DOI:
10.1039/C9SC03225H
(Edge Article)
Chem. Sci., 2019,
10, 8541-8546
Molybdenum and tungsten complexes with carbon dioxide and ethylene ligands†
Received
30th June 2019
, Accepted 12th August 2019
First published on 27th August 2019
Abstract
The first examples of stable metal complexes with coordinated ethylene and carbon dioxide ligands are reported. Reaction of tris(ethylene) complexes mer-M(C2H4)3(PNP) (M = Mo and W; PNP = 2,6-bis(diphenylphosphinomethyl)pyridine) with CO2 yields the corresponding, mixed cis-M(C2H4)2(CO2)(PNP) derivatives. X-ray studies reveal six-coordinate structures exhibiting η2-ethylene and κ2-C,O carbon dioxide coordination. Remarkably, the formation of the molybdenum CO2 adduct occurs also in the solid state at room temperature, under 4 bar of CO2, in a nearly quantitative manner.
Introduction
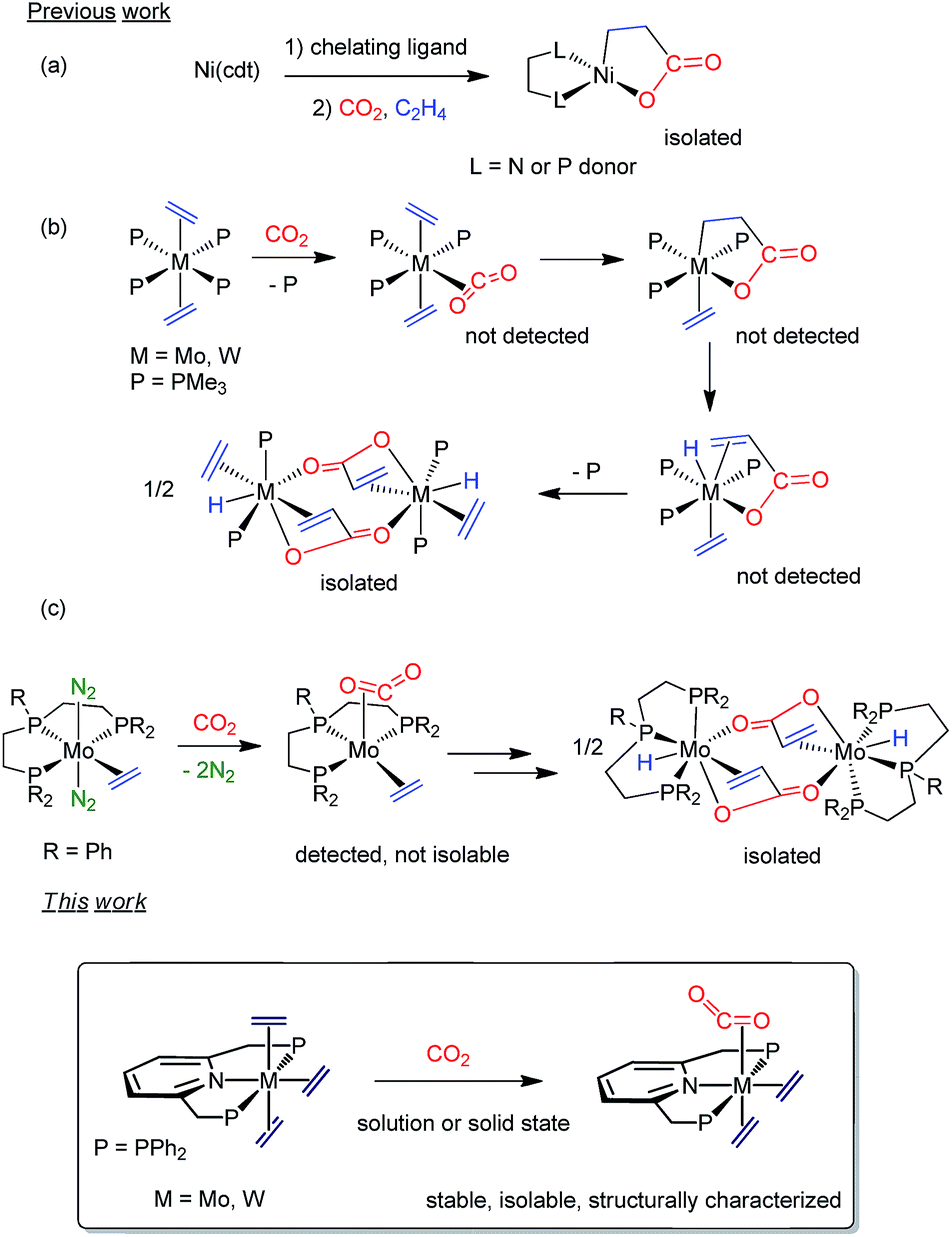
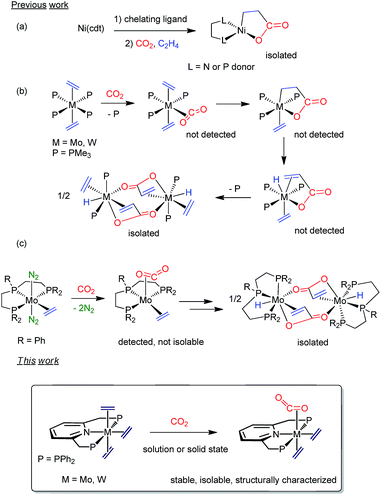
Since the twentieth century, organometallic chemistry has provided countless examples of molecules exhibiting two C-donor ligands possessing the potential to undergo metal-induced C–C coupling reactions.1 Yet to be discovered are, however, stable transition metal compounds with coordinated ethylene and carbon dioxide, which could be relevant in the context of acrylate formation2 by C–C bond formation. Notwithstanding decades of interest and intense research, catalytic conversion of C2H4 and CO2 into acrylate derivatives with the aim of replacing current non-environmentally friendly methods is still an elusive goal.3 In 1983, Hoberg4 achieved nickelalactone formation by means of a Ni(0)-mediated coupling of C2H4 and CO2 (Scheme 1a). This seminal discovery paved the way for the development of Ni- and Pd-catalyzed reactions, although productivity values were well below industrial needs.5,6 Only two years after Hoberg's findings, our parent laboratory disclosed that tertiary phosphine stabilized bis(ethylene) complexes of Mo(0) and W(0) promoted stoichiometric acrylate formation in the presence of CO2 under mild conditions (Scheme 1b).7 Though neither of the purported M(C2H4)(CO2) adduct and metallalactone intermediates could be detected, metal–hydrido-acrylate end-products derived from the latter by β–H elimination were isolated and characterized. Lately, Bernskoetter and coworkers described8 a modified system alike in reactivity but bearing a tridentate pincer PPP ligand (Scheme 1c). Solution studies suggested the intermediacy of a five-coordinate, sixteen-electron species with cis C2H4 and CO2 ligands, which could not be isolated.9 Although for the Mo and W systems CO2 and C2H4 coordination appears to be a prerequisite for acrylate formation,10 this proposal is at doubt for Ni.11 Related metallacycles have been reported for titanium,12 zirconium,13 vanadium,14 rhodium,15 cobalt16 and iron17 but corresponding species with the metal coordinated to both C2H4 and CO2 have escaped isolation. Chirik and coworkers have identified recently an iron-mediated C2H4–CO2 coupling to carboxylates of variable chain length up to 21 carbon atoms, and although they demonstrated control of the reaction outcome by the metallacyclic intermediates, evidence for the Fe(C2H4)(CO2) precursors was not provided.18
 |
| | Scheme 1 Transition metal complexes with ethylene and carbon dioxide as co-ligands. | |
