
Ultrafast and continuous-flow synthesis of AFX zeolite via interzeolite conversion of FAU zeolite†
Tatsushi
Yoshioka
a,
Zhendong
Liu
 *ab,
Kenta
Iyoki
a,
Anand
Chokkalingam
a,
Yasuo
Yonezawa
a,
Yuusuke
Hotta
c,
Ryohji
Ohnishi
c,
Takeshi
Matsuo
c,
Yutaka
Yanaba
d,
Koji
Ohara
e,
Takahiko
Takewaki
c,
Tsuneji
Sano
a,
Tatsuya
Okubo
a and
Toru
Wakihara
*ab
*ab,
Kenta
Iyoki
a,
Anand
Chokkalingam
a,
Yasuo
Yonezawa
a,
Yuusuke
Hotta
c,
Ryohji
Ohnishi
c,
Takeshi
Matsuo
c,
Yutaka
Yanaba
d,
Koji
Ohara
e,
Takahiko
Takewaki
c,
Tsuneji
Sano
a,
Tatsuya
Okubo
a and
Toru
Wakihara
*ab
aDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: liuzd@chemsys.t.u-tokyo.ac.jp; wakihara@chemsys.t.u-tokyo.ac.jp
bInstitute of Engineering Innovation, The University of Tokyo, 2-11-16 Yayoi, Bunkyo-ku, Tokyo 113-8656, Japan
cMitsubishi Chemical Corporation, Science and Innovation Center, Kamoshida-cho, Aoba-ku, Yokohama 227-8502, Japan
dInstitute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8505, Japan
eJapan Synchrotron Radiation Research Institute/SPring-8, 1-1-1 Kouto, Sayo-gun, Hyogo 679-5198, Japan
First published on 9th September 2020
Abstract
AFX zeolite is a small-pore zeolite that has attracted significant attention because of its potential as a high-performance catalyst for the selective catalytic reduction of NOx by ammonia (NH3-SCR). Long time and high cost for the synthesis of AFX zeolite, however, limit the practical applications of this small-pore zeolite. Herein, we report an ultrafast synthesis route that can yield highly crystalline AFX zeolite in as short as 12 min. The ultrafast synthesis was realized on the basis of interzeolite conversion, where FAU zeolite was employed as a source of silicon and aluminum. Moreover, the addition of acid-leached seeds was found to be crucial for promoting the crystallization of AFX zeolite while effectively eliminating the formation of byproducts. Detailed studies of the original and acid-leached seeds revealed that acid leaching could cause structural alterations to the original seeds, thereby significantly enhancing the seeding effect. Secondary nucleation occurred when the acid-leached seeds were added, which is assumed to be one of the primary reasons for the accelerated crystallization rate. Based on the ultrafast synthesis, continuous-flow synthesis of AFX zeolite in 10 min was also demonstrated. The ultrafast and continuous-flow synthesis of AFX zeolite is expected to facilitate the large-scale production and broad applications of AFX zeolite.
Introduction
Small-pore zeolites, which feature eight-membered-rings (8MR) as the largest pore, have been extensively studied in the zeolite community because of their applications as catalysts in the methanol-to-olefins (MTO) process and selective catalytic reduction of NOx by ammonia (NH3-SCR).1,2 Other reasons for exploring small-pore zeolites include their prospective applications, such as gas sorption and separation.3 While CHA is the most studied zeolite in this family, others have also been extensively researched for their unique features compared to those of CHA. AFX zeolite is among those that have attracted considerable attention. The AFX framework is a member of the ABC six-ring family of zeolite structures and is constructed by the AABBCCBB sequence of six-membered rings, which forms aft and gme cages (Fig. S1†).4 The first microporous material with AFX structure was synthesized in the silicoaluminophosphate (SAPO) form, named SAPO-56. Later (in 1993), Zones reported the aluminosilicate form of AFX, which was named SSZ-16.5 The aluminosilicate AFX is considered to be promising for NH3-SCR.6,7 For example, Priya et al. reported that copper-exchanged SSZ-16 exhibited a higher NO conversion in the NH3-SCR than did other copper-exchanged zeolites, even at high temperatures.8AFX zeolite was first synthesized with a heterocyclic compound, which is a derivative of quinuclidine, as the organic structure-directing agent (OSDA), and it took 6 days to yield the crystalline product.5 Thereafter, several synthesis routes for AFX zeolite have been reported.9–15 In general, a relatively long synthesis period (6–21 days) is required. Recently, the synthesis of small-pore zeolites using a pre-made zeolite (in most cases, an FAU type) as the starting material, instead of amorphous aluminum and silica sources, has gained much attention (called as interzeolite conversion).16,17 The FAU framework is composed of sod cages that are connected to each other via double 6-membered-rings (d6r). The AFX and FAU frameworks, albeit differing in cage type and pore size, are similar because both contain d6r as a common composite building unit.20 Interzeolite conversion has the potential to shorten the synthesis period, most likely because it represents a more kinetically favorable crystallization pathway.18,19 For example, Goel et al. proposed that the presence of common composite building units between the starting and