
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTracking the rearrangement of atomic configurations during the conversion of FAU zeolite to CHA zeolite†
Koki
Muraoka‡§
 a,
Yuki
Sada‡
a,
Atsushi
Shimojima
bc,
Watcharop
Chaikittisilp
d and
Tatsuya
Okubo
*a
a,
Yuki
Sada‡
a,
Atsushi
Shimojima
bc,
Watcharop
Chaikittisilp
d and
Tatsuya
Okubo
*a
aDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: okubo@chemsys.t.u-tokyo.ac.jp
bDepartment of Applied Chemistry, Waseda University, 3-4-1 Ohkubo, Shinjuku-ku, Tokyo 169-8555, Japan
cKagami Memorial Research Institute for Materials Science and Technology, Waseda University, 2-8-26 Nishiwaseda, Shinjuku-ku, Tokyo 169-0051, Japan
dResearch and Services Division of Materials Data and Integrated System (MaDIS), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
First published on 7th August 2019
Abstract
In order to realize designed synthesis, understanding the formation mechanism of zeolites at an atomic level has long been aspired, but remains challenging due to the fact that the knowledge of atomic configurations of the species formed during the process is limited. We focus on a synthesis system that crystallizes CHA zeolite from FAU zeolite as the sole source of tetrahedral atoms of Si and Al, so that end-to-end characterization can be conducted. Solid-state 29Si MAS NMR is followed by high-throughput computational modeling to understand how atomic configurations changed during the interzeolite conversion. This reveals that the structural motif commonly found in FAU and CHA is not preserved during the conversion; rather, there is a specific rearrangement of silicates and aluminates within the motif. The atomic configuration of CHA seems to be influenced by that of the starting FAU, considering that CHA synthesized without using FAU results in a random Al distribution. A Metropolis Monte-Carlo simulation combined with a lattice minimization technique reveals that CHA derived from FAU has energetically favorable, biased atomic locations, which could be a result of the atomic configurations of the starting FAU. These results suggest that by choosing the appropriate reactant, Al placement could be designed to enhance the targeted properties of zeolites for catalysis and adsorption.
Introduction
Understanding formation mechanisms at an atomic scale is essential for the rational design and synthesis of materials. Once established, this approach should be able to identify fundamental factors in controlling materials formation as well as the corresponding process parameters, ultimately enabling the synthesis of novel materials with desired and adjustable properties. This approach has been extensively used in organic synthesis to connect building blocks and has produced countless numbers of chemical entities.1,2 Although atomic-level understanding has been aspired to for years with zeolite synthesis,3 previous studies have been limited owing to the nature of hydrothermal synthesis, which is a standard technique to prepare these microporous materials. The reaction is generally mediated by amorphous aluminosilicates,4–8 which makes the elucidation of the formation mechanism difficult. In situ analyses of zeolite formation9–15 have been conducted to capture a detailed image of zeolite synthesis. Although these studies have deepened our understanding of the mechanism, the exact atomic rearrangement during this process is unknown, which makes it fundamentally challenging to link intermediate species with the formed zeolite structures.Atomic-level understanding has not even been achieved for the zeolite product itself. Zeolites are crystalline microporous materials built from tetrahedral atoms and bridging oxygen atoms. Depending on the topology of the chemical network, different three-letter codes are assigned, such as FAU and CHA. While it has been well accepted that Si or Al can occupy the tetrahedral sites of aluminosilicate zeolites, conventional diffraction techniques can hardly differentiate Si and Al; hence, the precise atomic arrangements of Si and Al within zeolites were barely understood until very recently.16 Current progress in characterization techniques, detailed catalytic studies, and theoretical studies have enabled the quantification of the location of Al in zeolites.17–26
Because the detailed structures of some zeolites have been studied extensively, employing them as a seed crystal in starting materials is an effective means to deduce zeolite formation mechanisms.27–29 Indeed, seed-directed zeolite syntheses by the authors have shown structural similarities between the seeds and products, implying that the atomic structures of aluminosilicate species formed before the completion of zeolite growth can influence the structure of the zeolite products.28 However, seed-directed methods generally entail additional Si and Al sources in the starting materials, which complicates the formation mechanism and makes it difficult to understand.
In this sense, zeolite synthesis using another zeolite as the sole Si and Al source—the so-called “true interzeolite conversion (IZC)”30—is an attractive system to study, because the atomic-level structural information for the reactant and product is available. As is the case with seed-directed methods, several studies have discussed the structural similarity in IZC. A previous study assessed IZC from *BEA to MFI and claimed that their common building unit, termed mor, played an important role during the synthesis.31 In addition to other studies emphasizing common structural motifs in IZC,32,33 Martín et al. noted the possibility that FAU in reactant mixtures supplies a building unit, termed a double-six-ring (d6r), to foster the formation of CHA.34 Interestingly, both FAU and CHA are solely constructed with d6r as shown in Fig. 1, although there is no evidence to prove that d6r in FAU is directly transferred to CHA with the chemical topology intact.35 Another interesting point is that true IZC generally proceeds without the formation of amorphous matter,30 implying that end-to-end solid characterization is achievable. These features motivated us to examine the atomic configuration of CHA synthesized from FAU without any additional Si or Al sources. The combined experimental and computational analyses showed that CHA derived from IZC had a biased, energetically favorable atomic configuration, which could be the result of the atomic configuration of the starting FAU, although the d6r structure did not remain intact during the conversion.
