
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A macrocyclic oligofuran: synthesis, solid state structure and electronic properties†
Sandip V.
Mulay‡§
 a,
Or
Dishi‡
a,
Yuan
Fang‡
b,
Muhammad R.
Niazi
b,
Linda J. W.
Shimon
c,
Dmitrii F.
Perepichka
*b and
Ori
Gidron
*a
a,
Or
Dishi‡
a,
Yuan
Fang‡
b,
Muhammad R.
Niazi
b,
Linda J. W.
Shimon
c,
Dmitrii F.
Perepichka
*b and
Ori
Gidron
*a
aInstitute of Chemistry, The Hebrew University of Jerusalem, Edmond J. Safra Campus, Jerusalem, Israel. E-mail: ori.gidron@mail.huji.ac.il
bDepartment of Chemistry, McGill University, Montreal, QC H3A 0B8, Canada. E-mail: dmitrii.perepichka@mcgill.ca
cChemical Research Support Unit, Weizmann Institute of Science, Rehovot, Israel
First published on 19th August 2019
Abstract
We report the first π-conjugated macrocyclic system with an oligofuran backbone. The calculated HOMO–LUMO gap is similar to that of the corresponding linear polymer, indicating a remarkable electron delocalization. The X-ray structure reveals a planar conformation, in contrast to the twisted conformation of macrocyclic oligothiophenes. The intermolecular π–π stacking distance is extremely small (3.17 Å), indicating very strong interactions. The macrocycle forms large π-aggregates in solution and shows a tendency toward highly ordered multilayer adsorption at the solid–liquid interface. The face-on orientation of molecules explains the higher hole mobility observed in the out-of-plane direction.
Introduction
π-Conjugated macrocycles with well-defined diameters have received significant attention in the last few decades, mainly because of their unique optical and electronic properties, their host–guest capabilities, and their potential as building blocks for supramolecular materials.1–6 Many π-conjugated macrocyclic systems aggregate into columnar structures,4,7–11 producing molecular channels that, with the appropriate inner-core functionalization, may give rise to ion channels, rod-like micellar aggregates, and even reaction chambers.12–15A particularly intriguing class of π-conjugated macrocycles is macrocyclic oligothiophenes (C-nT, Chart 1), introduced by Bäuerle's group in sizes ranging from C-8T to C-35T.16–18 These macrocycles demonstrate interesting electronic and optical properties,19 self-assembly at the solid–liquid interface,20–22 and stable redox behavior.23 Subsequently, Iyoda's group found that, when separated by acetylene spacers, thiophenes can form giant macrocyclic oligomers that arrange themselves into wire-like assemblies and form inclusion complexes with fullerenes.24–26 However, whereas the Iyoda's thienyleneethynylene macrocycles are planar, pure thiophene-based macrocycles, C-nT, in small diameters are distorted. Although no crystallographic information is available for C-8T, DFT calculations indicate that it adopts a spiderlike conformation27 and X-ray analysis of the larger C-10T reveals interring dihedral twisting of 26–34°.23,28 Such twisting may negatively affect both intramolecular π-conjugation and intermolecular interactions, which are crucial for optoelectronic applications.
 | ||
| Chart 1 Structures of the linear (L) and macrocyclic (C) α,α′-oligofurans (nBFI) and α,α′-oligothiophenes (nT) discussed in this work. R′ = n-butyl, R = 2-octyldodecyl. | ||
We have previously introduced linear α-oligofurans, which display high planarity/rigidity, good π-conjugation, and strong fluorescence compared with their thiophene analogs.29–32 Our calculations predicted that, in contrast to C-nT, small macrocyclic oligofurans (6- to 8-mers) should be planar, as the formed bisecting angle for each furan is 125° vs. 150° for thiophene, leading to low strain energies, lower HOMO–LUMO gaps, and stronger π-conjugation.33 Moreover, as macrocyclic oligofurans may be considered ‘conjugated crown ethers’, they are potentially interesting as host–guest systems or as components in supramolecular structures (rotaxenes, catenanes, etc.).34–37 However, despite the plethora of reported thiophene-based macrocycles,1,38 macrocyclic oligofurans are not known, possibly because the relative instability of oligofurans has hampered their development.39 To overcome this instability, we recently introduced 2,2′-bifuran-3,3′-dicarboximide oligomers and polymers (L-nBFI, Chart 1), which are significantly more stable than the parent furan.40 Since each BFI unit consists of two furans ‘locked’ in a syn orientation, and such preorganization is expected to favor macrocyclization by reducing strain energy,41 we envisioned that BFI could serve as a building unit for macrocyclic oligofuran scaffolds.
Here we describe the synthesis, structure, and properties of the first macrocycle having an oligofuran backbone, the α,α′-tetramer of 2,2′-bifuran-3,3′dicarboximide C-4BFI (Chart 1). In accordance with our calculations and crystallographic analysis, C-4BFI is planar and exhibits remarkably strong intermolecular interactions. In solution and as a solid, C-4BFI self-assembles via π-stacking. Scanning tunneling microscopy (STM) imaging reveals the formation of ordered multilayers at the solid–liquid interface. The face-on orientation of the molecules on the surface explains the observed charge transport anisotropy with higher mobility in out-of-plane direction.