resultant zeolites could help overcome the kinetic barrier during synthesis.21 Itakura et al. conducted the synthesis of RUT zeolite from different zeolites and found that faster synthesis could be achieved when zeolites as the starting material and product possessed common composite building units.22 The synthesis of AFX zeolite could be accelerated when FAU zeolite is used as the starting material. For instance, Yamada et al. reported the synthesis of AFX zeolite in 2 days by replacing the silica and alumina sources with a commercial FAU zeolite.23 Nakazawa et al. also reported synthesis in 43 h from a reaction mixture containing FAU zeolite and a rigid diquaternary ammonium compound, TEBOP (N,N,N′,N′-tetraethyl-exo,exo-bicyclo[2.2.2]oct-7-ene-2,3:5,6-dipyrrolidinium dihydroxide).24 Because the use of colloidal or fumed silica as a Si source under the same conditions led to the formation of the *BEA phase, the structural similarity between FAU and AFX was considered to be important for the efficient synthesis of AFX zeolite.25
Over the years, we have been developing a strategy for the ultrafast synthesis of zeolites using a tubular reactor (Fig. S2†) as a mini-autoclave with the feature of rapid thermal response and have succeeded in synthesizing a series of zeolites in minutes rather than hours or days, which is typically required in a normal protocol.26–31 Based on the ultrafast synthesis, the continuous-flow synthesis of zeolites has also been demonstrated.32–34 A general requirement for establishing continuous-flow synthesis of a zeolite is that the thermodynamics (in terms of the framework of the resultant product) and kinetics (in terms of the crystallization rate) of the hydrothermal synthesis and the hydrodynamics in the continuous-flow reactor should be correlated together. Assuming that the synthesis yields the desired phase, a shorter synthesis time is preferable for continuous-flow synthesis. Correspondingly, a shorter residence time is needed, thus facilitating a better reactor design and reducing the operation cost. Recently, our group reported the fast synthesis of AFX zeolite, which yielded highly crystalline AFX zeolite in as short as 2 h in a tubular reactor. The fast-synthesized AFX zeolite showed excellent hydrothermal stability and NH3-SCR activity.35 The 2 h synthesis time, however, was still too long to facilitate continuous-flow synthesis, because precipitation-related hydrodynamic failures could easily occur owing to the long residence time.
Herein, we report an ultrafast synthesis route that can yield highly crystalline AFX zeolite in as short as 12 min. The ultrafast synthesis was realized based on the so-called interzeolite conversion, where FAU zeolite was employed as a source of both silicon and aluminum. Moreover, the addition of acid-leached seeds was crucial for inducing the crystallization of the AFX zeolite while effectively eliminating the formation of the byproduct. Based on the ultrafast synthesis, continuous-flow synthesis of AFX zeolite in 10 min was also successfully demonstrated. The ultrafast and continuous-flow synthesis of AFX zeolite is expected to facilitate the large-scale production and broad applications of this small-pore zeolite.
Experimental section
Chemicals
Sodium hydroxide (50% aqueous solution; FUJIFILM Wako Pure Chemical; super special grade), aluminum nitrate nona-hydrate (FUJIFILM Wako Pure Chemical; guaranteed reagent), fumed silica (Aerosil® 200), FAU zeolite (Tosoh; HSZ-350HUA; Si/Al ratio = 5.5), 1 M sulfuric acid (FUJIFILM Wako Pure Chemical; for volumetric analysis), and ion-exchange resin (Dow Chemical; DOWEX™ MONOSPHERE™ 550A (OH)) were used as received.Synthesis of the AFX zeolite seeds
The AFX zeolite seeds were synthesized with N,N′-bis-triethyl-pentanediyldiammonium dibromide (Et6-diquat-5) as an OSDA, and the recipe was slightly modified from that reported in a previous work.13 Deionized water, sodium hydroxide solution, Et6-diquat-5, aluminum nitrate nonahydrate, and fumed silica were mixed in a 500 mL polypropylene bottle. The molar composition of the initial reaction mixture was 1 SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.017 Al2O3:1 NaOH:0.1 Et6-diquat-5:40 H2O. The mixture was stirred at 25 °C at 500 rpm overnight and then transferred to a Teflon®-lined stainless-steel autoclave, which was placed in an oven for synthesis at 150 °C for 7 days. The solid product was collected by filtration, washed with deionized water, and dried at 80 °C overnight. The crystalline zeolite was labeled the as-made seeds.
:0.017 Al2O3:1 NaOH:0.1 Et6-diquat-5:40 H2O. The mixture was stirred at 25 °C at 500 rpm overnight and then transferred to a Teflon®-lined stainless-steel autoclave, which was placed in an oven for synthesis at 150 °C for 7 days. The solid product was collected by filtration, washed with deionized water, and dried at 80 °C overnight. The crystalline zeolite was labeled the as-made seeds.
