
Photochlorination of toluene – the thin line between intensification and selectivity. Part 1: intensification and effect of operation conditions†
Ümit
Taştan
and
Dirk
Ziegenbalg
 *
*
Institute of Chemical Engineering, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: dirk.ziegenbalg@uni-ulm.de; Fax: +49 731 50 12 25700; Tel: +49 731 50 25703
First published on 7th October 2020
Abstract
This work reports on the potentials of intensifying the industrially applied side-chain photochlorinations of toluene. The application of microstructured reactors enabled an acceleration of the overall reaction up to 10 times compared to batch reactors and up to 100 times compared to the literature. Unexpectedly, this acceleration is accompanied by a decreased selectivity with respect to the desired side-chain chlorinated products. In contradiction to the in general well established theory of photochlorinations, significant amounts of ring chlorinated species were detected. By applying dynamic irradiation conditions, not only the formation of ring-chlorinated products was suppressed but also the energy consumption could be reduced significantly.
1 Introduction
Side-chain chlorinated species are important precursors for the production of a manifold of chemical compounds e.g. benzyl alcohol, benzyl butyl phthalate, benzaldehyde and benzoic acid. The chlorinated intermediates are typically produced by photochlorination of toluene in bubble columns with an annual production of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 t benzyl chloride ((chloromethyl)benzene) and 2000 t benzotrichloride ((trichloromethyl)benzene) – in Europe alone.1 Photochlorinations follow a radical-chain mechanism leading to a stepwise substitution of hydrogen by chlorine.2–5 Consequently, it is important to control the selectivity towards the desired degree of chlorination, even more since the generation of radicals can cause side reactions as well.
000 t benzyl chloride ((chloromethyl)benzene) and 2000 t benzotrichloride ((trichloromethyl)benzene) – in Europe alone.1 Photochlorinations follow a radical-chain mechanism leading to a stepwise substitution of hydrogen by chlorine.2–5 Consequently, it is important to control the selectivity towards the desired degree of chlorination, even more since the generation of radicals can cause side reactions as well.
Bubble columns with a toluene volume of about 100 L or higher are used industrially at temperatures above 70 °C to reduce the fraction of ring-chlorinated side-products.6 Mercury vapour lamps (MVL) are typically applied as light source. Since only a fraction of the total light emission of such lamps (λ < 420 nm) drives the photoreaction,7 the use of narrow-band monochromatic light sources becomes attractive. While the investment costs are rather high for LEDs, the combination of a narrow-band monochromatic emission with a long lifetime of the light source renders LEDs energetic and, thus, economically efficient.8 The feasibility of LEDs in the scope of photochlorinations was demonstrated recently.9 Consequently, new opportunities arise for process intensification through tuning the radiation profile and by adapting the reactor design to the emission characteristics of LEDs, potentially leading to compact high-performance reactors.
This work aims to intensify the photochlorination of toluene, see Fig. 1. Therefore, the impact of various reactor concepts and operation conditions on the reaction rate as well as on conversion and selectivity was examined. This contribution is the first of two articles on this topic and focuses primary on the intensification aspect. A detailed analysis and discussion of the selectivity effects is given in the second article.10
 | ||
| Fig. 1 Reaction pathways in the photochlorination of toluene (main reaction in green, ring-chlorination in blue), desired side-chain chlorination is highlighted. | ||
2 Experimental methods
Experiments were carried out with toluene (HPLC grade from Sigma-Aldrich®). For quantitative determination of product concentration, 1,2-dichlorobenzene (Sigma-Aldrich® 99%) was used as an internal standard for gas chromatographic measurements. The concentration of the internal standard was always 0.45 mol L−1. To ensure a constant concentration of the internal standard, a stock solution was prepared and used for the experiments. The gas chromatograph was calibrated prior to the experiments. A gas chromatograph (Agilent 7890A) with a HP5 column and a 7693 autosampler was used. A volume of 2 μL was injected with a split ratio of 200:1. The initial oven temperature was set to 60 °C. After 8 min the temperature was raised to 110 °C with 15 °C min−1 and hold for 10 min. Following this, the temperature was ramped to 250 °C at a heating rate of 25 °C min−1 and hold for 10 min.
Experimental setup “batch A” consisted of a triple-neck flask as reactor. A reflux cooler was installed on top to prevent loss of reaction solution (see Fig. 2a). The total internal volume of the reactor was 100 mL. The experiments were carried out at room temperature without tempering. For each experiment, the chlorine flow rate, the light source and the irradiation times were varied. All experiments were carried out in a dark room. For all batch setups, the light sources were installed centred below and with a distance of about 2 cm to the reaction vessel. The LEDs were attached to a CPU heat sink with active cooler to provide sufficient cooling. The positions are indicated in Fig. 2.
 | ||
| Fig. 2 (a) Schematic representation of the experimental setup of batch A. (b) Schematic representation of the experimental setup of batch B. | ||
Experimental setup “batch B” enabled experiments at different temperatures by tempering with an external thermostat/cryostat (see Fig. 2b). For these experiments, a modified reflux cooler was used as reactor. An additional glass plate was installed at the bottom side of the cooler. Temperature control of the reaction solution could be achieved with the cooling pipes of the reflux cooler. The total internal volume of the reactor was 50 mL. To install a gas supply for chlorine and nitrogen, two glass threads (GL-14) were fitted to the upper sides of the reactor with Teflon adapters for 1/8′′ capillaries. A second reflux cooler was installed on top to prevent loss of reaction solution. A photograph of the experimental setup is given in the ESI† (see. Fig. S2).