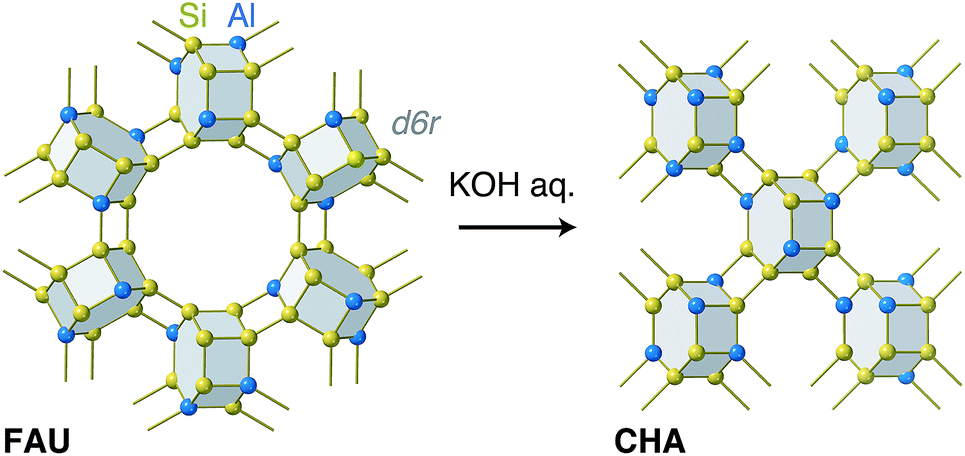 | ||
| Fig. 1 A schematic illustration of interzeolite conversion from FAU to CHA in the presence of a KOH aqueous solution without any additional Si or Al sources. Both FAU and CHA are constructed with a building unit, d6r (grey hexagonal prism). Yellow spheres are Si atoms, light blue spheres are Al atoms, and the yellow sticks are O atoms. | ||
Methods
Synthesis of zeolites
The starting Na-type FAU zeolite (HSZ-320NAA, Si/Al = 2.69) was purchased from Tosoh Corporation. Potassium chloride (99.9%) and potassium hydroxide (85%) were purchased from FUJIFILM Wako Pure Chemical Corporation. Ludox AS-40 colloidal silica (40%) and aluminum hydroxide were purchased from Sigma Aldrich. 1 g of the starting Na-type FAU and 4 g of potassium chloride were dispersed in 80 g of deionized water and agitated at 333 K for 2 h. The solid sample was recovered via filtration and thoroughly washed using deionized water. This ion-exchange procedure was repeated three times. The solid product was dispersed in a potassium hydroxide aqueous solution. This mixture had a chemical composition of 2.7 SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0 Al(OH)3:2.2 KOH:150H2O, and was heated at 423 K under autogenous pressure for 110 h in a Teflon reactor encapsulated in a stainless-steel autoclave tumbled at 20 rpm. The solid sample was recovered via filtration, thoroughly washed using deionized water, and dried at 353 K. For a control experiment, aluminum hydroxide was dissolved in a potassium hydroxide aqueous solution. After obtaining a clear colorless solution, colloidal silica was slowly added. The resulting mixture was agitated at ambient temperature and transferred to the Teflon reactor. The same heating and recovering procedures were applied.
:1.0 Al(OH)3:2.2 KOH:150H2O, and was heated at 423 K under autogenous pressure for 110 h in a Teflon reactor encapsulated in a stainless-steel autoclave tumbled at 20 rpm. The solid sample was recovered via filtration, thoroughly washed using deionized water, and dried at 353 K. For a control experiment, aluminum hydroxide was dissolved in a potassium hydroxide aqueous solution. After obtaining a clear colorless solution, colloidal silica was slowly added. The resulting mixture was agitated at ambient temperature and transferred to the Teflon reactor. The same heating and recovering procedures were applied.
Characterization
Powder X-ray diffraction (XRD) patterns were recorded on a diffractometer with CuKα radiation (λ = 1.54056 Å, Rigaku Ultima IV, 40 kV, 40 mA) at a scanning rate of 4° min−1 over a range of 3° to 50°. Thermogravimetric analyses were performed on a PU 4K (Rigaku) with a heating rate of 10 K min−1, using a mixture of 10% O2 and 90% He as the carrier gas. Samples dissolved in hydrofluoric acid were characterized using an inductively coupled plasma-atomic emission spectrometer (iCAP-6300, Thermo Scientific). Solid-state NMR spectra were recorded on a JEOL JNM ECA-400 spectrometer. 27Al magic-angle spinning (MAS) NMR spectra were recorded at 104.17 MHz with a relaxation delay of 1 s (30° pulse). 29Si MAS NMR spectra were recorded at 79.42 MHz with a relaxation delay of 100 s (45° pulse). Peak areas of the 29Si MAS NMR spectra were deconvoluted with Voigt functions using dmfit software.36 A field emission scanning electron microscope (SEM, Hitachi S-4700) was used for characterizing the morphology of the solid samples.Computation
Crystal structures of FAU and CHA were obtained from a database.37 After discarding crystal symmetries, crystal models of FAU with the chemical composition Si140Al52O384 (Si/Al = 2.69), without any Al–O–Al moieties, were generated by randomly introducing Al following a specific computational protocol.23,38 Models for CHA having the chemical composition Si198Al90O576 (Si/Al = 2.20) were created using the same procedure after expanding the unit cell by 2 × 2 × 2. The models closest to the experimental observations were identified using a method reported previously.23 Our in-house code was used to analyze the fraction of Q4(nAl) species. Models for CHA synthesized without using FAU were built through the same procedure with the reported chemical composition Si207Al81O576 (Si/Al = 2.56). The difference between the crystal models and experimentally observed 29Si MAS NMR spectra was defined as follows.FNMR(nAl) is the fraction of Q4(nAl) species by 29Si MAS NMR, while FModel(nAl) is that derived from the modeling.
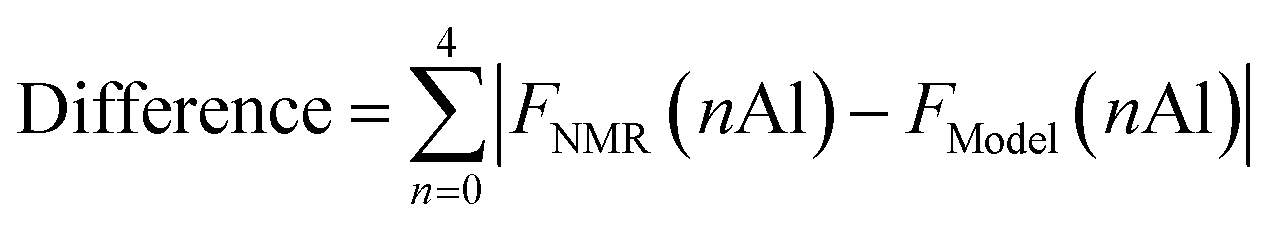
A Metropolis Monte-Carlo scheme was implemented and utilized based on the GULP package.39 Initial locations of counter cations were randomly decided based on geometrical considerations.23,38 After two counter cations were randomly selected, they were relocated based on randomly assigned oxygen atoms and the corresponding geometrical relations.23,38 After performing lattice minimization under constant pressure for 10 steps using a force field;40 this swapping was accepted if either the energy decreased, or with a probability of P = exp(−ΔE/kBT) if the energy increased. ΔE is the energy difference, kB is the Boltzman constant, and the temperature (T) was 423 K. After repeating the swapping 800 times, the final structure was optimized up to 1000 steps. Potassium cations were used for CHA, while sodium cations were used for FAU. Ten models with random Al distributions underwent the same Metropolis Monte-Carlo simulation for each case.