Beadmilling of the seeds
The as-made seeds (8 g) were dispersed in 300 mL of deionized water. The slurry was milled using a bead milling apparatus (LMZ015, Ashizawa Finetech Ltd., Japan) for 30 min using zirconia beads with a diameter of 300 μm. The milled zeolite was collected by centrifugation and dried at 80 °C overnight. The obtained zeolite was named the milled seeds.Acid-leaching of the seeds
The as-made seeds were dispersed in 0.5 M sulfuric acid (e.g., 0.9 g to 20 mL sulfuric acid), and the mixture was stirred for 27 h at 80 °C. Thereafter, the solid was recovered by centrifugation, washed with deionized water, and dried at 80 °C. The obtained zeolite was named the acid-leached seeds.Ultrafast synthesis of the AFX zeolite
TEBOP was synthesized following a procedure reported in previous works36,37 and exchanged by ion-exchange resin to obtain the OH− form. In a typical run of the ultrafast synthesis, deionized water, sodium hydroxide solution, TEBOP solution, and FAU zeolite were mixed in a 50 mL centrifuge tube. The chemical composition of the initial reaction mixture was 1 SiO2:0.092 Al2O3:0.25 NaOH:0.095 OSDA:30 H2O. The mixture was stirred at 90 °C for 18 h to ensure that it was fully homogenized, after which one of the types of seeds was added (or none were). Synthesis was conducted in a tubular reactor26 heated in an oil bath. A tubular reactor is an enclosed stainless-steel tube with a length of ca. 13.5 cm. Both sides of the tube were sealed with standard stainless-steel caps. A tubular reactor has a larger surface-to-volume ratio than conventional autoclaves and enables fast heating when combined with a preheated oil bath. Synthesis was conducted at 210 °C for 12 min. The obtained solid was collected by centrifugation, washed with deionized water, and dried at 80 °C. For comparison, a synthesis was conducted with an aging period and temperature of 2 h and 25 °C, respectively.
Continuous-flow synthesis of the AFX zeolite
A continuous-flow reactor was designed and built, which consisted of a core–shell structured flow reactor. A stainless-steel tube with an inner diameter of 3.2 mm was employed as the outer pipe, and a Teflon® tube (inner and outer diameter were 1 and 2 mm, respectively) was inserted as the inner tube; the two tubes formed a core–shell structure. Preheated hot water (240 °C, 6.2 cm3 min−1) passed through the shell, while the core contained the zeolite precursor (1.1 cm3 min−1), which was mixed with AFX seeds prior to the synthesis and stored in a tank. This design enabled the reaction mixture to be rapidly heated to the target temperature. Synthesis was conducted at 210 °C with a residence time of 10 min. The reactor was placed in an air-circulated oven (217 °C) to precisely maintain the synthesis temperature during the whole synthesis period. To avoid precipitation, the continuous-flow reactor was shaken with an air-driven vibrator during synthesis. The slurry flowing out of the reactor was cooled down to under 100 °C by cooling water (20 cm3 min−1). After shutting down the reactor, the sample was collected, recovered by centrifugation, washed with deionized water, and dried at 80 °C.Characterization
Powder X-ray diffraction (XRD) patterns of the samples were collected on an Ultima IV X-ray diffractometer (Rigaku, Japan) using CuKα radiation (λ = 0.15406 nm, 40 kV, 40 mA) at a scanning rate of 10° min−1. The relative crystallinity of the AFX phase was calculated based on the area of peaks at ca. 17.6, 21.9, 31.7, and 33.6°. The size and morphology of the products were observed by scanning electron microscopy (SEM, JSM-7000F, JEOL, Japan) with an accelerating voltage of 15 keV. Nitrogen adsorption–desorption measurements were carried out with an Autosorb-iQ instrument (Quantachrome Instruments) at −196 °C. Before the measurement, the samples were calcined in air at 550 °C for 5 h. Degassing was carried out at 150 °C for 30 min, followed by an additional 240 min at 350 °C. The solid state magic angle spinning nuclear magnetic resonance (MAS NMR) spectra were collected by a JNM-ECA 500 instrument (JEOL, Japan). The 29Si dipolar decoupling (DD) MAS NMR spectra were recorded by collecting 2048 scans at 99.37 MHz with a π/2 pulse length of 5.0 μs, a recycle delay of 60 s, and a spinning frequency of 10 kHz. 29Si cross polarization (CP) MAS NMR measurements were carried out with a recycle delay of 1.5 s and a spinning frequency of 6 kHz. The 27Al MAS NMR spectra were recorded at 130.33 MHz with a π/2 pulse length of 3.2 μs, a recycle delay of 5 s, and a spinning frequency of 15 kHz. Depending on the purpose of the analysis, the samples were measured in their as-made form or their protonated form, which were calcined in air, ion-exchanged by 1 M NH4NO3 solution three times, and calcined again. The elemental composition of the samples was determined by Thermo iCAP 6300 inductively coupled plasma atomic emission spectrometry (ICP-AES). High-energy X-ray total scattering (HEXTS) measurements were conducted at the BL04B2 high-energy XRD beamline (SPring-8, Japan). The scattering patterns of the samples were collected at room temperature under vacuum conditions. The obtained scattering patterns were corrected and normalized by various factors to calculate the total structure factor S(Q) (Q = 4πsinθ/λ).38 The reduced pair distribution functions, G(r), were obtained by implementing Fourier transformation, as expressed in the following equation:
The energy of the incident X-ray was 61.2 keV, and the maximum Q (Qmax) collected was 25.74 Å−1.