Results and discussion
To find the ideal candidate for macrocyclization, we first calculated the electronic structures (at the DFT/B3LYP/6-311G(d) level) of C-nBFI molecules of various sizes (Fig. 1a). We found that the tetramer, C-4BFI, has the lowest strain energy (2.4 kcal mol−1 per bifuran unit), being lower than that of the smaller C-3BFI (7.5 kcal mol−1) and of the larger C-5BFI (5.0 kcal mol−1). In addition, the backbone of C-4BFI is predicted to be planar which maximizes the π-conjugation.42 Calculations predicted that the HOMO–LUMO gap for C-4BFI (2.39 eV) is 0.3 eV lower than that of the linear tetramer (L-4BFI), and is almost identical to the bandgap of the linear polymer (Fig. 1b). The predicted HOMO–LUMO gap in C-3BFI is also smaller compared with the linear analogue L-3BFI (by 0.2 eV) but the gap of C-5BFI macrocycle is larger by 0.18 eV than the gap of L-5BFI, which is attributed to its non-planarity. Both the HOMO and LUMO resemble those of the parent macrocyclic furans, with a small contribution from the imide group to the LUMO (Fig. 1c). The computational results thus reveal C-4BFI as an optimum target for cyclization.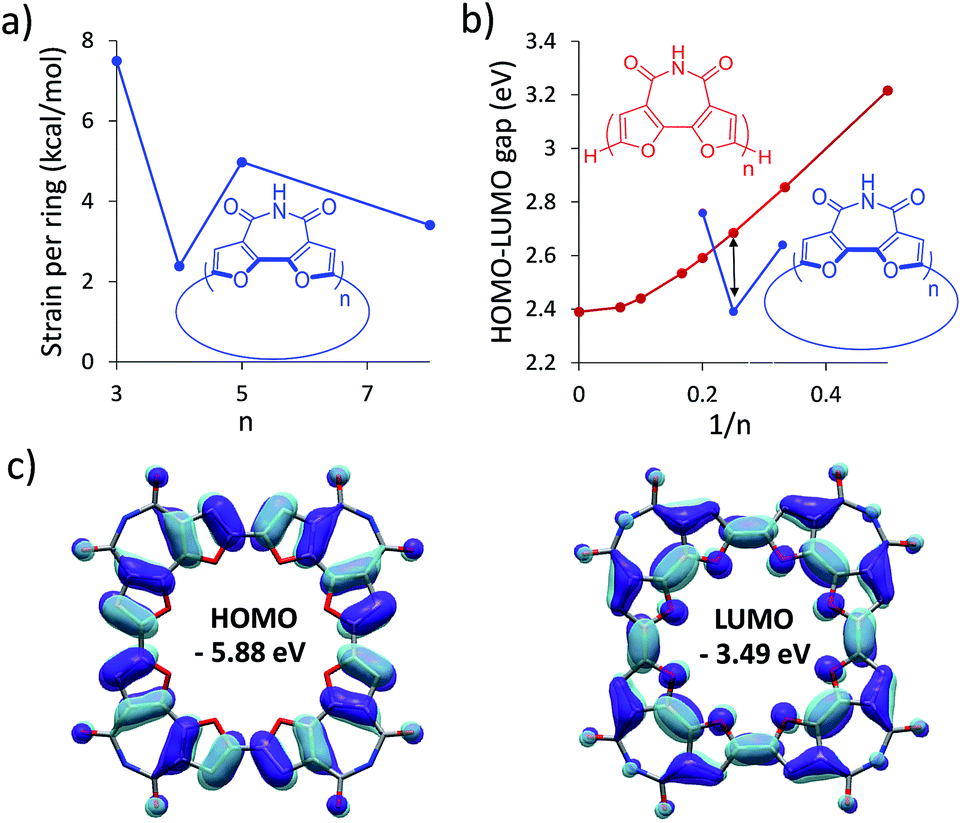 | ||
| Fig. 1 Calculated (B3LYP/6-311G(d)) (a) strain energy per unit for C-nBFI; (b) HOMO–LUMO gap for L-nBFI (red) and C-nBFI (blue), where the black arrow indicates the tetramer; and (c) the HOMO and LUMO surfaces of C-4BFI. | ||
The synthesis started with preparation of the linear tetramer L-4BFI by Stille coupling of stannane 1 with the dibrominated product of L-2BFI (2) (Scheme 1).40 The 2-octyldodecyl side chains were introduced for solubility (attempts to use n-hexyl side chain resulted in insoluble linear tetramer). The coupling product, L-4BFI, was then brominated using Br2 in the presence of FeCl3 to yield 3. Finally, macrocyclization was performed by adding a stoichiometric amount of Ni(COD)2 and 2,2′-bipyridine to a dilute solution (5 × 10−4 M) of 3 at 50 °C to yield C-4BFI (52%) after 48 h.43 The macrocycle was characterized using NMR and by MALDI-TOF (see ESI†). Cyclic voltammetry (CV) of C-4BFI shows a quasi-reversible oxidation peak at 0.78 V vs. Fc/Fc+, corresponding to HOMO level of −5.58 eV, and irreversible reduction at −1.50 V, corresponding to LUMO level of −3.30 eV, and resulting HLG value of 2.28 eV which is similar to the calculated value of 2.39 eV (see Section S9 in ESI†). While the current methodology was demonstrated for the cyclization of tetramer, it is plausible that larger macrocycles can also be prepared with BFI building block, as the strain energies decrease after reaching a maxima for the pentamer (Fig. 1a).
