
Photochlorination of toluene – the thin line between intensification and selectivity. Part 2: selectivity
Ümit
Taştan†
 a,
Phillip
Seeber†
a,
Stephan
Kupfer
*b and
Dirk
Ziegenbalg
*a
a,
Phillip
Seeber†
a,
Stephan
Kupfer
*b and
Dirk
Ziegenbalg
*a
aInstitute of Chemical Engineering, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: dirk.ziegenbalg@uni-ulm.de; Fax: +49 731 50 12 25700; Tel: +49 731 50 25703
bInstitut für Physikalische Chemie, Friedrich-Schiller-Universität Jena, Helmholtzweg 4, 07743 Jena, Germany. E-mail: stephan.kupfer@uni-jena.de; Tel: +49 3641 9 48334
First published on 7th October 2020
Abstract
Photochemical reactions, such as the light-driven side-chain chlorination of various aromatic compounds, are among the cornerstones of the chemical industry. Therefore, reoptimising reaction conditions using state-of-the-art reactor designs and irradiation techniques is economically and ecologically of uttermost importance. However, reaction rate enhancement needs to be accompanied by a high selectivity for the desired target compound. In part 1 of this work the potential of intensifying the side-chain photochlorination of toluene by various reactor setups and irradiation techniques was reported. While the reaction was accelerated by up to a factor of ten, its selectivity towards the desired side-chain chlorinated products decreased under certain conditions. Unexpectedly, significant amounts of ring chlorinated species were detected. High level ab initio molecular dynamic simulations allowed – in agreement with the experimental data – assessment of the underlying reaction mechanism leading to the undesired by-product. Photoexcitation of certain toluene–chlorine complexes, formed under highly intensified reaction conditions, yields charge-separated biradical species. In consequence, the pronounced driving force of charge- and radical-recombination leads to the formation of ring chlorinated species as derived by quantum chemical simulations. By applying dynamic irradiation conditions, not only was the formation of ring-chlorinated products suppressed but the energy consumption could also be reduced significantly.
1 Introduction
The production of side-chain chlorinated species at the industrial scale is of high relevance for the production of chemical compounds such as benzyl alcohol, benzyl butyl phthalate, benzaldehyde and benzoic acid. Several thousand tonnes of these substances are produced in bubble columns in Europe alone every year.2 The radical-chain mechanism of photochlorination possesses high reaction rates as well as highly reactive intermediates.3–6 Hence, controlling the selectivity towards side-chain chlorination is challenging but of high importance. This becomes even more important when the large bubble columns (100 L or higher) equipped with mercury vapour lamps (MVLs) that are used in industry are considered.7 Conventionally, temperatures above 70 °C are used to reduce the fraction of ring-chlorinated side-products.8 In a recent publication it was shown that the apparent reaction rate of toluene photochlorination can be increased by more than one order of magnitude if a sufficiently high chlorine supply can be realised technically, e.g. in microreactors.1 Furthermore, operating LEDs in a pulsed manner yielded a reduction of the required electrical energy of more than 90% without any reduction of the reaction rate and equally important showed an increase in selectivity towards benzyl chloride. For most experimental conditions, the selectivity was not influenced, except for extreme conditions such as very low temperatures or photon fluxes. Surprisingly, it was found that the selectivity was influenced by the applied type of light source triggering the photoreaction. In case a mercury vapour lamp was used, lower selectivities were observed (see Fig. 3(d)). Unexpectedly, the experimental results revealed high selectivity towards ring chlorination for intensified reaction conditions (Fig. 1). A comparison between different batch and microreactor setups revealed in general lower selectivity for the investigated microreactor setups. Furthermore, individual conversion–selectivity correlations were found for each setup. These selectivity observations are in contradiction to the established theory of photochlorination. Even more critical, some experimental results suggest contradictory interpretations.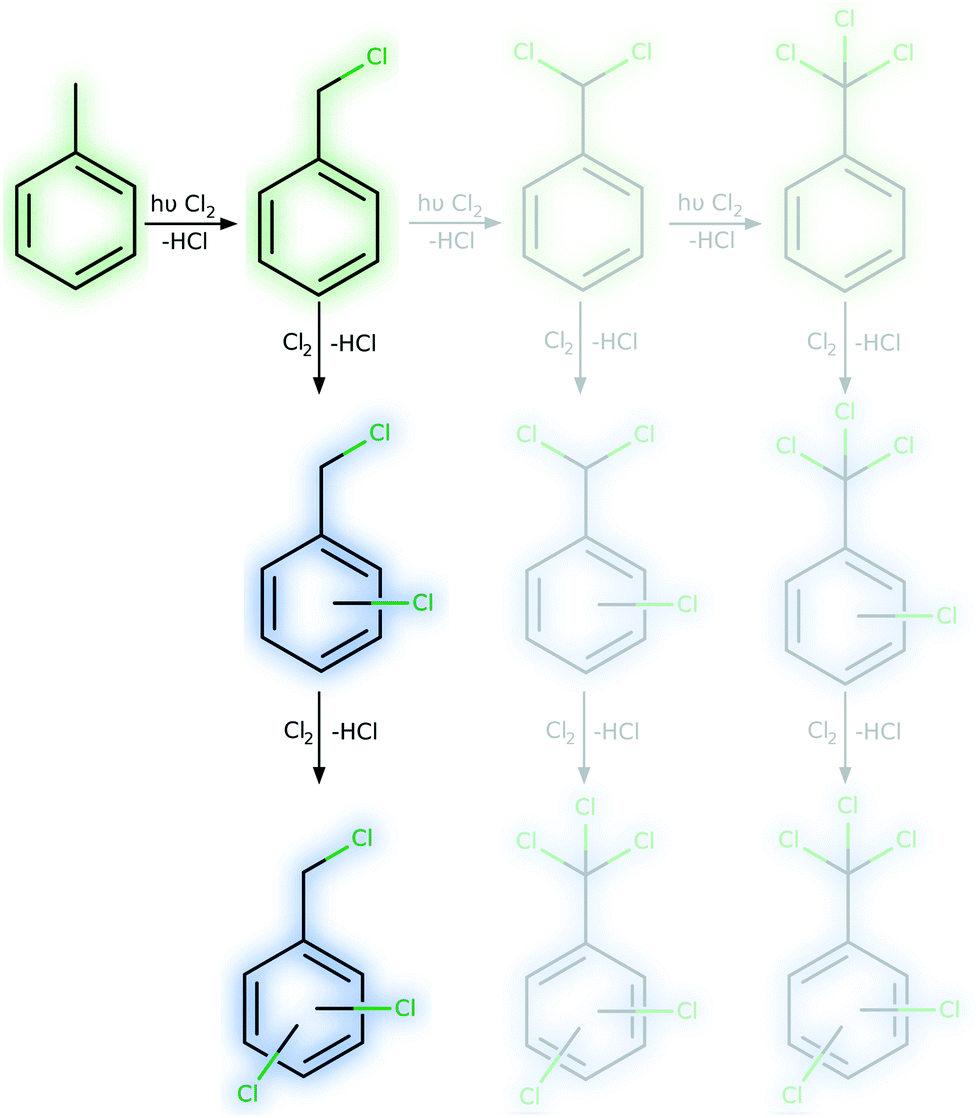 | ||
| Fig. 1 Reaction pathways in the photochlorination of toluene (main reaction in green, ring-chlorination in blue), selectivity aspects are highlighted. | ||
Consequently, this work elaborates on elucidating the aforementioned contradictions to the established theory of aromatic photochlorination as well as the apparent contradictory experimental results. In the past, numerous quantum chemical investigations9–14 have been carried out to evaluate the mechanism of various aromatic substitution reactions. Most recently, Šakić and Vrček9 revealed that manual specification and preselection of initial structures easily misses important low energy structures, as shown by their extensive stochastic search in the scope of various arene chlorinations performed at the density functional theory (DFT) level of theory. The authors highlighted the importance of an accurate description of long-range dispersion interactions that potentially even changes the energetics from antibinding to binding interactions. Furthermore, solvent effects were considered by the polarizable continuum model (PCM)15,16 approach, i.e. exclusively accounting for dielectric properties of the solvent. However, to the best of our knowledge, no efforts were made to unravel light-triggered aromatic substitution reactions, e.g. for the present chlorination of toluene. Therefore, we applied, in close collaboration with the experimental investigations ab initio molecular dynamics simulations to understand the origin of the highly unexpected experimental findings, and ultimately to assess the mechanism underlying the present photoreaction. With this synergistic approach, the reasons leading to the – at first – contradicting results could be identified at the molecular level and the reaction conditions for side-chain photochlorinations of aromatic compounds could be optimised.