Results and discussion
To realize faster crystallization of AFX, the composition of the initial precursor was further optimized. Synthesis with the modified recipe was attempted at 210 °C without the addition of seeds but in the presence of OSDA (TEBOP). Owing to the employment of an appropriate starting material, high temperature, and fast heating, the synthesis period was shorter than that previously reported. According to the XRD patterns in Fig. 1(a), the crystallization proceeded very quickly, and the conversion of FAU was completed in just 60 min. However, the product was not a pure AFX phase but contained a considerable amount of the ANA phase, as indicated by both the XRD patterns and the SEM image in Fig. 1(b). ANA is often observed as a co-crystallizing phase in the synthesis of AFX zeolite.39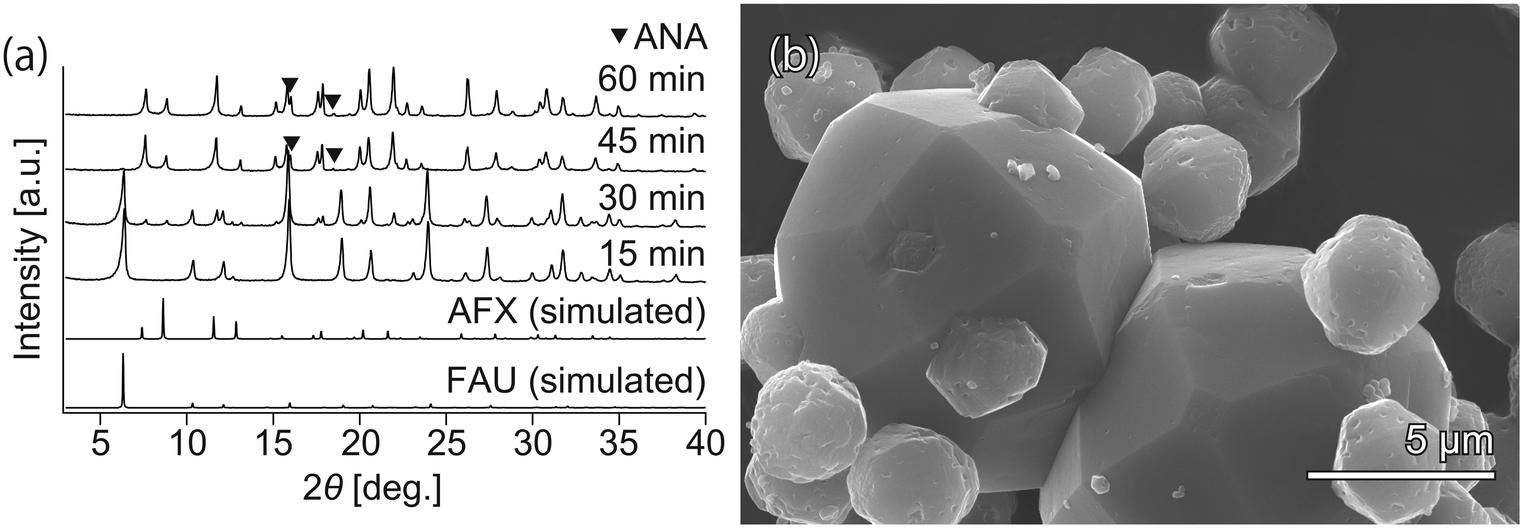 | ||
| Fig. 1 (a) Powder XRD patterns and (b) SEM image of the samples synthesized without seeds. The small crystals (3–5 μm) are AFX zeolite, while the large crystals (8–15 μm) are ANA zeolite. | ||
To shorten the synthesis period further, as well as to avoid the formation of byproducts, a seeding method was employed. AFX seeds were synthesized using Et6-diquat-5 as OSDA, according to a previous study,13 and used in the as-synthesized form. No apparent enhancement of crystallization was observed when the seeds were added (Fig. 2(a) and S3(a)†), possibly due to their large particle size (ca. 5–8 μm, Fig. S4(a)†). Thus, mechanical milling was introduced to reduce the particle size of the seeds. As shown in Fig. S4(b),† after milling for 30 min, the particle size of the seeds was reduced to ca. 200 nm, and the relative crystallinity to the as-made seeds was ca. 46% (Fig. S4(c)†). Although mechanical milling could cause a certain degree of amorphization, the milled seeds could be effective in promoting crystallization, as our group has previously discussed.28 In the present work, the milled AFX seeds provided a faster crystallization rate, resulting in complete FAU conversion in only 20 min. Nevertheless, ANA was still found to be the dominant phase in the product (Fig. 2(b) and S3(b)†).