Results and discussion
In conventional FAU-to-CHA conversion, dealuminated FAU with many defects41 in the framework is frequently employed;32,34,42 still, it is not desirable for analyzing atomic configurations.23 Here, we started with Al-rich FAU to crystallize CHA in a KOH aqueous solution without any organics as shown in Fig. 1. The XRD patterns in Fig. 2 show that the peaks resulting from FAU decrease as the diffraction derived from CHA increases, as the hydrothermal treatment progresses.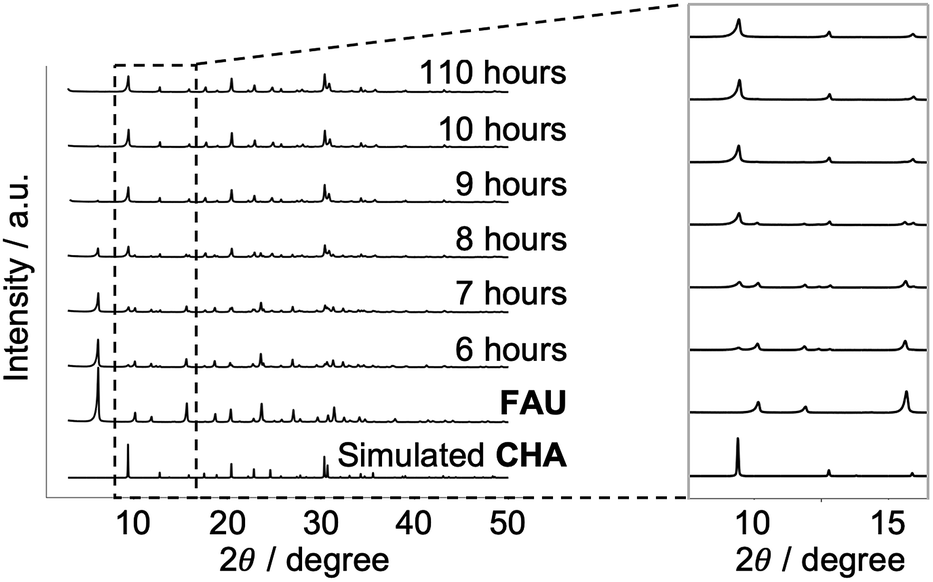 | ||
| Fig. 2 Powder X-ray diffraction patterns of the starting FAU and recovered samples with different heating times together with the simulated patterns for CHA. | ||
After 110 hours, all FAU was completely consumed and fully crystallized CHA was obtained. By examining the XRD patterns (Fig. 2) and SEM images (Fig. S1†), no amorphous matter can be observed throughout the conversion process, which is a common occurrence in other IZC systems,30 although the resolution of these techniques cannot confirm complete absence. Elemental analysis showed that the Si/Al molar ratio decreased from 2.69 to 2.17 via the FAU-to-CHA conversion. The decrease in Si/Al under alkaline synthesis conditions is a common phenomenon because the Si–O–Si bond is more easily cleaved by hydroxyl ions than the Si–O–Al bond.43 Such a change in the chemical composition is one of the possible factors that drive the conversion. Another plausible driving force in IZC is the formation of zeolites with a higher framework density,44,45 which holds for the conversion of FAU with a framework density of 13.3 T nm−3 to CHA with a framework density of 15.1 T nm−3.37
The peaks centered at approximately 60 ppm in the 27Al NMR spectra of FAU and CHA indicated that all Al species were incorporated into the zeolite frameworks with tetrahedral coordination (Fig. S2†), which is consistent with the high crystallinity of both zeolites. Because FAU and CHA have only one crystallographic tetrahedral site each, highly symmetric and sharp peaks were observed. The fact that FAU and CHA have only one crystallographic tetrahedral site is advantageous for analyses using 29Si MAS NMR.23 It is well accepted, particularly for zeolites with a single crystallographic tetrahedral site, that peaks in the 29Si MAS NMR spectra are ascribed to Si(–O–Si)4−n(–O–Al)n, i.e. Q4(nAl) species. From the peak areas in the 29Si MAS NMR spectra (Fig. 3a and e), the Si/Al ratios in the frameworks of FAU and CHA were calculated to be 2.74 and 2.20, respectively. These values are remarkably consistent with those from elemental analyses, presumably owing to the scarcity of defects (i.e., Q3; Si(–O–H)(–O–Si)3−n(–O–Al)n, n = 0–3) as confirmed from the NMR spectra. This is also supported by the crystal shapes and sizes (Fig. S1†).
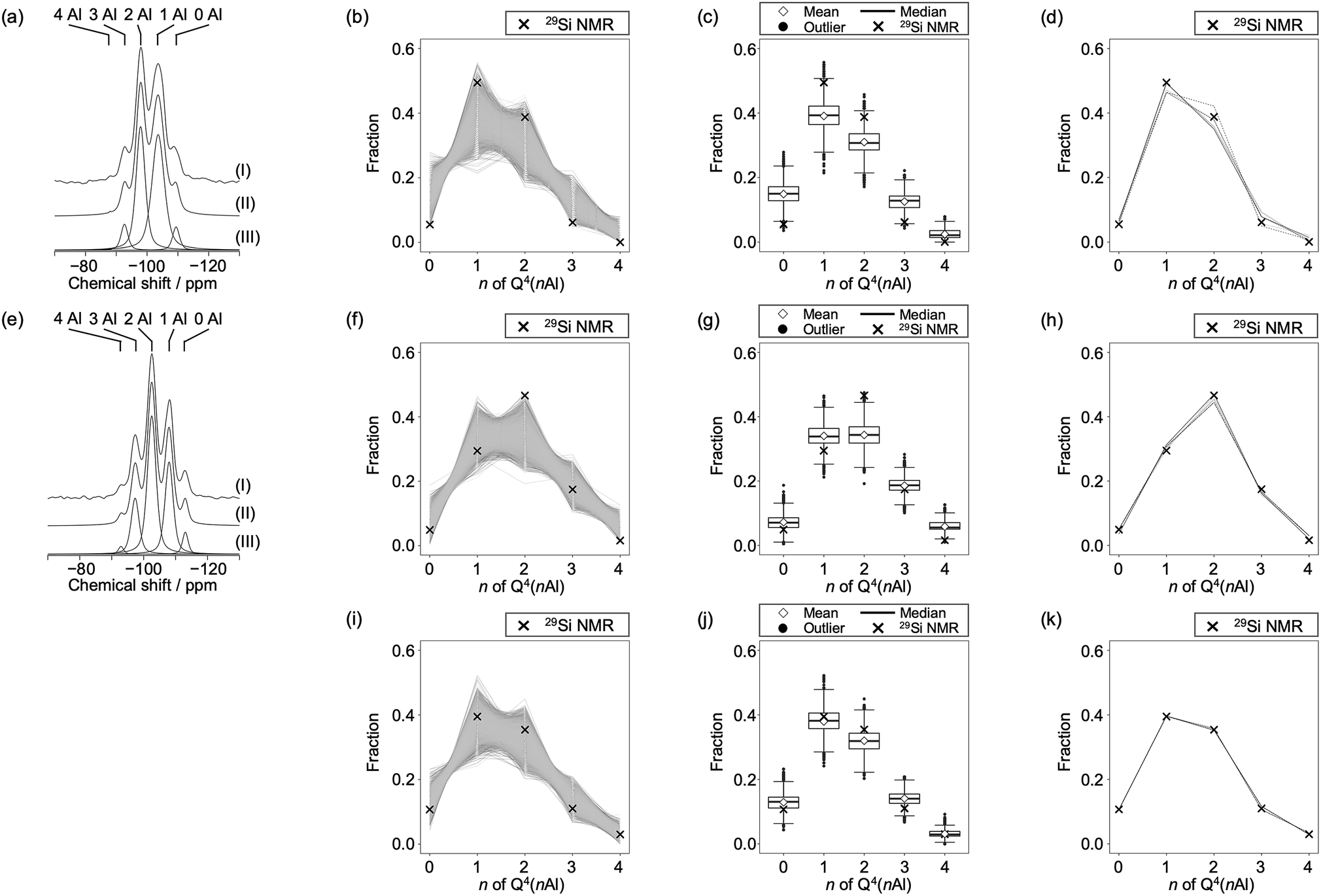 | ||
| Fig. 3