Inspired by the work19 of Nishibayashi, Beller or Kichner, among others, about the capacity of Mo(PNP) linkages to promote different stoichiometric or catalytic transformations with low reactive molecules, we envisioned replacing our original ligand scaffold based on monodentate phosphines7 with the simple, –PPh2 bearing, pyridine-based pincer ligand 2,6-bis(diphenylphosphinomethyl)pyridine (from now on PNP in shorthand notation; see Scheme 1, bottom). Besides providing kinetic stability to M(0) complexes, the tridentate PNP unit would additionally thwart the mer-to-fac coordination change documented for the PPP tridentate phosphine ligand in Scheme 1c, and favor M(C2H4)(CO2) adducts. Herein, we report that the said tridentate ligand allows the synthesis of tris(ethylene) complexes, mer-M(C2H4)3(PNP) (M = Mo, 1a; W, 1b), as well as their conversion into the target mixed adducts, M(C2H4)2(CO2)(PNP) (M = Mo, 3a; W, 3b). Notably, formation of CO2 adducts, 3, proceeds readily in solution and the solid state for Mo in a quantitative manner. In spite of the presence of C2H4 and CO2cis ligands in the molecules of 3, these complexes are stable toward the formation of metallalactone and acrylate products. Computational support for the above observations is also reported.
Results and discussion
Synthesis of tris(ethylene) Mo(0) and W(0) complexes 1a and 1b
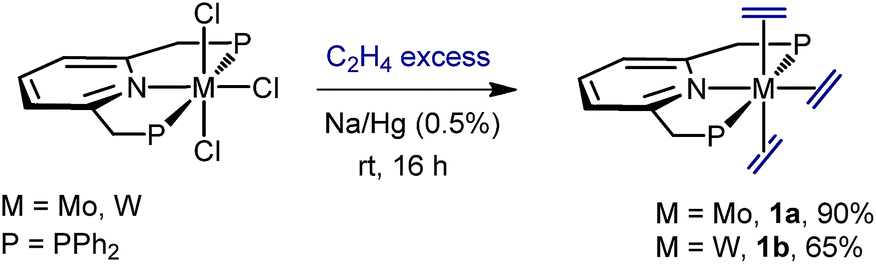
We found that sodium amalgam reduction of MCl3(PNP) under ethylene, at room temperature, yielded the tris(ethylene) complexes mer-M(C2H4)3(PNP) (M = Mo, 1a; W, 1b), which were isolated as dark red crystalline solids in good yields (Scheme 2). Attempts to prepare analogous complexes of PNP ligands with other –PR2 termini (R = i-Pr, Cy and t-Bu) proved fruitless. Spectroscopic data are in good agreement with the proposed structure (see the ESI†). In particular, the C2H4 ligands of complexes 1 give rise to two 13C{1H} NMR resonances at about 40 (two trans C2H4) and 49 ppm, both showing 2JC–P values close to 5 Hz. Comparison with the δ value found for free C2H4 of 123 ppm reveals Δδ shifts of 83 and 74 ppm to lower frequencies, consistent with increased electronic density due to strong back-donation.20 To our knowledge, complexes 1 are the first isolated tris(ethylene) complexes of molybdenum and tungsten, though Bernskoetter and co-workers detected in solution a species of this type when monitoring the reaction of a Mo(H)4(PPP) complex with C2H4.9
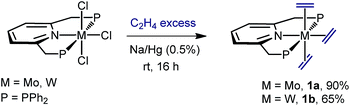 |
| | Scheme 2 Synthesis of the tris(ethylene) complexes 1a and 1b. | |
X-ray crystallography21 led to unequivocal structure authentication (Fig. 1) and confirmed the meridional arrangement of the ethylene molecules. The two in trans are staggered with respect to one another, whereas the two carbon atoms of the third, which occupy the coordination site trans to the pyridine N atom, are essentially co-planar to the PNP donor atoms. No significant difference exists among the Mo–Calkene distances to all C2H4 ligands, despite their diverse geometrical distribution. Thus, for 1a the Mo–C distances have values of 2.26–2.27 Å. Likewise, the three coordinated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds feature similar lengths, for instance 1.416(2) (C22–C23) and 1.425(2) Å (C24–C25). These metrical parameters, and those disclosed for 1b (see the ESI†), are significantly longer than the 1.34 Å distance corresponding to free C2H4,22 denoting the existence of strong electronic interactions between the electron-rich M(0) centers of complexes 1 and the π–C2H4 ligands.