This contribution is the second of two articles on this topic and focuses primarily on the selectivity aspect as well as on the mechanism associated with the formation of the undesired by-product. A detailed analysis and discussion of the intensification is given in the first article.1
2 Methods
2.1 Experimental methods
Experimental investigations were carried out with three types of reactors: two different batch reactors and one recycle reactor. Basically, the first batch reactor (referred to as “batch A”) was a simple round-bottom flask with a reflux cooler on top. For the second batch reactor (referred to as “batch B”), an additional reflux cooler was sealed at the bottom, yielding a batch reactor with enhanced cooling capacity. The effect of separating reaction and mass transfer on the selectivity was assessed by means of a micro-scale recycle reactor (referred to as a “microreactor”). The separation of reaction and transport processes was realised by altering the position of the LED (see Fig. 2). When the LED was installed at the point of contact among chlorine and the liquid reaction mixture, the gas–liquid-system was irradiated while chlorine is absorbed in the liquid (referred to as “irradiation during absorption – IDA”). The reaction and absorption of chlorine could be separated by installing the LED at the end of the capillary used as an absorber (referred to as “irradiation after absorption – IAA”). To quantify the availability of chlorine, the chlorine feed rate (Q) was defined as: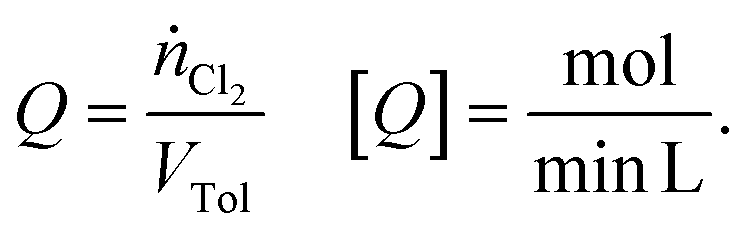 | (1) |
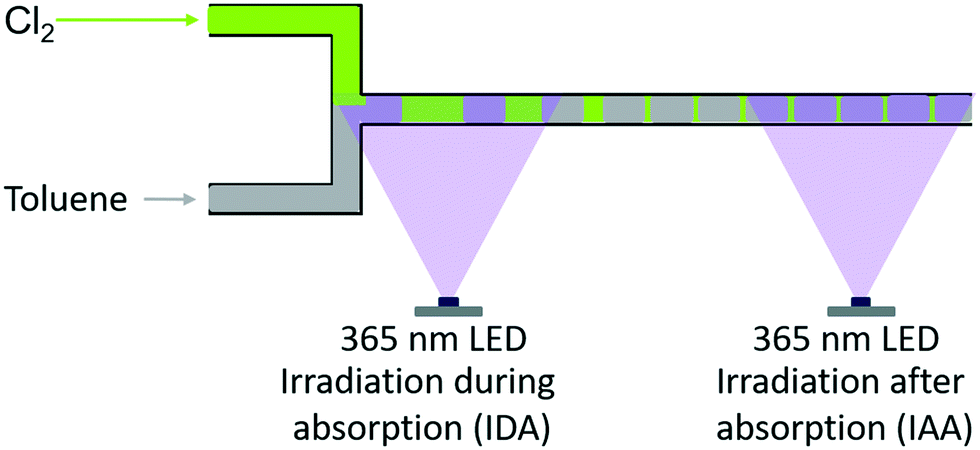 | ||
| Fig. 2 Schematic representation of the different irradiation positions used with the microreactor setup, i.e. irradiation during (IDA) and after absorption (IAA) of chlorine.1 | ||
2.2 Computational details
Ab initio molecular dynamics (AIMD) simulations were performed with CP2K 6.1 (ref. 17) software in the framework of fully periodic hybrid Gaussian and plane wave density functional theory (DFT).18 The TPSS functional19 in conjunction with polarised short-range double ζ valence basis sets (DZVP-MOLOPT-SR-GTH)20 describing the valence electrons was applied, while furthermore GTH-PBE pseudopotentials21 were utilised. A 250 Ry planewave cutoff and a relative cutoff of 50 Ry on five real space grids were used. To account for long-range interactions, i.e. dispersion, Grimme's D3 correction22 with Becke–Johnson damping23–25 was employed.Initially, a cubic cell with an edge length of 1.9 nm comprising 35 toluene and 15 dichlorine molecules, respectively, was created using Packmol 18.169.26 In the following, the edge length was increased to 2.0 nm for the periodic DFT simulations performed in CP2K to force a minimum inter-molecular distance of 200 pm among the repetition units (at t = 0) and to reproduce the experimental density of ρ = 0.89 g mL−1. This system was equilibrated for 7.5 ps with a time-step of 0.5 fs in a canonical (NVT) ensemble at T = 350 K, enforced by a strongly coupling Nosé–Hoover thermostat (τ = 10 fs). Subsequently, the time evolution of the equilibrated system was simulated for 25 ps using the same setup as described above, but with a weaker coupling constant of the Nosé–Hoover thermostat (τ = 150 fs). The resulting trajectory was analysed with Travis (release 02.10.2018)27 and several distribution functions were calculated: i) the spatial distribution function of chlorine atoms in the vicinity of toluene and two combined distribution functions, ii) the radial/radial distribution function describing the distance between one chlorine atom of each dichlorine molecule and the toluene with respect to the distance between the second chlorine atom of each dichlorine molecule and the toluene and iii) the radial/angular distribution function, correlating the radial distance between each chlorine atom and the toluene with the angle between the toluene ring plane and the dichlorine bond.