29Si MAS NMR analyses combined with computational modeling for (a–d) the starting FAU, (e–h) CHA synthesized from FAU, and (i–k) CHA synthesized without using FAU. Note that the 29Si MAS NMR spectrum of CHA synthesized without using FAU is not shown because data are taken from the literature.46 (a) and (e) show 29Si MAS NMR spectra of zeolites showing peaks derived from Si species coordinated with n O–Al bonds and 4 − n O–Si bonds where n = 0, 1, 2, 3, and 4 for (I) observed, (II) simulated, and (III) deconvoluted spectra. (b), (f), and (i) show fractions of Q4(nAl) species of 10000 generated models with random atomic configurations as lines, together with the experimentally observed values as cross symbols. (c), (g), and (j) show box plots of the fractions of Q4(nAl) species of 10000 models with random atomic configurations. The top and the bottom of the box are the 75th and 25th percentiles, respectively. The bold bar inside the box indicates the median, while the diamond indicates the mean. The upper and lower bars are the most extreme data points within 1.5 times the interquartile range. Outliers are shown as black dots. Experimental values are shown as cross symbols. (d), (h), and (k) show the fractions of Q4(nAl) Si species of the top five model structures (solid lines) that possess Si speciation closest to 29Si NMR data (cross symbols). | ||
To obtain the possible atomic configurations, we employed previously reported methods25 to compute 10000 aluminosilicate models for both FAU and CHA with the observed respective chemical compositions, but with random Al distributions.25 We then calculated the fraction of Q4(nAl) for each crystalline model and compared it to the experimentally observed values. As shown in Fig. 3b and f, this comparison shows that experimental values of both the starting FAU and resulting CHA were not statistically average but biased, as observed for siliceous FAU obtained via direct synthesis.23 The statistical plots in Fig. 3c and g confirm this, as the experimental values are within the range of outliers for both cases. For comparison, we performed the same computational analyses for CHA with similar Si/Al values, synthesized by hydrothermally treating a mixture of colloidal silica, aluminum hydroxide, potassium hydroxide, and water with the chemical composition 2.5SiO2:1.0Al(OH)3:2.5KOH:300H2O.46 Interestingly, the Q4(nAl) speciation of CHA prepared without using FAU was near the statistical average as shown in Fig. 3i and j. This indicates that the Al distribution of CHA derived from IZC is influenced by FAU.
To clarify the relationship between the Al distributions of FAU and CHA, we selected five models that are closest to the experimental Q4(nAl) values as shown in Fig. 3d, h, and k. The crystal structures are shown in Fig. S3–S5.† To consistently analyze FAU and CHA, we focused on the atomic configuration of their common building unit, d6r. As previously reported,23 there are only 19 topologically distinct atomic configurations for d6r as shown in Fig. 4, because Al–O–Al bonding is avoided in synthetic zeolites, which constitutes Löwenstein's rule.47 As shown in Fig. 4, the starting FAU possessed d6r with three Al atoms (3Al), particularly 3Al-1, 3Al-2, and 3Al-3, as dominant species (see the first row in Fig. 5). After the FAU-to-CHA conversion, d6r with 2Al and d6r with 3Al decreased, while the fraction of d6r with 4Al increased, which is consistent with the decrease in Si/Al via the IZC. This result is significantly different from a hypothetical scheme in which FAU directly supplies d6r to crystallize CHA. It is likely that a more intensive rearrangement is involved, such as the formation of oligomers smaller than d6r via dissolution, and substitutions between Si and Al—a prism break.