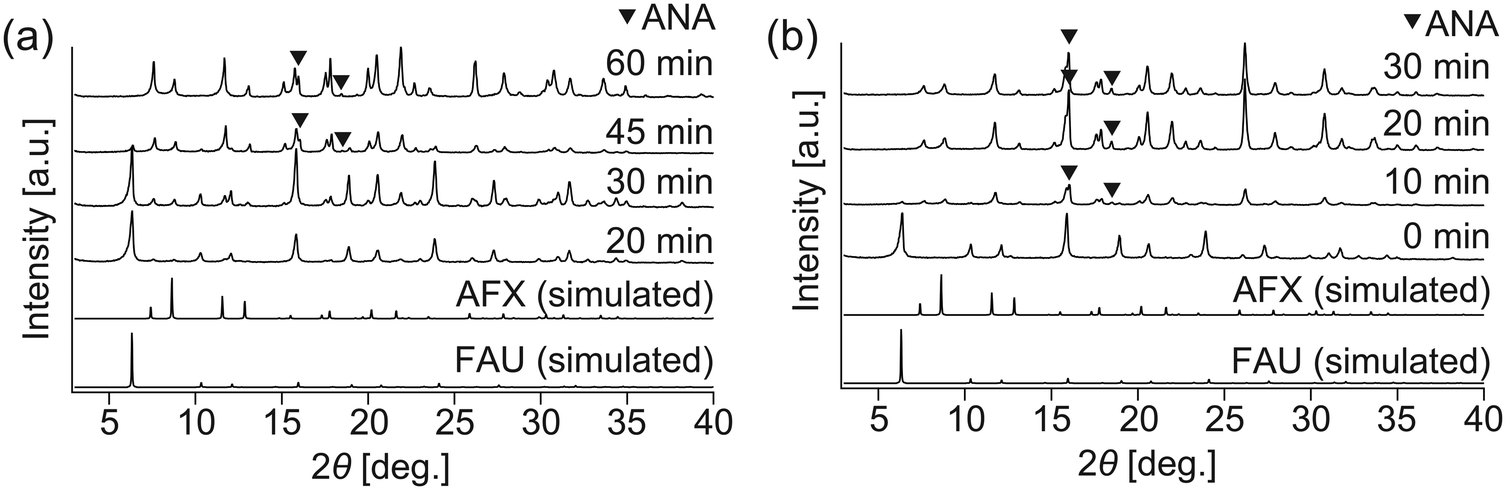 | ||
| Fig. 2 Powder XRD patterns of the samples synthesized with the (a) as-made and (b) milled seeds. | ||
In previous studies, it was demonstrated that the addition of seeds exerts a positive effect on the crystallization of the same phase because spontaneous nucleation can be bypassed.28 In this study, however, formation of the undesired phase was observed in the presence of seeds. In particular, the amount of ANA phase in the product increased even when milled seeds were used. We believe that this result might be due to the damage of the crystal structure from mechanical milling. In the case of interzeolite conversion, the presence of common composite building units in starting and product zeolite plays an important role.17 Since ANA structure does not contain any common composite building units with FAU nor AFX, the formation of ANA from fully crystalline FAU or AFX zeolite should be less kinetically favorable. Nevertheless, partial amorphization of the seed could lead to its partial dissolution at a faster pace; and the disordered aluminosilicate species released from the amorphized seed could have initiated the crystallization of ANA phase. The importance of crystallinity of the starting materials was supported by the synthesis results where a mechanically milled FAU zeolite was used. As seen in Fig. S5,† crystallization was much slower, with low crystallinity observed even after 60 min.
The above results stimulated us to think about how to alter the properties of the AFX seeds and improve their effectiveness in promoting the crystallization of the pure AFX phase. To this end, acid leaching of the original seeds was carried out at 80 °C for 27 h in a 0.5 M H2SO4 solution. The acid-leached seeds could significantly accelerate the crystallization rate of AFX while eliminating the formation of any undesired phases. As shown in Fig. 3, ultrafast conversion from FAU to AFX was observed, and after a synthesis of 12 min, a pure AFX phase was obtained. Fig. 4(a) shows the 29Si DD MAS NMR spectrum of the product of the ultrafast synthesis. The framework Si/Al ratio calculated from the spectrum was 4.5, which is consistent with the bulk Si/Al ratio (4.7) measured by ICP-AES. The nitrogen adsorption–desorption isotherm exhibited a typical reversible type I isotherm (Fig. 4(b)), indicating the absence of mesopores in the product. The micropore volume calculated from the nitrogen adsorption isotherm was 0.24 cm3 g−1, which is in good agreement with values reported in the literature,24,35 confirming the high crystallinity of the product.
 | ||
| Fig. 3 Powder XRD pattern of the samples synthesized with the acid-leached seeds. At 12 min, all diffraction peaks are due to AFX zeolite. | ||
 | ||
| Fig. 4 (a) 29Si MAS NMR spectrum and (b) nitrogen adsorption–desorption isotherms of the sample synthesized with acid-leached seeds for 12 min. | ||
Pair distribution function analysis, obtained from the HEXTS measurement, was carried out to monitor the structural evolution during the ultrafast synthesis of AFX zeolite (Fig. S6 and S7†). The peaks at 1.6, 2.6, and 3.1 Å are due to the first T–O, the first T–T, and the first O–O correlations, respectively (T = Si or Al). These peaks observed during synthesis are almost identical, indicating that the structural changes cannot be distinguished from the correlations of neighboring atoms. In contrast, the peaks at 3.8 and 4.3 Å, which are assigned to the second T–O correlations in 4-membered-rings (4MR) and in larger rings (>6MR), respectively, were considerably dissimilar. Peak shifts observed during the synthesis, which is consistent with the rapid conversion from FAU to AFX.
