk lattice planes of the Ag substrate. The reflected beam is not only present at the Bragg condition, but extends over a certain range of either incident angle or photon energy (referred to as the Darwin width), and can be understood by dynamical diffraction theory.48 The phase of this standing wave, i.e. the height of the nodal and anti-nodal planes parallel to the respective Bragg diffraction planes,49 varies continuously across the entire Darwin width.47 Thus, adsorbate atoms immersed within this standing wave will experience varying photon intensity, as one scans across the Darwin width. By utilizing photoemission as a probe of photon intensity, not only elemental sensitivity can be attained, but atoms in different chemical environments can be distinguished by means of their respective core level shifts. As NIXSW is insensate to surface relaxations, the discussion below assumes a negligible (≤2%) surface relaxation of the Ag(111).50 Notably, excluding graphene and related 2D monolayers,51,52 NIXSW has only been sparingly used to study on-surface synthesized covalent organic networks. The only study known to the authors was published recently by Saywell et al. on organometallic and covalent polyphenylene wires and networks on Ag(111).44NIXSW experiments were conducted for all previously discussed sample stages using the Bragg reflection of the (111) lattice planes parallel to the surface. The large chemical shift of C 1s between Ctriazine and Cphenyl of ∼2.5 eV allows the two moieties to be safely discriminated, thereby allowing the network's vertices and spokes to be measured separately. In addition, for the organometallic intermediates, the adsorption height of CAg could also be separately measured, as this species represents a sizable fraction of ∼17% of the carbon atoms in the phenyl rings and exhibits a sufficiently large chemical shift of 0.8 eV.
The NIXSW photoelectron yield curves of Cphenyl, Ctriazine and CAg (only for organometallic intermediates) for all three successive sample stages and the respective fits are summarized in Fig. 4. For fitting, the multidimensional parameter space is normally reduced to only two parameters, i.e. the coherent fraction fH and the coherent position PH,47 where H stands for the specific hkl indices of the measured reflection (here 111). Often, fH is considered a measure for the uniformity of adsorption heights, whereas PH is considered the mean adsorption height in units of the bulk lattice plane spacing dAg(111) = 2.354 Å. Table 1 summarizes all deduced f111 and P111, and Fig. 5 illustrates the corresponding molecular conformations. The experiments were challenged by the relatively low signal intensity due to the intentionally low coverage of the porous networks. Adsorption height measurements for nitrogen atoms in the triazine rings were compromised by the interference of the N 1s core level with a loss feature of Ag(111) that also changed upon iodination in a non-trivial way. Nevertheless, we find Ntriazine adsorption heights to be consistent with those of Ctriazine (ESI†), but refrain from interpreting minute differences in terms of triazine ring deformation.
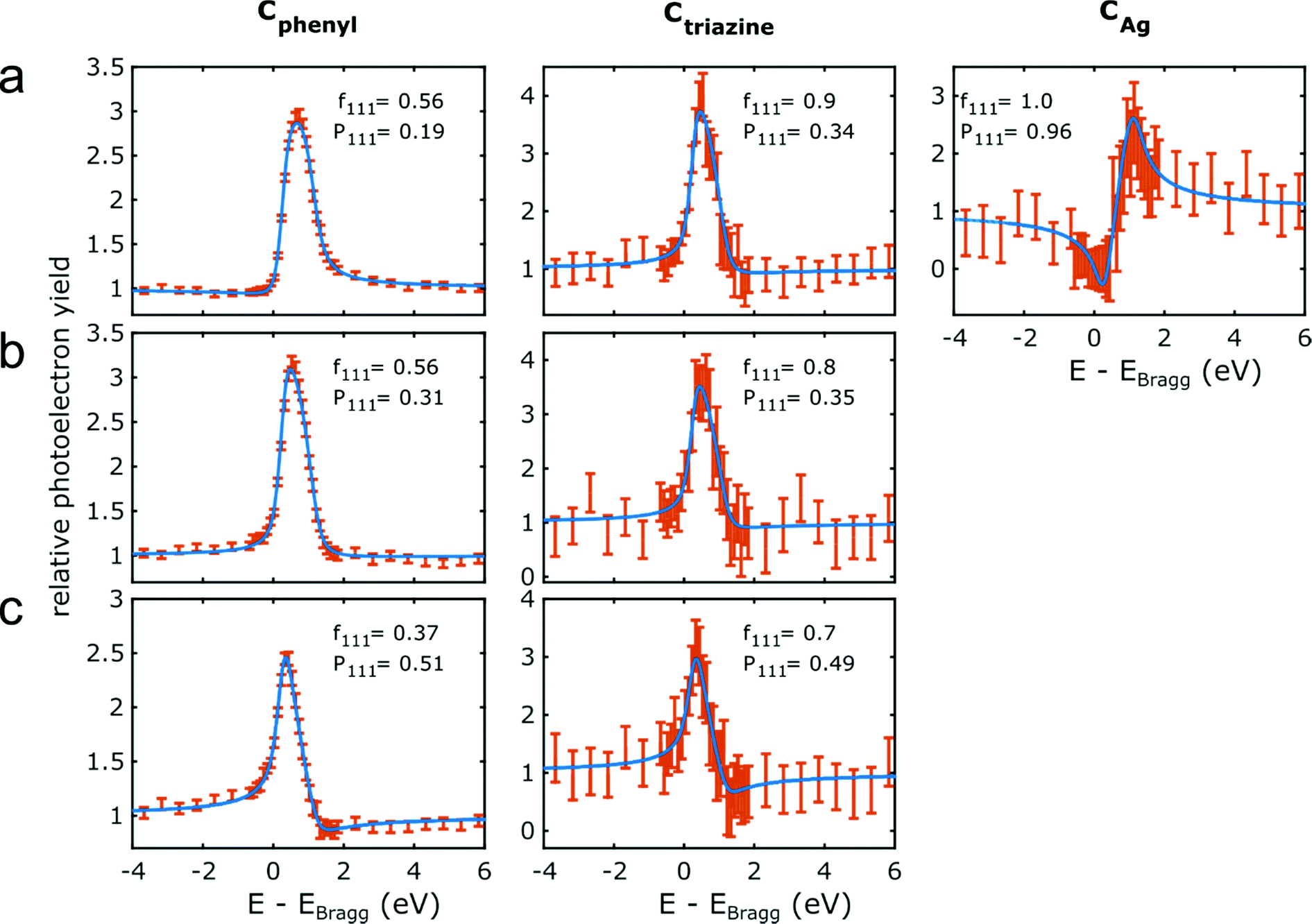 | ||