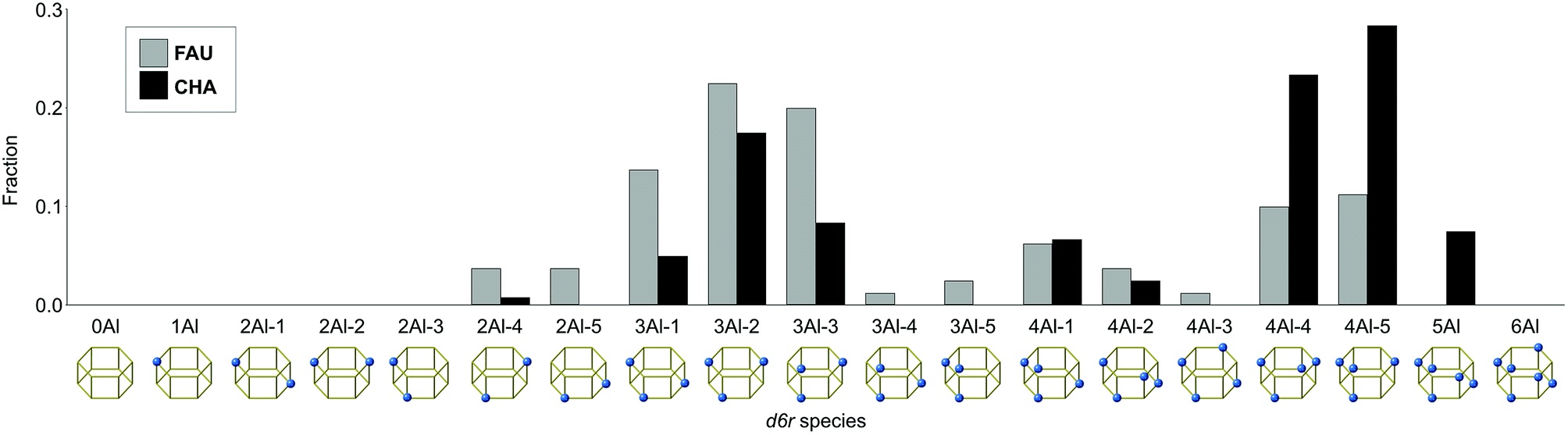 | ||
| Fig. 4 Fractions and configurations of Al in the d6r units found in the starting FAU and resulting CHA. | ||
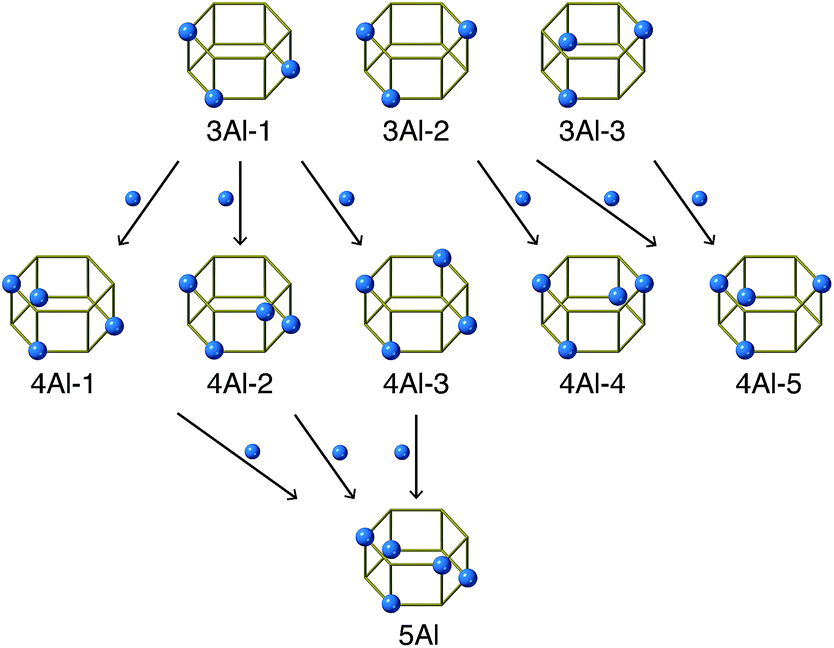 | ||
| Fig. 5 Relationships between d6r species. The first row is the most abundant species in FAU; 3Al-1, 3Al-2, and 3Al-3. Arrows connect pairs of d6r species that can be transformed via isomorphic substitution between Si and Al. See Fig. S6† for complete relationships. | ||
As opposed to the view that IZC can proceed by selectively dissolving Si without mobilizing Al,30 the current system likely involves mobile Al species owing to the highly alkaline conditions. Although the overall fraction of d6r with 4Al increased via the FAU-to-CHA conversion, the change in 4Al-1, 4Al-2, and 4Al-3 was relatively small. In the CHA derived from FAU, however, 4Al-4 and 4Al-5 markedly increased and were the dominant species. Among all d6r species with 4Al, 4Al-5 possesses the smallest number of Al–O–Si–O–Al moieties, which should be avoided as much as possible according to Dempsey's rule.23,48 Because the Al–O–Al moiety is not allowed by Löwenstein's rule, 4Al-5 lacks a Si site that can be replaced with Al. Similarly, 4Al-4 disallows the substitution of Al for Si without violating Löwenstein's rule. Fig. 5 maps the relationships that would occur by isomorphically substituting Si with Al. While 4Al-4 and 4Al-5 are saturated with Al, the other species, namely, 4Al-1, 4Al-2, and 4Al-3, have replaceable Si sites, which can form d6r with 5Al directly (Fig. 5 and S6†). This could explain why only 4Al-4, 4Al-5, and 5Al significantly increased while 3Al-1, 3Al-2, and 3Al-3 decreased during the FAU-to-CHA conversion.
To confirm that these findings were not trends inherent in the current chemical composition, we compared the average abundance ratios of d6r species with the experimental results, and for randomly generated models with the same chemical compositions. The plot in Fig. S7† shows that 4Al-4, 4Al-5, and 3Al-2 are remarkably abundant in the models consistent with the experiments, confirming that the effects are likely derived from the specific Al distribution present in the synthesized CHA. Notably, the actual process of atomic rearrangement during the FAU-to-CHA conversion is likely to be more complicated than that shown in Fig. 5. Nevertheless, considering that CHA synthesized without using FAU had a random Al distribution (see Fig. 3j), it is reasonable to think that the Al distribution of the starting FAU influenced the resulting CHA.
Possible factors that can lead to such biased atomic configurations are charge balancing, the local environment of atoms, and energy stability.21–23,25,38,49 To examine how energy stability affects the present system, we performed computational modeling based on the crystal models obtained from 29Si MAS NMR. The crystal models in Fig. S3–S5† lack the information of counter cations, which are potassium or sodium cations for the synthesis of the present CHA or the typical synthesis of FAU,23 respectively. Brute-force modeling of all locations is feasible when the number of Al for each cell is small50 but is unrealistic for the current Al-rich models with a myriad of possible combinations. Instead, we employed the Metropolis Monte-Carlo method51 to obtain the location of counter cations within a reasonable amount of computational time. A cycle of the computational protocol starts with updating the location of cations and subsequently optimizing the structure. The application of this scheme for a CHA model consistent with 29Si MAS NMR yielded the crystal structure shown in Fig. 6. As suggested in previous studies, the sites for potassium were the sites inside an eight-membered-ring and sites above d6r.37,50 An identical computational protocol was applied for ten CHA models with the same chemical compositions but with random Al locations. As shown in Fig. 6c, CHA derived from FAU was energetically preferred with an energy difference of −328 kJ per mol per unit cell (U.C.) on average. On the other hand, the models consistent with CHA synthesized without FAU showed statistically similar energy to the counterparts with random Al locations (Fig. 6f).