From this trajectory (25 ps), 100 frames – in which one toluene and dichlorine molecule are nearest neighbours – were (randomly) selected using Travis.27 In the following the system was treated by a mixed quantum mechanics/molecular mechanics (QM/MM) approach, where this one pair of toluene–dichlorine neighbours was described in a quantum mechanical fashion and the remaining (34 toluene and 14 dichlorine) molecules were assessed by molecular mechanics. Therefore, each of these 100 frames was treated by the following computational workflow:
1. Detect covalent bonds.
2. Assign AMOEBA09 (ref. 28) force field types.
3. Find the dichlorine and toluene molecules (QM) that were selected using Travis.
4. Replicate the system to obtain a 3 × 3 × 3 super cell, with the toluene–dichlorine pair (QM) in the central unit cell.
5. Detect molecules based on bonds in the super cell.
6. Wrap molecules outside the super cell back to the super cell.
7. Export all structural data, the assigned QM/MM scheme and the respective force field types in a Tinker-based29 format.
The AMOEBA09 force field contains parameters for toluene, but lacks parameters for dichlorine. Therefore, the parameters of the dichlorine force field were derived according to the literature,30 while the quantum chemical calculations were performed using Gaussian 16,31 multipole analysis was carried out via GMDA 2.2.11 (ref. 32) and the resulting force field parameters were obtained using Tinker from the quantum chemical calculations. Finally, QM/MM calculations were performed using LICHEM33,34 in conjunction with Psi4 1.2.1 (ref. 35) and Tinker 8.4.4, whereas the AMOEBA09 force fields (MM region) polarise the QM region described at the MP2/cc-pVTZ36–38 level of theory and vice versa with point charges representing the MM-multipoles.
Subsequently, the polarised QM region was further treated at the reduced-cost-ACD(2)/aug-cc-pVTZ39 level of theory using the MRCC program40,41 to elucidate the nature of the 20 lowest excited singlet states contributing to the UV-vis absorption spectrum of the reactants within each of the 100 frames. The electronic character of these excitations was analysed using charge density differences neglecting crossterms.
3 Results and discussion
3.1 Experimental results on the selectivity towards benzyl chloride
A comprehensive experimental investigation showed that photochlorinations can be intensified by using microreactors to overcome technical constraints of the chlorine supply to the reaction solution.1 This approach was found to be associated with a drop in selectivity towards benzyl chloride which cannot be explained by established theories on photochlorination.A comparison of the conversion–selectivity correlation towards benzyl chloride for the microreactor setup and the two irradiation positions with the results from batch A is given in Fig. 3(a). This chart indicates the results of almost all conducted experiments irrespective of the operating conditions. Only extreme conditions such as very low temperatures (T = 30 °C), very low chlorine feed rates (Q < 0.03 mol L−1 min−1) or very low electric currents driving the LED (I < 100 mA) were excluded. It is obvious that the selectivity correlates with the experimental setup. The highest selectivities were obtained with the batch setup. For the microreactor setup, irradiation during absorption led to higher selectivities than irradiation after absorption. It should be noted that the trends are remarkably consistent over a wide range of reaction conditions.
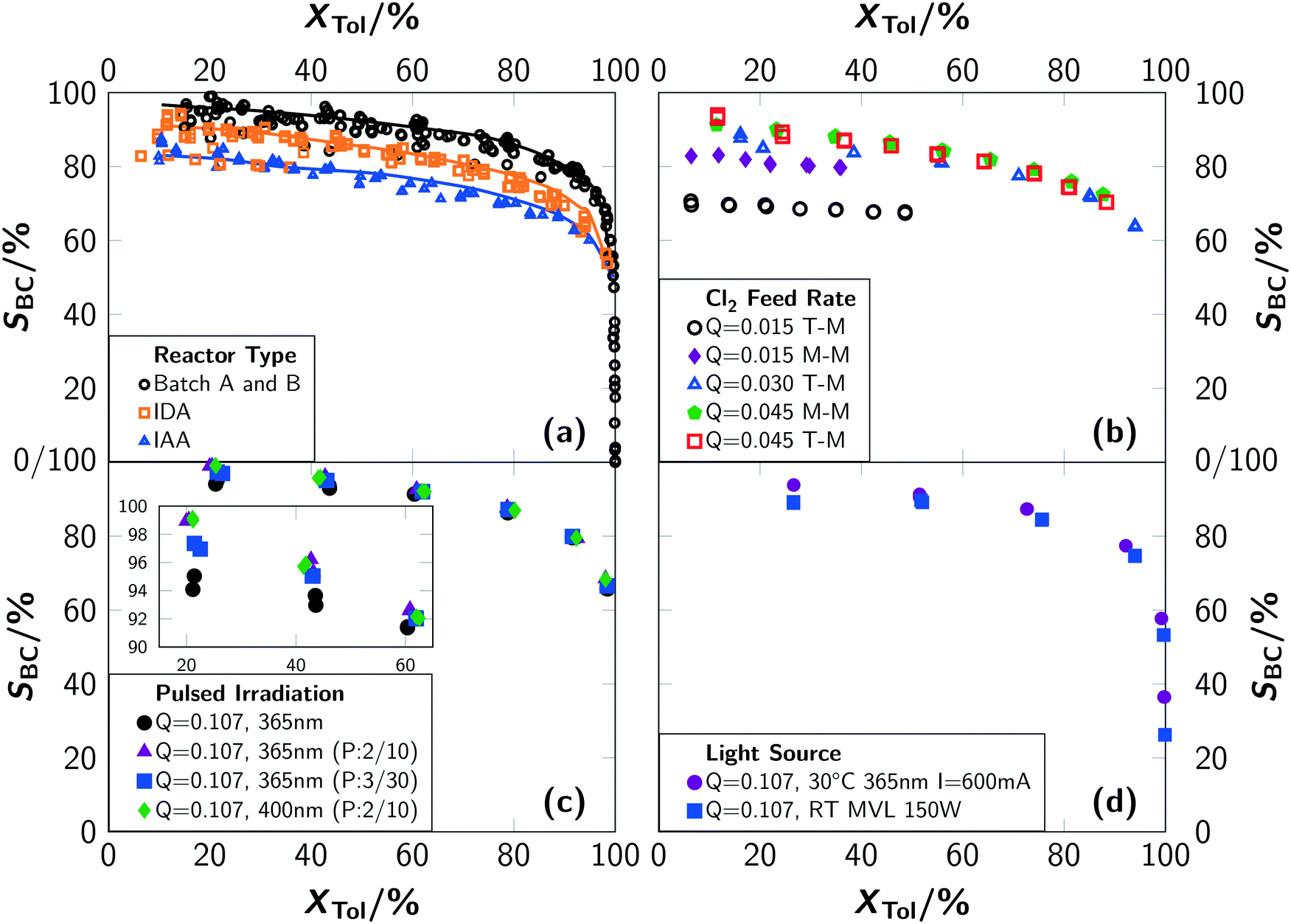 | ||
| Fig. 3 Conversion–selectivity correlation of toluene, with the selectivity towards benzyl chloride denoted as SBC. (a) Summary of experiments in batch setups (batch A and batch B) and in the microreactor setup with irradiation during absorption (IDA) and irradiation after absorption (IAA). The different lines indicate the moving average of the selectivities for either batch experiments, the IDA or IAA microreactor experiments for both mixers. (b) For different chlorine feed rates Q. The experiments were carried out at 30 °C using two 365 nm LEDs at 600 mA and the microreactor. “T-M” indicates the T-mixer and “M-M” indicates the micromixer. (c) For continuous and pulsed operation of LEDs. Q is given in mol L−1 min−1. (d) In batch B for different light sources. | ||
This finding is in contradiction to the well established concept of photochlorination, where the selectivity is exclusively governed by the temperature.8 As side-products ring-chlorinated species are formed. Due to the lower reactivity of the aromatic ring, the formation of ring-chlorinated substances is less likely. Commonly, the ring has to be activated with the help of Lewis acids such as FeCl3. Since metal contaminations were excluded by the design of the reactor setups and analytically verified by measurements with inductively coupled plasma mass spectrometry (ICP-MS), a straight-forward reason for the increasing ring activation could not be identified in the first instance.