| Fig. 4 C 1s NIXSW data for the three successive sample stages. Photoelectron yield curves for the chemically distinct carbon species as noted above for (a) organometallic networks on Ag(111); covalent networks (b) directly on Ag(111) and (c) after decoupling by iodine intercalation. Data points are plotted in orange and the error bars correspond to the standard error in the fit of the individual XP spectra at two standard deviations. The blue lines represent fits with the corresponding f111 and P111 indicated. Fits of the simultaneously acquired reflectivity curves are presented in the ESI.† | ||
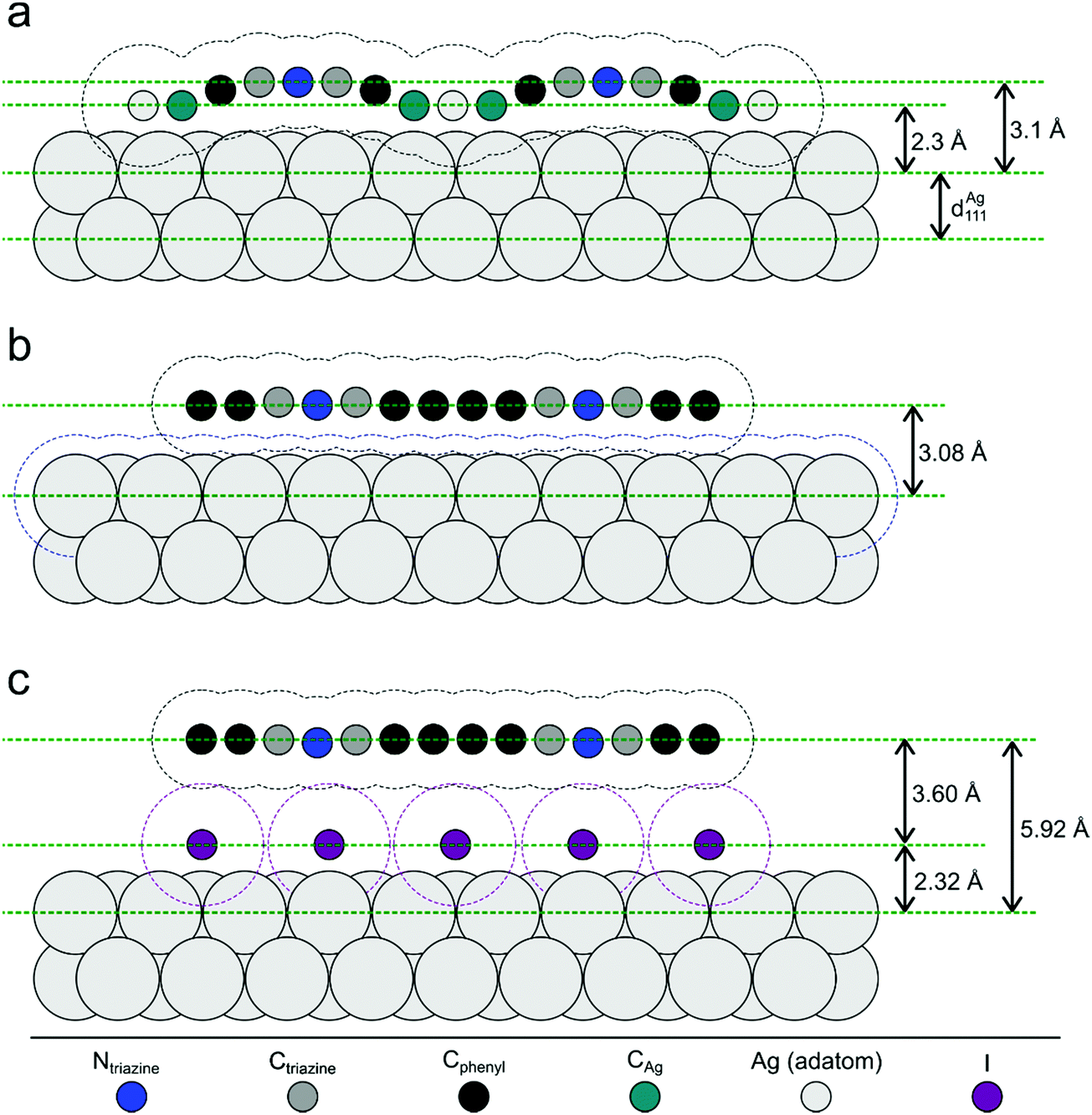 | ||
| Fig. 5 Illustration of the NIXSW results. Side views of the three sample stages. (a) Organometallic networks; covalent networks (b) directly on Ag(111) and (c) after decoupling by iodine intercalation; vertical distances are true to scale, whereas the horizontal distances of the atoms are drawn equidistantly for demonstrative purposes. Hydrogen atoms are omitted for clarity. The dashed contour lines indicate the van der Waals surfaces as defined by the van der Waals radii of the respective elements.54 For the organometallic Ag atom the DFT-calculated adsorption height was used. Tilting of the phenyl rings in the covalent networks, as consistently indicated by low f111 and DFT calculations, was omitted for clarity. | ||
Organometallic intermediates
For the organometallic intermediates NIXSW yields a notably smaller adsorption height of CAg of (2.3 ± 0.2) Å as compared to the average adsorption height of Cphenyl of (2.82 ± 0.04) Å, which is again lower than the adsorption height of Ctriazine of (3.1 ± 0.2) Å. This indicates strong binding of CAg to the surface with an extended effect on the entire phenyl ring as illustrated in Fig. 5a. By contrast, the adsorption height of Ctriazine lies in the usual range for aromatic molecules on Ag(111),53 and is corroborated by the similar adsorption height measured for Ntriazine. This suggests that the binding of CAg to the surface does not affect the central triazine rings anymore. We modeled the organometallic Ag atom as an adatom as the commonly considered configuration rather than as a surface atom. To avoid distortions induced by the periodic boundary conditions, we performed the DFT calculations with the organometallic networks modeled as dispersed Ag-terminated organometallic dimers based on adatoms as shown in Fig. 2a. Thereby all Ag adatoms could be adsorbed close to the preferred three-fold hollow sites. For the organometallic phase, experimental and DFT-calculated adsorption heights coincide for all carbon species within the experimental error (cf.Table 1) and also with the NIXSW results of Saywell,44 thereby corroborating the adatom model. The DFT-optimized structure shows how the organometallic bond to the low lying Ag adatom pulls down the entire phenyl ring. The adsorption height of the organometallic Ag atom itself could not be directly measured by NIXSW against the vast substrate background, but according to DFT its adsorption height amounts to 2.37 Å. This value almost coincides with the lattice plane spacing d111(Ag) = 2.354 Å, suggesting that Ag adatom adsorption is hardly influenced by C–Ag–C bonding. A semi-quantitative determination by AFM resulted in a comparable Ag adsorption height.19 The associated molecular deformations give rise to a distribution of carbon adsorption heights in the phenyl rings, which also explains the relatively low f111 for Cphenyl of 0.55 ± 0.03. On the other hand, notably larger f111 of 0.9 ± 0.2 and 0.8 ± 0.2 were obtained for Ctriazine and Ntriazine, respectively. This implies more uniform