The reaction solutions in both setups were purged continuously with nitrogen. Therewith, HCl and unreacted chlorine were separated from the liquid phase and passed to the gas scrubbing unit. The gas scrubbing part consists of three wash bottles. For safety reasons, an empty bottle was installed directly after the reactor. The second bottle contained a potassium hydroxide (KOH) solution for neutralisation of HCl. The third bottle was filled with sodium sulfite solution and used to remove unreacted chlorine.
A certain amount of toluene was given in the reactor and after degassing the solution with nitrogen, chlorine was bubbled into the reaction solution (100 mL for batch A and 20 mL for batch B). The chlorine flow rate was adjusted by a rotameter (DK47/PV, KROHNE Messtechnik GmbH). The light source was placed at the bottom of the reactor unit and switched on simultaneously with the chlorine gas. After starting the experiments, samples were taken at specific time intervals, purged with nitrogen and analysed via gas chromatography without further purification.
The reaction was stopped after reaching a defined conversion and the chlorine gas was switched to nitrogen. Therewith, chlorine was completely removed from the plant.
The potentials of process intensification were assessed by means of a micro-scale recycle reactor (referred to “microreactor”). The experimental setup of the microreactor is shown in Fig. 3a and described in detail elsewhere.9,11 For each experiment, a certain amount of toluene was placed in the phase separator and continuously pumped in a cycle with a micro annular gear pump (mzr-6355, HNP Mikrosysteme GmbH, Germany). Both fluids, toluene and chlorine, were contacted by using a T-mixer (referred to “T-M”) or a micromixer (referred to “M-M”, see Fig. 3b).
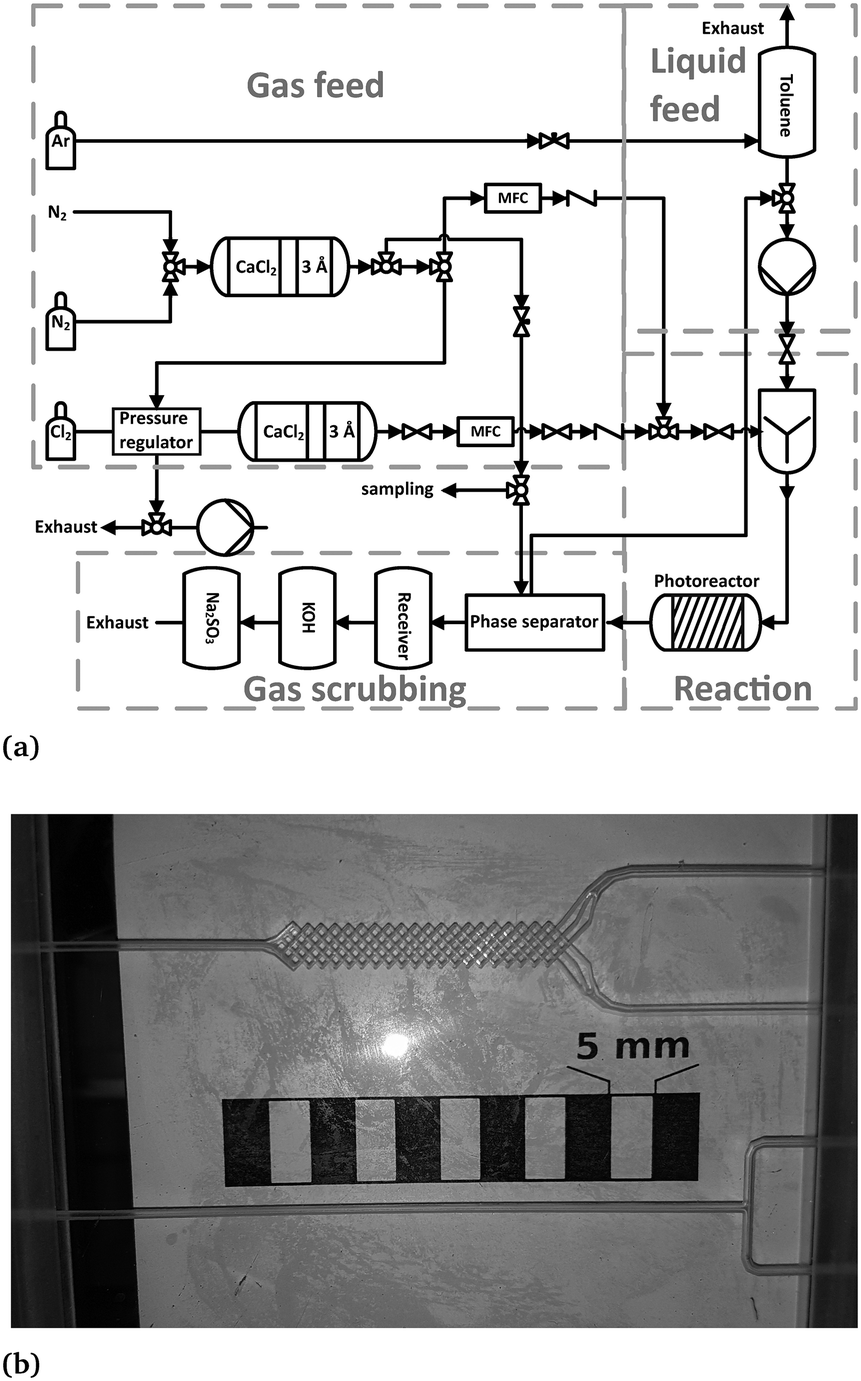 | ||
| Fig. 3 (a) Flow sheet diagram of the microreactor setup.9 (b) Picture of the used mixers. Top: Micromixer, bottom: T-mixer. | ||
The application of such microreactor further allowed altering the position – and thus the timing – of irradiation (see Fig. 4). The LED was installed at the point of contact among chlorine and the liquid reaction mixture. Thus, the gas–liquid-system is irradiated while chlorine is absorbed in the liquid (referred to “irradiation during absorption – IDA”). Alternatively, physical absorption of the reactive gas and initiation of the reaction can be separated by irradiating the reaction solution exclusively after the absorption is completed (referred to “irradiation after absorption – IAA”). This is realised by installing the LED at the end of the capillary used as absorber.