In contrast to the batch experiments, the selectivity in the microreactor was significantly influenced by the operating conditions (see Fig. 3(b)). The selectivity decreased with a decreasing chlorine feed rate. This effect is correlated with the increased degree of backmixing for low chlorine feed rates. Reasoned by the stoichiometric ratio, the number of recycles – required to reach a certain degree of conversion – is larger for these conditions and with this the degree of backmixing.
Liquid-sided mass transport can be an additional parameter influencing the selectivity of consecutive reactions. To address this process, two different mixers were installed in the microreactor setup. First, a simple T-mixer with a channel width of 1 mm was installed. By utilising a dedicated micromixer instead of a T-mixer, the influence of liquid mixing can be elucidated. Therefore, a split-and-recombine mixer was chosen that continuously shrinks the diffusion distances to accelerate mixing.
Experiments with the T-mixer yielded lower selectivities compared to the micromixer for the same chlorine feed rates (see Fig. 3(b)). This effect is most pronounced for low chlorine feed rates. At higher values, the selectivity is similar for both devices. These observations are correlated to a more homogeneous distribution of chlorine, toluene and chlorinated products in the liquid phase when using the micromixer. This mixer provides better mixing of the liquid phase even at low volumetric flow rates. On the other hand, high local concentrations of chlorine and chlorinated toluenes are present in the T-mixer, leading to an increase of side reactions. Analysis of the product spectrum revealed ring-chlorinated benzyl and benzal chlorides. The dependency of the selectivity on the chlorine feed rate is in line with the speed of mixing provided by the respective device. Higher chlorine feed rates lead to higher superficial velocities of the reaction solution and with this to an improved mixing.
The differences among the three sets of experiments performed are: i) differing chlorine feed rates and ii) the conditions under which the reaction is initiated. The results of batch A stem from low chlorine feed rates and simultaneous irradiation. In comparison, for the microreactor experiments with irradiation during absorption, the chlorine feed rate is up to one order of magnitude higher. Hence, the steady state concentration of chlorine in solution is higher for the microreactor setup. For the microreactor experiments with irradiation after absorption, the concentration of chlorine in solution is even higher since no reaction occurs before the educt mixture enters the irradiation section. Conclusively, high (local) chlorine concentrations are detrimental for achieving high selectivity towards benzyl chloride. Furthermore, it was found that the reaction rates are typically lower when the irradiation occurs after the absorption. The opposite was expected because a higher photon flux can be absorbed as a result of the high chlorine concentration. Again, both observations are in strong contrast to the common understanding of photochlorination.8
The reaction rate is faster with higher chlorine concentrations, however a higher concentration of chlorine radicals leads to a pronounced occurrence of consecutive reactions towards benzal chloride and benzotrichloride and even more important side reactions (e.g. ring chlorinations). While the formation of consecutive products is to be expected, the ratio between different consecutive products should be invariant, independent of the apparent reaction rate of toluene and according to the relative reactivity. This is not the case for IAA conditions, where an increased formation of benzyl chloride with respect to benzal chloride is found compared to IDA conditions (see Fig. 4(a)). An influence of the type of mixer on the ratio of benzyl chloride and benzal chloride selectivity for IDA conditions was only observed for conversions below 40%, where the ratio for the micromixer slightly varies (see Fig. 4(b)). A comparison of the side-product formation showed a pronounced formation of side-products for IAA conditions (see Fig. 4(c) and (d)).
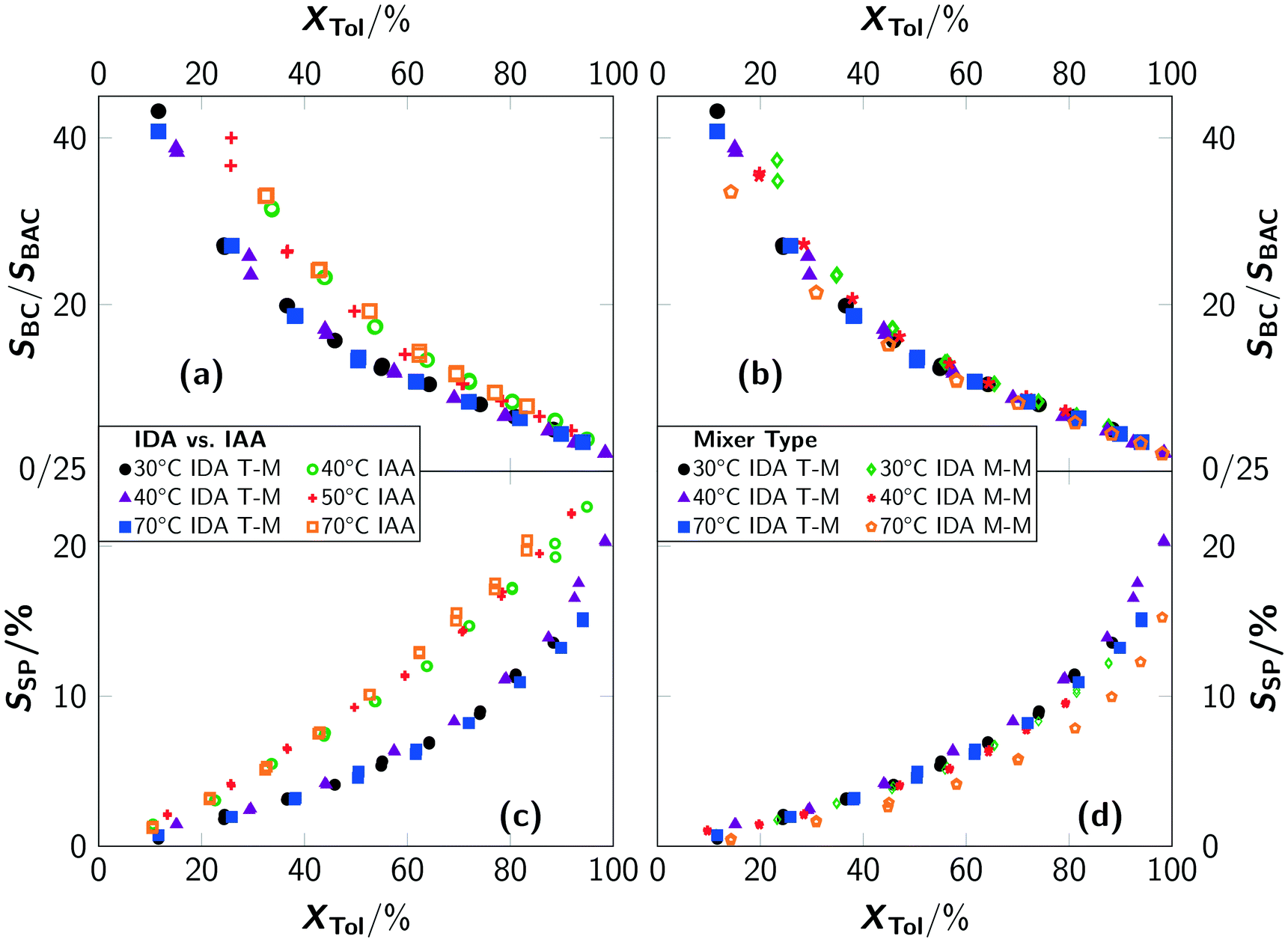 | ||
| Fig. 4 Conversion–selectivity ratio correlation for (a) different irradiation conditions and reaction temperatures and (b) different mixer types and temperatures. Conversion–selectivity of side-product correlations for (c) different irradiation conditions and reaction temperatures and (d) different mixer types and temperatures. Experiments were conducted with a chlorine feed rate of Q = 0.045 mol min−1 L−1. SBC/SBAC denotes the ratio of the selectivity towards benzyl chloride (SBC) and the selectivity towards benzal chloride (SBAC), respectively. SSP is the selectivity to side-products. “IDA” indicates irradiation during absorption and “IAA” indicates irradiation after absorption. “T-M” indicates the T-mixer and “M-M” indicates the micromixer. | ||
Consequently, these side-products affect the ratio of benzyl and benzal chloride. Evaluation of the side-product spectrum revealed an increased formation of ring-chlorinated products, thus the overall selectivity of benzyl chloride decreased with irradiation after absorption. Compared to the batch setup, very high local chlorine concentrations occur for IAA conditions. This leads to significant over-exposure to chlorine, and in consequence, to increased formation of higher-chlorinated products. In contrast, the accelerated formation of ring-chlorinated products cannot be explained by high local chlorine concentrations, i.e. activation of the aromatic ring is required. Hence, the well-known reaction mechanism of photochlorination needs to be re-evaluated for the present reaction conditions.