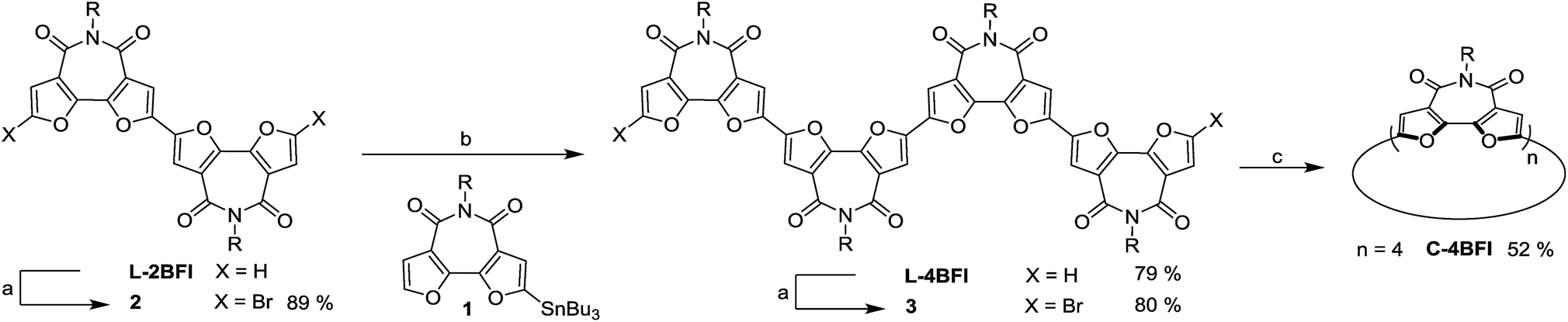 | ||
| Scheme 1 Synthesis of C-4BFI Conditions: (a) Br2, FeCl3, CH2Cl2, rt, dark; (b) Pd(PPh3)4, toluene, 90 °C; (c) Ni(COD)2, 2,2′-bipyridine, THF, 50 °C, 48 h. | ||
Single crystals of C-4BFI were grown by slow diffusion of acetone into dichloromethane solution, yielding red needles. The single crystal X-ray analysis shows that the macrocycle is planar (Fig. 2a). This contrasts calculated and experimental data for C-nT: the DFT predicts a spiderlike up and down conformation for C-8T,27,33 while the X-ray crystal structure for C-10T (the smallest structure available for marocyclic thiophenes) shows that the thiophene rings twist with dihedral angles of 26–34°.23 In order to compare the net heteroatom effect, we have calculated the thiophene analog of C-4BFI, C-4BTI. We found that while C-4BFI is nearly planar, with a planarization energy of 0.2 kcal mol−1, C-4BTI is distorted out of planarity, with planarization energy as large as 17 kcal mol−1 (Fig. S34, see ESI†). Thus, the heteroatom (sulfur or oxygen) plays a crucial role in the backbone planarity. The interring bond length between the adjacent BFI units in C-4BFI is 1.43 Å, which is shorter than that in C-4BFI is an indication an enhanced conjugation (see Fig. 56 ESI†). The inner diameter of C-4BFI macrocycles is 7.33 Å, corresponding to a van der Waals cavity of 4.3 Å. This is similar to the predicted inner cavity for 24-crown-8 ether (4–4.5 Å),44 yet achieved in a highly rigid π-conjugated structure that is interesting as a host for supramolecular engineering of optoelectronic materials.34
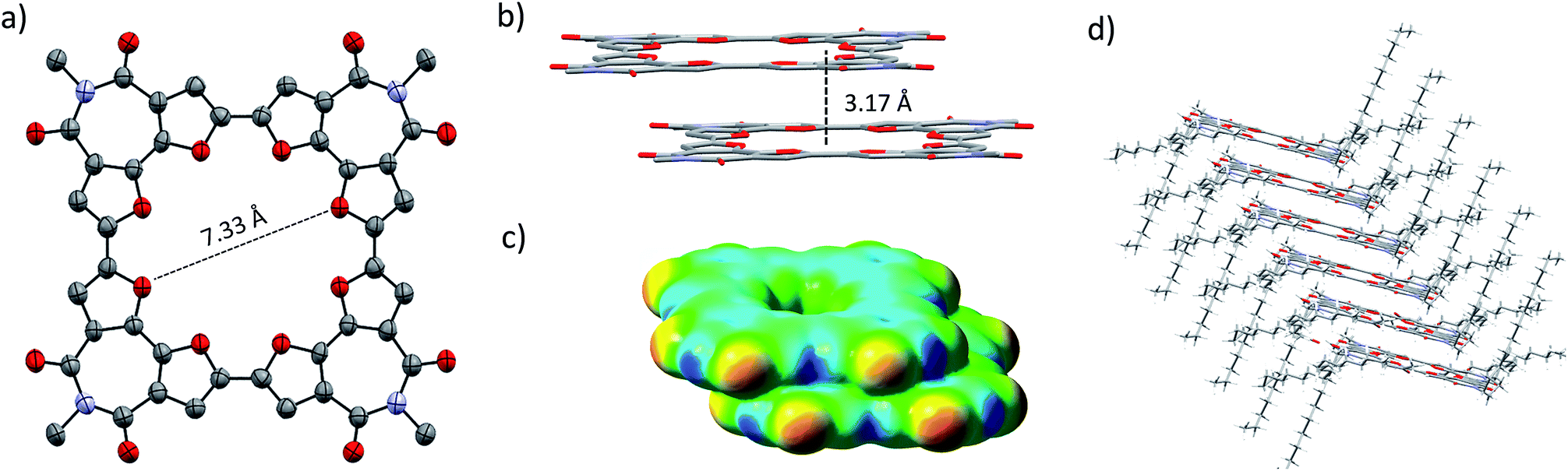 | ||
| Fig. 2 X-ray structure of C-4BFI. (a) Ellipsoid representation and (b) packing in stick representation excluding the 2-octyldodecyl groups. Hydrogens and solvent molecules are omitted for clarity. (c) Electrostatic potential map of C-4BFI dimer, calculated at the B3LYP/6-311G(d) level. (d) Stick representation of C-4BFI column, including 2-octyldodecyl side groups, showing slip-stack packing arrangement. | ||
The macrocycles pack in a slip-stacked motif (Fig. 2b), with a very short intermolecular π–π stacking distance of 3.17 Å (closest C⋯C distance 3.26 Å). The short packing distances contrast sharply with those found in thiophene–acetylene macrocycles (3.7 Å)24 or in C-10T (>8 Å),23 and is even closer than the interplane distance of graphite (3.35 Å). This tight packing is a result of the planarity of the macrocycle and the quadrupole moment brought about by four imide groups, which induce strong π–π interactions. The calculated electrostatic potential (ESP) map of the dimer, extracted from the X-ray structure, explains the slippage as resulting from interactions between the electron-poor imide groups and the electron-rich bifuran units (Fig. 2c). The large 2-octyldodecyl groups protrude up and down on four sides of the macrocycle, forming isolated π-channels with strong but one-dimensional interactions (Fig. 2d).