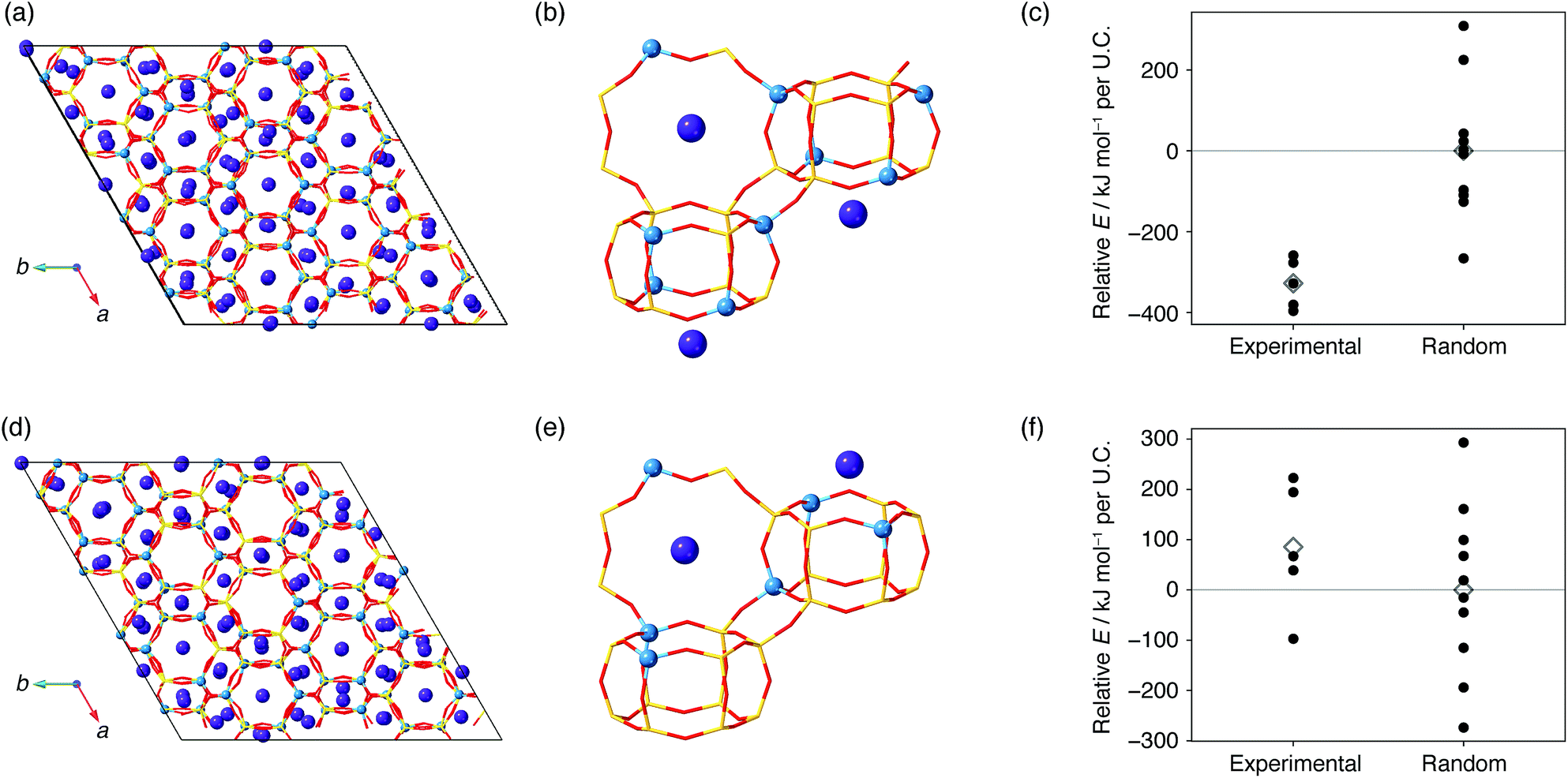 | ||
| Fig. 6 Results of the Metropolis Monte-Carlo simulation for CHA synthesized from FAU (a–c) and CHA synthesized without using FAU (d–f).46 (a), (b), (d), and (e) show the crystal structures derived from the Metropolis Monte-Carlo simulation of the models closest to 29Si MAS NMR. Yellow sticks are Si atoms, red sticks are O atoms, light blue spheres are Al atoms, and purple spheres are K atoms. (c) and (f) show the framework energies of the final structures of the Metropolis Monte-Carlo simulation (closed circles) for the five models consistent with 29Si MAS NMR and ten randomly selected models. Average energies are shown as open diamonds. Energies were offset by the average energy of models with random Al locations. | ||
Why did CHA synthesized from FAU have an energetically favorable, biased atomic configuration, while FAU-free synthesis produced CHA with random Al locations? Dusselier and Davis mentioned a low OH− content, required for the successful crystallization of zeolites with even membered-rings, as a characteristic of IZC.30 Conventional synthesis generally requires a high OH/Si ratio to promote the formation of zeolites with even membered-rings, but this could have been substituted with the 4 membered-ring, 6 membered-ring, and d6r found in FAU which could act as a substitute for the highly alkaline medium that favors even membered-rings.30 Indeed, the synthesis of CHA by IZC requires less hydroxide (OH/Si = 0.82) than FAU-free synthesis (OH/Si = 1.0). When FAU was replaced with conventional Si and Al sources in an otherwise identical procedure, CHA did not crystallize, but formed a dense phase as shown in Fig. S8,† which implies that FAU influences formation pathways. The highly alkaline conditions in the FAU-free route may intensively dissociate and reunite Si–O and Al–O bonds in starting materials to form monomers and very small oligomers that result in random atomic configurations, while lower alkalinity in the IZC route would maintain part of the atomic configurations of the starting FAU to construct CHA with spatially biased Al locations. This hypothesis should be confirmed by other characterization methods such as solution-state NMR techniques, which merits investigation in future work.
If the atomic configuration of FAU helps achieve a structure with low energy in the resulting CHA, it is natural to anticipate that the starting FAU also has an energetically stable Al distribution. To validate this hypothesis, we performed Metropolis Monte-Carlo analysis on FAU zeolite models. As shown in Fig. 7, the models consistent with the starting FAU showed more negative energies than the counterparts with random Al locations, which has also been reported for laboratory-prepared FAU.23 Since detailed methods to reproduce commercial FAU have not been disclosed, it is of interest to investigate such laboratory-prepared FAU zeolites as starting materials in the future.