To clarify the critical role of the acid-leached seeds in the ultrafast synthesis, seeds with and without acid leaching were carefully characterized. Fig. 5(a) compares the XRD patterns of the original and acid-leached seeds. After acid leaching, the relative crystallinity decreased to ca. 73% of the original seeds. The SEM images in Fig. 5(b) and (c) suggest that no apparent changes in bulk morphology and particle size were observed. The framework Si/Al ratio of the acid-leached seeds, calculated from the 29Si MAS NMR spectrum in Fig. S8,† was 7.0, and the bulk Si/Al ratio calculated from ICP-AES was 6.8, whereas those for the original seeds were 4.0 and 4.3, respectively. These results indicate that some Al atoms detached from the framework and were extracted into the liquid phase.40 In spite of dealumination, the micropore volume of the acid-leached seeds was 0.23 cm3 g−1, which was almost identical to that of the original seeds (0.21 cm3 g−1), suggesting that neither blockage of micropores by extra-framework Al nor excessive degradation of the zeolite structure proceeded during the acid leaching. The 27Al MAS NMR spectra in Fig. 5(d) illustrate that most aluminum remained in tetrahedral coordination after acid leaching although a small amount of octahedral aluminum appeared.
 | ||
| Fig. 5 (a) Powder XRD patterns, (b) and (c) SEM images, (d) 27Al MAS NMR spectra, and (e) 29Si MAS NMR spectra of the as-made and acid-leached seeds. The 29Si MAS NMR measurements were conducted on the protonated form of the samples. The pink lines correspond to CP MAS NMR, while the black lines represent DD MAS NMR. | ||
From the 29Si MAS NMR measurement of the protonated seeds in Fig. 5(e), important information about the coordination environment of silicon atoms in the seeds was obtained. As seen from the 29Si DD MAS NMR spectra, both seeds featured two peaks at ca. 111 and 105 ppm, which can be assigned to resonances from Q4 (OSi)4 and Q4 (OAl)(OSi)3 species, respectively.41,42 To confirm the structural difference, 1H–29Si CP MAS NMR was performed. The acid-leached seeds featured a peak at ca. 100 ppm, which can be assigned to Q3 (OH)(OSi)3 species; however, this peak was less pronounced in the original seeds. This result suggests that the acid-leached seeds contained hydroxyl defects (silanol groups Si–OH), which might be the key factor that helped trigger secondary nucleation of the AFX phase and facilitate its further crystal growth.
To understand the evolution of acid-leached seeds during the ultrafast synthesis, morphological changes that occurred during the ultrafast synthesis were captured and analyzed. Fig. 6 shows SEM images of the solid products synthesized over different periods of time. The SEM images were obtained at two different resolutions to better demonstrate the features. At 0 min, only FAU crystals (Fig. 6(a)) and acid-leached seeds (Fig. 6(b)) can be observed. The FAU crystals were irregular polyhedrons with ca. 100–300 nm particle sizes, while the particle size of the seeds was 4–7 μm.
 | ||
| Fig. 6 SEM images of the products synthesized with acid-leached seeds for different periods of time: (a) and (b), 0 min; (c) and (d), 4 min; (e) and (f), 6 min; (g) and (h), 8 min; and (i) and (j), 12 min. The upper and lower panels correspond to the images of different resolutions of the same sample. | ||
It is worth noting that both large and small particles were observed throughout the synthesis (Fig. 6 and S9†). The large particles are supposed to be due to the acid-leached seeds whose bulk morphology was primarily preserved during the synthesis, as seen from the high-resolution images in the lower panel of Fig. 6. The small particles in the beginning were caused by the FAU crystals, while the newly formed AFX zeolite also exhibited a similar particle size. At the early stages of the synthesis, the surface of the large FAU crystals became uneven and mesopores started to develop (Fig. 6(c), (e) and (g)), indicating the instability of FAU zeolite under the synthesis conditions.43 After the synthesis for 6 min, smaller AFX crystals were clearly observed (Fig. 6(e)). The FAU particles completely disappeared after 12 min, and the final AFX product featured both newly formed small particles and large particles. The larger particles still resembled the acid-leached seeds in morphology although their surface was covered with small AFX crystals (Fig. 6(j)).
These results suggest that secondary nucleation occurred and contributed to the ultrafast synthesis of AFX.44–46 As discussed above, the acid-leached seeds underwent dealumination and, thus, contained a considerable amount of silanol defects. Compared with the original seeds, the acid-leached seeds contained fewer aluminum atoms, which made it easier to partially dissolve them in an alkaline solution and release microcrystalline units into the liquid phase. These microcrystalline units could have better access to the liquid phase, as well as the FAU zeolite, to form nuclei by secondary nucleation and then grew independently into the newly formed AFX crystals. At the same time, the defects initially present in the acid-leached seeds as well as those formed to release the microcrystalline units during the synthesis could be restored through a solution-mediated mechanism. Meanwhile, the amount of large crystals would be less than 30%, even if the seed had not dissolved nor lost in the synthesis, which would not significantly affect the overall activity of the product.
In addition to seeding, aging is an important factor affecting the synthesis outcome. To achieve ultrafast synthesis and obtain a pure AFX phase, the initial precursor was aged at 90 °C for 18 h prior to the high-temperature synthesis. In contrast, a shorter aging period at a lower temperature (2 h at 25 °C) would lead to slower crystallization and promote the formation of the ANA phase, as shown in Fig. S10.† It was reported that the interaction between inorganic and organic species could be enhanced during aging, which creates a suitable environment for the subsequent crystallization.47 In this study, adequate aging is believed to be indispensable for the creation of a microenvironment that, in combination with the acid-leached seeds, enabled secondary nucleation through surface breeding.