Fig. 3(c) shows the selectivity towards benzyl chloride for experiments with pulsed LEDs. Surprisingly, the selectivity increases with pulsed operation. For instance, a selectivity of 99% at a conversion of 20% was recorded for sequence A for both LEDs. Continuous irradiation resulted in a selectivity of 95% at the same conversion.
The investigations showed that the selectivity towards benzyl chloride and the rate of side-product formation strongly depend on the operating conditions and the reactor setup. Higher selectivities were found for batch operation compared to continuous operation. A micromixer performs better than a T-mixer for low chlorine feed rates and pulsed operation yields higher selectivities. Irradiation during absorption leads to higher selectivities in comparison to irradiation after absorption.
Impacts on the selectivity when using continuously operated light sources can be summarized as follows: i) very high local chlorine concentrations in the reaction solution correlate with enhanced formation of ring-chlorinated toluenes and ii) a high degree of backmixing that occurs for low chlorine feed rates due to the required recycling causes primarily the formation of ring-chlorinated benzyl and benzal chlorides.
Despite this, the results obtained for pulsed operation of LEDs are in strong contradiction to the results obtained in the microreactor. During the dark phase, chlorine dissolves in the reaction solution, resulting in high chlorine concentrations. In principle, this makes the (local) reaction conditions in the batch similar to those in the microreactor when irradiation is conducted after absorption. Hence, it is unclear why similar reaction conditions yield strongly diverging selectivities. However, these similar local or rather molecular reaction conditions cannot be assessed solely by the applied experimental techniques.
Furthermore, the formation of ring-chlorinated products cannot be explained with conventional mechanistic concepts. Considering previous results that indicated an intense electronic interaction between chlorine and aromatic compounds, it seemed reasonable that this interaction might be the reason for the contradictory results.42 To elucidate the reaction mechanism underlying the unexpected ring-chlorination, a series of AIMD and quantum chemical simulations has been performed to assess the photophysical and photochemical properties at the molecular level.
3.2 Quantum mechanical studies
To assess photo-induced reactions, i.e. the present photochlorination of toluene, firstly a thorough and accurate description of the ground state properties of the reactants is indispensable. As the computational modelling of mixtures is difficult with the PCM as merely implicit solvent effects are considered, and cooperative interactions of multiple dichlorine molecules with a single toluene molecule cannot be excluded at high dichlorine concentrations, a full quantum mechanical approach was employed in the present study.Furthermore, as highlighted by Šakić and Vrček,9 a biased preselection of the initial structures may lead to an improper computational description of the reaction thermodynamics. Therefore, AIMD simulations of a highly concentrated solution of dichlorine in toluene were performed with the dispersion corrected TPSS functional.19 This setup is capable of addressing the mentioned challenges of previous theoretical investigations:9,15,16 i) by sampling a broad range of molecular configurations of the reactants and ii) by addressing inter-molecular interactions explicitly, i.e. among 35 toluene and 15 dichlorine molecules. The AIMD simulations performed were stable after an initial equilibration at T = 350 K. Analysis of the trajectory by several distribution functions (see Fig. 5) clearly reveals that dichlorine–toluene-based π-complexes are the most favourable configurations in solution. The combined radial distribution function in Fig. 5(a) features intense and narrowly confined regions around (R(Tol–1Cl) ≈ 300 pm, R(Tol–2Cl) ≈ 500 pm and vice versa) – attributed to a dichlorine molecule being positioned normal to the aromatic ring. Furthermore, Fig. 5(a) indicates a typical Cl–Cl distance of ≈200 pm by the difference ΔR(Tol–1/2Cl), when the dichlorine molecule is normal to the toluene ring. This value is very similar to the chlorine–chlorine bond length of dichlorine in the gas phase and much shorter than the required Cl–Cl bond length of approximately 230 pm in σ-complexes as discussed for Lewis acid catalysed interactions.11 Eventually, the regions around R(Tol–1Cl) ≈ 400 pm (or R(Tol–2Cl) ≈ 400 pm) and R(Tol–1Cl) ≈ 500 pm (or R(Tol–2Cl) ≈ 500 pm) are assigned to less frequent configurations with dichlorine molecules in proximity and parallel to the toluene ring as well as T-shaped complexes in the long-range domain, respectively. The radial angular distribution function, Fig. 5(b), provides an even more pronounced perspective on the perpendicular π-complexes with maxima at 90°. Finally, the cross sections of the spatial distribution function, Fig. 5(c), clearly show oval ring structures above and below the ring plane of toluene for chlorine atoms. The presence of these highly localised (secondary) rings, in combination with a very low probability density of dichlorine in their centres, indicates a strong preference for π-complexes.