The UV-Vis absorption spectrum of the linear tetramer, L-4BFI in chloroform, displays the expected π–π* transition, with two vibronic peaks at 457 nm and 489 nm corresponding to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C backbone stretch of 0.178 eV (1432 cm−1; Fig. 3, red trace). The emission spectrum shows the similarly structured S1 → S0 transition, with a Stokes shift of 0.09 eV. For the macrocyclic oligofuran C-4BFI, the absorption maximum measured in chloroform is at 401 nm (Fig. 3, blue trace), corresponding to the S0 → S2 transition (extinction coefficient of 1.1 × 105 cm−1 M−1). The shoulder that appears in the ∼480–520 nm range is likely associated with the S0 → S1 transition, which is Laporte forbidden due to a conservation of orbital symmetry in the centrosymmetric macrocycle. The emission spectrum shows a similar pattern for the macrocycle and the linear tetramer, corresponding to the S1 → S0 transition. However, the emission maximum is bathochromically shifted by 0.142 eV for C-4BFI compared with L-4BFI, which is in-line with the calculated difference in their HOMO–LUMO gap. The fluorescence quantum yield for the macrocycle measured in chloroform at 450 nm excitation is 18%, which is smaller than that of the linear oligomer (61%), in line with the symmetry-forbidden S1 → S0 transition. However, it is larger than the fluorescence quantum yield of macrocyclic oligothiophenes.24
C backbone stretch of 0.178 eV (1432 cm−1; Fig. 3, red trace). The emission spectrum shows the similarly structured S1 → S0 transition, with a Stokes shift of 0.09 eV. For the macrocyclic oligofuran C-4BFI, the absorption maximum measured in chloroform is at 401 nm (Fig. 3, blue trace), corresponding to the S0 → S2 transition (extinction coefficient of 1.1 × 105 cm−1 M−1). The shoulder that appears in the ∼480–520 nm range is likely associated with the S0 → S1 transition, which is Laporte forbidden due to a conservation of orbital symmetry in the centrosymmetric macrocycle. The emission spectrum shows a similar pattern for the macrocycle and the linear tetramer, corresponding to the S1 → S0 transition. However, the emission maximum is bathochromically shifted by 0.142 eV for C-4BFI compared with L-4BFI, which is in-line with the calculated difference in their HOMO–LUMO gap. The fluorescence quantum yield for the macrocycle measured in chloroform at 450 nm excitation is 18%, which is smaller than that of the linear oligomer (61%), in line with the symmetry-forbidden S1 → S0 transition. However, it is larger than the fluorescence quantum yield of macrocyclic oligothiophenes.24
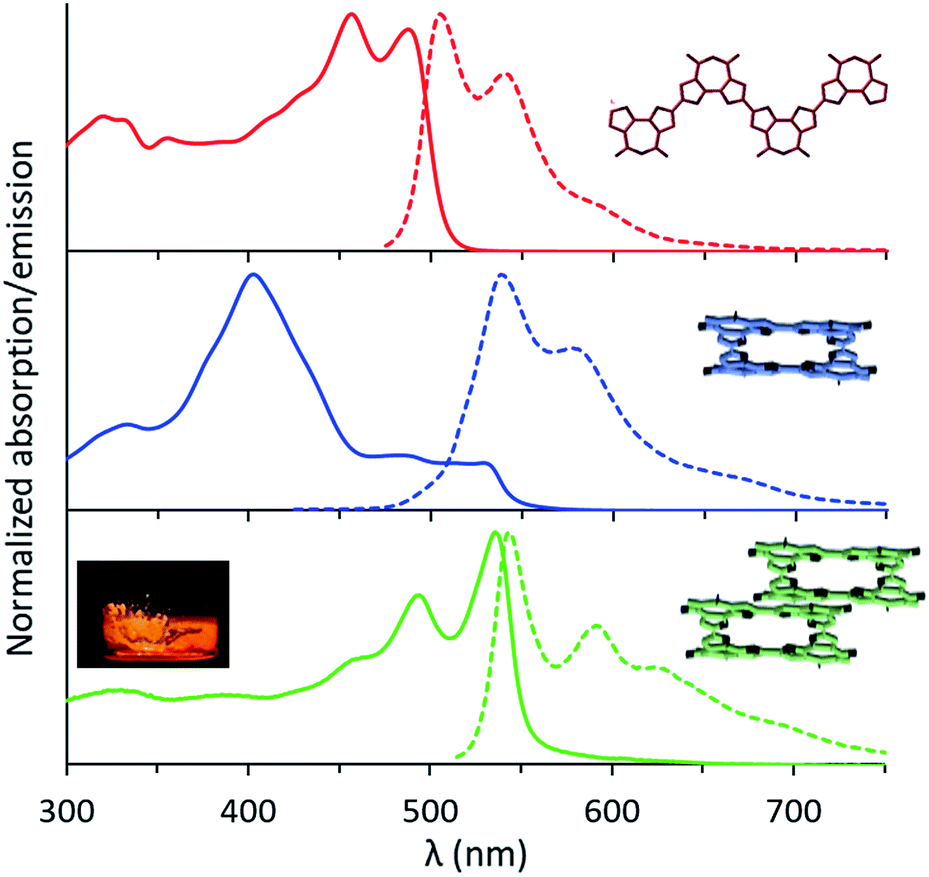 | ||