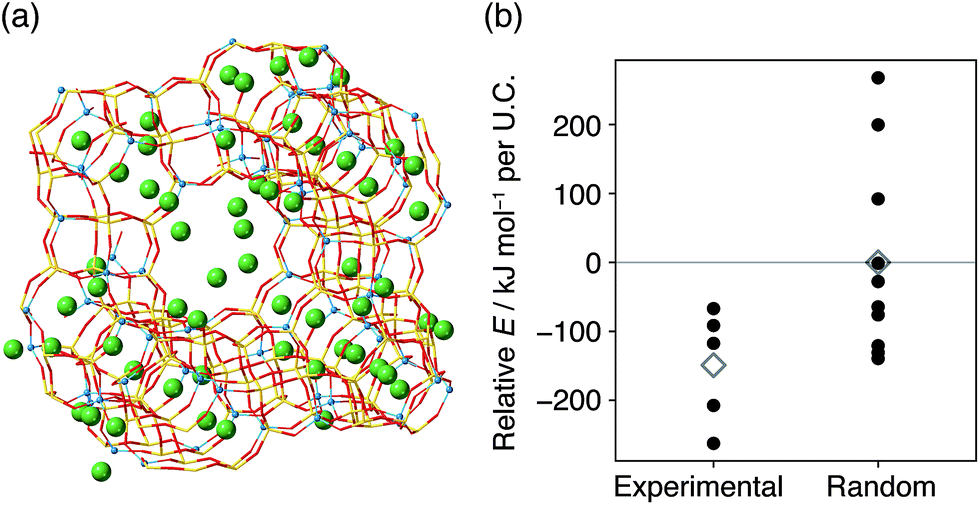 | ||
| Fig. 7 (a) The crystal structure derived from the Metropolis Monte-Carlo simulation of the model closest to 29Si MAS NMR of the starting FAU. Yellow sticks are Si atoms, red sticks are O atoms, light blue spheres are Al atoms, and green spheres are Na atoms. (b) Framework energies of the final structures of the Metropolis Monte-Carlo simulation (closed circles) for the five models consistent with 29Si MAS NMR and ten randomly selected models. Average energies are shown as open diamonds. Energies were offset by the average energy of models with random Al locations. | ||
The findings reported here suggest that CHA synthesized via IZC can be energetically influenced by the atomic configuration of the starting FAU. A simple transfer of d6r cannot explain this phenomenon, which leads us to think that a more complicated breaking and reuniting of T–O bonds is involved. This reconstruction of aluminosilicate frameworks was not likely enough to completely depolymerize the energetically favorable atomic configuration in FAU, which could result in the crystallization of CHA with stable Al locations.
Conclusions
To the best of our knowledge, we performed the first end-to-end characterization of atomic configurations during FAU-to-CHA conversion. It is a relevant system for this purpose because both zeolites have only one tetrahedral site and are constructed with only d6r. Our synthesis employs FAU as the sole Si and Al source and avoids amorphous matter formation. The results suggested that a specific rearrangement of silicates and aluminates occurred rather than a simple transfer of d6r. However, the atomic configuration of CHA was influenced by the starting FAU. Considering that the location of Al in the zeolite framework is essential for the most industrially important applications of CHA, namely, in methanol-to-olefin reactions and in the reduction of NOx,30,52–54 the atomic-level insight obtained in this study will contribute to the development of catalysts with improved performance. In addition, the approach used in this study which applies the spectroscopic technique and high-throughput computational modeling based on theoretical calculations would be applicable to other materials with complex formation mechanisms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Computational resources were provided by the Supercomputer Center at the Institute for Solid State Physics (ISSP), The University of Tokyo. K. M. is a Japan Society for the Promotion of Science (JSPS) research fellow (DC1) and is supported by a JSPS Research Fellowship for Young Scientists (16J10484). The authors acknowledge the help of Dr T. Shibue (Materials Characterization Central Laboratory at Waseda University) for NMR measurements.55References
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- M. B. Park, Y. Lee, A. Zheng, F.-S. Xiao, C. P. Nicholas, G. J. Lewis and S. B. Hong, J. Am. Chem. Soc., 2013, 135, 2248–2255 CrossRef CAS PubMed.
- M. E. Davis and R. F. Lobo, Chem. Mater., 1992, 4, 756–768 CrossRef CAS.
- T. M. Davis, T. O. Drews, H. Ramanan, C. He, J. Dong, H. Schnablegger, M. A. Katsoulakis, E. Kokkoli, A. V. McCormick, R. L. Penn and M. Tsapatsis, Nat. Mater., 2006, 5, 400–408 CrossRef CAS PubMed.
- V. P. Valtchev and K. N. Bozhilov, J. Am. Chem. Soc., 2005, 127, 16171–16177 CrossRef CAS PubMed.
- S. Mintova, N. H. Olson and T. Bein, Angew. Chem., Int. Ed., 1999, 38, 3201–3204 CrossRef CAS PubMed.
- R. Li, N. Linares, J. G. Sutjianto, A. Chawla, J. Garcia-Martinez and J. D. Rimer, Angew. Chem., Int. Ed., 2018, 57, 11283–11288 CrossRef CAS PubMed.
- A. I. Lupulescu and J. D. Rimer, Science, 2014, 344, 729–732 CrossRef CAS PubMed.
- I. I. Ivanova, Y. G. Kolyagin, I. A. Kasyanov, A. V Yakimov, T. O. Bok and D. N. Zarubin, Angew. Chem., Int. Ed., 2017, 56, 15344–15347 CrossRef CAS PubMed.
- M. Khaleel, W. Xu, D. A. Lesch and M. Tsapatsis, Chem. Mater., 2016, 28, 4204–4213 CrossRef CAS.
- Q. Liu and A. Navrotsky, Geochim. Cosmochim. Acta, 2007, 71, 2072–2078 CrossRef CAS.
- A. Aerts, C. E. A. Kirschhock and J. A. Martens, Chem. Soc. Rev., 2010, 39, 4626–4642 RSC.
- S. Kumar, T. M. Davis, H. Ramanan, R. L. Penn and M. Tsapatsis, J. Phys. Chem. B, 2007, 111, 3398–3403 CrossRef CAS PubMed.
- M. Kumar, M. K. Choudhary and J. D. Rimer, Nat. Commun., 2018, 9, 2129 CrossRef PubMed.
- J. Dědeček, Z. Sobalík and B. Wichterlová, Catal. Rev., 2012, 54, 135–223 CrossRef.
- R. Gounder and E. Iglesia, J. Am. Chem. Soc., 2009, 131, 1958–1971 CrossRef CAS PubMed.
- R. Gounder and E. Iglesia, Acc. Chem. Res., 2012, 45, 229–238 CrossRef CAS PubMed.