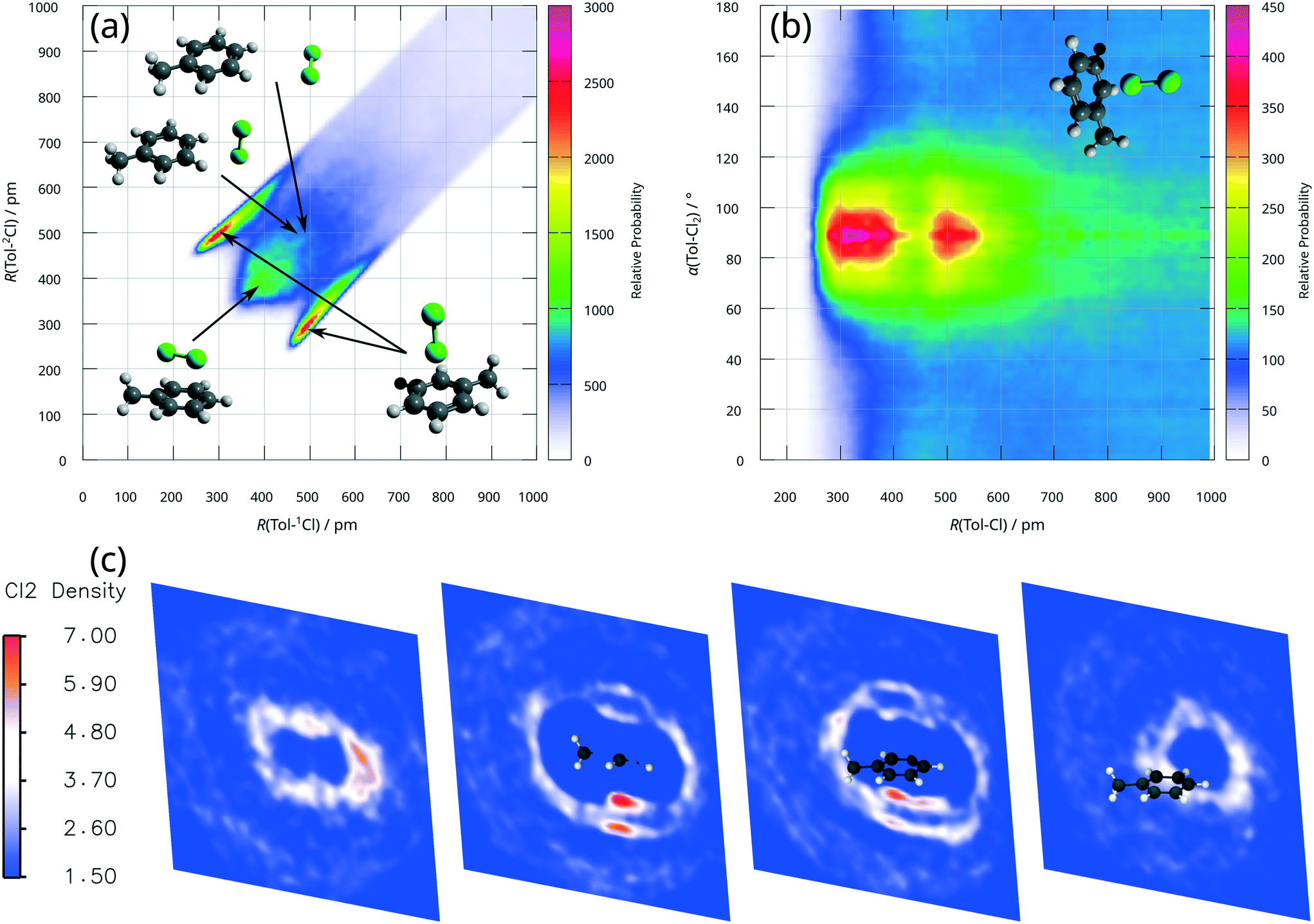 | ||
| Fig. 5 Analysis of the AIMD trajectory. (a) Radial–radial combined distribution function, correlating the radial distribution function of toluene to 1Cl respective 2Cl of a dichlorine molecule. (b) Radial–angular combined distribution function, correlating the radial distribution function of the chlorine atom–toluene distance with the angle between the toluene ring plane and the Cl–Cl bond vector. (c) Slices through the spatial distribution function of chlorine atoms around a toluene molecule. | ||
Subsequently, excited state properties of the dichlorine–toluene mixture were simulated to elucidate the photo-induced chlorination of the aromatic core versus the aliphatic chain. Therefore, electronic absorption spectra for one hundred arbitrarily selected AIMD snapshots were calculated at the ADC(2) level of theory.39 The resulting UV/vis spectrum (Fig. 6(a)) – averaged over these one hundred configurations – coincides with the experimental reference obtained in the available transmission window of the photoreactor.42 The charge density differences (CDDs), illustrating the nature of the electronic transitions underlying the respective absorption bands obtained for these AIMD frames, reveal a pronounced dependency of the excited state landscape with respect to the relative orientation of the toluene and the dichlorine molecules, i.e. for the different types of dichlorine–toluene complexes, see Fig. 6(c). Typically, π-complexes, e.g. frames 16 and 18 in Fig. 6(b) and (c), feature bright charge-transfer states of π(toluene) → σ*(Cl2) character. These charge-transfer excitations are exclusively optically accessible (large oscillator strength fosc) if dichlorine is positioned vertically above the ring such as in frame 16, while in all other configurations, charge-transfer excitations, e.g. in frame 17, are dark. The η2 π-complex of frame 16 has a large oscillator strength fosc = 0.18, while the less strongly interacting coordination in frame 17 provides only a small contribution to the absorption band around 280 nm with an oscillator strength of merely fosc = 3.0 × 10−4. The strong absorption bands at ca. 280 nm and 230 nm are thus caused by strongly interacting π-complexes at high dichlorine concentrations. Further relative orientations of dichlorine and toluene molecules, as in frame 21 or frame 76 are also described by Šakić and Vrček,9 but contrary to the π-complexes, such configurations do not contribute strongly to the electronic absorption in this spectral region. Rather, the excited states of these configurations are mainly governed by the intra-molecular excitations of the isolated species. Exemplarily, frame 21 shows an excitation of π → σ* character localised on the dichlorine, which is only slightly shifted compared to a free dichlorine molecule, while frame 76 is an example of a high lying π → π* excited state of the toluene around 200 nm.