Ultrafast synthesis in as short as 12 min provides a solid basis for establishing continuous-flow synthesis. Because faster heating could be achieved in the continuous-flow reactor (Fig. 7(a)) than that in the batch-operated tubular reactor, a relatively shorter synthesis period was needed to obtain fully crystalline AFX zeolite. After 10 min in the continuous-flow reactor, the product exhibited a high crystallinity, as seen in Fig. 7(b). The yield was 4.2 g h−1. The SEM image in Fig. 7(c) demonstrates that the morphology of the crystals was similar to that obtained from the batch synthesis. For comparison, the space time yield for continuous-flow synthesis was 4560 kg m−3 d−1, while it was 1.7 kg m−3 d−1 for the conventional batch synthesis, showing that continuous-flow synthesis has a significantly higher productivity.
 | ||
| Fig. 7 (a) Flowchart of the continuous-flow reactor. (b) Powder XRD pattern and (c) SEM image of the product from continuous-flow synthesis. | ||
In summary, we reported that AFX zeolite can be synthesized in as short as 12 min through interzeolite conversion from FAU. With acid-leached seeds, highly crystalline AFX zeolite was synthesized within 12 min in the batch operation. According to SEM observations, the obtained samples were composed of newly formed small crystals and the remaining large seed particles whose size and morphology was unchanged during synthesis. From the analysis of different types of seeds, the acid-leached seeds showed a higher Si/Al ratio and more silanol defects. These differences could have contributed to the detachment of microcrystalline units from the seed particles and initiated crystallization of AFX zeolite. Based on the recipe for the batch operation, continuous-flow synthesis of AFX zeolite was successfully conducted with a residence time of 10 min. The success of ultrafast continuous-flow synthesis will contribute to the industrial application of AFX zeolite.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z. L. acknowledges the Japan Society for the Promotion of Science (JSPS) for the financial support (a Grant-in-Aid for Young Scientists: 18K14049). HEXTS measurements conducted at beam line 04B2, SPring-8 were approved by the Japan Synchrotron Radiation Research Institute under proposal number 2019B0155. This work was supported by the Process Science Project of MEXT, grant number JPMXP0219192801.References
- M. Moliner, C. Martínez and A. Corma, Chem. Mater., 2014, 26, 246–258 CrossRef CAS.
- M. Dusselier and M. E. Davis, Chem. Rev., 2018, 118, 5265–5329 CrossRef CAS.
- R. B. Eldridge, Ind. Eng. Chem. Res., 1993, 32, 2208–2212 CrossRef.
- IZA Structure Database, http://www.iza-structure.org/ Search PubMed.
- S. I. Zones, U.S. Pat., No. 4508837, 1985 Search PubMed.
- D. W. Fickel, E. D'Addio, J. A. Lauterbach and R. F. Lobo, Appl. Catal., B, 2011, 102, 441–448 CrossRef CAS.
- U. Deka, I. Lezcano-Gonzalez, B. M. Weckhuysen and A. M. Beale, ACS Catal., 2013, 3, 413–427 CrossRef CAS.
- S. V. Priya, T. Ohnishi, Y. Shimada, Y. Kubota, T. Masuda, Y. Nakasaka, M. Matsukata, K. Itabashi, T. Okubo, T. Sano, N. Tsunoji, T. Yokoi and M. Ogura, Bull. Chem. Soc. Jpn., 2018, 91, 355–361 CrossRef CAS.
- S. I. Zones, U.S. Pat., No. 5194235, 1993 Search PubMed.
- R. F. Lobo, S. I. Zones and R. C. Medrud, Chem. Mater., 1996, 8, 2409–2411 CrossRef CAS.
- S. I. Zones, Y. Nakagawa, L. T. Yuen and T. V. Harris, J. Am. Chem. Soc., 1996, 118, 7558–7567 CrossRef CAS.
- D. W. Fickel and R. F. Lobo, J. Phys. Chem. C, 2010, 114, 1633–1640 CrossRef CAS.
- S.-H. Lee, C.-H. Shin, G. J. Choi, T.-J. Park, I.-S. Nam, B. Han and S. B. Hong, Microporous Mesoporous Mater., 2003, 60, 237–249 CrossRef CAS.
- Y. Bhawe, M. Moliner-Marin, J. D. Lunn, Y. Liu, A. Malek and M. E. Davis, ACS Catal., 2012, 2, 2490–2495 CrossRef CAS.
- T. M. Davis, U.S. Pat., No. 9908108B2, 2018 Search PubMed.
- T. Sano, M. Itakura and M. Sadakane, J. Jpn. Pet. Inst., 2013, 56, 183–197 CrossRef CAS.
- S. Goel, S. I. Zones and E. Iglesia, Chem. Mater., 2015, 27, 2056–2066 CrossRef CAS.
- H. Jon, S. Takahashi, H. Sasaki, Y. Oumi and T. Sano, Microporous Mesoporous Mater., 2008, 113, 56–63 CrossRef CAS.
- H. Sasaki, H. Jon, M. Itakura, T. Inoue, T. Ikeda, Y. Oumi and T. Sano, J. Porous Mater., 2009, 16, 465–471 CrossRef CAS.
- K. Itabashi, Y. Kamimura, K. Iyoki, A. Shimojima and T. Okubo, J. Am. Chem. Soc., 2012, 134, 11542–11549 CrossRef CAS.
- S. Goel, S. I. Zones and E. Iglesia, J. Am. Chem. Soc., 2014, 136, 15280–15290 CrossRef CAS.