adsorption heights in the triazine rings, hence a fairly planar adsorption geometry that is additionally enforced by the three-fold symmetric bonding. Moreover, also CAg features a high f111 of 1.0 ± 0.2, corroborating highly uniform adsorption heights of the organometallic carbon atoms. We propose that this is imparted by the preferred adsorption of Ag adatoms in bridge or hollow sites,19,44 which is consistent with the relatively low FWHM in the C 1s XP spectra (vide supra). Our measured carbon adsorption heights agree well with those of a recent study on organometallic polyphenylene networks,44 suggesting a minor influence of the triazine moiety on the overall conformation of the organometallic networks. The adsorption height of the dissociated bromine atoms chemisorbed on the surface is (2.19 ± 0.03) Å, and thus in close agreement with DFT calculations of monolayers (2.13 Å, ESI†). It also remains unaltered for all subsequent sample stages.Covalent networks
The conversion of organometallic into covalent networks by reductive elimination of the organometallic Ag atoms not only has profound effects on the network structure, but also on its conformation as inferred from adsorption heights. The average adsorption height of Cphenyl increased markedly to (3.08 ± 0.05) Å. It is similar to that of Ctriazine and Ntriazine within the experimental error, indicating uniform average adsorption heights throughout the covalent networks. Yet, for Cphenyl the coherent fraction f111 of 0.56 ± 0.04 remains notably below those of Ctriazine and Ntriazine, which are close to unity. Again this originates in a broader distribution of carbon adsorption heights in the phenyl rings, arising from tilting due to steric hindrance between ortho-hydrogen atoms at the newly established C–C bonds (central biphenyl linkage). An even lower coherent fraction f111 of 0.43 ± 0.06 was reported for polyphenylene networks,44 possibly due to enhanced steric hindrance in the phenyl–phenyl bonds between vertices and spoke that is absent here for the central triazine moiety. By assuming rigid phenyl rings with their para-carbon atoms defining the tilt axis, a tilt angle of 27° ± 2° would conform to the experimental f111, while a slightly larger tilt angle of 30° was deduced for the polyphenylene networks.44 On the contrary, a more direct assessment of phenyl tilt angles by Near-Edge X-ray Absorption Fine Structure (NEXAFS) yielded a significantly smaller tilt angle of 15° ± 5° for polyphenylene networks on Ag(111).11 Nevertheless, all experimental results agree in so far as steric hindrance in the central biphenyl linkage is sufficiently strong that it cannot be overcome by molecule–surface interactions to yield fully planar structures. Yet, DFT calculations performed for covalent networks adsorbed on Ag(111) indicate an almost planar geometry with a negligible phenyl tilt angle of ∼4° (Fig. 2b). The discrepancy to the experiment may be related to the optimized phenyl adsorption sites in the commensurate DFT model, resulting in a systematic overestimation of network–surface interactions. An alternative explanation is offered by thermally excited motions of the phenyl rings that would reduce f111, but are not taken into account by DFT. The intensity of the remaining CAg at the periphery (vide supra) was too low to separately measure their adsorption heights.Decoupled covalent networks