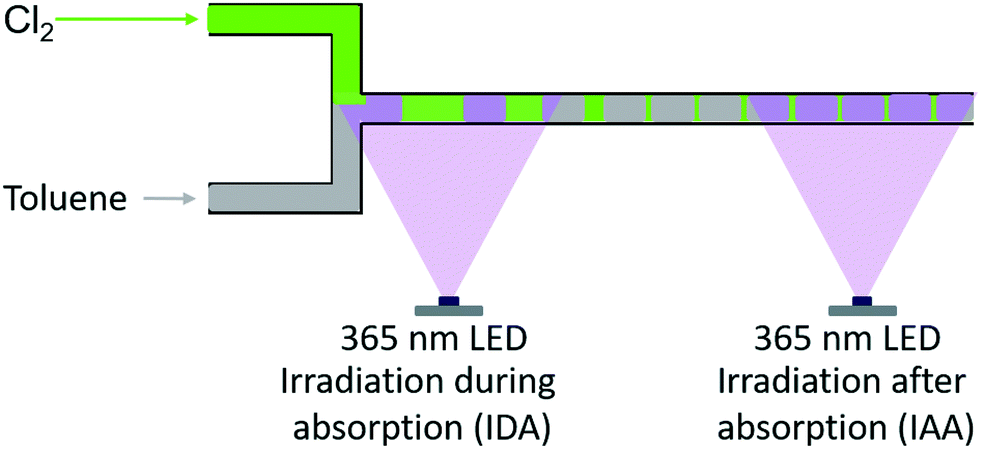 | ||
| Fig. 4 Schematic representation of the different irradiation positions used with the microreactor setup, i.e. irradiation during (IDA) and after absorption (IAA) of chlorine. | ||
Unless stated otherwise, the reaction mixture was irradiated with a LED at the contacting point of both fluids. After leaving the reaction section, the mixture was separated in the phase separator from HCl by purging with N2 and pumped in cycle again. The flow rate of toluene was set to ![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) Tol = 1 mL min−1 if not stated otherwise and the chlorine flow rate was varied between Cl2 = 10 mL min−1 and Cl2 = 25 mL min−1. Table S1 in the ESI† summarises the experimental conditions. The total volume of toluene was VTol = 25 mL, which was continuously recycled through the setup. After starting the experiments, samples were taken at specific time intervals and analysed via gas chromatography without further purification. Experiments with very high chlorine rates were realised through dissolving chlorine at once and subsequent irradiation with two 365 nm LEDs at 600 mA at the contacting point of both fluids. The reaction solution was not circulated in the system. The flow rate of toluene was set to Tol = 1 mL min−1. The flow rate of chlorine was varied between Cl2 = 20 mL min−1 (Q = 0.9 mol min−1 L−1) and Cl2 = 200 mL min−1 (Q = 8.5 mol min−1 L−1).
Tol = 1 mL min−1 if not stated otherwise and the chlorine flow rate was varied between Cl2 = 10 mL min−1 and Cl2 = 25 mL min−1. Table S1 in the ESI† summarises the experimental conditions. The total volume of toluene was VTol = 25 mL, which was continuously recycled through the setup. After starting the experiments, samples were taken at specific time intervals and analysed via gas chromatography without further purification. Experiments with very high chlorine rates were realised through dissolving chlorine at once and subsequent irradiation with two 365 nm LEDs at 600 mA at the contacting point of both fluids. The reaction solution was not circulated in the system. The flow rate of toluene was set to Tol = 1 mL min−1. The flow rate of chlorine was varied between Cl2 = 20 mL min−1 (Q = 0.9 mol min−1 L−1) and Cl2 = 200 mL min−1 (Q = 8.5 mol min−1 L−1).
If not described otherwise, a 365 nm UV LED (NVSU233A, NICHIA Corp., Japan) was used as light source with a constant electrical current of 100 mA (photonflux q = 3.2 × 10−7 mol s−1) or 600 mA (q = 1.9 × 10−6 mol s−1) to initiate the reaction, while the reactions were carried out at room temperature. The photonflux was calculated from the data sheet and represents the photonflux emitted by the LED. With batch A, a blue 400 nm LED (LZ4-00UA00, q = 1.9 × 10−6 mol s−1 @ I = 500 mA, LED ENGIN, USA) was used as well. Both LEDs have a full width at half height (FWHM) of about 20 nm (see Fig. S4 in the ESI†). The influence of a polychromatic emission was evaluated with a mercury vapour lamp (Peschl Ultraviolet GmbH, medium pressure 150 W).