C bonds feature similar lengths, for instance 1.416(2) (C22–C23) and 1.425(2) Å (C24–C25). These metrical parameters, and those disclosed for 1b (see the ESI†), are significantly longer than the 1.34 Å distance corresponding to free C2H4,22 denoting the existence of strong electronic interactions between the electron-rich M(0) centers of complexes 1 and the π–C2H4 ligands.
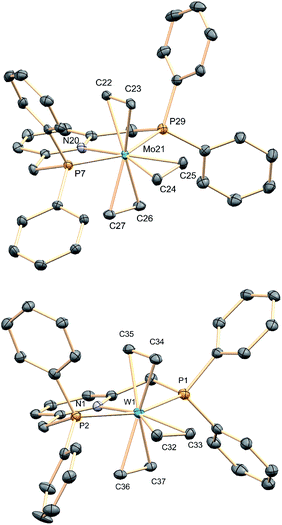 |
| | Fig. 1 Solid-state structures of tris(ethylene) complexes 1a (top) and 1b (bottom). Hydrogen atoms have been omitted for clarity. | |
Reactivity of complexes 1a an 1b toward carbon monoxide
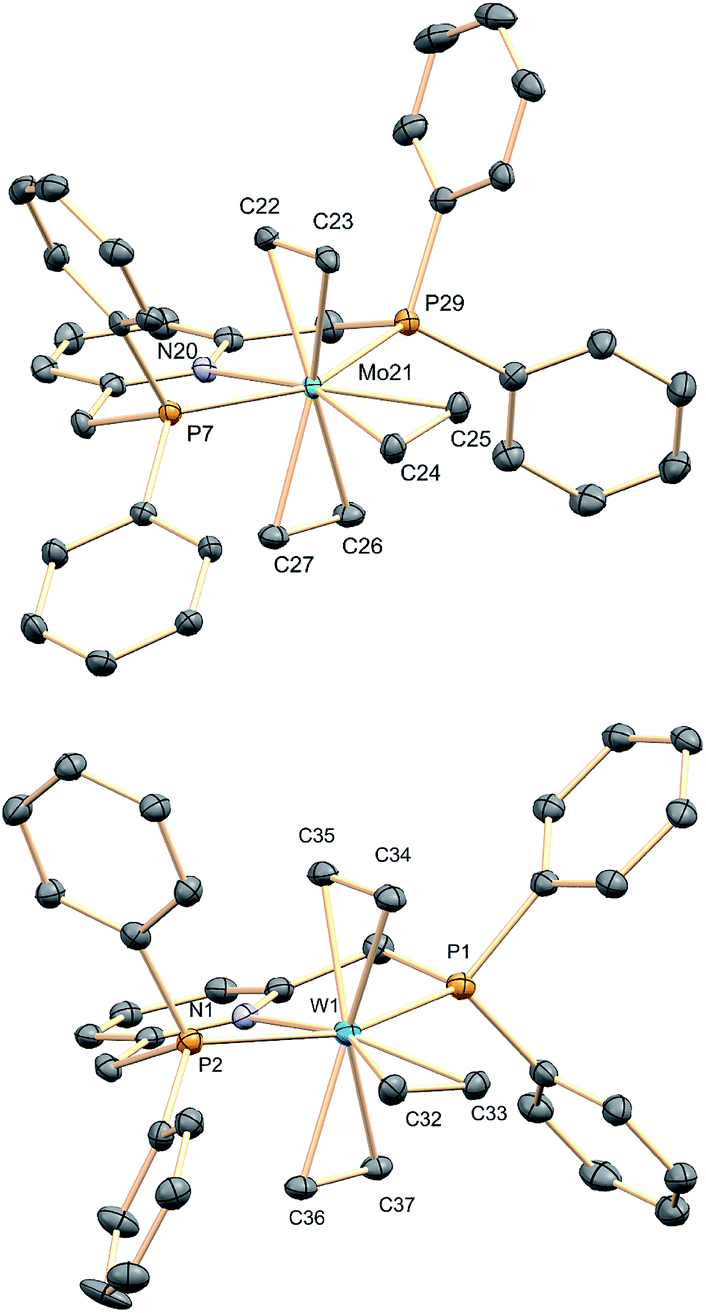
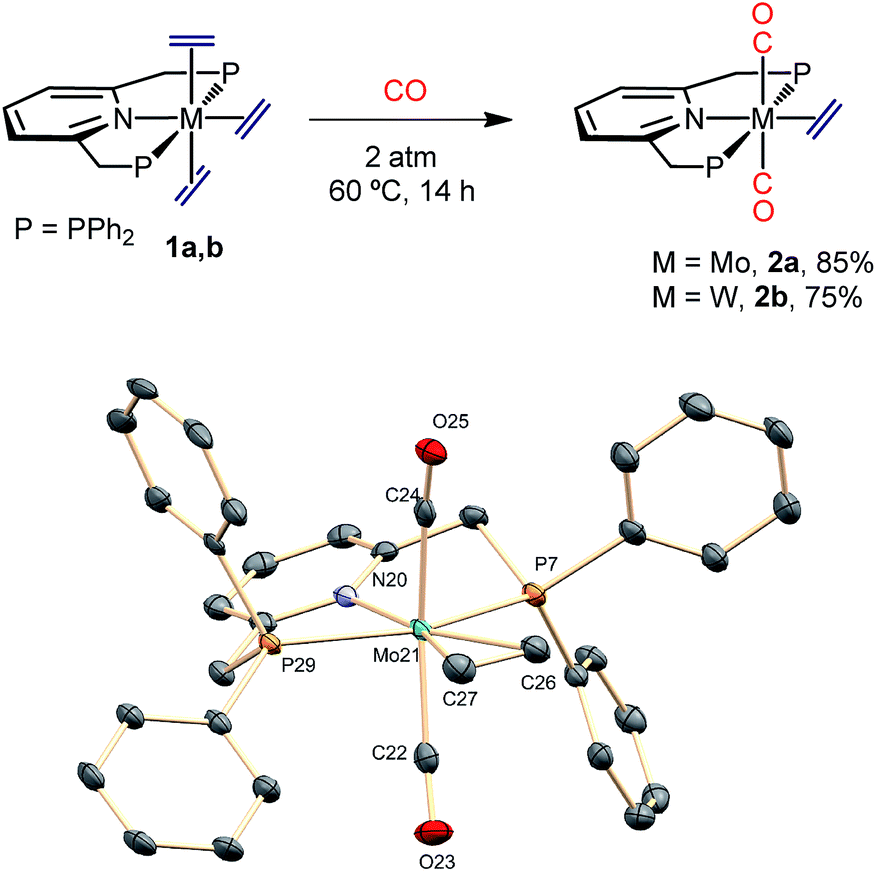
In contrast to results reported for trans-M(C2H4)2(PMe3)4 complexes (M = Mo and W),23 which are known to react with CO to give the mono-carbonyls trans,mer-M(C2H4)2(CO)(PMe3)3, exposure of complexes 1 to an atmosphere of CO originated the trans-M(C2H4)(CO)2(PNP) dicarbonyls (2a, Mo; 2b, W) as depicted in Scheme 3. The complexes were fully characterized by microanalysis, IR data [νasym(C–O) is 1807 and 1781 cm−1 for 2a and 2b, respectively], NMR spectroscopy and X-ray studies21 (see Scheme 3). The exchange of the axial ethylene ligands instead of the equatorial one provides a singular feature of compounds 1 when compared to the related, previously described bis(ethylene) complexes of Mo and W. This difference in behavior must be a consequence of the use of the PNP ancillary ligand instead of the monodentate PMe3 ligands, a fact that should also affect the reactivity toward carbon dioxide (see below).
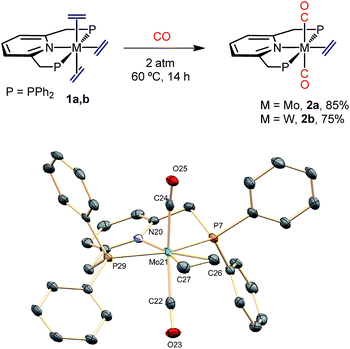 |