- J. Dedecek, M. J. Lucero, C. Li, F. Gao, P. Klein, M. Urbanova, Z. Tvaruzkova, P. Sazama and S. Sklenak, J. Phys. Chem. C, 2011, 115, 11056–11064 CrossRef CAS.
- A. Vjunov, J. L. Fulton, T. Huthwelker, S. Pin, D. Mei, G. K. Schenter, N. Govind, D. M. Camaioni, J. Z. Hu and J. A. Lercher, J. Am. Chem. Soc., 2014, 136, 8296–8306 CrossRef CAS PubMed.
- T. Yokoi, H. Mochizuki, S. Namba, J. N. Kondo and T. Tatsumi, J. Phys. Chem. C, 2015, 119, 15303–15315 CrossRef CAS.
- J. R. Di Iorio and R. Gounder, Chem. Mater., 2016, 28, 2236–2247 CrossRef CAS.
- M. D. Oleksiak, K. Muraoka, M. F. Hsieh, M. T. Conato, A. Shimojima, T. Okubo, W. Chaikittisilp and J. D. Rimer, Angew. Chem., Int. Ed., 2017, 56, 13366–13371 CrossRef CAS PubMed.
- B. C. Knott, C. T. Nimlos, D. J. Robichaud, M. R. Nimlos, S. Kim and R. Gounder, ACS Catal., 2018, 8, 770–784 CrossRef CAS.
- K. Muraoka, W. Chaikittisilp, Y. Yanaba, T. Yoshikawa and T. Okubo, Angew. Chem., Int. Ed., 2018, 57, 3742–3746 CrossRef CAS PubMed.
- J. E. Schmidt, L. Peng, J. D. Poplawsky and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2018, 57, 10422–10435 CrossRef CAS PubMed.
- Y. Kamimura, W. Chaikittisilp, K. Itabashi, A. Shimojima and T. Okubo, Chem.–Asian J., 2010, 5, 2182–2191 CrossRef CAS PubMed.
- K. Itabashi, Y. Kamimura, K. Iyoki, A. Shimojima and T. Okubo, J. Am. Chem. Soc., 2012, 134, 11542–11549 CrossRef CAS PubMed.
- T. Ikuno, W. Chaikittisilp, Z. Liu, T. Iida, Y. Yanaba, T. Yoshikawa, S. Kohara, T. Wakihara and T. Okubo, J. Am. Chem. Soc., 2015, 137, 14533–14544 CrossRef CAS PubMed.
- M. Dusselier and M. E. Davis, Chem. Rev., 2018, 118, 5265–5329 CrossRef CAS PubMed.
- S. Goel, S. I. Zones and E. Iglesia, Chem. Mater., 2015, 27, 2056–2066 CrossRef CAS.
- M. Itakura, T. Inoue, A. Takahashi, T. Fujitani, Y. Oumi and T. Sano, Chem. Lett., 2008, 37, 908–909 CrossRef CAS.
- T. Sano, M. Itakura and M. Sadakane, J. Jpn. Pet. Inst., 2013, 56, 183–197 CrossRef CAS.
- N. Martín, M. Moliner and A. Corma, Chem. Commun., 2015, 51, 9965–9968 RSC.
- C. Li, M. Moliner and A. Corma, Angew. Chem., Int. Ed., 2018, 57, 15330–15353 CrossRef CAS PubMed.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- Database of Zeolite Structures, http://www.iza-structure.org/databases Search PubMed.
- K. Muraoka, W. Chaikittisilp and T. Okubo, J. Am. Chem. Soc., 2016, 138, 6184–6193 CrossRef CAS PubMed.
- J. D. Gale and A. L. Rohl, Mol. Simul., 2003, 29, 291–341 CrossRef CAS.
- R. A. Jackson and C. R. A. Catlow, Mol. Simul., 1988, 1, 207–224 CrossRef.
- C. Schroeder, M. R. Hansen and H. Koller, Angew. Chem., Int. Ed., 2018, 57, 14281–14285 CrossRef CAS PubMed.
- Y. Ji, M. A. Deimund, Y. Bhawe and M. E. Davis, ACS Catal., 2015, 5, 4456–4465 CrossRef CAS.
- R. M. Ravenelle, F. Schüβler, A. D'Amico, N. Danilina, J. A. van Bokhoven, J. A. Lercher, C. W. Jones and C. Sievers, J. Phys. Chem. C, 2010, 114, 19582–19595 CrossRef CAS.
- M. Maldonado, M. D. Oleksiak, S. Chinta and J. D. Rimer, J. Am. Chem. Soc., 2013, 135, 2641–2652 CrossRef CAS PubMed.
- W. Qin, R. Jain, F. C. Robles Hernández and J. D. Rimer, Chem.–Eur. J., 2019, 25, 5893–5898 CrossRef CAS PubMed.
- D. E. Akporiaye, I. M. Dahl, H. B. Mostad and R. Wendelbo, J. Phys. Chem., 1996, 100, 4148–4153 CrossRef CAS.
- W. Lowenstein, Am. Mineral., 1954, 39, 92–96 Search PubMed.
- E. Dempsey, G. H. Kuehl and D. H. Olson, J. Phys. Chem., 1969, 73, 387–390 CrossRef CAS.
- Y. Román-Leshkov, M. Moliner and M. E. Davis, J. Phys. Chem. C, 2011, 115, 1096–1102 CrossRef.
- R. E. Fletcher, S. Ling and B. Slater, Chem. Sci., 2017, 8, 7483–7491 RSC.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, J. Chem. Phys., 1953, 21, 1087–1092 CrossRef CAS.
- M. Yoshioka, T. Yokoi and T. Tatsumi, ACS Catal., 2015, 5, 4268–4275 CrossRef CAS.
- T. Nishitoba, N. Yoshida, J. N. Kondo and T. Yokoi, Ind. Eng. Chem. Res., 2018, 57, 3914–3922 CrossRef CAS.
- B. E. R. Snyder, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Chem. Rev., 2018, 118, 2718–2768 CrossRef CAS PubMed.
- C. Izutani, D. Fukagawa, M. Miyasita, M. Ito, N. Sugimura, R. Aoyama, T. Gotoh, T. Shibue, Y. Igarashi and H. Oshio, J. Chem. Educ., 2016, 93, 1667–1670 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: SEM images, crystal structures closest to 29Si MAS NMR spectra, complete relationship of d6r species, fraction of d6r species, and an XRD pattern of a control experiment. See DOI: 10.1039/c9sc02773d |
| ‡ These authors contributed equally. |
| § Present address: Energy Technologies Area, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. |
| This journal is © The Royal Society of Chemistry 2019 |