For typical reaction conditions, the availability of chlorine is the limiting parameter.2 To describe the influence of this parameter objectively across all setups, the chlorine feed rate Q, defined as:
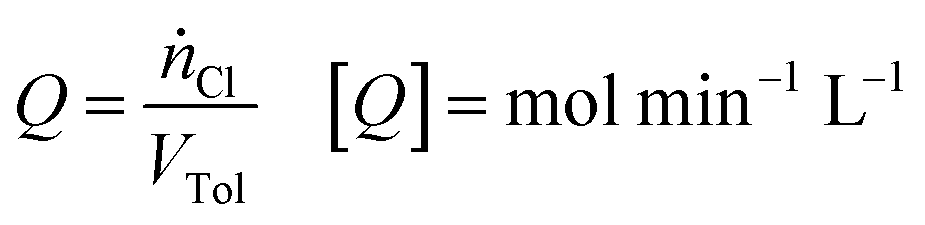 | (1) |
3 Kinetic considerations
By applying the steady-state hypothesis, the effective rate of photochlorinating toluene re can be described for radical species by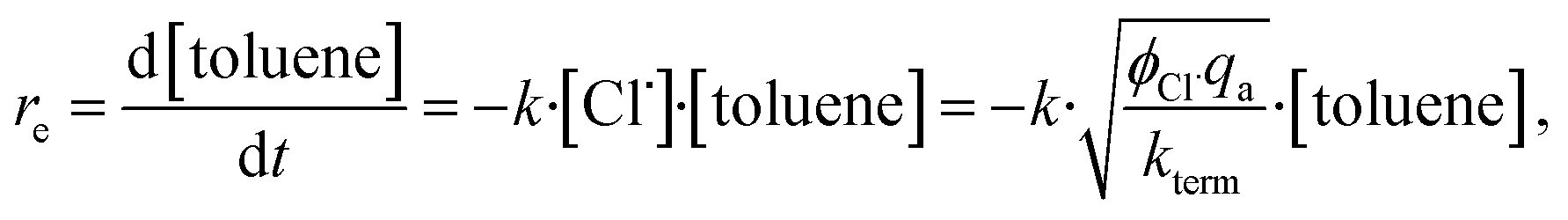 | (2) |
| re = −k′·qa1/2, | (3) |
4 Results and discussion
Acceleration of the apparent reaction rate is the most prominent aspect of intensifying reactions and can be realised by microstructured devices as one approach to maximise the effectiveness of inter-molecular events.12 Consequently, the potentials of intensification of the photochlorination were evaluated by comparing results from the batch setups with the microreactor setup. The impact of the most prominent parameters governing the reaction rate, i.e. the chlorine feed rate Q, the reaction temperature T, the photon flux q, the wavelength λ of the incident photons and pulsed irradiation, was thoroughly evaluated.For all investigated chlorine feed rates in the respective batch reactors (batch A and batch B), increasing the chlorine feed rate Q led to a faster overall reaction rate and a linear increase of toluene conversion was observed (see Fig. 5(a)). Furthermore, a linear correlation between the apparent pseudo zeroth-order reaction rate with Q was found (see section 3). In previous reports,2 chlorine feed rates exceeding Q = 0.034 mol min−1 L−1 were found to lead to conditions where the reaction rates are independent of Q. In contrast, even for Q ≈ 0.45 mol min−1 L−1, representing the technical limits of the batch setup, the apparent reaction rate correlates linear with the chlorine feed rate (see Fig. 6). Investigations with the microreactor revealed a persistent linear increase of the apparent rate constant k′ up to a chlorine feed rate of Q ≈ 4 mol min−1 L−1 (see Fig. 6). For larger feed rates, the apparent rate constant k′ still increases but with a smaller slope, indicating the presence of a rate limiting step. It has to be emphasised that the correlation between the chlorine feed rate Q and the apparent rate constant k′ is linear for such a large parameter