Finally, adsorption heights were measured after decoupling of the covalent networks from Ag(111) by intercalation of an iodine monolayer. We first measured iodine adsorption heights in the monolayer, resulting in (2.32 ± 0.03) Å in excellent agreement with DFT calculations (2.31 Å for a four layer Ag(111) slab, converging to 2.32 Å for more than seven layers in the slab, ESI†). The large f111 of 0.96 ± 0.03 corroborates a highly uniform adsorption of iodine in fcc three-fold hollow sites.55 In the decoupled covalent network, P111 for Cphenyl and Ctriazine are 0.51 ± 0.02 and 0.49 ± 0.07, respectively, and thus almost similar. Converting P111 into absolute adsorption heights remains ambiguous with regard to an integer multiple of the bulk lattice spacing (i.e. parameter n in Table 1). The covalent network is assumed to adsorb above the iodine monolayer with an adsorption height significantly greater than 1 Å. Thus, for the intercalated covalent network, the only plausible value for n is 2 (in contrast to the networks prior to iodine intercalation, where n was assumed to be 1 for Cphenyl and Ctriazine). With this assumption in mind, the mean adsorption height of Cphenyl is found to be (5.92 ± 0.04) Å, with respect to the Ag(111) surface or (3.60 ± 0.05) Å with respect to the iodine monolayer. Comparing the latter to the adsorption height of Cphenyl of (3.08 ± 0.05) Å above Ag(111) clearly confirms notably weaker network–surface interactions after decoupling. Measured adsorption heights of Ctriazine and Ntriazine are both similar to that of Cphenyl within the experimental error, indicating a constant average adsorption height across the covalent networks above the iodine monolayer. Yet, a further reduced coherent fraction f111 of 0.37 of Cphenyl hints toward enlarged tilts of the phenyl rings. Applying the same model as above results in an increased phenyl tilt angle of 35° ± 2° with respect to Ag(111) that is consistent with the NEXAFS measured tilt angle of 35° ± 5° in polyphenylene networks after iodine intercalation.11 Unexpectedly, f111 of Ctriazine and Ntriazine also become smaller upon iodination. A possible explanation is offered by a stronger influence of adsorption site on the highly corrugated iodine monolayer. An independent second experimental run at an approximately doubled coverage of ∼30% yielded similar adsorption heights within the experimental error for all three sample stages (ESI†).Comparison of experiment and theory
Lastly, we would like to compare experimental and theoretical adsorption heights. Average adsorption heights of both the organometallic and covalent networks on pristine Ag(111) are well reproduced by DFT calculations, but less so the phenyl tilt angles. Yet, upon iodine intercalation the calculated phenyl tilt angle increases markedly to 14.4°. Even though it still remains below the experimental value, the trend meets the expectations for weaker network–surface interactions. This is also reflected in the DFT-calculated adsorption heights of the networks after iodine intercalation of 5.68 Å with respect to Ag(111) or 3.38 Å with respect to iodine. However, the agreement of adsorption heights on the iodine monolayer with experimental results is inferior. We also used DFT to calculate the change of adsorption energy as a more direct gauge of the interaction strength between networks and surface, resulting in −1.09 eV for the covalent networks directly on Ag(111) as compared to −0.58 eV on the iodine monolayer per monomeric unit (as depicted in the lower right of Fig. 3).Discussion and summary