| | Scheme 3 Synthesis of the dicarbonyls 2a and 2b and the molecular structure of 2a. See the ESI† for the structure of the tungsten analogue 2b. Hydrogens are omitted for clarity. | |
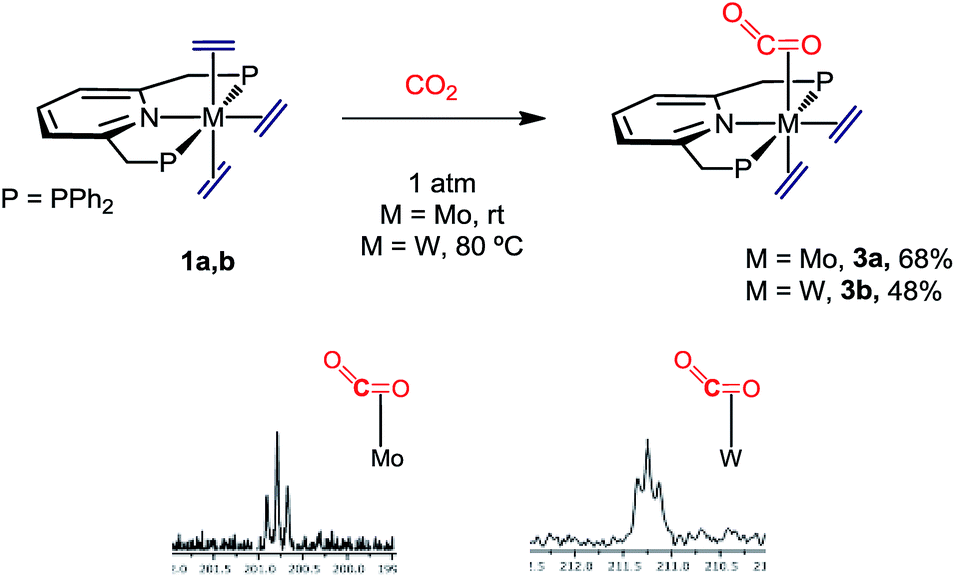
The mixed complexes M(C2H4)2(CO2)(PNP) (3a–3b)
When complexes 1 were submitted to a carbon dioxide atmosphere only one of the trans C2H4 ligands underwent ligand substitution upon exposure to CO2 (1 bar, toluene solution). For molybdenum, CO2 coordination took place at room temperature, but formation of the tungsten analogue needed heating at 80 °C for 4 h (Scheme 4). The new compounds were isolated as brown and yellow-brown crystalline solids, respectively, in 68% and 48% yields. With reference to the molybdenum complex 3a an IR absorption (nujol mull) centered at ca. 1700 cm−1 can be taken as an indication of κ2-C,O coordination of the molecule of carbon dioxide.24 Additional support for this proposal comes from the observation of a triplet 13C{1H} resonance with δ 200.8 ppm and 2JC–P = 14 Hz (Scheme 4), the two parameters resembling closely those reported for trans-Mo(CO2)2(PMe3)4.25 The two ethylene ligands seem to undergo fast rotation around the respective Mo–C2H4 bond axis at room temperature, so that only two 13C{1H} resonances are recorded (43.8, br s, and 50.6 ppm, t, 2JC–P = 4 Hz). Although the two 31P nuclei of these complexes are equivalent and resonate at 46.9 ppm in the 31P{1H} NMR (datum for 3a), the reduced symmetry brought about by the C2H4-by-CO2 replacement becomes apparent from the observation of a 1H AB spin system (δA = 4.75, δB = 4.65 ppm; 2JAB = 18 Hz) for the now diastereotopic methylene protons of the PNP ligand. Signature 