| Fig. 3 Normalized UV-Vis absorption (solid line) and emission (dashed line) spectra for L-4BFI (red), C-4BFI (blue) in chloroform, and C-4BFI film (green). Inset: photograph of solid C-4BFI fluorescing upon illumination with UV light (365 nm). | ||
The absorption spectrum of C-4BFI in hexane at high dilution is similar to that observed in chloroform, with a strong S0 → S2 transition and a weak S0 → S1 tail. Increasing the concentration leads to the emergence of new transitions at 535 nm and 489 nm, until a solid film is formed in which only these new transitions can be observed (Fig. 3, green trace). The emission spectrum is nearly a mirror image of the absorption, with an extremely small Stokes shift of 0.034 eV, indication of a rigid structure. Aggregation is also easily observed in hexane solutions by dynamic light scattering measurements (DLS): at a concentration of 10−5 M, C-4BFI forms aggregates with a hydrodynamic diameter ∼45 nm; these grow to large particles (∼90 nm) as the concentration increases to 10−4 M (Fig. 4, blue trace). With time, these aggregates form a red film on the container walls. Atomic Force Microscopy (AFM) imaging of this film shows particles up to ∼100 nm size, in line with DLS measurements, and reveals their crystalline nature (see ESI†). By comparison, the linear tetramer L-4BFI shows a much lower tendency to aggregate, with no observable aggregates at a concentration of 10−5 M and only small (5 nm) aggregates appearing at 10−4 M (Fig. 4, red trace). 1H-NMR shows the concentration dependence of the chemical shift of the β-proton up from 10−5 M, which supports strong intermolecular interactions. The estimated association constant for dimer formation in chloroform-d is 725 ± 134 M−1 (see Section S9 in ESI†). Samples dissolved in toluene display higher solubility compared with hexane, and no change in spectra even at 10−4 M concentration, supporting that π–π interactions of the macrocyclic core is the main cause for the observed aggregation.
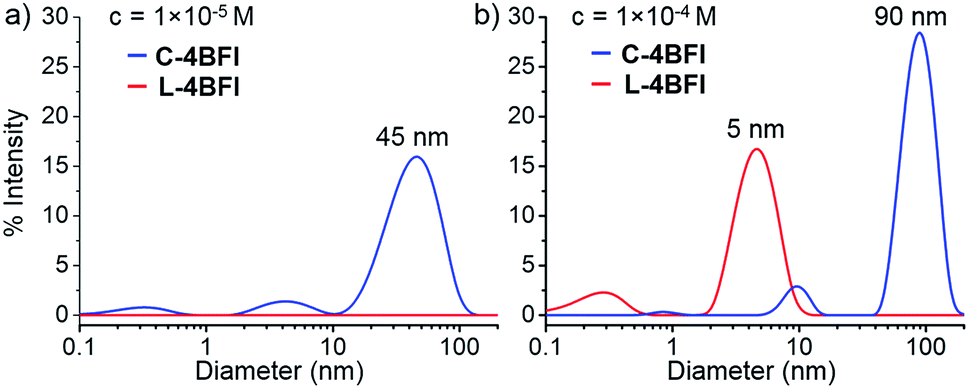 | ||
| Fig. 4 Dynamic light scattering (DLS) of C-4BFI and L-4BFI in hexane. (a) c = 1 × 10−5 M, and (b) c = 1 × 10−4 M. | ||
The self-assembly of C-4BFI at the solid–liquid interface was imaged using STM. The macrocycles adopt a lamellar arrangement with an oblique unit cell (a = 1.7 ± 0.1 nm, b = 2.4 ± 0.1 nm, γ = 84 ± 1°, Fig. 5a). The conjugated macrocyclic core appears bright and the darker region between the macrocycle rows corresponds to interdigitated alkyl chains. The central cavities of the macrocycles are clearly resolved. Molecular modelling of the observed pattern suggests that, within the rows, the macrocycles are linked by two-point hydrogen bonding involving furan CH donors and carbonyl