We experimentally studied the evolution of adsorption heights for all successive sample stages of the on-surface synthesis and decoupling of covalent triazine–phenylene networks on Ag(111) from TBPT precursors. For the organometallic intermediates the surface exerts a particularly strong influence on the network's conformation by forming C–Ag–C bonds to low lying Ag adatoms. Thereby, the directly bonded phenyl rings are pulled down, whereas there is no measurable influence on the adsorption height of the central triazine vertices. By contrast, average adsorption heights become highly uniform across the covalent networks obtained by further annealing. Yet, the relatively low f111 for the phenyl carbon atoms indicate a tilt of the phenyl rings due to the steric hindrance in the newly formed phenyl–phenyl linkages that cannot be fully overcome by interactions with the Ag(111) surface. Upon intercalation of an iodine monolayer the average carbon adsorption heights increase markedly from 3.1 Å to 5.9 Å with respect to the Ag(111) surface. Yet, also the increased adsorption height above iodine of 3.60 Å, i.e. 0.50 Å higher than above Ag(111), indicates diminished interactions between networks and their support as corroborated by DFT-calculated adsorption energies. After decoupling, the influence of steric hindrance within the networks becomes stronger due to the weaker network–surface interactions. This is expressed in enlarged phenyl tilt angles as estimated from f111 and in qualitative agreement with DFT calculations. An analogous structural relaxation was previously found by NEXAFS in isostructural polyphenylene networks,11 and was also qualitatively reproduced by DFT calculations. The two independent experimental NIXSW runs resulted in similar adsorption heights for all sample stages. Since in both runs overall coverages still remained low, we presume that the observed deviations do not necessarily indicate a coverage dependence, but rather provide an impression of the experimental accuracy. After the various heating steps in the sample preparation, we find consistent chemical shifts and changes of FWHM in XPS. Their dependence on chemical species may indicate specific interactions with the surface as preferred adsorption sites or bond formation between nitrogen in the triazine rings and the Ag substrate.For both the organometallic and the covalent networks directly on Ag(111), the agreement between experimental and calculated mean adsorption heights is excellent, whereas phenyl tilt angles are underestimated by DFT. For the iodine intercalated networks, DFT consistently computes lower adsorption heights than measured experimentally, whereas the adsorption height of iodine itself shows excellent agreement. The systematic underestimation of molecular adsorption heights on the iodine monolayer by DFT could be related to the optimized adsorption of the commensurately modelled regular networks which only serve as proxy of the less regular networks in reality. Overestimation of van der Waals interactions between the iodine monolayer and covalent networks in DFT can also not be excluded, because the used parameters have not been evaluated for iodinated surfaces. Yet, this issue could be settled by future combined NIXSW and DFT studies of commensurate self-assembled monolayers on iodine-passivated Ag(111).