13C{1H} resonances for the tungsten complex 3b were recorded at 210.2 (W–CO2, t, 2JC–P = 13 Hz), 45.4 (W–C2H4, t, 2JC–P = 4 Hz) and 44.0 ppm (W–C2H4, br s), and are also comparable to literature data for related complexes (see the ESI† for full description).23,26
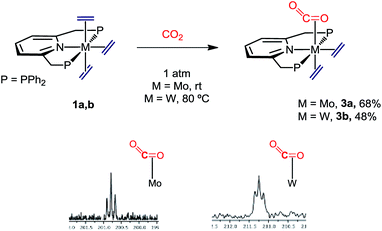 |
| | Scheme 4 Synthetic route to M(C2H4)2(CO2)(PNP) (M = Mo, 3a; M = W, 3b) and region of their 13C{1H} NMR spectra containing the CO2 resonances. | |
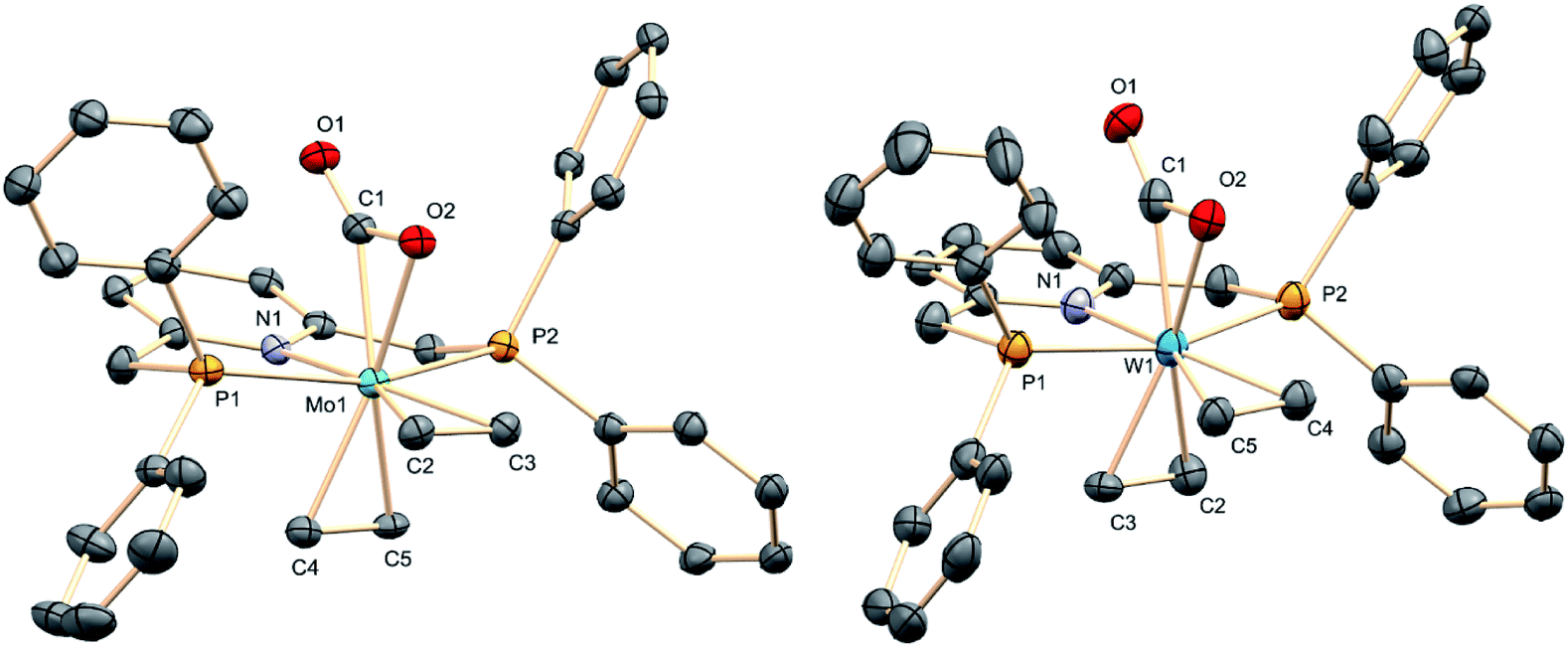
The molecular complexity proposed for complexes 3 on the basis of microanalytical and spectroscopic data was confirmed by X-ray crystallography.21 These studies provided the solid-state molecular geometries presented in Fig. 2, which possess the two C2H4 ligands in a cis distribution, hence a cis,mer-M(C2H4)2(CO2)(PNP) formulation. The molecule of carbon dioxide is bound to the metal in a κ2 fashion through one of the CO bonds, in a staggered orientation with respect to the trans ethylene ligand. The coordinated CO bond eclipses the M–N bond, while the two CC bonds eclipse the trans P–M–P vector. The four Mo–C2H4 distances in 3a have similar magnitude, close to 2.25 Å. The Mo1–C1 bond within the Mo–CO2 linkage is nonetheless significantly shorter at 2.107(4) Å, and it is also shorter than the associated Mo1–O2 separation of 2.215(3) Å. These metrics (see the ESI† for corresponding X-ray data for 3b) along with the elongation of the coordinated CO bond relative to the non-coordinated one (C1–O2 = 1.261(4) and C1–O1 = 1.213(4) Å; 1.243(12) and 1.248(13) Å in the tungsten analog, 3b) hint at the existence of strong M–CO2 electronic interactions in these complexes.