acceptors (Fig. 5b and d). Such assembly allows for interactions between only half of the alkyl chains (4 out of 8) and the HOPG surface. Presumably, the other alkyl chains are adsorbed on top of themselves (Fig. 5b) as has been observed for other alkylated molecules.45 We also note a pronounced tendency of the macrocycle to form multilayers at the liquid–solid interface, in line with its aggregation in solution.46Fig. 5c shows the multilayer, with molecules missing from the upper layer giving the appearance of holes in which a lower contrast molecule (of the lower layer) can still be identified.47
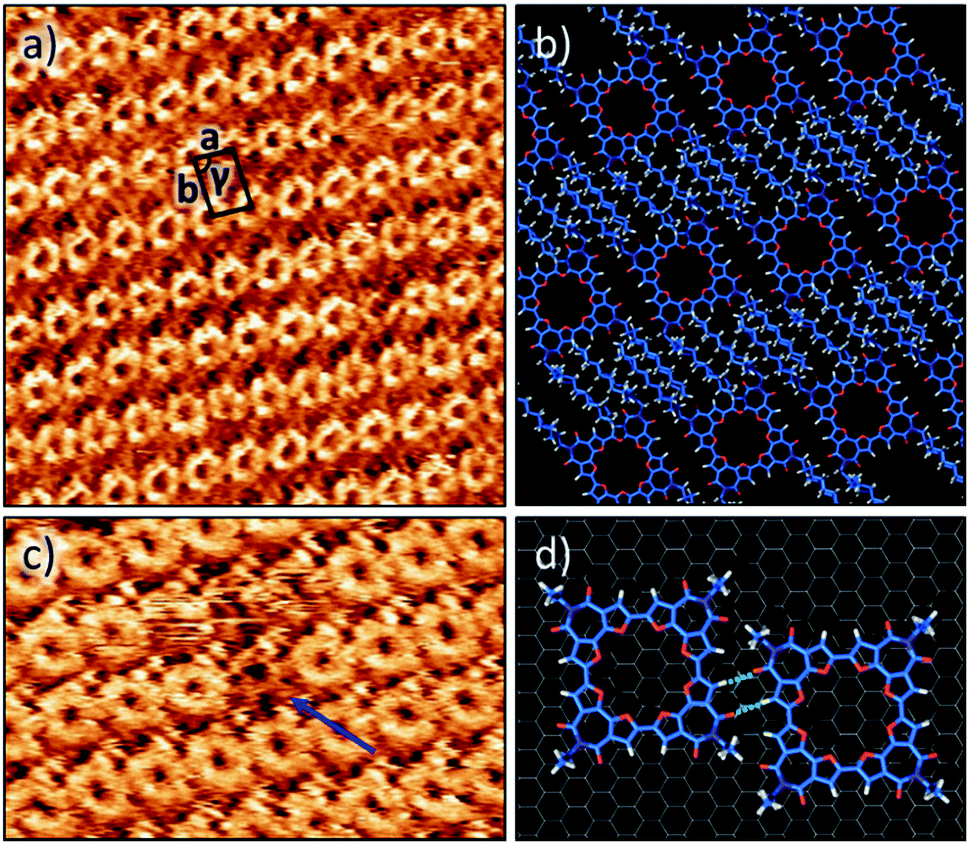 | ||
| Fig. 5 (a) STM image of C-4BFI (5 × 10−2 M) at the 1,2,4-trichlorobenzene–graphite interface. Image scale: 19.3 × 19.3 nm2. Imaging conditions: Iset = 1 pA, and Vbias = −1.2 V. (b) Molecular modelling unit cell parameters: a = 1.7 ± 0.1 nm, b = 2.4 ± 0.1 nm and γ = 84 ± 1°. (c) STM image showing the multilayer. Image scale: 13.6 × 13.6 nm2. Blue arrow shows the vacancy defect in the upper layer through which the STM contrast still shows a molecular structure (from the layer below). (d) Molecular model displaying CO⋯H hydrogen bonding between two adjacent macrocycles. | ||
The out-of-plane charge transport properties of C-4BFI films were investigated in a diode configuration. The log–log plots of current-density/voltage (J–V) of the hole-only diodes (Fig. 6a) display ohmic, trap-limited space charge limited current (SCLC) and trap filling regimes. The charge carrier mobility (μ) in SCLC regime was determined using the Mott-Gurney's law to be 2 × 10−4 cm2 V−1 s−1, which is within the typical range for organic (light-emitting, photovoltaic) diode applications. On the other hand, the field-effect transistor (FET) measurements for the same films did not display any field-effect current modulation, which at least in part can be explained by the very low HOMO of C-4BFI (−5.88 eV in gas phase) that leads to easy hole trapping. We were able to fabricate functional p-type FET devices by blending C-4BFI with a nitrofluorene acceptor (2,5,7-trinitro-4-(2,2,3,3,4,4,5,5,6,6-decafluorohexoxycarbonyl)-9-dicyanomethylenefluorene), in order to annihilate the hole traps (i.e. impurities and interfacial states with HOMO higher than that of bulk C-4BFI, Fig. 6b).48 Nevertheless, the measured field-effect hole mobility (5 × 10−6 cm2 V−1 s−1) for in-plane transport is almost two orders of magnitude lower than that measured by SCLC for out-of-plane transport. This difference points to the preferential face-on molecular orientation of the molecules (and therefore out-of-plane π-stacking direction), already seen in the STM experiments (Fig. 5).