Finally, it is compelling to compare iodine intercalation to other prevalent means for decoupling organic adsorbates from metal surfaces. In this regard, adsorption heights of cobalt porphine (CoP)17 and 3,4,9,10-perylene tetracarboxylic dianhydride (PTCDA)18 were measured by NIXSW both on pristine and hBN passivated Cu(111). The molecular adsorption heights of 2.25 Å (CoP) and 2.66 Å (PTCDA) on the strongly interacting Cu(111) surface are both comparatively low. On the hBN monolayer, the adsorption heights with respect to Cu(111) increased markedly to 6.45 Å (CoP) and 6.61 Å (PTCDA), i.e. significantly larger than the adsorption height after iodine intercalation of 5.92 Å above Ag(111). This is partly related to the relatively large mean adsorption height of 3.38 Å17 (3.24 Å18) of the hBN layer above Cu(111) as compared to 2.32 Å for the iodine layer above Ag(111). However, when using the decoupling layer as height reference, the adsorption height above iodine of 3.60 Å is even larger than the 3.07 Å (CoP) and 3.27 Å (PTCDA) above hBN. This may indicate weaker adsorption on the former, but the larger van der Waals radius of iodine will also have its bearing. In addition, the hBN layer itself also shows a pronounced height modulation, and adsorbates may preferably accumulate at lower regions with enhanced molecule–surface interactions. However, also the relatively small adsorption heights directly on Cu(111) indicate a stronger interaction before decoupling so that a direct comparison between different metals could be misleading. More importantly, hBN passivation layers are grown under harsh conditions that are generally incompatible with organic adsorbates,56 hence it cannot be employed for post-synthetic decoupling of on-surface synthesized nanostructures by intercalation.
Despite the low structural perfection of on-surface synthesized porous covalent networks, NIXSW measured adsorption heights provide unprecedented information on the network's out-of-plane structure with detailed insights into conformations by virtue of its chemical sensitivity. The direct comparison to DFT calculations of commensurately modelled networks is useful to substantiate the results and to support the interpretation, in particular for assigning low coherent fractions to a broader distribution of adsorption heights due to relaxations of the molecular networks. Future studies should address the sizable deviations between experimentally deduced and calculated phenyl tilt angles to enhance the accurateness of DFT.
Author contributions
M. L., L. G. and W. M. H. designed and conceived the study. S. D. and M. S. synthesized, characterized and purified the TBPT compound. L. G. performed the STM experiments. L. G., D. A. D., S. P. J., R. G. J. and M. L. carried out the XPS and NIXSW experiments. L. G. and D. A. D. analyzed and fitted the experimental data. J. B. and J. R. conducted and analysed the DFT calculations. M. L. co-wrote the manuscript, with contributions from all authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We would like to thank Diamond Light Source for the award of beam time (SI15729) and the project CALIPSOplus under Grant Agreement 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020. The research leading to this result has been supported by the Deutsche Forschungsgemeinschaft (grant LA 1842/9-1). J. R. and J. B. acknowledges support from the Swedish Government Strategic Research Area in Materials Science on Functional Materials at Linköping University (Faculty Grant SFO Mat LiU No. 2009 00971) and the Swedish Research Council. Computer resources were allocated by the Swedish National Infrastructure for Computing and carried out at the National Supercomputer Centre, Sweden.References
- S. Schlögl, W. M. Heckl and M. Lackinger, On-surface radical addition of triply iodinated monomers on Au(111)—the influence of monomer size and thermal post-processing, Surf. Sci., 2012, 606, 999–1004 CrossRef
.
- F. Klappenberger,
et al., On-Surface Synthesis of Carbon-Based Scaffolds and Nanomaterials Using Terminal Alkynes, Acc. Chem. Res., 2015, 48, 2140–2150 CrossRef CAS
- M. Lackinger, On-surface polymerization – a versatile synthetic route to two-dimensional polymers, Polym. Int., 2015, 64, 1073–1078 CrossRef CAS
- Q. T. Fan, J. M. Gottfried and J. F. Zhu, Surface-Catalyzed C-C Covalent Coupling Strategies toward the Synthesis of Low-Dimensional Carbon-Based Nanostructures, Acc. Chem. Res., 2015, 48, 2484–2494 CrossRef CAS PubMed