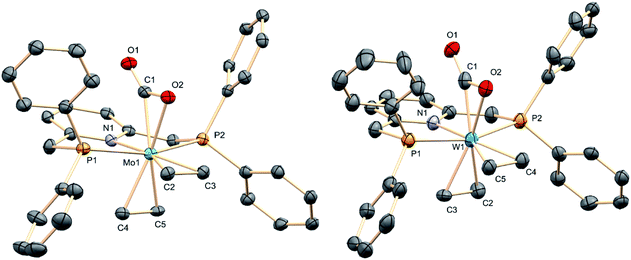 |
| | Fig. 2 The structures in the solid state of M(C2H4)2(CO2)(PNP) (M = Mo, 3a, left; M = W, 3b, right). Hydrogen atoms have been omitted for clarity. | |
Solid state ethylene replacement with carbon dioxide
Most notably, the reaction of crystalline samples of 1a with CO2 occurred also in the solid state at room temperature. Loss of crystallinity and a color change to brown (Fig. 3) were observed after 24 hours under 4 bar of carbon dioxide. NMR analysis of the resulting powdery brown solid revealed clean conversion into complex 3a, though minor amounts (<2%) of unreacted 1a were still present. For the tungsten case, such mild conditions were not enough to promote the exchange. However, heating solid 1b under 4 bar of CO2 for 24 h at 80 °C led to the formation of 3b in 20% yield, with 70% of 1b yet unreacted and 10% of free PNP ligand, formed upon decomposition, being also observed in the 31P{1H} NMR spectrum (see the ESI†).
 |
| | Fig. 3 Left: solid, crystalline sample of 1a before being submitted to 4 bar of CO2. Right: the same sample after 12 h, containing >98% of complex 3a. | |
The observation of the formation of these compounds in the solid state constitutes an uncommon observation linking transition metal carbon dioxide chemistry and solid-state molecular organometallic chemistry. The latter is a highly important, though still underdeveloped field of research.27
Stability of the complexes M(C2H4)2(CO2)(PNP) (3a,b)
The presence of both ethylene and carbon dioxide ligands in the coordination sphere of the M(C2H4)2(CO2)(PNP) complexes resembles the intermediate previously proposed (Scheme 1b) en route to their coupling, although in that case, which was also verified by DFT studies,10 the two ethylene ligands were in mutually trans disposition at variance with the cis geometry shown in 3a,b. Notwithstanding the presence of cis C2H4 and CO2 ligands in the coordination polyhedron of complexes 3, the putative C–C coupling reaction leading to metallalactone or acrylate derivatives could not be ascertained. Heating toluene solutions of compounds 3 under argon, in the absence or in the presence of C2H4 and CO2, with added base or with Lewis acids, resulted only in gradual decomposition with slow liberation of the PNP ligand, and no observation of acrylate formation.
Computational studies
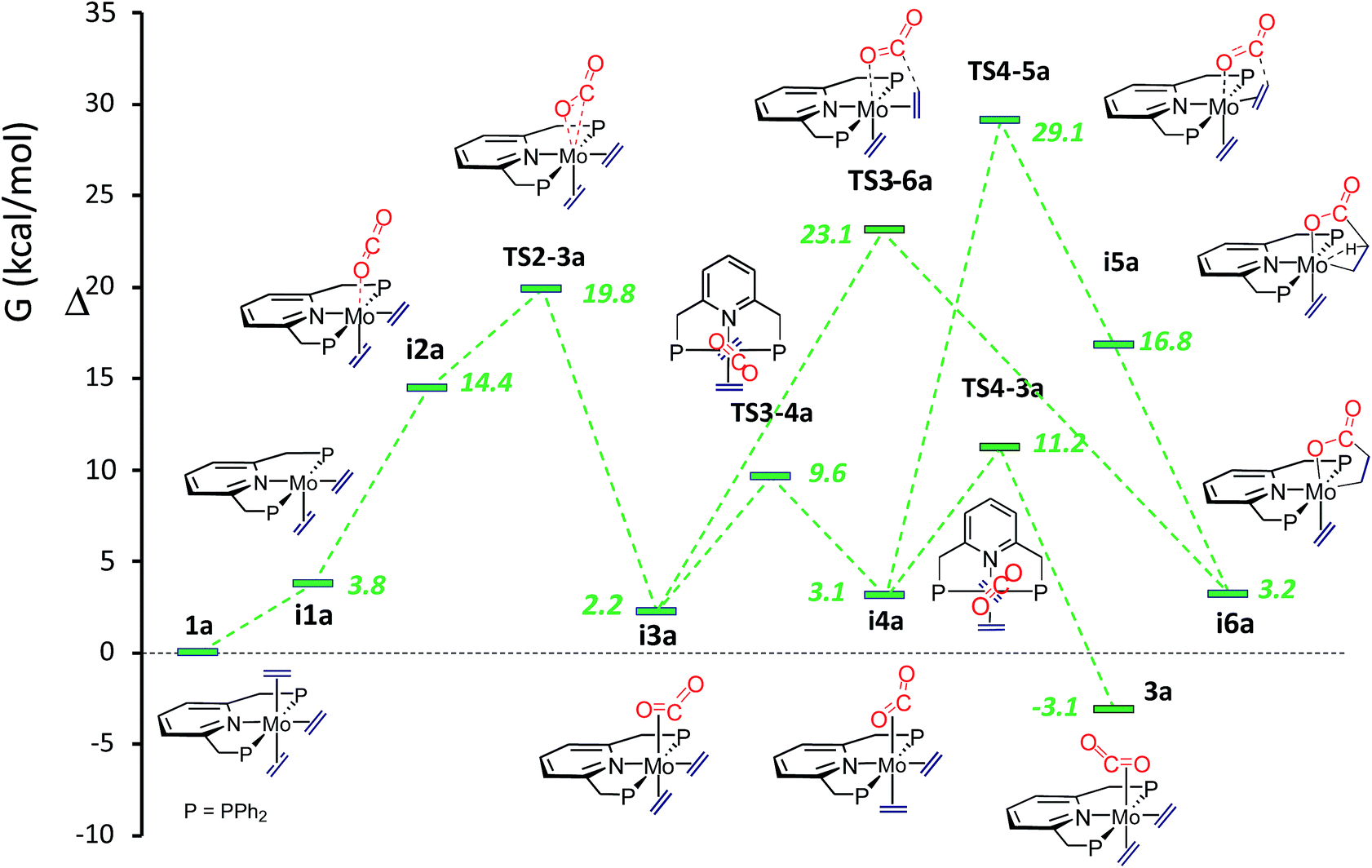
To gain a deeper understanding of all the findings reported in this contribution, computational studies were undertaken. Calculations revealed that conversion of the tris(ethylene) molybdenum complex into the carbon dioxide adduct 3a is a facile process, in agreement with the already described experimental findings. Furthermore, two intermediate isomeric rotamers, i3a and i4a (Fig. 4), were identified and were shown to interconvert by means of a concerted synchronous rotation28 of the trans CO2 and C2H4 ligands. Not unexpectedly, the 1a-to-3a conversion requires the dissociation of one of the trans ethylene molecules of the 18-electron complex 1, and subsequent coordination of CO2. The initially formed κ1-O adduct isomerizes to the κ2-C,O rotamer i3a, which is only 2.2 kcal mol−1 above the tris(ethylene) complex 1a, through TS2-3a, with energy just below 20 kcal mol−1 above 1a (see Fig. 4). Rotamers i3a and i4a have