range and for three different reactor setups. Considering the operation conditions where the slope of the apparent rate constant change as the point of maximum possible intensification for the investigated setups, an intensification by a factor of 10 was achieved by switching from a batch reactor to a microreactor. Compared to literature, an intensification factor of over 100 was realised.
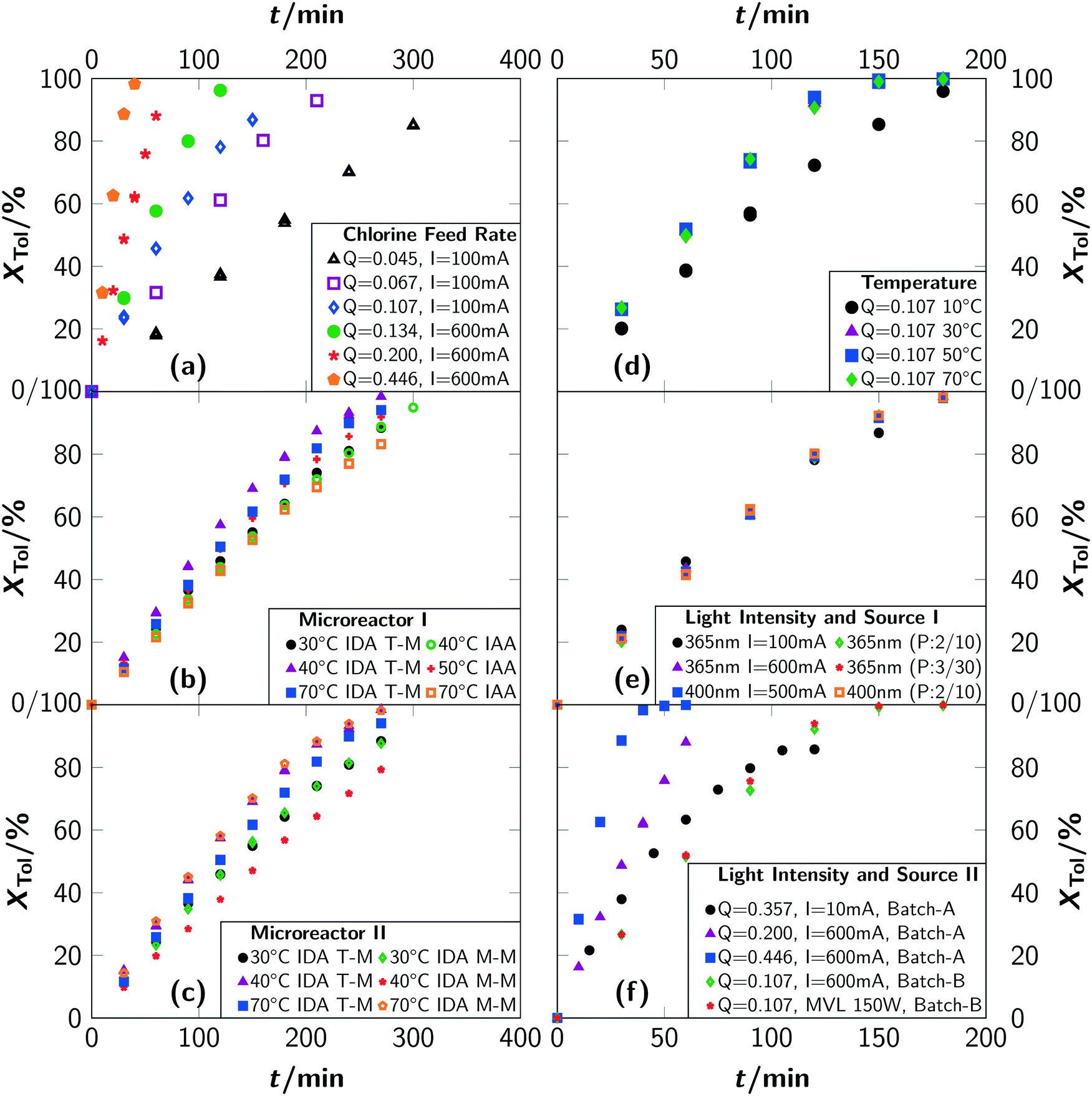 | ||
| Fig. 5 Conversion of toluene for different reaction conditions: (a) chlorine feed rate Q dependencies for batch A (I = 100 mA) and batch B (I = 600 mA) at room temperature. (b) and (c) Temperature dependencies in different microreactor setups. Experiments were carried out with a constant chlorine feed rate of Q = 0.045 mol min−1 L−1 with two 365 nm LEDs at 600 mA. “T-M” indicates the T-mixer and “M-M” indicates the micromixer. “IDA” indicates irradiation during absorption and “IAA” indicates irradiation after absorption. (d) Temperature dependencies for experiments in batch B with a 365 nm LED at 600 mA. (e) Photon flux dependencies for experiments in batch B for continuous and pulsed operation of LEDs. A 365 nm or a 400 nm LED was used as light source and operated at different driving currents. “P:x/y” indicates an irradiation for x seconds with a pause between irradiations of y seconds. The chlorine feed rate was set to Q = 0.107 mol L−1 min−1. (f) Influence of the type and operation of the light source in batch A and batch B. “MVL” indicates mercury vapour lamp. Q is given in mol L−1 min−1. | ||
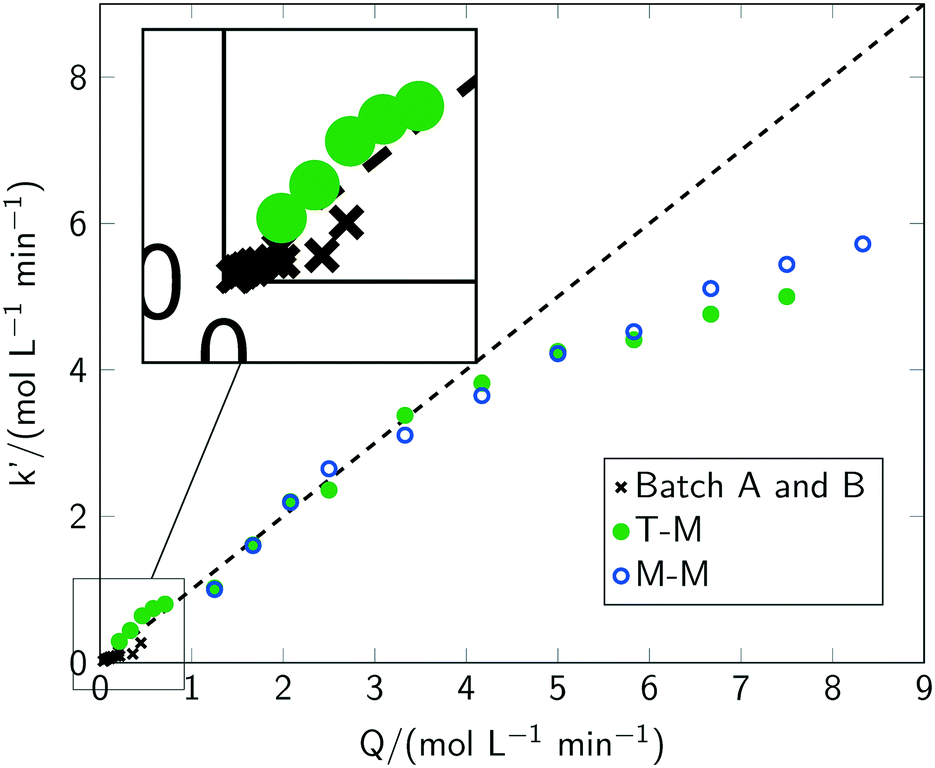 | ||
| Fig. 6 Apparent rate constant for toluene conversion as a function of the chlorine feed rate Q for batch A and two versions of the microreactor. “T-M” indicates the T-mixer and “M-M” indicates the micromixer. The insert highlights the results for the batch setup. | ||
An analysis of the temperature dependency in batch B yielded no influence for a temperature range of T = 30 °C to 70 °C. For T = 10 °C, a lower reaction rate as well as a drop in selectivity towards benzyl chloride was observed (see Fig. 5(d) and 7(d)). Gas chromatographic analysis allowed to unambiguously associate the lower selectivity to the formation of ring-chlorinated side-products at lower reaction temperatures.