- S. Clair and D. G. de Oteyza, Controlling a Chemical Coupling Reaction on a Surface: Tools and Strategies for On-Surface Synthesis, Chem. Rev., 2019, 119, 4717–4776 CrossRef CAS
- L. Grill and S. Hecht, Covalent on-surface polymerization, Nat. Chem., 2020, 12, 115–130 CrossRef CAS
- G. Galeotti, M. Fritton and M. Lackinger, Carbon-Carbon Coupling on Inert Surfaces by Deposition of En Route Generated Aryl Radicals, Angew. Chem., Int. Ed., 2020, 59, 22785–22789 CrossRef CAS PubMed
- L. Grossmann,
et al., Synthesis of mesoscale-ordered two-dimensional polymer by on-surface photopolymerization, Nat. Chem., 2021, 13, 730–736 CrossRef CAS
- M. Lackinger, Synthesis on inert surfaces, Dalton Trans., 2021, 50, 10020–10027 RSC
- K. W. Sun, Y. Fang and L. F. Chi, On-Surface Synthesis on Nonmetallic Substrates, ACS Mater. Lett., 2021, 3, 56–63 CrossRef CAS
- A. Rastgoo-Lahrood,
et al., Post-Synthetic Decoupling of On-Surface-Synthesized Covalent Nanostructures from Ag(111), Angew. Chem., Int. Ed., 2016, 55, 7650–7654 CrossRef CAS
- O. Deniz,
et al., Revealing the Electronic Structure of Silicon Intercalated Armchair Graphene Nanoribbons by Scanning Tunneling Spectroscopy, Nano Lett., 2017, 17, 2197–2203 CrossRef CAS
- S. Y. Wang,
et al., On-surface synthesis and characterization of individual polyacetylene chains, Nat. Chem., 2019, 11, 924–930 CrossRef CAS PubMed
- C. Bombis,
et al., Single Molecular Wires Connecting Metallic and Insulating Surface Areas, Angew. Chem., Int. Ed., 2009, 48, 9966–9970 CrossRef CAS PubMed
- P. Ruffieux,
et al., On-surface synthesis of graphene nanoribbons with zigzag edge topology, Nature, 2016, 531, 489–492 CrossRef CAS
- A. Rastgoo-Lahrood,
et al., Reversible intercalation of iodine monolayers between on-surface synthesised covalent polyphenylene networks and Au(111), Nanoscale, 2017, 9, 4995–5001 RSC
- M. Schwarz,
et al., Quantitative determination of a model organic/insulator/metal interface structure, Nanoscale, 2018, 10, 21971–21977 RSC
- C. Brulke,
et al., Quantitative analysis of the electronic decoupling of an organic semiconductor molecule at a metal interface by a monolayer of hexagonal boron nitride, Phys. Rev. B, 2019, 99, 121404 CrossRef
- S. Kawai,
et al., Organometallic Bonding in an Ullmann-Type On-Surface Chemical Reaction Studied by High-Resolution Atomic Force Microscopy, Small, 2016, 12, 5303–5311 CrossRef CAS PubMed
- Q. T. Fan,
et al., Precise Monoselective Aromatic C-H Bond Activation by Chemisorption of Meta-Aryne on a Metal Surface, J. Am. Chem. Soc., 2018, 140, 7526–7532 CrossRef CAS
- M. Zugermeier,
et al., On-surface synthesis of heptacene and its interaction with a metal surface, Nanoscale, 2017, 9, 12461–12469 RSC
- Y. Q. Zhang,
et al., Unusual Deprotonated Alkynyl Hydrogen Bonding in Metal-Supported Hydrocarbon Assembly, J. Phys. Chem. C, 2015, 119, 9669–9679 CrossRef CAS
- F. Hanke, S. Haq, R. Raval and M. Persson, Heat-to-Connect: Surface Commensurability Directs Organometallic One-Dimensional Self-Assembly, ACS Nano, 2011, 5, 9093–9103 CrossRef CAS
- D. P. Woodruff, Quantitative determination of molecular adsorption structures: STM and DFT are not enough, Jpn. J. Appl. Phys., 2019, 58, 100501 CrossRef CAS
- A. Michaelides, K. Reuter and M. Scheffler, When seeing is not believing: Oxygen on Ag(111), a simple adsorption system?, J. Vac. Sci. Technol., A, 2005, 23, 1487–1497 CrossRef CAS
- V. G. Ruiz, W. Liu, E. Zojer, M. Scheffler and A. Tkatchenko, Density-Functional Theory with Screened van der Waals Interactions for the Modeling of Hybrid Inorganic-Organic Systems, Phys. Rev. Lett., 2012, 108, 146103 CrossRef
- C. Bürker,
et al., Exploring the bonding of large hydrocarbons on noble metals: Diindoperylene on Cu(111), Ag(111), and Au(111), Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 165443 CrossRef
- J. Björk and S. Stafstrom, Adsorption of Large Hydrocarbons on Coinage Metals: A van der Waals Density Functional Study, ChemPhysChem, 2014, 15, 2851–2858 CrossRef
- M. Matena,
et al., On-surface synthesis of a two-dimensional porous coordination network: Unraveling adsorbate interactions, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 125408 CrossRef
- K. Berland,
et al., van der Waals forces in density functional theory: a review of the vdW-DF method, Rep. Prog. Phys., 2015, 78, 066501 CrossRef
- M. Di Giovannantonio,
et al., Mechanistic Picture and Kinetic Analysis of Surface-Confined Ullmann Polymerization, J. Am. Chem. Soc., 2016, 138, 16696–16702 CrossRef CAS