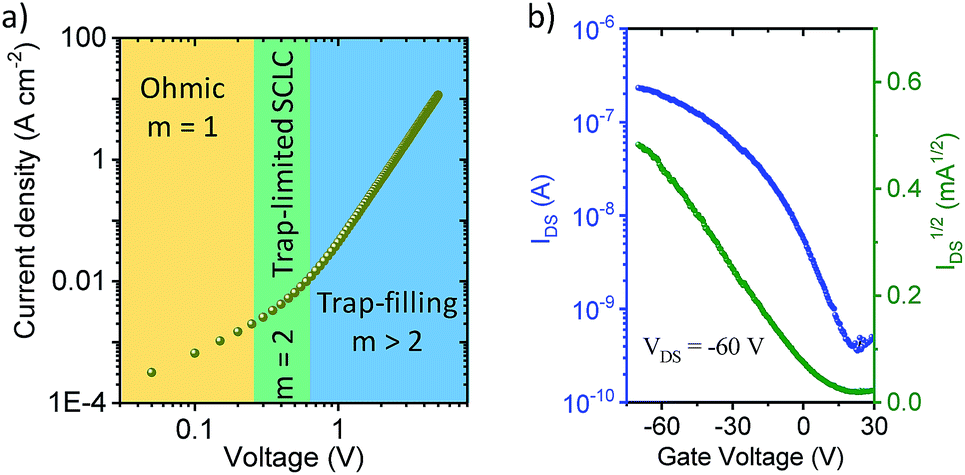 | ||
| Fig. 6 (a) Hole-only diode SCLC characteristics of C-4BFI, where m is the slope; (b) transfer characteristics of C-4BFI films blended with nitrofluorene acceptor. | ||
Conclusions
In summary, we have introduced C-4BFI as the first macrocyclic oligofuran. Its X-ray structure reveals a planar backbone, with very short interplanar distances, and a small inner cavity whose dimensions are similar to those of 24-crown-8 ether. The macrocycle shows a tendency to self-aggregate in solution, with ordered multilayers forming at the solid–liquid interface. It exhibits distinct absorption and emission spectra, and fluoresces in the solid state. C-4BFI displays a p-type behavior with anisotropic mobility of up to 2 × 10−4 cm2 V−1 s−1 in thin films. We are currently investigating the capabilities of oligofuran macrocycles to form inclusion complexes with its oxygen-containing cavities, as well as supramolecular aggregates with different side groups.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is dedicated to François Diederich on the occasion of his retirement. The work is supported by the German-Israeli Foundation for Scientific Research and Development. The work in Canada was supported by NSERC Strategic and Discovery grants. We thank Dr Mark M. Karpasas for MALDI measurements and Dr Igor F. Perepichka for donating the sample of nitrofluorene acceptor.Notes and references
- M. Iyoda, J. Yamakawa and M. J. Rahman, Angew. Chem., Int. Ed., 2011, 50, 10522–10553 CrossRef CAS PubMed.
- Y. Xu, R. Kaur, B. Wang, M. B. Minameyer, S. Gsänger, B. Meyer, T. Drewello, D. M. Guldi and M. von Delius, J. Am. Chem. Soc., 2018, 140, 13413–13420 CrossRef CAS PubMed.
- J. D. Cojal González, M. Iyoda and J. P. Rabe, Angew. Chem., 2018, 130, 17284–17288 CrossRef.
- Y. Zhong, Y. Yang, Y. Shen, W. Xu, Q. Wang, A. L. Connor, X. Zhou, L. He, X. C. Zeng, Z. Shao, Z.-l. Lu and B. Gong, J. Am. Chem. Soc., 2017, 139, 15950–15957 CrossRef CAS PubMed.
- D. Lorbach, A. Keerthi, T. M. Figueira-Duarte, M. Baumgarten, M. Wagner and K. Müllen, Angew. Chem., Int. Ed., 2016, 55, 418–421 CrossRef CAS PubMed.
- H. Gregolińska, M. Majewski, P. J. Chmielewski, J. Gregoliński, A. Chien, J. Zhou, Y.-L. Wu, Y. J. Bae, M. R. Wasielewski, P. M. Zimmerman and M. Stępień, J. Am. Chem. Soc., 2018, 140, 14474–14480 CrossRef PubMed.
- W. Zhang and J. S. Moore, Angew. Chem., Int. Ed., 2006, 45, 4416–4439 CrossRef CAS PubMed.
- U. H. F. Bunz, Chem. Rev., 2000, 100, 1605–1644 CrossRef CAS PubMed.
- A. S. Shetty, J. Zhang and J. S. Moore, J. Am. Chem. Soc., 1996, 118, 1019–1027 CrossRef CAS.
- Y. Tobe, N. Utsumi, K. Kawabata, A. Nagano, K. Adachi, S. Araki, M. Sonoda, K. Hirose and K. Naemura, J. Am. Chem. Soc., 2002, 124, 5350–5364 CrossRef CAS PubMed.
- S. Höger, K. Bonrad, A. Mourran, U. Beginn and M. Möller, J. Am. Chem. Soc., 2001, 123, 5651–5659 CrossRef PubMed.
- J. M. W. Chan, J. R. Tischler, S. E. Kooi, V. Bulović and T. M. Swager, J. Am. Chem. Soc., 2009, 131, 5659–5666 CrossRef CAS PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- M. Fischer, G. Lieser, A. Rapp, I. Schnell, W. Mamdouh, S. De Feyter, F. C. De Schryver and S. Höger, J. Am. Chem. Soc., 2004, 126, 214–222 CrossRef CAS PubMed.
- G.-B. Pan, X.-H. Cheng, S. Höger and W. Freyland, J. Am. Chem. Soc., 2006, 128, 4218–4219 CrossRef CAS PubMed.
- J. Krömer, I. Rios-Carreras, G. Fuhrmann, C. Musch, M. Wunderlin, T. Debaerdemaeker, E. Mena-Osteritz and P. Bäuerle, Angew. Chem., Int. Ed., 2000, 39, 3481–3486 CrossRef.
- G. Fuhrmann, T. Debaerdemaeker and P. Bäuerle, Chem. Commun., 2003, 948–949 RSC.
- F. Zhang, G. Götz, H. D. F. Winkler, C. A. Schalley and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 6632–6635 CrossRef CAS PubMed.
- A. Bhaskar, G. Ramakrishna, K. Hagedorn, O. Varnavski, E. Mena-Osteritz, P. Bäuerle and T. Goodson, J. Phys. Chem. B, 2007, 111, 946–954 CrossRef CAS PubMed.
- E. Mena-Osteritz and P. Bäuerle, Adv. Mater., 2001, 13, 243–246 CrossRef CAS.