similar energies but the experimentally observed rotamer 3a is about 5 kcal mol−1 more stable, making the overall reaction exergonic by −3.1 kcal mol−1 (toluene solvent). Similar ΔG values have been computed for the tungsten system (Scheme S2 and Fig. S2 in the ESI†), with the principal difference with respect to the molybdenum analogue being a significantly higher transition state TS2-3b at 27.1 kcal mol−1 (also in toluene) relative to complex 1b. This is in excellent agreement with the slower formation of 3b, which requires heating at 80 °C for 4 h.
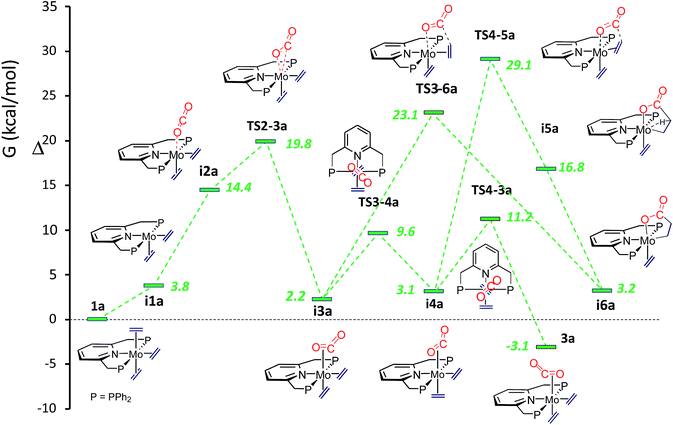 |
| | Fig. 4 Comparative ΔG profiles for the formation of CO2-complex 3a and lactone-complex i6a in toluene. | |
In accordance with expectations, metallalactone product formation was computed to be endergonic (3.2 and 2.1 kcal mol−1, in toluene, for the Mo and W systems, respectively). As shown in Fig. 4, two routes were calculated for the formation of the metallalactone species i6a starting from rotamers i3a and i4a. They are closely related to those described by the group of Papai for the M(C2H4)2(PMe3)4-plus-CO2 reactions10 (see the ESI†). A transition state for the purported formation of a hydrido-acrylate product from i6a could not be found, probably due to the rigidity of the Mo–PNP19c and Mo–metallated lactone structures. The putative hydrido acrylate structure has been computed as lying 23.6 kcal mol−1 above i6a (25.7 kcal mol−1 for i6b).
Conclusions
In conclusion, a suitable choice of ancillary ligands has allowed for the isolation of the first stable complexes bearing both C2H4 and CO2 ligands in the metal (Mo and W) coordination sphere. We have also shown that metallalactone formation by C–C coupling of the coordinated ligands is slightly endergonic, while the subsequent rearrangement to a hydrido-acrylate isomeric structure is strongly hindered kinetically by the rigidity of the M–PNP chelating structure, besides being thermodynamically uphill. We believe these results will be useful to design suitable catalytic systems for the conversion of C2H4 and CO2 into acrylate derivatives.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank MINECO for support with Grants CTQ2017-82893-C2-1-R and CTQ2017-82893-C2-2-R and a FPI fellowship (MA). A. G. thanks the Centro de Servicios de Informática y Redes de Comunicaciones (CSIRC), Universidad de Granada, for providing the computing time. We thank Francisco Molina for X-ray structure determination.
Notes and references
-
J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science books, 2009 Search PubMed.
-
(a) M. Limbach, Adv. Organomet. Chem., 2015, 63, 175–202 CrossRef CAS;
(b) X. Wang, H. Wang and Y. Sun, Chem, 2017, 3, 211–228 CrossRef CAS.
-
H. J. Arpe, Industrial Organic Chemistry, Wiley-VCH, 5th edn, 2010 Search PubMed.
-
(a) H. Hoberg and D. Schaefer, J. Organomet. Chem., 1983, 251, C51–C53 CrossRef CAS;
(b) H. Hoberg, Y. Peres, C. Krüger and Y.-H. Tsay, Angew. Chem., Int. Ed., 1987, 26, 771–773 CrossRef.
-
(a) N. Huguet, I. Jevtovikj, A. Gordillo, M. L. Lejkowski, R. Lindner, M. Bru, A. Y. Khalimon, F. Rominger, S. A. Schunk, P. Hofmann and M. Limbach, Chem.–Eur. J., 2014, 20, 16858–16862 CrossRef CAS PubMed;
(b) C. Hendriksen, E. A. Pidko, G. Yang, B. Schäffner and D. Vogt, Chem.–Eur. J., 2014, 20, 12037–12040 CrossRef CAS PubMed.