- K. A. Simonov,
et al., Synthesis of armchair graphene nanoribbons from the 10,10′-dibromo-9,9′-bianthracene molecules on Ag(111): the role of organometallic intermediates, Sci. Rep., 2018, 8, 3506 CrossRef CAS
- M. Fritton,
et al., The Role of Kinetics versus Thermodynamics in Surface-Assisted Ullmann Coupling on Gold and Silver Surfaces, J. Am. Chem. Soc., 2019, 141, 4824–4832 CrossRef CAS PubMed
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, van der Waals density functional for general geometries, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed
- I. Hamada, van der Waals density functional made accurate, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 121103 CrossRef
- J. Eichhorn,
et al., On-surface Ullmann polymerization via intermediate organometallic networks on Ag(111), Chem. Commun., 2014, 50, 7680–7682 RSC
- M. Lischka,
et al., On-Surface Polymerization of 1,6-Dibromo-3,8-diiodpyrene-A Comparative Study on Au(111) Versus Ag(111) by STM, XPS, and NEXAFS, J. Phys. Chem. C, 2018, 122, 5967–5977 CrossRef CAS
- M. Lischka,
et al., Remote functionalization in surface-assisted dehalogenation by conformational mechanics: organometallic self-assembly of 3,3′,5,5′-tetrabromo-2,2′,4,4′,6,6′-hexafluorobiphenyl on Ag(111), Nanoscale, 2018, 10, 12035–12044 RSC
- M. Lackinger, Surface-assisted Ullmann coupling, Chem. Commun., 2017, 53, 7872–7885 RSC
- H. Walch, R. Gutzler, T. Sirtl, G. Eder and M. Lackinger, Material- and Orientation-Dependent Reactivity for Heterogeneously Catalyzed Carbon-Bromine Bond Homolysis, J. Phys. Chem. C, 2010, 114, 12604–12609 CrossRef CAS
- A. Migani and F. Illas, A systematic study of the structure and bonding of halogens on low-index transition metal surfaces, J. Phys. Chem. B, 2006, 110, 11894–11906 CrossRef CAS PubMed
- L. Guo, M. Wang, X. F. Zeng and D. P. Cao, Luminescent porous organic polymer nanotubes for highly selective sensing of H2S, Mater. Chem. Front., 2017, 1, 2643–2650 RSC
- Q. T. Fan,
et al., Surface Adatom Mediated Structural Transformation in Bromoarene Monolayers: Precursor Phases in Surface Ullmann Reaction, ACS Nano, 2018, 12, 2267–2274 CrossRef CAS
- C. J. Judd,
et al., Structural characterisation of molecular conformation and the incorporation of adatoms in an on-surface Ullmann-type reaction, Commun. Chem., 2020, 3, 166 CrossRef CAS
- P. L. Lalaguna,
et al., Determination of the preferred reaction pathway of acetophenone on Si(001) using photoelectron diffraction, J. Phys.: Condens. Matter, 2021, 33, 214002 CrossRef CAS
- A. Rastgoo Lahrood, J. Björk, W. M. Heckl and M. Lackinger, 1,3-Diiodobenzene on Cu(111) - an exceptional case of on-surface Ullmann coupling, Chem. Commun., 2015, 51, 13301–13304 RSC
- D. P. Woodruff, Surface structure determination using x-ray standing waves, Rep. Prog. Phys., 2005, 68, 743–798 CrossRef CAS
- B. W. Batterman, Effect of Dynamical Diffraction in X-Ray Fluorescence Scattering, Phys. Rev., 1964, 133, A759–A764 CrossRef
- M. J. Bedzyk and G. Materlik, Two-beam dynamical diffraction solution of the phase problem: A determination with x-ray standing-wave fields, Phys. Rev. B: Condens. Matter Mater.
Phys., 1985, 32, 6456–6463 CrossRef CAS
- P. Statiris, H. C. Lu and T. Gustafsson, Temperature-Dependent Sign Reversal of the Surface Contraction of Ag(111), Phys. Rev. Lett., 1994, 72, 3574–3577 CrossRef CAS
- C. Busse,
et al., Graphene on Ir(111): Physisorption with Chemical Modulation, Phys. Rev. Lett., 2011, 107, 036101 CrossRef PubMed
- J. Sforzini,
et al., Approaching Truly Freestanding Graphene: The Structure of Hydrogen-Intercalated Graphene on 6H-SiC(0001), Phys. Rev. Lett., 2015, 114, 106804 CrossRef CAS PubMed
- S. Duhm,
et al., Pentacene on Ag(111): Correlation of Bonding Distance with Intermolecular Interaction and Order, ACS Appl. Mater. Interfaces, 2013, 5, 9377–9381 CrossRef CAS
- S. S. Batsanov, van der Waals radii of elements, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS
- F. Forstmann, W. Berndt and P. Büttner, Determination of the Adsorption Site by Low-Energy Electron Diffraction for Iodine on Silver (111), Phys. Rev. Lett., 1973, 30, 17–19 CrossRef CAS
- K. K. Kim,
et al., Synthesis of Monolayer Hexagonal Boron Nitride on Cu Foil Using Chemical Vapor Deposition, Nano Lett., 2012, 12, 161–166 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nh00486g |
| This journal is © The Royal Society of Chemistry 2022 |