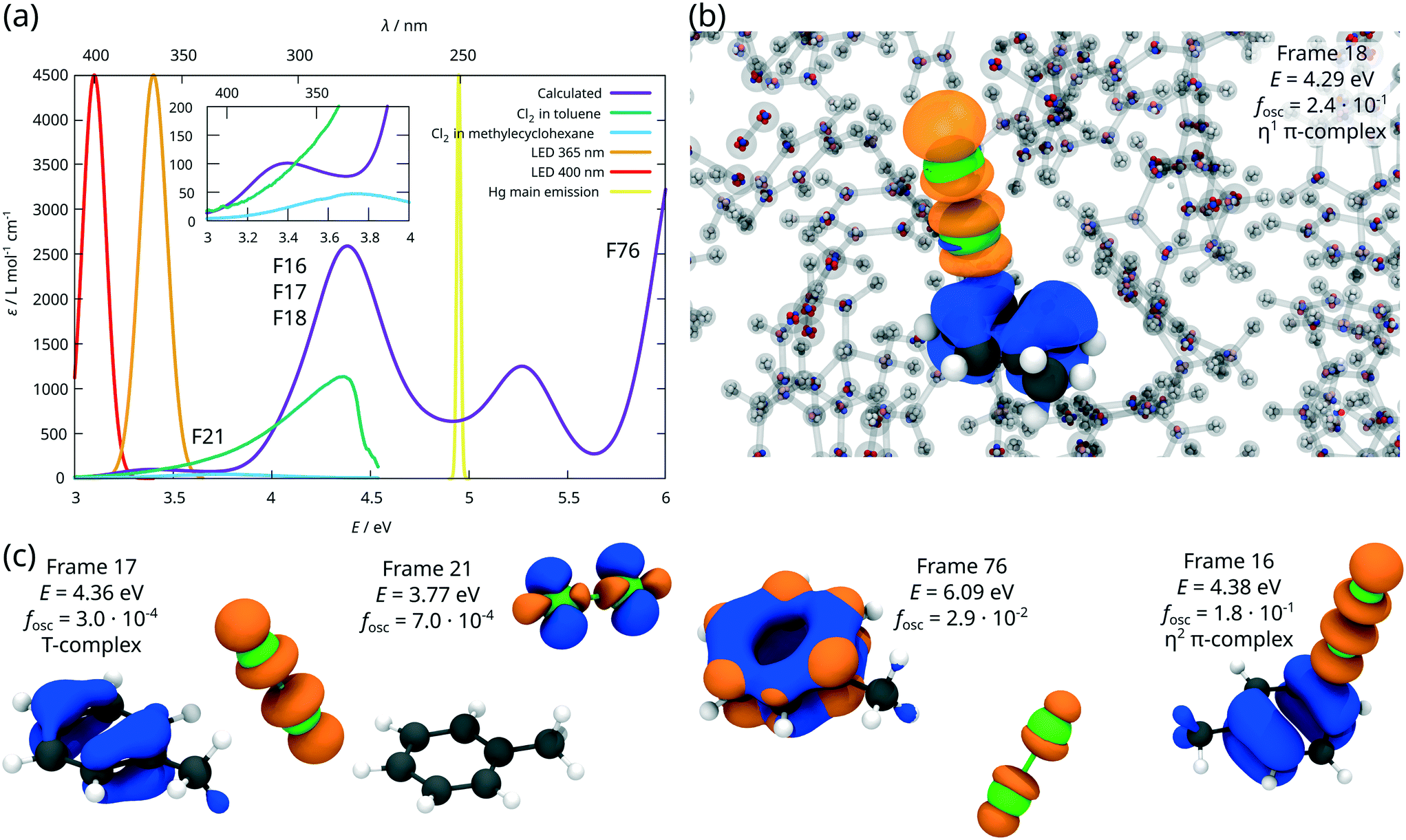 | ||
| Fig. 6 ADC(2) calculations in a polarisable QM/MM setup. (a) Calculated and experimental electronic absorption spectra as well as approximated emission spectra of the respective light sources.1,42 (b) QM/MM ADC(2) setup shown exemplarily for frame 18. The QM atoms are drawn as large solid spheres. A slice of the MM system is shown as transparent molecules and the multipoles of the MM atom, which are represented as point charges, are shown as small colour-coded spheres on the MM atoms (red = negative charge, blue = positive charge). Frame 18 is an η1 π-complex with a very bright charge transfer excitation at E = 4.29 eV and an oscillator strength of fosc = 2.4 × 10−1. The corresponding charge density difference is shown as isosurface (blue = negative, orange = positive). (c) CDDS and electronic properties for selected complexes from the AIMD simulation. | ||
The intense absorption caused by bright charge-transfer excitations from toluene to dichlorine leads in consequence to a light-driven charge separation, yielding a toluenyliumyl radical cation (C7H8˙+) and a dichloridyl radical anion (Cl2˙−), see Fig. 7. These highly reactive species are not only in close proximity due to the initial requirement for this excitation of forming a π-complex, but they will also be stabilised and drawn closer as a result of Coulomb attraction in this excited state. As the positive charge resides in the π-system of toluene, this induces a strong preference for a negatively charged chlorine species to attack the ring selectively, while the bond order of the dichlorine molecule is reduced due the singly-occupied σ*-orbital. Consequently, a strong driving force for the photochemical reaction via charge-recombination processes and radical recombinations is anticipated, while the ring-chlorinated photoproduct is obtained upon excited state relaxation back to the electronic ground state. However, a detailed computational investigation regarding the various relaxation cascades of the excited states, forming the ring-chlorinated toluene species, exceeds the present scope of investigations. Future theoretical studies will focus on assessing the excited state relaxation cascades leading to the formation of the ring-chlorinated by-product.
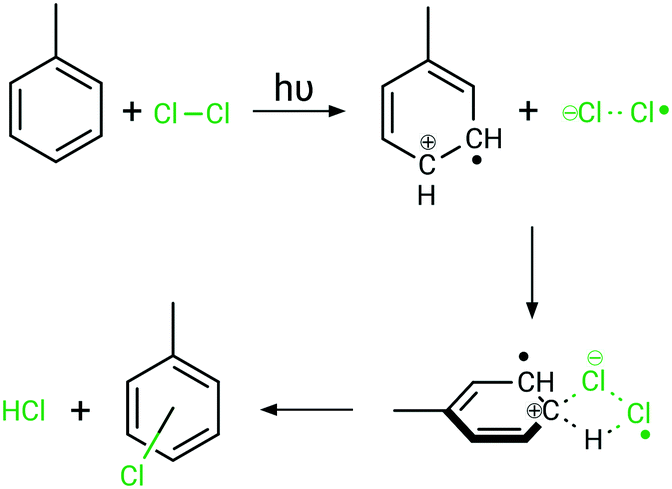 | ||
| Fig. 7 Proposed mechanism for selective ring-photochlorination at high dichlorine concentrations under irradiation. The Coulomb attraction enables selective reactions on the toluene ring. | ||
The computational results clearly show that indeed a change in the reaction mechanism occurs when high chlorine concentrations can be realised during the reaction. Instead of a homolytic cleavage of the Cl–Cl bond, inter-molecular electron transfer occurs and the aromatic ring is activated. Consequently, the aromatic ring can be chlorinated under such reaction conditions. This mechanism not only explains the occurrence of ring-chlorinated side-products but also the contradictory results that were obtained for the microreactor and pulsed irradiation. Continuous irradiation of the microreactor causes permanent excitation of the bright toluene–Cl2-charge-transfer and subsequently yields ring chlorinations. In contrast, this reaction pathway is only available for a short time frame when pulsed irradiation is used. During dark phases, the reaction follows the common radical reaction pathway, yielding a significant reduction of ring chlorinations. A similar argumentation reasons the differences observed between irradiation during and after absorption. When the reaction solution is irradiated during absorption, chlorine is present in lower concentrations and with this, the probability of ring chlorination is lower than for the irradiation after absorption where high chlorine concentrations are present.
The spectral dependency of the selectivity can be explained by the computational findings as well. The absorption coefficients of the dichlorine–toluene-moiety are significantly larger in the deeper UV-region as in the UV-A or visible wavelength region. Since the mercury vapour lamp emits light in this deeper UV-region, the probability of exciting the toluene–Cl2-charge-transfer increases and the selectivity towards benzyl chloride decreases.