-
(a) S. Mazini, A. Cadu, A.-C. Schimdt, N. Huguet, O. Trapp, R. Paciello and R. Schaub, ChemCatChem, 2017, 9, 2269–2274 CrossRef;
(b) S. C. E. Stieber, N. Huguet, T. Kageyama, I. Jevtovikj, P. Ariyananda, A. Gordillo, S. A. Schunk, F. Rominger, P. Hofmann and M. Limbach, Chem. Commun., 2015, 51, 10907–10909 RSC.
-
(a) R. Álvarez, E. Carmona, D. J. Cole-Hamilton, A. Galindo, E. Gutiérrez-Puebla, A. Monge, M. L. Poveda and C. Ruíz, J. Am. Chem. Soc., 1985, 107, 5529–5531 CrossRef;
(b) R. Álvarez, E. Carmona, A. Galindo, E. Gutiérrez-Puebla, J. M. Marín, A. Monge, M. L. Poveda, C. Ruíz and J. M. Savariault, Organometallics, 1989, 8, 2430–2439 CrossRef;
(c) A. Galindo, A. Pastor, P. J. Pérez and E. Carmona, Organometallics, 1993, 12, 4443–4451 CrossRef CAS;
(d) C. Collazo, M. M. Conejo, A. Pastor and A. Galindo, Inorg. Chim. Acta, 1998, 272, 125–130 CrossRef CAS.
- W. H. Bernskoetter and B. T. Tyler, Organometallics, 2011, 30, 520–527 CrossRef CAS.
- Y. Zhang, B. S. Hanna, A. Dineen, P. G. Williard and W. H. Bernskoetter, Organometallics, 2013, 32, 3969–3979 CrossRef CAS.
- G. Schubert and I. Papai, J. Am. Chem. Soc., 2003, 125, 14847–14858 CrossRef CAS PubMed.
- M. Al-Ghamdi, S. V. C. Vummaleti, L. Falivene, F. A. Pasha and D. J. Beetstra, Organometallics, 2017, 36, 1107–1112 CrossRef CAS.
- S. A. Cohen and J. E. Bercaw, Organometallics, 1985, 4, 1006–1014 CrossRef CAS.
- H. G. Alt and C. E. Denner, J. Organomet. Chem., 1990, 390, 53–60 CrossRef CAS.
- B. Hessen, A. Meetsma, F. van Bolhuis, J. H. Teuben, G. Helgesson and S. Jagner, Organometallics, 1990, 9, 1925–1936 CrossRef CAS.
- M. Aresta and E. Quaranta, J. Organomet. Chem., 1993, 463, 215–221 CrossRef CAS.
- I. Knopf, M.-A. Courtemanche and C. C. Cummins, Organometallics, 2017, 36, 4834–4843 CrossRef CAS.
- H. Hoberg, K. Jenni, K. Angermund and C. Krüger, Angew. Chem., Int. Ed., 1987, 26, 153–155 CrossRef.
- S. M. Rummelt, H. Zhong, I. Korobkov and P. J. Chirik, J. Am. Chem. Soc., 2018, 140, 11589–11593 CrossRef CAS PubMed.
-
(a) K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120–125 CrossRef CAS PubMed;
(b) T. Leischner, A. Spannenberg, K. Junge and M. Beller, Organometallics, 2018, 37, 4402–4408 CrossRef CAS;
(c) O. Öztopcu, C. Holzhacker, M. Puchberger, M. Weil, K. Mereiter, L. F. Veiros and K. Kirchner, Organometallics, 2013, 32, 3042–3052 CrossRef PubMed.
- L. Cavallo, A. Macchioni, C. Zuccaccia, D. Zuccaccia, I. Irabona and F. Ruffo, Organometallics, 2004, 23, 2137–2145 CrossRef CAS.
- CCDC 1911725 (1a), 1911727 (1b), 1911730 (2a), 1911733 (2b), 1911735 (3a), and 1911736 (3b) contain the supplementary crystallographic data for this paper.†.
- H. J. Bernstein, Trans. Faraday Soc., 1961, 57, 1649–1656 RSC.
-
(a) E. Carmona, J. M. Marín, M. L. Poveda, J. L. Atwood and R. D. Rogers, J. Am. Chem. Soc., 1983, 105, 3014–3022 CrossRef CAS;
(b) E. Carmona, A. Galindo, M. L. Poveda and R. D. Rogers, Inorg. Chem., 1985, 24, 4033–4039 CrossRef CAS.
- A. Pastor, F. Montilla and A. Galindo, Adv. Organomet. Chem., 2017, 68, 1–91 CrossRef.
-
(a) R. Álvarez, E. Carmona, M. L. Poveda and R. Sánchez-Delgado, J. Am. Chem. Soc., 1984, 106, 2731–2732 CrossRef;
(b) R. Álvarez, E. Carmona, E. Gutiérrez-Puebla, J. M. Marín, A. Monge and M. L. Poveda, J. Chem. Soc., Chem. Commun., 1984, 1326–1327 RSC.
- Related concerted synchronous rotation of CO2 groups was demonstrated in trans,mer-Mo(CO2)2(PMe3)3(CNR) and related compounds. See: E. Carmona, A. K. Hughes, M. A. Muñoz, D. M. O'Hare, P. J. Perez and M. L. Poveda, J. Am. Chem. Soc., 1991, 113, 9210–9218 CrossRef CAS.
- S. D. Pike and A. S. Weller, Philos. Trans. R. Soc., A, 2015, 373, 20140187 CrossRef PubMed.
- Computed barriers for ethylene rotation in 3a are lower than 15.4 kcal mol−1.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of new compounds, crystallographic data and DFT calculations. CCDC 1911725, 1911727, 1911730, 1911733, 1911735 and 1911736. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03225h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Pedro J.
Pérez
*b,
Pedro J.
Pérez