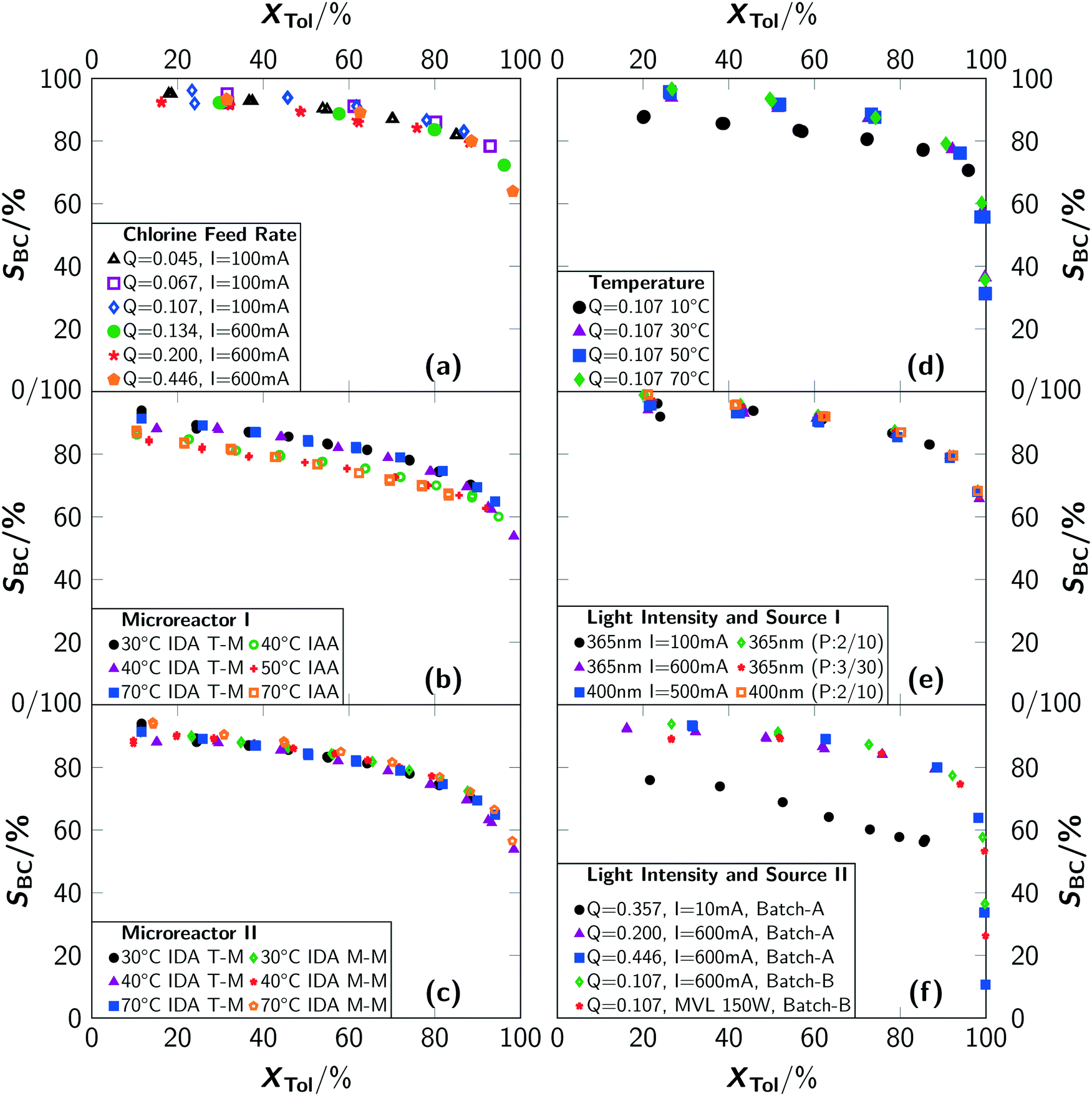 | ||
| Fig. 7 Conversion of toluene to selectivity of benzyl chloride correlations for different reaction conditions: (a) chlorine feed rate Q dependencies for batch A (I = 100 mA) and batch B (I = 600 mA) at room temperature. (b) and (c) Temperature dependencies in different microreactor setups. Experiments were carried out with a constant chlorine feed rate of Q = 0.045 mol min−1 L−1 with two 365 nm LEDs at 600 mA. “T-M” indicates the T-mixer and “M-M” indicates the micromixer. “IDA” indicates irradiation during absorption and “IAA” indicates irradiation after absorption. (d) Temperature dependencies for experiments in batch B with a 365 nm LED at 600 mA. (e) Photon flux dependencies for experiments in batch B for continuous and pulsed operation of LEDs. A 365 nm or a 400 nm LED was used as light source and operated at different driving currents. “P:x/y” indicates an irradiation for x seconds with a pause between irradiations of y seconds. The chlorine feed rate was set to Q = 0.107 mol L−1 min−1. (f) Influence of the type and operation of the light source in batch A and batch B. “MVL” indicates mercury vapour lamp. Q is given in mol L−1 min−1. | ||
For the microreactor, the reaction progress was partially influenced by the temperature but no reliable correlation between temperature and reaction rate could be determined, as the registered differences were of stochastic character (see Fig. 5(b) and (c)). An influence of the temperature on the selectivity towards benzyl chloride was not found. Despite this, operation of the microreactor setup in irradiation-after-absorption-mode (IAA) always yielded lower reaction rates and selectivities as when operated in irradiation-during-absorption-mode (IDA). A significant difference between the two types of mixer could not be observed with respect to conversion. The selectivity was found to be slightly higher when using the micromixer M-M instead of the T-mixer (T-M) (see Fig. 7(c)).
In general, the selectivity towards benzyl chloride was found to be independent of the chlorine feed rate (see Fig. 7(a)) for the same reactor type. A comparison between the reactor types showed the highest selectivities for the batch reactor (see Fig. 7(a)–(c)). It is surprising that independent conversion–selectivity-correlations exist for each reactor type, when extreme conditions (e.g. very high T or very low q) are excluded. The reasons for this observation are elaborated in the second article.10
Under typical reaction conditions, limitation by the photonflux is unlikely since quantum yields exceeding 105 are reported for photochlorinations. This assessment was verified by varying the irradiation conditions. For typical experiments, which were conducted with electrical currents to drive the LED of I = 100 mA and more, no chlorine was detected at the outlet of batch B, proofing full conversion of chlorine. The reaction progress was independent of the used electrical current (see Fig. 5(e) and (f)). Only for very low photon fluxes, residual chlorine leaving the reactor was detected. For a current of I = 10 mA, which was the lowest value that still resulted in a light emission of the LED, it was impossible to convert all chlorine fed to the reactor (see Fig. 5(f)). This resulted in a low reaction rate even at a high chlorine feed rate together with a reduced selectivity towards benzyl chloride (see Fig. 7(f)). As side-products, ring-chlorinated toluenes (2- and 4-chlorotoluene) were observed. Since such an effect was not observed for higher electrical currents, it can be concluded that the photonflux is the limiting parameter when operating the LED with a very low electrical current but not when operating at currents of I = 100 mA or above.
Obviously and besides the photon flux, the excitation wavelength can influence the reaction progress severely either by altering the reaction path or the actually absorbed photon flux. With batch A, the effect of a blue 400 nm LED (q = 9.1 × 10−6 mol s−1) instead of the 365 nm LED was assessed. No changes to the reaction rate or the selectivity were observed (see Fig. 5(e) and 7(e)). This finding further supports the interpretation that the photonflux is not the limiting parameter even when the fraction of absorbed photons is lower for the blue LED due to the lower spectral absorption coefficient of chlorine (see Fig. S1 in the ESI†).13
Narrow-band monochromatic light sources such as LEDs increase the energy efficiency of photochemical processes – translating into economic benefits.8 Furthermore, LEDs offer the opportunity to easily pulse the irradiation. While pulsation would lead to a fast ageing of e.g. mercury vapour lamps, the performance of LEDs does not change when operated in pulse mode. In contrast, it is even possible to realise larger light outputs during a single pulse in comparison to continuous operation. During dark phases, heat can easily dissipate from the chip, allowing higher electrical driving currents during pulses.
Pulsed operation can be highly beneficial for photochemically initiated radical reactions such as photochlorinations since it potentially allows a further improvement of the energy efficiency. During dark phases, no energy is consumed while the reaction is still ongoing. Consequently, the effect of a pulsed operation was investigated employing batch A. Two different pulse sequences were investigated with a 365 nm LED and a 400 nm LED, respectively. Sequence A consisted of a 2 s irradiation and a 10 s dark phase, thus the light source was active only during roughly 17% of the total sequence duration. In contrast, for sequence B, the light pulse had a duration of 3 s and the dark phase lasted 30 s. Hence, the LED was switched on for about 9% of the time. The results are summarised in Fig. 5(e).
Independent of the light source and the pulse sequence, the reaction progresses with the same apparent rate. This leads to the conclusion that for continuous operation of LEDs either the radical chain length/the apparent quantum yield is lower or a significant fraction of photons does not trigger the homolytic Cl–Cl bond cleavage. A pulsed operation of LEDs can indeed improve the energetic efficiency of photochlorinations. Depending on the sequence, 83% or even 91% of the electrical energy used to drive the LEDs can be saved.
Using a pulsed irradiation sequence increases the selectivity towards benzyl chloride as well. A selectivity of 99% was found at a conversion of 20% for the 365 nm and 400 nm LED when pulsed with sequence A. For sequence B and the 365 nm LED, a selectivity of 97% was recorded. Continuous operation of this LED yielded only selectivities of 95%.
Due to the missing tempering possibilities in batch A, it was impossible to ensure a constant reaction temperature when using a mercury vapour lamp as light source. Hence, the effect of a mercury vapour lamp was evaluated with batch B; the results are shown in Fig. 5(f). The reaction rate does not change for the mercury vapour lamp when compared to the 365 nm LED.
Interestingly, the selectivity towards benzyl chloride SBC decreases for low conversions when using the mercury vapour lamp (see Fig. 7(f)). This lower selectivity stems from an increased formation of side-products (see Fig. 8), which were analysed to be mainly ring-chlorinated benzyl and benzal chlorides.
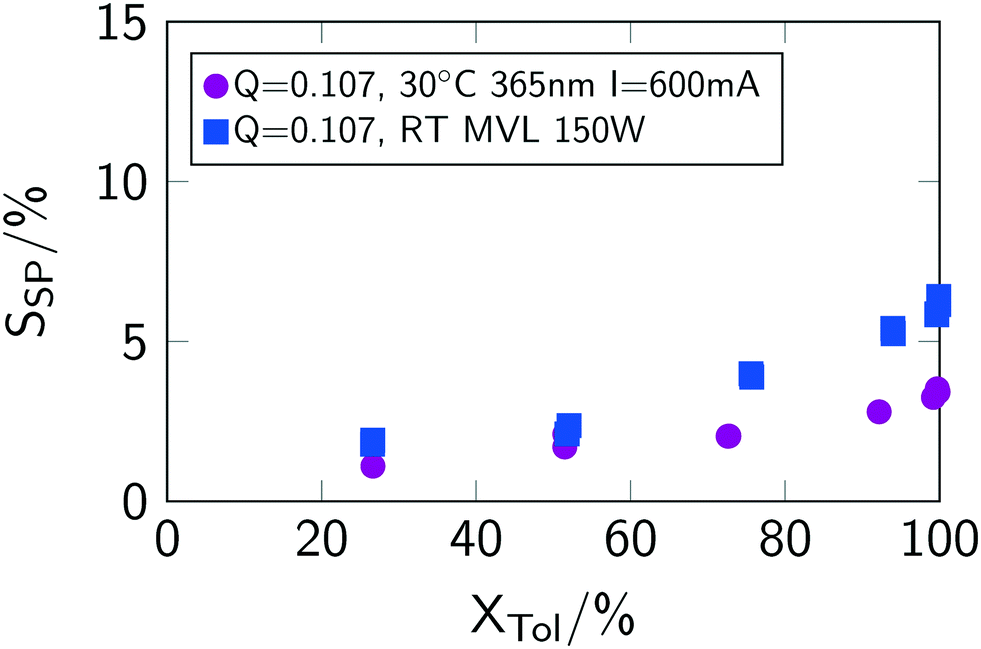 | ||
| Fig. 8 Conversion–selectivity of side-products correlation for different light sources. Q is given in mol L−1 min−1. “MVL” indicates mercury vapor lamp. | ||
The observations made with respect to the selectivity towards benzyl chloride for different reactor setups, light sources and the pulsed irradiation can not be explained with the established theory of photochlorinations, since only the generation of chlorine radicals would be relevant in this theory. The subsequent chain reaction should be independent of these parameters. Except from impurities, the theory on photochlorinations considers only the temperature as a relevant reaction parameter that influences the selectivity.6 Considering the homolytic cleavage of the chlorine bond, none of the identified influencing parameters has an impact. Furthermore, the reduced selectivity was found to be correlated to the formation of ring-chlorinated species that are not observed for other reaction conditions in this work. Elaboration of the reasons for this unexpected selectivity changes is out of scope of this work and therefore content of part 2 of this work.10
5 Conclusions
The presented results show that an intensification of the photochlorination of toluene by a factor of more than 10 with respect to the reaction rate is possible. Compared to published data, a 100 times faster reaction rate could be achieved. It can be concluded that the supply of chlorine to the reaction solution is the rate limiting parameter which has to be addressed to gain intensification. The use of a microreactor is a viable option to realise this. In most cases, the reaction rate is insensitive to other reaction parameters such as the temperature or the light intensity. Operating LEDs in pulsed mode allows for energy savings of more than 90% and leads as a second benefit to higher selectivities.For most batch experiments, the operation conditions did not influence the selectivity. However, a decrease of the selectivity towards benzyl chloride was registered at extreme conditions (very low T and q) and – even more interestingly – in case of the mercury vapour lamp. A comparison between the batch setups and the two microreactor setups revealed individual conversion–selectivity correlations for each setup. Highest selectivities could be observed in the batch reactors, followed by the microreactor with irradiation during absorption and with irradiation after absorption.
In conclusion, the benefit of intensification strongly depends on the operation conditions and might be restricted by an increased formation of ring-chlorinated side-products. Formation of these species is in contradiction to the established theory of photochlorinations. Furthermore, there is no obvious explanation for the drop in selectivity for the microreactor setups or the increased selectivities found for a pulsed operation of LEDs. Due to the high relevance of the selectivity, the reasons for these correlations are elaborated in part 2 of this work.10
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support provided by the German Research Foundation within the Priority Program “Reactive Bubbly Flows”, SPP 1740 (ZI 1502/1-1) as well as within the TRR CATALIGHT – Projektnummer 364549901 – TRR234 (project C6). Dirk Ziegenbalg thanks Prof. Dr.-Ing. Elias Klemm (ITC, University of Stuttgart) for continuous support.References
- INEOS AG, INCH Magazine, 2014, vol. 7, p. 30 Search PubMed.
- H. G. Haring and H. W. Knol, Chem. Process Eng., 1964, 560–567 Search PubMed
.
- H. G. Haring and H. W. Knol, Chem. Process Eng., 1964, 619–623 CAS
- H. G. Haring and H. W. Knol, Chem. Process Eng., 1964, 691–693 Search PubMed
-
K.-A. Lipper, E. Löser and O. Brücher, in Ullmann's Encyclopedia of Industrial Chemistry, 2017, pp. 1–22 Search PubMed
-
A. M. Braun, M.-T. Maurette and E. Oliveros, Photochemical Technology, John Wiley & Sons Ltd, New York, 1991 Search PubMed
- D. J. Seery and D. Britton, J. Phys. Chem., 1964, 68, 2263–2266 CrossRef CAS
- M. Sender and D. Ziegenbalg, Chem. Ing. Tech., 2017, 89, 1159–1173 CrossRef CAS
- Ü. Taştan, F. Guba and D. Ziegenbalg, Chem. Eng. Technol., 2017, 40, 1418–1424 CrossRef
- Ü. Taştan, P. Seeber, S. Kupfer and D. Ziegenbalg, React. Chem. Eng., 2020 10.1039/d0re00366b
-
Ü. Taştan, PhD thesis, Ulm University, Institute of Chemical Engineering, 2018 Search PubMed
- T. V. Gerven and A. Stankiewicz, Ind. Eng. Chem. Res., 2009, 48, 2465–2474 CrossRef
- Ü. Taştan, J. Dollinger and D. Ziegenbalg, Flow Meas. Instrum., 2018, 59, 211–214 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00263a |
| This journal is © The Royal Society of Chemistry 2021 |