- M. Itakura, K. Ota, S. Shibata, T. Inoue, Y. Ide, M. Sadakane and T. Sano, J. Cryst. Growth, 2011, 314, 274–278 CrossRef CAS.
- H. Yamada, T. Iida, Z. Liu, Y. Naraki, K. Ohara, S. Kohara, T. Okubo and T. Wakihara, Cryst. Growth Des., 2016, 16, 3389–3394 CrossRef CAS.
- N. Nakazawa, S. Inagaki and Y. Kubota, Adv. Porous Mater., 2016, 4, 219–229 CrossRef.
- N. Martín, C. Paris, P. N. Vennestrøm, J. R. Thøgersen, M. Moliner and A. Corma, Appl. Catal., B, 2017, 217, 125–136 CrossRef.
- Z. Liu, T. Wakihara, D. Nishioka, K. Oshima, T. Takewaki and T. Okubo, Chem. Commun., 2014, 50, 2526–2528 RSC.
- Z. Liu, T. Wakihara, C. Anand, S. H. Keoh, D. Nishioka, Y. Hotta, T. Matsuo, T. Takewaki and T. Okubo, Microporous Mesoporous Mater., 2016, 223, 140–144 CrossRef CAS.
- J. Zhu, Z. Liu, A. Endo, Y. Yanaba, T. Yoshikawa, T. Wakihara and T. Okubo, CrystEngComm, 2017, 19, 632–640 RSC.
- J. Zhu, Z. Liu, S. Sukenaga, M. Ando, H. Shibata, T. Okubo and T. Wakihara, Microporous Mesoporous Mater., 2018, 268, 1–8 CrossRef CAS.
- Z. Liu, J. Zhu, T. Wakihara and T. Okubo, Inorg. Chem. Front., 2019, 6, 14–31 RSC.
- T. Yoshioka, Z. Liu, K. Iyoki, T. Sano, M. Ando, S. Sukenaga, H. Shibata, T. Okubo and T. Wakihara, Chem. Lett., 2020, 49, 1006–1008 CrossRef CAS.
- Z. Liu, T. Wakihara, K. Oshima, D. Nishioka, Y. Hotta, S. P. Elangovan, Y. Yanaba, T. Yoshikawa, W. Chaikittisilp, T. Matsuo, T. Takewaki and T. Okubo, Angew. Chem., Int. Ed., 2015, 54, 5683–5687 CrossRef CAS.
- Z. Liu, J. Zhu, C. Peng, T. Wakihara and T. Okubo, React. Chem. Eng., 2019, 4, 1699–1720 RSC.
- A. Deneyer, Q. Ke, J. Devos and M. Dusselier, Chem. Mater., 2020, 32, 4884–4919 CrossRef CAS.
- A. Chokkalingam, W. Chaikittisilp, K. Iyoki, S. H. Keoh, Y. Yanaba, T. Yoshikawa, T. Kusamoto, T. Okubo and T. Wakihara, RSC Adv., 2019, 9, 16790–16796 RSC.
- S. Inagaki, Y. Tsuboi, Y. Nishita, T. Syahylah, T. Wakihara and Y. Kubota, Chem. – Eur. J., 2013, 19, 7780–7786 CrossRef CAS.
- D. C. Calabro, J. C. Cheng, R. A. Crane, Jr., C. T. Kresge, S. S. Dhingra, M. A. Steckel, D. L. Stern and S. C. Weston, U.S. Pat., No. 6049018, 2000 Search PubMed.
- S. Kohara, M. Itou, K. Suzuya, Y. Inamura, Y. Sakurai, Y. Ohishi and M. Takata, J. Phys.: Condens. Matter, 2007, 19, 506101 CrossRef.
- E. Mitani, Y. Yamasaki, N. Tsunoji, M. Sadakane and T. Sano, Microporous Mesoporous Mater., 2018, 267, 192–197 CrossRef CAS.
- Y. Ji, M. A. Deimund, Y. Bhawe and M. E. Davis, ACS Catal., 2015, 5, 4456–4465 CrossRef CAS.
- C. J. Heard, L. Grajciar, C. M. Rice, S. M. Pugh, P. Nachtigall, S. E. Ashbrook and R. E. Morris, Nat. Commun., 2019, 10, 1–7 CrossRef CAS.
- K. Iyoki, K. Kikumasa, T. Onishi, Y. Yonezawa, A. Chokkalingam, Y. Yanaba, T. Matsumoto, R. Osuga, S. P. Elangovan, J. N. Kondo, A. Endo, T. Okubo and T. Wakihara, J. Am. Chem. Soc., 2020, 142, 3931–3938 CrossRef CAS.
- Y. Wang, X. Li, Z. Xue, L. Dai, S. Xie and Q. Li, J. Phys. Chem. B, 2010, 114, 5747–5754 CrossRef CAS.
- L.-Y. Hou and R. W. Thompson, Zeolites, 1989, 9, 526–530 CrossRef CAS.
- P. K. Dutta and J. Bronic, Zeolites, 1994, 14, 250–255 CrossRef CAS.
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1–78 CrossRef CAS.
- Z. Liu, K. Okabe, C. Anand, Y. Yonezawa, J. Zhu, H. Yamada, A. Endo, Y. Yanaba, T. Yoshikawa, K. Ohara, T. Okubo and T. Wakihara, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14267–14271 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00219d |
| This journal is © The Royal Society of Chemistry 2021 |