- E. Mena-Osteritz, Adv. Mater., 2002, 14, 609–616 CrossRef CAS.
- E. Mena-Osteritz and P. Bäuerle, Adv. Mater., 2006, 18, 447–451 CrossRef CAS.
- F. Zhang, G. Götz, E. Mena-Osteritz, M. Weil, B. Sarkar, W. Kaim and P. Bäuerle, Chem. Sci., 2011, 2, 781–784 RSC.
- K. Nakao, M. Nishimura, T. Tamachi, Y. Kuwatani, H. Miyasaka, T. Nishinaga and M. Iyoda, J. Am. Chem. Soc., 2006, 128, 16740–16747 CrossRef CAS PubMed.
- H. Shimizu, J. D. Cojal González, M. Hasegawa, T. Nishinaga, T. Haque, M. Takase, H. Otani, J. P. Rabe and M. Iyoda, J. Am. Chem. Soc., 2015, 137, 3877–3885 CrossRef CAS PubMed.
- M. Williams-Harry, A. Bhaskar, G. Ramakrishna, T. Goodson, M. Imamura, A. Mawatari, K. Nakao, H. Enozawa, T. Nishinaga and M. Iyoda, J. Am. Chem. Soc., 2008, 130, 3252–3253 CrossRef CAS PubMed.
- J. Fabian and H. Hartmann, J. Phys. Org. Chem., 2007, 20, 554–567 CrossRef CAS.
- We note that based on semiempirical calculations, C-12T adsorbed on graphite was conclude to be “almost planar”. See ref. 22.
- O. Gidron, Y. Diskin-Posner and M. Bendikov, J. Am. Chem. Soc., 2010, 132, 2148–2150 CrossRef CAS PubMed.
- O. Gidron and M. Bendikov, Angew. Chem., Int. Ed., 2014, 53, 2546–2555 CrossRef CAS PubMed.
- O. Gidron, A. Dadvand, Y. Sheynin, M. Bendikov and D. F. Perepichka, Chem. Commun., 2011, 47, 1976–1978 RSC.
- A. Hayoun Barak, G. de Ruiter, M. Lahav, S. Sharma, O. Gidron, G. Evmenenko, P. Dutta, M. Bendikov and M. E. van der Boom, Chem.–Eur. J., 2013, 19, 8821–8831 CrossRef CAS PubMed.
- O. Dishi and O. Gidron, J. Org. Chem., 2018, 83, 3119–3125 CrossRef CAS PubMed.
- P. R. Ashton, P. T. Glink, C. Schiavo, J. F. Stoddart, E. J. T. Chrystal, S. Menzer, D. J. Williams and P. A. Tasker, Angew. Chem., Int. Ed., 1995, 34, 1869–1871 CrossRef CAS.
- Y.-L. Zhao, L. Liu, W. Zhang, C.-H. Sue, Q. Li, O. Š. Miljanić, O. M. Yaghi and J. F. Stoddart, Chem.–Eur. J., 2009, 15, 13356–13380 CrossRef CAS PubMed.
- G. Götz, X. Zhu, A. Mishra, J.-L. Segura, E. Mena-Osteritz and P. Bäuerle, Chem.–Eur. J., 2015, 21, 7193–7210 CrossRef PubMed.
- M. Ammann, A. Rang, C. A. Schalley and P. Bäuerle, Eur. J. Org. Chem., 2006, 2006, 1940–1948 CrossRef.
- M. Iyoda and H. Shimizu, Chem. Soc. Rev., 2015, 44, 6411–6424 RSC.
- E. Vogel, N. Jux, J. Dörr, T. Pelster, T. Berg, H.-S. Böhm, F. Behrens, J. Lex, D. Bremm and G. Hohlneicher, Angew. Chem., Int. Ed., 2000, 39, 1101–1105 CrossRef CAS PubMed.
- S. V. Mulay, B. Bogoslavky, I. Galanti, E. Galun and O. Gidron, J. Mater. Chem. C, 2018, 6, 11951–11955 RSC.
- Modern supramolecular chemistry; Strategies for macrocycle synthesis, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed.
- While calculated optimized structure for C-4BFI was slighly bent out-of planarity, the planarization energy was in the range of 0.2 kcal mol−1, which is within the accepted inaccuracy for this computational level.
- V. Martí-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736–8834 CrossRef PubMed.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef CAS.
- Y. Fang, M. Cibian, G. S. Hanan, D. F. Perepichka, S. De Feyter, L. A. Cuccia and O. Ivasenko, Nanoscale, 2018, 10, 14993–15002 RSC.
- S. Lee, B. E. Hirsch, Y. Liu, J. R. Dobscha, D. W. Burke, S. L. Tait and A. H. Flood, Chem.–Eur. J., 2016, 22, 560–569 CrossRef CAS PubMed.
- Z. Yuan, S.-L. Lee, L. Chen, C. Li, K. S. Mali, S. De Feyter and K. Müllen, Chem.–Eur. J., 2013, 19, 11842–11846 CrossRef CAS PubMed.
- M. Nikolka, I. Nasrallah, B. Rose, M. K. Ravva, K. Broch, A. Sadhanala, D. Harkin, J. Charmet, M. Hurhangee, A. Brown, S. Illig, P. Too, J. Jongman, I. McCulloch, J.-L. Bredas and H. Sirringhaus, Nat. Mater., 2016, 16, 356 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1892379. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03247a |
| ‡ These authors contributed equally. |
| § Current address: Artificial Photosynthesis Research Group, Korea Research Institute of Chemical Technology (KRICT), Daejeon, Republic of Korea. |
| This journal is © The Royal Society of Chemistry 2019 |