These results clearly show that intensification of photochlorination requires careful consideration of the reaction conditions. It is of paramount importance to avoid the activation of the aromatic ring. For this, photons or in other words energy, must be delivered to dichlorine in a controlled manner. Pulsed irradiation of light sources emitting far outside the absorption region proved to be a viable option to realise the required reaction control.
4 Conclusions
The present joint experimental–theoretical contribution aimed to address the selectivity of the photochlorination of toluene, a reaction of tremendous industrial importance. The combined expertise allowed identification of the reasons for changing the selectivities of photochlorination under intensified reaction conditions that were in contradiction to the established theory of photochlorination. The formation of a toluene–chlorine complex that activates the aromatic ring upon irradiation was perceived as the cause of ring activation. This insight not only allows an in-depth understanding of the relevant reaction parameters but also represents a new pathway for activating aromatic rings.To intensify photochlorination it is essential to suppress the excitation of the toluene–Cl2-charge-transfer. This can be easily realised by pulsed irradiation and the use of light sources emitting outside the absorption window of the toluene–chlorine complex at approximately 290 nm, e.g. in the range of 350 to 420 nm. The application of forced dynamic irradiation conditions is an attractive but yet unexplored tool to gain reaction control for photochemical reactions. It not only allows for direct intervention in the activation of photochemical steps but also for the decoupling of transport processes and reactions. Furthermore, dynamic irradiation leads to significant energy savings. This makes the dynamic steering of the irradiation field a powerful option for the development of highly selective and efficient photochemical processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support provided by the German Research Foundation within the Priority Program “Reactive Bubbly Flows”, SPP 1740 (ZI 1502/1-1) as well as within the TRR CATALIGHT – Projektnummer 364549901 – TRR234 (projects C5 and C6). Dirk Ziegenbalg thanks Prof. Dr.-Ing. Elias Klemm (ITC) for continuous support.References
- Ü Taştan and D. Ziegenbalg, React. Chem. Eng., 2020 10.1039/d0re00263a.
- INEOS AG, INCH Magazine, 2014, vol. 7, p. 30 Search PubMed.
- H. G. Haring and H. W. Knol, Chem. Process Eng., 1964, 560–567 Search PubMed.
- H. G. Haring and H. W. Knol, Chem. Process Eng., 1964, 619–623 CAS.
- H. G. Haring and H. W. Knol, Chem. Process Eng., 1964, 691–693 Search PubMed.
- K.-A. Lipper, E. Löser and O. Brücher, in Ullmann's Encyclopedia of Industrial Chemistry, 2017, pp. 1–22 Search PubMed.
- Ü. Taştan, F. Guba and D. Ziegenbalg, Chem. Eng. Technol., 2017, 40, 1418–1424 CrossRef.
- A. M. Braun, M.-T. Maurette and E. Oliveros, Photochemical Technology, John Wiley & Sons Ltd, New York, 1991 Search PubMed.
- D. Šakić and V. Vrček, J. Phys. Chem. A, 2012, 116, 1298–1306 CrossRef.
- S. R. Spencer, M. Zhang and C. R. F. Lund, J. Phys. Chem. A, 2003, 107, 10335–10345 CrossRef CAS.
- Y. Osamura, K. Terada, Y. Kobayashi, R. Okazaki and Y. Ishiyama, J. Mol. Struct.: THEOCHEM, 1999, 461–462, 399–416 CrossRef CAS.
- Y. Wei, B.-W. Wang, S.-W. Hu, T.-W. Chu, L.-T. Tang, X.-Q. Liu, Y. Wang and X.-Y. Wang, J. Phys. Org. Chem., 2005, 18, 625–631 CrossRef CAS.
- A. K. Croft and H. M. Howard-Jones, Phys. Chem. Chem. Phys., 2007, 9, 5649–5655 RSC.
- M.-L. Tsao, C. M. Hadad and M. S. Platz, J. Am. Chem. Soc., 2003, 125, 8390–8399 CrossRef CAS.
- S. Miertus and J. Tomasi, Chem. Phys., 1982, 65, 239–245 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CAS.
- G. Lippert, M. Parrinello and J. Hutter, Mol. Phys., 2010, 92, 477–488 CrossRef.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef.
- M. Krack, Theor. Chem. Acc., 2005, 114, 145–152 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 123, 154101 CrossRef.
- E. R. Johnson and A. D. Becke, J. Chem. Phys., 2006, 124, 174104 CrossRef.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2006, 124, 221101 CrossRef.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef.
- M. Brehm and B. Kirchner, J. Chem. Inf. Model., 2011, 51, 2007–2023 CrossRef CAS.
- J. W. Ponder, C. Wu, P. Ren, V. S. Pande, J. D. Chodera, M. J. Schnieders, I. Haque, D. L. Mobley, D. S. Lambrecht, R. A. DiStasio, M. Head-Gordon, G. N. I. Clark, M. E. Johnson and T. Head-Gordon, J. Phys. Chem. B, 2010, 114, 2549–2564 CrossRef CAS.
- J. A. Rackers, Z. Wang, C. Lu, M. L. Laury, L. Lagardère, M. J. Schnieders, J.-P. Piquemal, P. Ren and J. W. Ponder, J. Chem. Theory Comput., 2018, 14, 5273–5289 CrossRef CAS.
- P. Ren, C. Wu and J. W. Ponder, J. Chem. Theory Comput., 2011, 7, 3143–3161 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision B.01, Gaussian Inc. Wallingford CT, 2016 Search PubMed.
- A. J. Stone, J. Chem. Theory Comput., 2005, 1, 1128–1132 CrossRef CAS.
- E. G. Kratz, A. R. Walker, L. Lagardère, F. Lipparini, J.-P. Piquemal and G. Andrés Cisneros, J. Comput. Chem., 2016, 37, 1019–1029 CrossRef CAS.
- E. G. Kratz, R. E. Duke and G. A. Cisneros, Theor. Chem. Acc., 2016, 135, 166 Search PubMed.
- J. Turnkey, A. Simmonet, R. Parrish, E. Hohenstein, F. Evangelista, J. Fermann, B. Mintz, L. Burns, J. Wilke, M. Abrams, N. Russ, M. Leininger, C. Janssen, E. Seidl, W. Allen, H. Schaefer, R. King, E. Valeev, C. Sherrill and T. Crawford, WIREs Comput. Mol. Sci., 2012, 2, 556 Search PubMed.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358 CrossRef CAS.
- D. Mester, P. R. Nagy and M. Kállay, J. Chem. Phys., 2018, 148, 094111 CrossRef.
- M. Kállay, P. R. Nagy, Z. Rolik, D. Mester, G. Samu, J. Csontos, J. Csóka, B. P. Szabó, L. Gyevi-Nagy, I. Ladjánszki, L. Szegedy, B. Ladóczki, K. Petrov, M. Farkas, P. D. Mezei and B. Hégely, MRCC, a quantum chemical program suite, 2019, www.mrcc.hu Search PubMed.
- P. R. Nagy, G. Samu and M. Kállay, J. Chem. Theory Comput., 2018, 14, 4193–4215 CrossRef CAS.
- Ü. Taştan, J. Dollinger and D. Ziegenbalg, Flow Meas. Instrum., 2018, 59, 211–214 CrossRef.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |