
Elastomeric nanocomposite scaffolds made from poly(glycerol sebacate) chemically crosslinked with carbon nanotubes
Akhilesh K.
Gaharwar†‡
 *ab,
Alpesh
Patel‡
cd,
Alireza
Dolatshahi-Pirouz§
bcd,
Hongbin
Zhang
cd,
Kaushik
Rangarajan
cd,
Giorgio
Iviglia
cd,
Su-Ryon
Shin
bcd,
Mohammad Asif
Hussain
e and
Ali
Khademhosseini
*bcdfg
*ab,
Alpesh
Patel‡
cd,
Alireza
Dolatshahi-Pirouz§
bcd,
Hongbin
Zhang
cd,
Kaushik
Rangarajan
cd,
Giorgio
Iviglia
cd,
Su-Ryon
Shin
bcd,
Mohammad Asif
Hussain
e and
Ali
Khademhosseini
*bcdfg
aDavid H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
bWyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA
cBiomaterials Innovation Research Center, Division of Biomedical Engineering, Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Cambridge, MA 02139, USA
dHarvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
eDepartment of Electrical and Computer Engineering (Biomedical Engineering Option), King Abdulaziz University, Jeddah 21589, Saudi Arabia
fDepartment of Maxillofacial Biomedical Engineering and Institute of Oral Biology, School of Dentistry, Kyung Hee University, Seoul 130-701, Republic of Korea
gDepartment of Physics, King Abdulaziz University, Jeddah 21569, Saudi Arabia. E-mail: alik@rics.bwh.harvard.edu; gaharwar@tamu.edu
First published on 1st September 2014
Abstract
Carbon nanotube (CNT)-based nanocomposites often possess properties such as high stiffness, electrical conductivity, and thermal stability and have been studied for various biomedical and biotechnological applications. However, the current design approaches utilize CNTs as physical fillers, and thus, the true potential of CNT-based nanocomposites has not been realized. Here, we introduce a general approach to fabricating stiff, elastomeric nanocomposites from poly(glycerol sebacate) (PGS) and CNTs. The covalent crosslinking between the nanotubes and polymer chains resulted in novel property combinations that are not observed in conventional nanocomposites. The addition of 1% CNTs resulted in a five-fold increase in the tensile modulus and a six-fold increase in compression modulus compared with PGS alone, which is far superior to the previously reported studies for CNT-based nanocomposites. Despite a significant increase in mechanical stiffness, the elasticity of the network was not compromised and the resulting nanocomposites showed more than 94% recovery. This study demonstrates that the chemical conjugation of CNTs to a PGS backbone results in stiff and elastomeric nanocomposites. Additionally, in vitro studies using human mesenchymal stem cells (hMSCs) indicated that the incorporation of CNTs into the PGS network significantly enhanced the differentiation potential of the seeded hMSCs, rendering them potentially suitable for applications ranging from scaffolding in musculoskeletal tissue engineering to biosensors in biomedical devices.
Introduction
Carbon nanotubes (CNTs) have shown exceptional structural, electrical, mechanical, and thermal properties.1–3 CNT-based nanocomposites possess a unique property combination and have been explored for numerous technological applications such as actuators,4,5 body armor,6 conductive tapes,7 flame retardants,8 energy storage,9 tissue engineering,10,11 delivery devices,12,13 biosensors,14–16 and biomedical devices.17–19 Despite interesting physical and chemical properties, the true potential of CNT-based nanocomposites is yet to be realized.20–22 This is due to strong π–π stacking interactions between CNTs that limit the dispersion of CNTs within the polymeric matrix and decrease their ability to improve the structural, chemical and biological properties of the nanocomposite network.To overcome this shortcoming, numerous techniques are used to augment the dispersion of CNTs within the polymeric network, such as surface functionalization.23–26 The surfaces of CNTs are modified with different polar groups including carboxyl,27 hydroxyl,28 and amine29 to facilitate their uniform distribution within the polymeric matrix. Other strategies to enhance the solubility of CNTs in aqueous and non-aqueous solutions include the use of surfactants,30 proteins,31 and single-stranded deoxyribonucleic acid (ssDNA).32 The uniform dispersion of CNTs within the polymeric network results in enhanced surface interactions and significant increases in the stiffness of the nanocomposites.33
Grafting polymeric chains onto nanotube surfaces can modify the surface of CNTs.34 In this approach, polymeric chains shield the surface of CNTs, and the adjoining polymeric network simply recognizes the surface-grafted polymer. This shielding method improves CNT distribution within the polymeric matrix due to an increase in physical interactions between polymer chains and surface-grafted CNTs. Two different strategies that are currently employed to covalently graft polymers onto CNT surfaces are “grafting from” and “grafting to” strategies. The “grafting to” strategy showed limitations in obtaining dense grafting density on the CNT surfaces due to the steric hindrance of reactive groups present on the polymer backbone and a relatively small number of functional groups available on the surface of CNTs. However, the “grafting from” strategy showed the formation of dense grafting of polymers onto the CNT surfaces. In the last decade, these strategies were extensively used to physically reinforce polymeric networks with CNTs to obtain mechanically superior nanocomposites.21,22 In a recent study, the CNT surfaces were grafted with poly(cyclohexyl methacrylate) by surface-initiated atom transfer radical polymerization. The grafting of polymer onto CNTs significantly enhanced the electrical and mechanical properties of resulting nanocomposites.35
Despite the increase in interaction between CNTs and the polymeric network, the utility of CNTs as a reinforcing agent is yet to be fully exploited as most studies have used functionalized CNTs as a physical reinforcer.27,29 By chemically conjugating CNT surfaces with the polymeric network, CNTs can be used as a crosslinking agent. To our knowledge, limited efforts are directed towards utilizing CNTs as a crosslinking agent. It is expected that if CNTs are covalently conjugated within the polymeric network, novel property combinations may be achieved.
To design CNT-reinforced composite, poly(glycerol sebacate) (PGS),36,37 a polyester, is selected to chemically conjugate with CNTs. PGS is an elastomeric polyester synthesized by polycondensation of sebacic acid and glycerol without the presence of any toxic catalyst.37 The availability of additional hydroxyl groups hydrates the PGS surface and provides cells with a tissue-like environment. Due to the elastomeric properties of PGS, it has been used to engineer soft tissues such as cardiac, renal, and heart valve tissues.38,39 When implanted in vivo, PGS is readily metabolized under physiological conditions since both glycerol and sebacic acid are endogenous components. Moreover, the degradation of PGS follows the surface erosion mechanism and can be tailored to follow the ECM biosynthesis without exhibiting any sudden change in structural, physical and chemical properties.37
Here, we developed chemically crosslinked PGS–carbon nanotube (PGS–CNT) nanocomposites. The additional hydroxyl groups present on the PGS backbone esterify with the carboxylic groups present on CNT surfaces during the thermal curing process, which is an advantage over other polyester-based nanocomposites. The chemical conjugation of CNTs with the polyester backbone considerably amplifies the physical and chemical properties of the nanocomposites. By covalently conjugating PGS with CNTs, we expect to obtain mechanically stiff nanocomposites that can be used for a range of biomedical applications, including as bone scaffolds, cardiac patches, nerve conduits, as well as for a range of other biomedical devices.
Materials and methods
Synthesis of poly(glycerol sebacate) (PGS)
Sebacic acid (Mw = 202.25 g mol−1, HO2C(CH2)8CO2H) and glycerol (Mw = 92.09 g mol−1, HOCH2CH(OH)CH2OH) were obtained from Sigma-Aldrich (USA). PGS was synthesized by polycondensation reaction using a previously reported method.39,40 Briefly, glycerol and sebacic acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) were mixed in a 250 mL two necks round bottom flask and heated to 130 °C in a silicon bath (under argon atmosphere for 2 hours). The pressure was then decreased to 50 mTorr in 5 hours, and the reaction was continued for 24 hours at 130 °C. The PGS prepolymer solution was then cooled to room temperature and its molecular weight distribution was analyzed using gel permeation chromatography (GPC) (Mn = 3960 g mol−1; PDI = 1.6). Since the initial molar ratio of carboxylic acid to hydroxyl groups was 2:3, additional hydroxyl groups were available on the PGS backbone for further crosslinking.
:1 molar ratio) were mixed in a 250 mL two necks round bottom flask and heated to 130 °C in a silicon bath (under argon atmosphere for 2 hours). The pressure was then decreased to 50 mTorr in 5 hours, and the reaction was continued for 24 hours at 130 °C. The PGS prepolymer solution was then cooled to room temperature and its molecular weight distribution was analyzed using gel permeation chromatography (GPC) (Mn = 3960 g mol−1; PDI = 1.6). Since the initial molar ratio of carboxylic acid to hydroxyl groups was 2:3, additional hydroxyl groups were available on the PGS backbone for further crosslinking.
Synthesis of PGS–CNT nanocomposites
Carboxyl functionalized multi-walled CNTs (50–250 μm in length; ∼9 ± 8 nm in diameter; 95% purity) were obtained from NanoLab Inc. (MA, USA). The nanocomposites were prepared by mixing PGS prepolymer in tetrahydrofuran (THF) (50% w/v) and adding a specific amount of CNTs (0.25, 0.5, and 1%) to the solution. CNTs were suspended in the PGS solution using ultrasonication. A substantial increase in the viscosity of PGS solution was detected due to strong interactions between PGS and CNTs. The solution was then poured in Teflon-based flat Petri dish and the THF was evaporated overnight. The dried PGS–CNTs prepolymer films were cured at 130 °C for 40 hours under vacuum. The nanocomposites were named as PGS-0.25%CNTs, PGS-0.5%CNTs and PGS-1%CNTs, respectively, depending on the final concentration of CNTs with respect to PGS.Chemical characterizations
An Alpha FTIR Bruker spectrometer was used to determine Fourier Transform Infrared (FTIR) spectra of the samples. 48 scans at a resolution of 4 cm−1 were obtained for each sample. Raman spectra of the samples were recorded using a R200-L SENTERRA Raman microscope on a Olympus BX51 microscope stand using excitation: 785 nm and 532 nm (He–Ne laser, with a power of 5 mW as the excitation source). The optical spectra of the samples were measured using an UV/vis spectrophotometer (Epoch Biotech Instruments).Sol content analysis
The crosslinking degree of the nanocomposite network was evaluated by determining sol (uncrosslinked polymer or nanotubes) and gel (crosslinked network) contents. The nanocomposite samples (8 mm diameter and 2 mm thick, and initial weight (Wi)) were swollen in THF for 24 hours (n = 5) (swollen weight (Ws)). The swollen samples were washed with THF and dried overnight (final weight (Wd)). The percentage of sol contents and degree of swelling were calculated by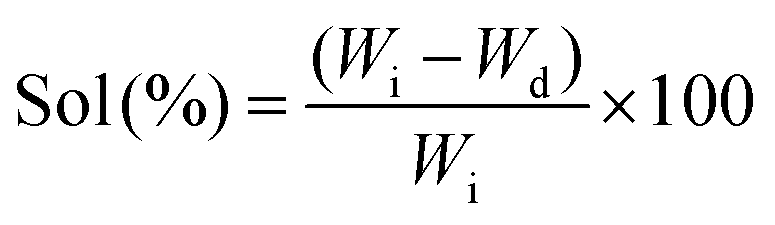 | (1) |
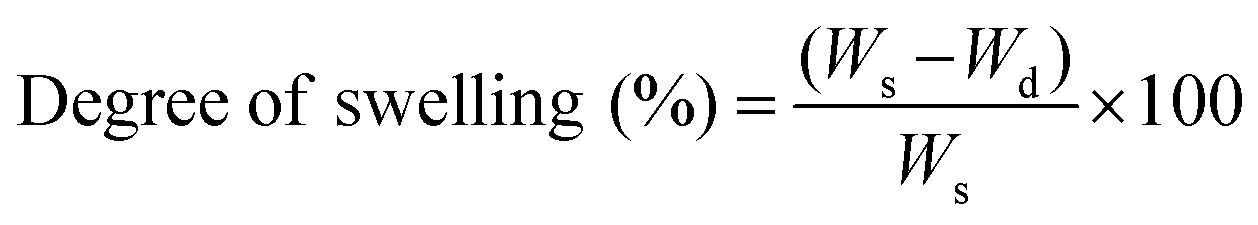 | (2) |
Thermal analysis
The thermal properties of the nanocomposite network were investigated by differential scanning calorimetry (DSC) (DSC8500, Perkin-Elmer, USA) and thermogravimetry analysis (TGA) (Pyris 1 TGA, Perkin-Elmer, USA). The samples were dried at 35 °C overnight under vacuum. For TGA, samples (∼10 mg) were subjected to a heating cycle of 50 °C to 600 °C at a heating rate of 10 °C min−1. Initial degradation temperature (TDI) and peak degradation temperature (TDP) were determined using the first derivative curve to determine the onset of thermal degradation. For DSC, samples (∼3–5 mg) were heated from −70 °C to 100 °C under N2 at a rate of 10 °C min−1, held at 100 °C for 5 min, and cooled down to −70 °C at a rate of 10 °C min−1. The 1st cooling cycles and 2nd heating cycles were used to determine the thermal properties (glass transition temperature (Tg), melting point (Tm), enthalpy of the composites (ΔH)). Considering the thermal decomposition as a first-order reaction, the activation energy (Ea) was calculated using the Horowitz–Metzger method41,42 as shown in eqn (3):| ln(ln(1/y)) = −Eaθ/RTd2 | (3) |
Here, Ea is the activation energy (kJ mol−1); y is the weight loss in fraction; Td is the reference temperature (K), R is the universal gas constant and θ = (T − Td) (K). The slope obtained from linear plots of ln(ln(1/y)) vs. θ was used to calculate the activation energy.
Mechanical analysis
The mechanical properties of nanocomposite samples were evaluated by uniaxial tensile, and unconfined compression testing using the Instron 5943 Materials Testing System (Norwood, MA, USA) loaded with 50 N load cell. For uniaxial tensile, nanocomposite samples (2 mm wide, 5 mm long × 1.5 mm thick) were used. For compression testing, nanocomposite samples with 5 mm diameter, and 2 mm thickness were used. The crosshead speed of 10 mm min−1 was used for both tensile and compression testing. For cyclic testing, 5 cycles consisting of a loading and an unloading cycle were implemented. The maximum strain was limited to 20%. Mechanical properties such as compressive modulus, ultimate compressive stress, ultimate compressive strain, toughness, and energy absorbed were calculated from the stress–strain curve. The elastic modulus was determined from the linear stress–strain region by fitting a straight line between 5 and 20% strain.Scanning electron microscopy
The structural morphologies of the nanocomposite surface were evaluated using a scanning electron microscope (JSM 5600LV, JEOL USA Inc., MA). The samples were freeze-fractured using liquid nitrogen to visualize the distribution of CNTs within the PGS network. The samples were allowed to dry in a desiccator for 24 hours before imaging. The nanocomposite samples were coated with Au/Pd for 2 minutes using a Hummer 6.2 sputter coater (Ladd Research, Williston, VT). The fractured surfaces of the nanocomposite samples from the mechanical testing were also analyzed using this method.Surface analysis, swelling and degradation study
The surface hydrophilicity of nanocomposite films (n = 5) was determined by the drop shape analysis method. A drop of water was poured onto the nanocomposite surface using a 21-gauge flat needle and optical images were collected after 10 seconds of contact. The contact angle of water was determined from the optical images. The swelling degree and degradation of nanocomposite samples (6 mm diameter, 2 mm thick and dried weight (Wi)) were determined in phosphate buffer saline (PBS) at 37 °C. For swelling study, samples were collected after 24 hours, the excess surface water was blotted and the samples were weighed (Ws). The water-swelling ratio was calculated using eqn (2). For degradation study, samples were obtained at a predetermined time from PBS, and dried weight (Wd) was determined. The related mass losses were calculated from eqn (4) from their initial and final dry weights: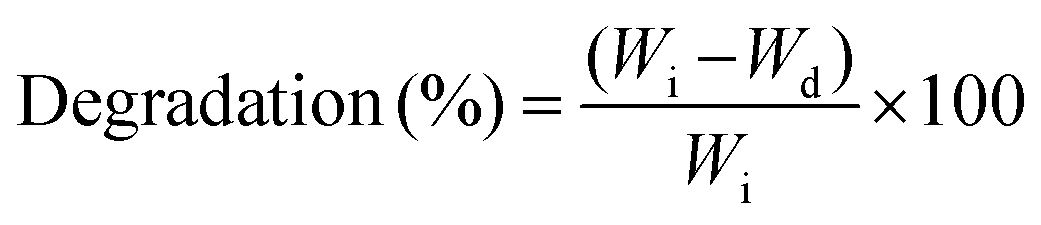 | (4) |
Protein adsorption
The adsorption of protein on nanocomposite surfaces was determined by soaking the samples (6 mm diameter and 2 mm thick) in PBS overnight and then in 10% fetal bovine serum (FBS) for 24 hours at 37 °C. The samples were washed three times with PBS for 10 minutes to remove non-specifically adsorbed proteins. Then samples were subjected to 2% sodium dodecyl sulfate (SDS) solution under shaking conditions (50 rpm, 37 °C) for 6 hours to collect adhered proteins. The supernatant was collected and analyzed using bicinchoninic acid (BCA) protein assay reagent (Pierce BCA, Thermo Scientific). The bovine serum albumin was used as a standard and the supernatant was quantified using an UV/vis spectrophotometer (Epoch Biotech Instruments) at 562 nm.In vitro study
Bone marrow-derived hMSCs (PT-2501, Lonza, USA) were cultured in normal growth media (α-MEM), containing 10% of heat-inactivated fetal bovine serum (HiFBS, Gibco, USA) and 1% Pen/Strep (penicillin/streptomycin, 100 U/100 μg mL−1, Gibco, USA) at 37 °C, under a humidified atmosphere with 5% CO2. Before cell seeding, the nanocomposite scaffolds were washed using 80–90% ethanol solution and subjected to UV light, followed by thorough washing with PBS. The cells were cultured until 70–75% confluence and were used before passage 4 for all the experiments. The cells were trypsinized (CC-3232, Lonza, USA) and seeded on nanocomposite scaffolds (6 mm cylindrical samples) at the density of 10,000 cells per sample in normal growth media. Cell proliferation over 7 days of culture was evaluated by Alamar Blue assay (Invitrogen, USA) following the standard manufacturer's protocol. For osteogenic differentiation studies, osteogenic media (PT-3924 & PT-4120, Lonza, USA) supplemented with Pen/Strep, L-glutamine, ascorbate, β-glycerolphosphate, and dexamethasone were used. Alkaline phosphatase (ALP) activity of hMSCs seeded on the nanocomposite surface was quantified on days 7, 14 and 21 using an ALP Colorimetric Assay Kit (ab83369 Abcam, USA) following the standard manufacturer's protocol. The amount of calcium was quantified using a calcium quantification assay kit (ab112115 Abcam, USA) following the standard manufacturer's protocol. The ALP activity and calcium content from each sample were normalized with the DNA content. The DNA content was determined using a Quant-iT PicoGreen kit (Invitrogen, USA) following the standard manufacturer's protocol.Statistics
The experimental results are represented as mean ± standard deviation (n = 3 to 5). One-way ANOVA with Tukey post-hoc analysis was used to determine the statistical differences between the groups. Statistical significance was represented as *p < 0.05, **p < 0.01, ***p < 0.001.Results and discussion
Covalently reinforced PGS–CNT nanocomposites
The covalently reinforced PGS–CNT nanocomposites were obtained by a two-step process. In the first step, a low molecular weight PGS prepolymer (Mn = 3960 g mol−1 and PDI = 1.6) was prepared by the polycondensation method. Then the PGS prepolymer was dissolved in THF (50% w/v) and the carboxylic functionalized CNTs were incorporated in the PGS prepolymer solution using ultrasonication. The addition of CNTs to PGS pre-polymer solution increased its viscosity. This implies that the low molecular weight PGS prepolymer strongly interacts with carboxyl functionalized CNTs. The resulting PGS–CNT solution was found to be stable with no phase separation or segregation. This PGS–CNT prepolymer mixture was then allowed to dry in a vacuum to evaporate the solvent (THF). In the second step, the dried PGS–CNT prepolymer film was subjected to 130 °C for 40 hours to obtain covalently crosslinked PGS–CNT nanocomposites (Fig. 1a). During this curing step, hydroxyl groups that are present on the PGS chains esterify with the carboxyl groups present on the CNT surfaces to form a covalently crosslinked network.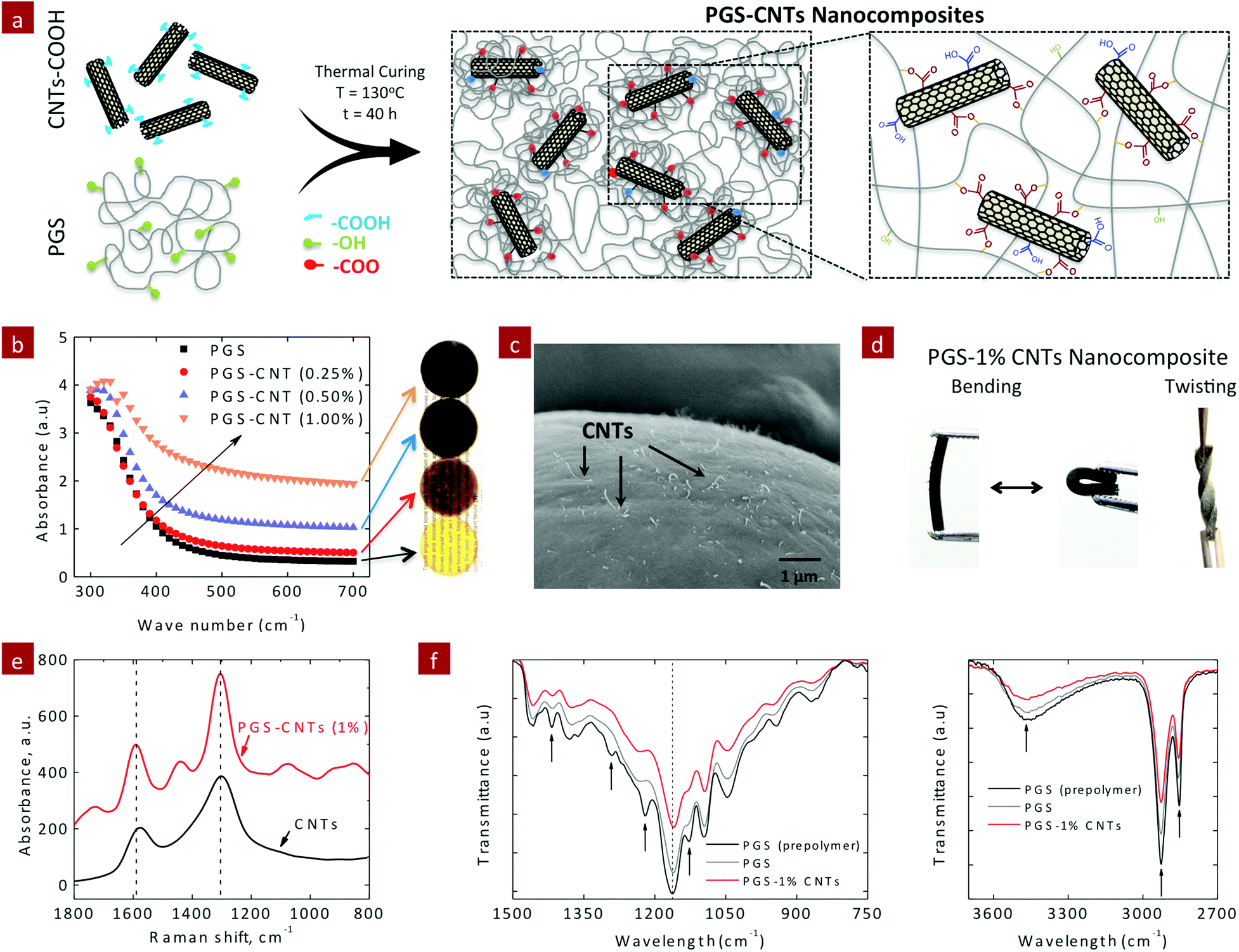 | ||
| Fig. 1 Synthesis and fabrication of PGS–CNT nanocomposites. (a) Schematic representation of PGS–CNT nanocomposite synthesis. COOH-functionalized CNTs act as both physical and covalent crosslinkers. The presence of hydroxyl groups on the PGS backbone esterifies with the carboxylic groups present on CNT surfaces during the thermal curing process, resulting in a densely crosslinked network. (b) UV-vis spectra of PGS–CNT nanocomposites showing decrease in optical transmittance due to addition of CNTs. (c) SEM image of the freeze-fractured surface of PGS-1% CNT nanocomposites shows uniform distribution of CNTs on the surface. (d) PGS–CNT nanocomposites show high flexibility and can sustain bending and twisting. (e) Raman spectra of COOH–CNT and PGS–CNT nanocomposites. The COOH-functionalized CNTs show a stronger G-band (1580 cm−1) compared to a D-band (1350 cm−1). After thermal crosslinking of PGS with CNTs, the intensity of the D-band was greater than the G-band. This indicates that the structure of CNTs was distorted due to chemical conjugation of CNTs with PGS chains. (f) FTIR spectra of PGS–CNT nanocomposites. Reduction in the hydroxyl peak upon curing in the presence of CNTs supports the covalent interaction of CNTs within the PGS network. | ||
The optical images indicate uniform distribution of CNTs within the PGS network. As the concentration of CNT increases, the transparency (Fig. 1b) of the nanocomposite film decreases. This is also evident by the increase in the absorbance value in the visible spectra (400–700 cm−1). At a macroscopic length scale, uniform dispersion of CNTs within the PGS matrix was observed. The dispersion of CNTs at micro- and nano-length scales was observed by electron microscopy. The electron microscopy images of the fractured nanocomposite surface (Fig. 1c) indicate the uniform distribution of CNTs within a polymeric matrix. All the nanocomposite films show high flexibility and can sustain bending and twisting (Fig. 1d).
The Raman spectra of the PGS–CNT nanocomposites were obtained to determine the interactions between PGS and CNTs (Fig. 1e). The Raman spectra of multiwall CNTs display two characteristic peaks – 1580 cm−1 (graphite or G-band) and 1360 cm−1 (distorted or D-band). The G-band derives from the graphite-like in-plane mode (also known as tangential mode), whereas the D-band corresponds to intrinsic impurities or defects of nanotubes. For carboxylic functionalized CNTs, the G-band is stronger compared to the D-band. After thermal crosslinking of PGS with CNTs, the intensity of the D-band was greater than that of the G-band. This indicates that the structure of CNTs was distorted due to chemical conjugation of CNTs with PGS chains. Earlier studies have also shown that covalent conjugation of the polymer to CNT surfaces results in an increase in D-band intensity.43,44 The infrared (IR) spectra also indicated fabrication of chemically crosslinked nanocomposite networks after the thermal crosslinking process. The peaks at 1350 cm−1 (–COOH), 1100 cm−1 (–OH) and 3450 cm−1 (–OH) decreased after the crosslinking process (Fig. 1f). The addition of 1% CNTs further resulted in the reduction of these peaks, indicating a higher degree of crosslinking compared to pure PGS.
Another method to ascertain the covalent crosslinking between PGS and CNTs is to determine the change in crosslinking density due to the addition of CNTs. The crosslinking density of the nanocomposite network was determined by evaluating the amount of sol content (uncrosslinking macromer) within the network. It is expected that the esterification of CNTs with excess hydroxyl groups available on the PGS backbone provides additional crosslinking sites, and significantly reduces the amount of sol content (Fig. 2a). The amount of sol within the nanocomposites was determined using THF. PGS readily swells in THF, and thus the sol can be easily leached out and the amount of gel (crosslinked network) can be determined (Fig. 2b). The addition of CNTs to the PGS network results in a lower swelling degree in THF (Fig. 2c). For example, pure PGS swells to 987 ± 17%, whereas the addition of 0.25, 0.5 and 1% CNTs reduces the swelling degree to 557 ± 36%, 462 ± 56% and 357 ± 20%, respectively (Fig. 2c). The amount of sol for PGS was determined to be 20.63 ± 0.67%, similar to the previously reported literature.45 The addition of CNTs as a chemical crosslinker significantly decreases the amount of sol content due to enhanced crosslinking between CNTs and PGS (Fig. 2d). The sol contents for PGS nanocomposites containing 0.25, 0.5, and 1% CNTs were 19.33 ± 0.19%, 13.67 ± 0.86%, and 8.36 ± 0.77%, respectively. The addition of 1% CNTs reduces the sol content by 60% to that of pure PGS. These results indicate that CNTs acted as a multifunctional crosslinker and covalently crosslinked with PGS chains.
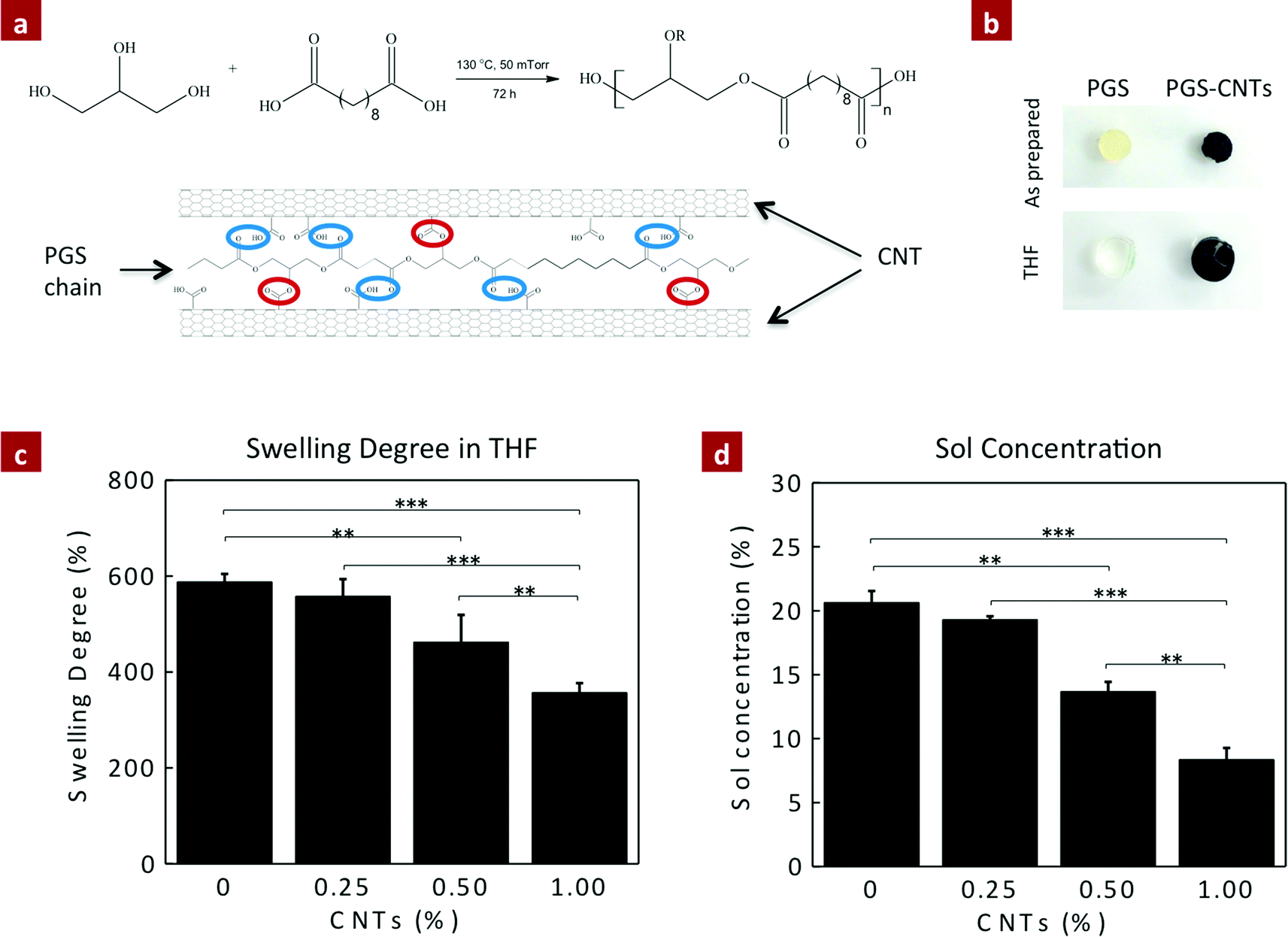 | ||
| Fig. 2 Effect of crosslinking between CNTs and PGS on the sol–gel content. (a) Schematic presentation of synthesis of the PGS prepolymer and physical and chemical interaction of CNTs upon curing. (b) PGS and PGS-1%CNT samples before and after swelling in THF. Analysis of (c) swelling degree, and (d) soluble contents in PGS and PGS–CNT nanocomposites determined. Addition of CNTs within PGS demonstrated more than two-fold reduction in sol fraction. (One-way ANOVA with Tukey post-hoc analysis, **p < 0.01, ***p < 0.001.) | ||
Thermal stability of PGS–CNT nanocomposites
The chemical infusion of CNTs within the PGS network had improved thermal stability compared to the pure PGS network. The weight loss characteristic of PGS and PGS–CNT nanocomposites was evaluated by thermal gravimetric analysis (TGA) (Fig. 3a). The derivative curve of the thermal decomposition spectra of PGS and PGS–CNT nanocomposites indicates a multiple-step degradation profile. These steps are introduced due to the polydispersity of the PGS macromer. The first derivative curve, used to evaluate the initial degradation temperature (TDI), indicates the onset of degradation and the peak degradation temperature (TDP). For PGS, TDI was ∼320 °C and TDP was ∼439 °C (Fig. 3a). The infusion of CNTs (0.25%) within the PGS network enhances the thermal stability of nanocomposites (TDI ∼ 350 °C and TDP ∼ 467 °C) due to strong covalent interactions between CNTs with PGS. The improved thermal stability of the nanocomposites was attributed to covalent crosslinking between CNTs and PGS that constrains the segmental motion of the PGS backbone. However, no effect on TDI and TDP was observed due to a further increase in CNTs. Similar trends of improving the thermal stability of polymers after reinforcing with CNTs were reported previously.46,47 The effect of CNT addition on the kinetics of thermal decomposition of the nanocomposite network was also investigated. The activation energy (Ea) of PGS and PGS–CNT nanocomposites is shown in Fig. 3c. The addition of CNTs to PGS results in an increase in activation energy and the data implied uniform dispersion of CNTs within the PGS network.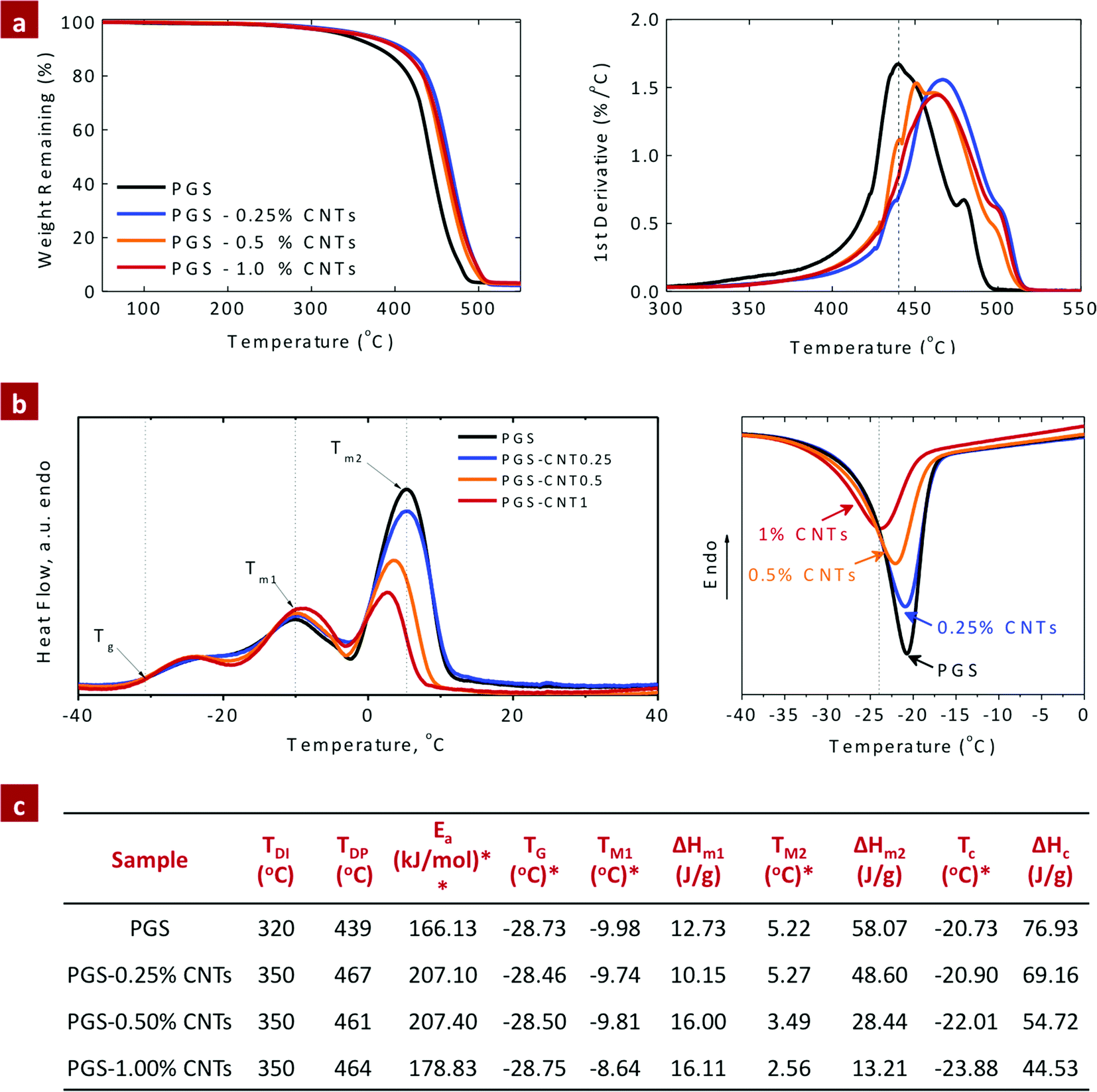 | ||
| Fig. 3 Effect of CNTs on the thermal properties of the PGS network. (a) TGA thermograph of PGS–CNT nanocomposites indicates that the addition of CNTs to the PGS network results in significantly enhanced thermal stability of the network. (b) DSC thermograph indicates decrease in crystallization temperature and enthalpy of crystallization due to the addition of CNTs to the PGS network. This might be attributed to increase in number of nucleation sites and enhanced interactions between CNTs and PGS. (c) Summary of thermal properties obtained from TGA and DSC thermographs. TDI and TDP are the initial and peak thermal decomposition temperatures, respectively obtained from TGA thermographs. *The glass transition temperature (Tg), melting temperature (Tm), crystallization temperature (TC) and enthalpy of the composites (ΔH) were obtained from the heating cycle of DSC endotherms. **The activation energy of thermal decomposition was calculated using the Horowitz–Metzger method using eqn (3). | ||
The effect of CNTs on phase transformation of PGS was investigated using DSC. As shown in DSC endotherms (Fig. 3b), the glass transition temperature (Tg) of PGS was observed around −28.5 °C. A weak endothermic peak at −10 °C (Tm1), followed by a strong endothermic peak at 5.5 °C (Tm2), was due to the melting points of PGS, similar to previously reported literature.48 The addition of CNTs had no effect on Tg; however, the melting points were decreased due to the disruption of the polymeric network by CNTs. During the cooling cycle, an exothermic crystalline peak was observed in PGS, similar to the previously reported literature.39 As shown in the cooling cycle, the presence of CNTs within the PGS network decreases both the crystallization temperature and the enthalpy of crystallization (Fig. 3c). The addition of CNTs increases the number of nucleation sites and the strong interactions between CNTs and PGS results in a decrease in crystallization degree and the enthalpy of recrystallization. Similar results were reported by Liang et al. on PGS–bioglass composites.49 They reported a decrease in recrystallization energies and recrystallization temperature due to the addition of bioglass to the PGS network. Overall, the results indicate that PGS–CNT nanocomposites are semicrystalline elastomers below their melting point, and amorphous at physiological temperature.
Mechanical properties of PGS–CNT nanocomposites
The covalent crosslinking of PGS and CNTs significantly improved the mechanical properties of the polymeric network. Due to covalent crosslinking of CNTs within the PGS network, it is expected that higher load is required to deform the network compared to the pure PGS network (Fig. 4a). The mechanical stiffness of PGS–CNT nanocomposites was evaluated by uniaxial tensile testing (Fig. 4b). The elastic modulus and ultimate tensile strength (UTS) of pure PGS were 198 ± 20 kPa and 122 ± 16 kPa, respectively. The addition of CNTs (up to 1%) to PGS resulted in more than a two-fold increase in UTS (275 ± 44 kPa) and in more than a five-fold increase in tensile modulus compared to the PGS network (Fig. 4c). For example, the nanocomposites containing 0.25, 0.5, and 1% of CNTs resulted in an increase in elastic modulus to 287 ± 13, 732 ± 77, and 1014 ± 103 kPa, respectively. While the addition of CNTs to the PGS network resulted in decrease in the total elongation of nanocomposites, pure PGS showed a maximum elongation of 122 ± 15%. The addition of 0.25, 0.5, and 1% CNTs reduced elongation to 99 ± 10, 38 ± 4, and 43 ± 5%, respectively. The decrease in elongation is expected due to an increase in crosslinking density with an addition of CNTs. We further analyzed the fracture mode of the nanocomposites by electron microscopy (Fig. 4d). PGS generated an elastomeric scaffold and the fractured surface of PGS indicates the formation of stress concentration that ultimately leads to failure indicating ductile fracture. As the amount of CNT increases, the fracture mode changes from a ductile fracture to a brittle fracture. Nanocomposites containing 0.5 and 1%, CNTs showed minimum surface deformation and the fracture surface displayed a relatively smooth surface morphology typically observed in a brittle fracture.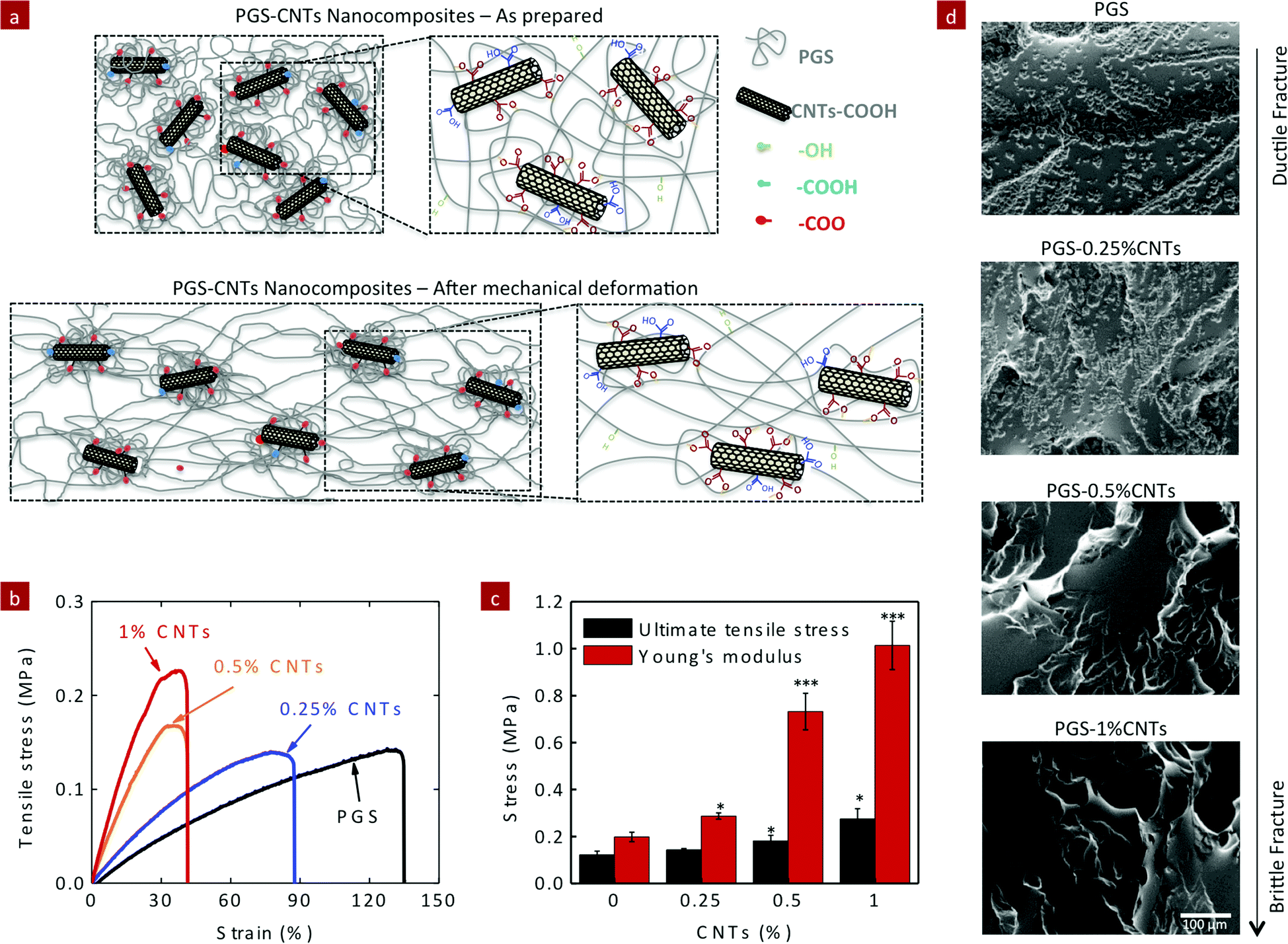 | ||
| Fig. 4 Effect of CNTs on the tensile properties of PGS–CNT nanocomposites. (a) CNTs act as a physical and covalent crosslinker, and result in increase in mechanical stiffness of the PGS network by increasing the crosslinking density. (b) The addition of CNTs (0.25, 0.5 and 1%) to PGS results in significant increase in the Young's modulus and ultimate tensile stress of the nanocomposite network. (c) By addition of only 1% CNT, almost a five-fold increase in Young's modulus was observed, whereas the ultimate tensile stress increased more than two-fold. (d) SEM images of fractured surfaces of PGS–CNT nanocomposites after tensile testing show the fracture mechanism dominating the nanocomposite network. PGS is an elastomeric polymer and shows ductile fracture. The addition of CNTs reinforces the PGS network due to covalent crosslinking and results in brittle fracture. (One-way ANOVA with Tukey post-hoc analysis, *p < 0.05, ***p < 0.001.) | ||
Elastomeric properties of PGS–CNT nanocomposites
The elastomeric properties of PGS–CNT nanocomposites were investigated using unconfined cyclic compression (Fig. 5a). The compressive modulus of pure PGS was determined to be 0.66 ± 0.07 MPa. The addition of 0.25%, 0.50%, and 1% CNTs significantly increases (*p < 0.05) the compressive modulus of the nanocomposites to 1.73 ± 0.09, 3.34 ± 0.06, and 4.06 ± 0.11 MPa, respectively. The addition of 1% CNTs to PGS results in more than a six-fold increase in the compressive modulus, which is far superior to the mechanical properties previously reported for CNT-based nanocomposites.43,50–61 This increase may be attributed to the strong covalent interactions between PGS and CNTs that restricts polymer chain movement during mechanical deformation.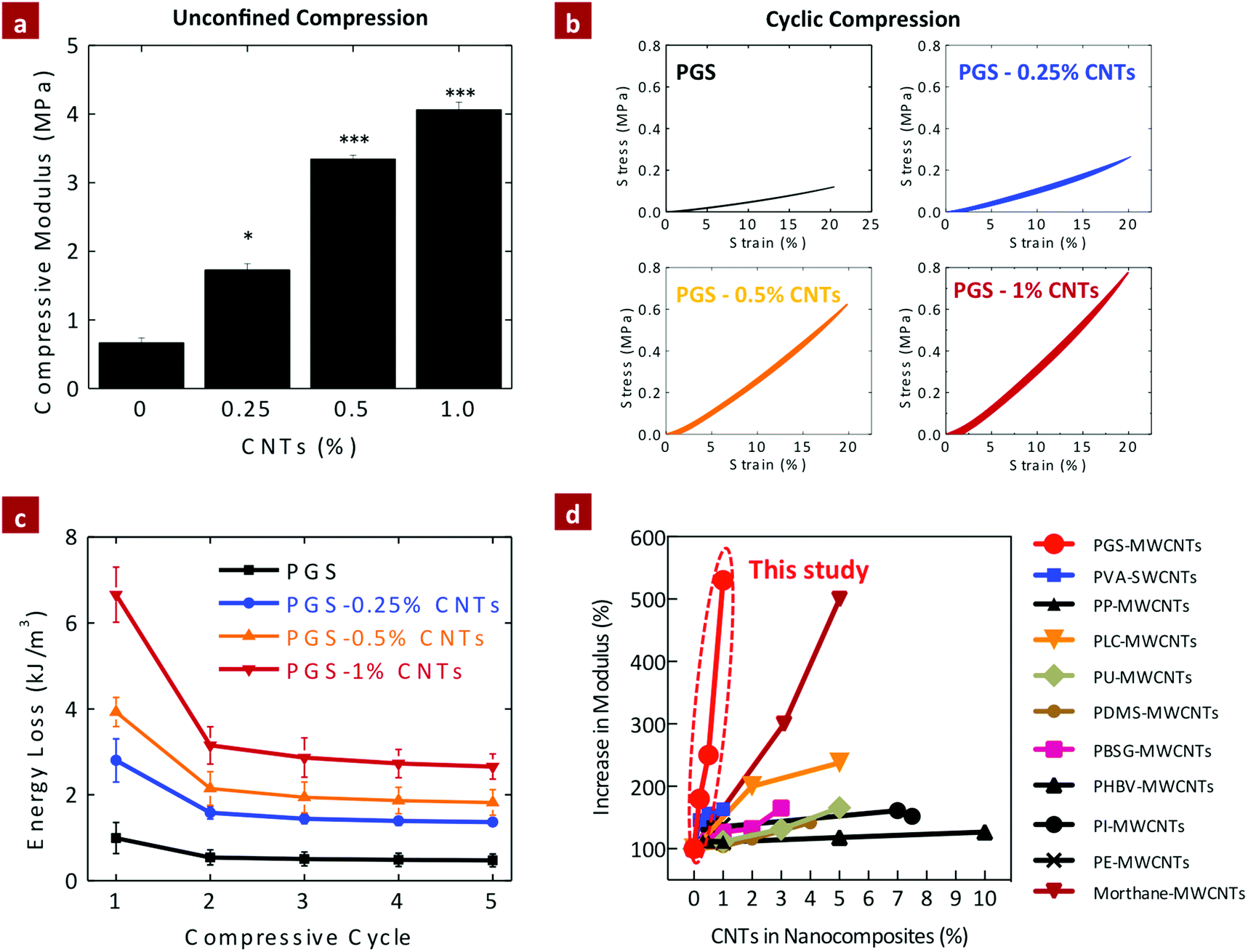 | ||
| Fig. 5 Effect of CNTs on the compression properties of PGS–CNT nanocomposites. (a) The addition of CNTs to the PGS network results in significant increase in compressive modulus of the nanocomposites. (b) Cyclic compression of PGS and PGS–CNTs indicates that elastomeric properties of the PGS network is preserved. (c) The energy absorbed by the network during cyclic deformation of the network indicates maximum loss in energy during the first cycle. In the subsequent cycle the energy loss was constant for the network. (d) The increase in mechanical stiffness due to addition of CNTs to different polymeric matrices is summarized (PVA,50 PLGA,51 PP,52 PLC,53 PU,54 PDMS,55 PBSG,56 PHBV,57 PI,58 PE,59 Morthane60). These data have been re-plotted from published reports. Most of the earlier studies have reported a moderate 20–300% increase in modulus due to the addition of 1 to 10% of CNTs. Current study (PGS–CNTs) showed more than 500% increase in modulus of polymeric networks by the addition of 1% CNTs, which is not reported previously. (One-way ANOVA with Tukey post-hoc analysis, *p < 0.05, ***p < 0.001.) | ||
The effect of CNTs on energy absorbed by the network during the cyclic compression was also evaluated (Fig. 5b and c). The nanocomposite scaffolds were subjected to five loading and unloading curves until 20% strain. The energy absorbed by the network during deformation and the recovery of the network after the deformation was determined. Pure PGS showed energy absorption of 0.99 ± 0.36 kJ m−3 (recovery ∼95 ± 1.4%) during the first cycle and subsequent cycles (2–5 cycles) showed energy absorption of 0.5 ± 0.03 kJ m−3 (recovery ∼99.2 ± 0.3%). The addition of CNTs significantly enhances (*p < 0.05) the amount of energy absorbed without compromising elastic recovery of the network. For example, nanocomposites containing 1% CNTs showed energy absorption of 6.66 ± 0.64 kJ m−3 (recovery ∼94.9 ± 1.2%) for the first cycle and the subsequent cycles (2–5 cycles) showed energy absorption of 2.85 ± 0.22 kJ m−3 (recovery ∼98.9 ± 0.3%). Thus the incorporation of CNTs into the PGS network results in a significant increase in mechanical stiffness without compromising the elasticity of the nanocomposite networks. This is mainly attributed to the esterification of carboxyl groups of CNTs with the PGS backbone that results in improved load transfer efficiency within the crosslinked network.
Overall, the mechanical stiffness due to addition of CNTs in both compression and tension has been shown to increase by five-fold. This is an improvement compared to earlier studies43 that have reported a moderate 20–300% increase in modulus due to the addition of 1 to 10% of CNTs as summarized in Fig. 5d. For example, So et al. showed a 30% increase in modulus due to the addition of 5% of COOH-modified-CNTs to polyimide (PI).58 Cha et al. showed an ∼260% increase in modulus due to the addition of 10% CNTs to copper nanocomposites.61 In a similar study, Koerner et al. reinforced a thermoplastic elastomer (Morthane) with 1–5% CNTs and observed a 200–500% increase in modulus.60 They attributed the significant increase in mechanical properties in the formation of a percolation network.60 In another study, Lahiri et al. showed that the addition of 2 and 5% CNTs to poly(L-lactide-co-caprolactone) (PLC) results in two-fold and 2.4-fold increases in modulus, respectively.53 Jell et al. developed CNT-reinforced polyurethane (PU) scaffolds using a thermally induced phase separation method and was able to enhance the compressive modulus by two-fold by adding 5% CNTs.54 In a similar study, Armentano et al. synthesized poly(DL-lactide-co-glycolide) (PLGA 50:50) nanocomposite by incorporating pristine and carboxyl functionalized single-walled CNTs.51 The elastic modulus of PLGA improved more than 300% due to the addition of 1% CNT.51 The addition of 1% multiwalled CNTs to polyethylene (PE) films also resulted in moderate increase in tensile strength (125%).59 Hou et al. also showed relatively moderate increase (∼132%) in the mechanical stiffness of polyvinyl alcohol (PVA) due to addition of single-walled and multiwalled CNTs.50 Other polymers such as poly(dimethylsiloxane) (PDMS),55 polypropylene (PP),52 poly(butylene succinate-co-ethylene glycol) (PBSG),56 poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV)57 also showed moderate increase in mechanical properties as shown in Fig. 5d. The results reported here (PGS–CNTs) are quite unique as more than 500% modulus of polymeric networks can be enhanced by just the addition of 1% CNTs. Moreover, the synthesis technique to obtain PGS–CNT nanocomposites is relatively simple and does not require extensive post-processing.
In vitro stability of PGS–CNT nanocomposites
For biomedical and biotechnological applications, it is important to investigate the in vitro stability of nanocomposite biomaterials. The surface characteristic and in vitro degradation of PGS–CNT nanocomposites were determined under a physiological environment. It is expected that the addition of CNTs to PGS should increase the in vitro stability of nanocomposites due to the increase in the crosslinking density.The surface characteristic of PGS and PGS–CNT nanocomposites was determined by optical tensiometry (goniometry). The esterification of carboxyl groups of CNTs with hydroxyl groups of PGS reduces the hydrophilic nature of the nanocomposites. The contact angle of a sessile water drop on the nanocomposite surface was used to determine the hydrophilicity of the material (Fig. 6a and b). The contact angle of the PGS surface was observed to be 69.78 ± 1.39°, similar to the previously reported literature.40 Incorporation of CNTs into the PGS network significantly enhances the contact angle of water. The addition of 0.25%, 0.5%, and 1% CNTs to PGS increases the contact angle of water to 71.01 ± 1.26°, 78.54 ± 1.65°, and 79.63 ± 1.10°. This result was further supported by evaluating hydration degree of nanocomposites (Fig. 6c). The addition of CNTs significantly reduces water uptake ability of the nanocomposites. It is expected that this may also retard degradation of the polyester backbone and also alter the interaction of nanocomposites with biological entities such as proteins and cells.
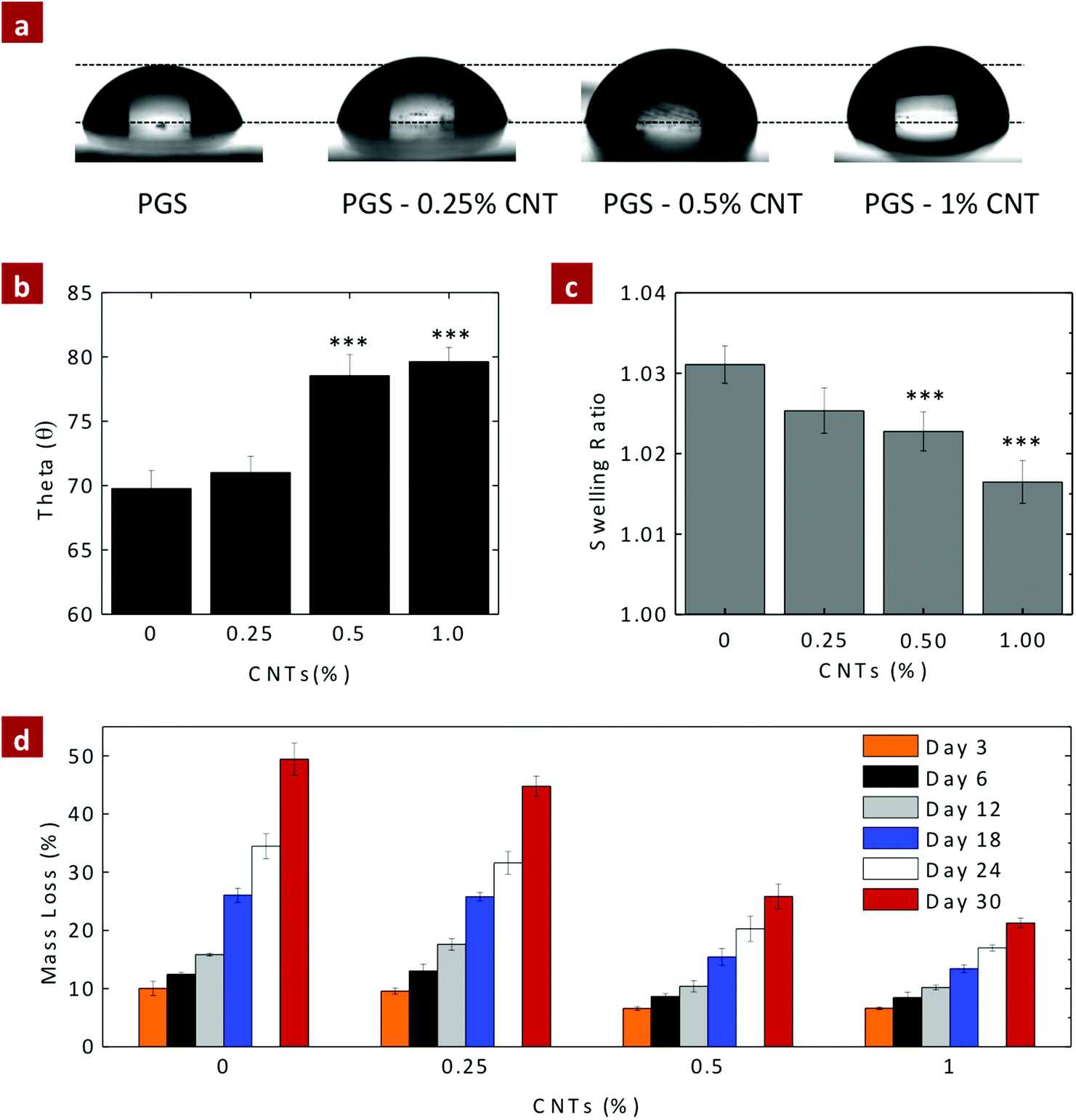 | ||
| Fig. 6 Effect of crosslinking on surface characteristic of nanocomposites. (a) & (b) The effect of addition of CNTs to PGS on hydrophobicity of the nanocomposite surface was analyzed using water contact angle measurement (drop shape analysis). The pure PGS surface was observed to have a water contact angle of 69.78 ± 1.39°. The addition of 0.25, 0.5 and 1% of CNTs, increases the water contact angle to 71.01 ± 1.26°, 78.54 ± 1.65°, and 79.63 ± 1.10°, indicating an increase in hydrophobicity. (c) The swelling study was performed to determine water uptake ability of the nanocomposite network at 37 °C. The addition of CNTs reduces the water uptake ability of nanocomposite networks due to increase in hydrophobicity of the nanocomposite. (d) The mass loss data indicate that with an increase in CNT concentration, the rate of degradation decreases under alkaline-catalyzed conditions. The mass loss for pure PGS after 30 days was 49.4 ± 2.8% (degradation rate ∼11.5% per week), addition of 0.25%, 0.50%, and 1% of CNTs results in mass loss (degradation rate) of 44.8 ± 1.7% (∼10.5% per week), 25.8 ± 2.2% (∼6% per week), and 21.3 ± 0.8% (∼5% per week), respectively. (One-way ANOVA with Tukey post-hoc analysis, *p < 0.05, **p < 0.01, ***p < 0.001.) | ||
PGS degrades via the surface erosion mechanism and displays linear degradation behavior under in vitro and in vivo conditions.36,45 A linear degradation behavior is preferable for tissue engineered scaffolds to follow, with the ECM biosynthesis rate. The degradation properties of PGS and PGS–CNT nanocomposites were determined under physiological conditions (PBS, 37 °C). From the contact angle measurement results, it was observed that the addition of CNTs decreases the hydrophilicity of the nanocomposite network; thus, it was expected that the addition of CNTs should result in a decrease in the degradation rate of the nanocomposites.
Under in vivo conditions, PGS degraded relatively fast compared to in vitro conditions. The fast in vivo degradation of PGS is attributed to the presence of esterases in the surrounding microenvironment that accelerates the degradation process. In order to closely mimic the in vivo degradation, PGS and PGS–CNT nanocomposites were subjected to 0.01 M NaOH, which also accelerates the rate of degradation through alkaline-catalyzed conditions. The mass loss of PGS and PGS–CNT nanocomposites in 0.01 M NaOH was monitored over 30 days (Fig. 6d). All samples follow a linear degradation without compromising the structural integrity. After 30 days, the mass loss of PGS was 49.4 ± 2.8% (degradation rate ∼11.5% per week), addition of 0.25%, 0.50%, and 1% of CNTs results in mass loss (degradation rate) of 44.8 ± 1.7% (∼10.5% per week), 25.8 ± 2.2% (∼6% per week), and 21.3 ± 0.8% (∼5% per week), respectively. The mass loss data indicate that with an increase in CNT concentration, the rate of degradation decreases.
Compared to earlier studies with polyester-carboxyl functionalized CNT nanocomposites, the covalently crosslinked PGS–CNT nanocomposites reported here showed opposite trends in degradation behavior. For example, Armentano et al. analyzed the in vitro degradation behavior of PLGA–CNT nanocomposites where it was observed that the presence of carboxyl functionalized CNTs induced faster degradation of nanocomposites.62 Zhao et al. also observed similar trends in the PLLA–CNT nanocomposite system.63 In both of these studies, the functionalized CNTs were physically entrapped within the polyester structures and no covalent interactions of CNTs with the polymer backbone were observed.62,63 Moreover, due to the hydrophilic nature of carboxyl groups present on functionalized CNTs, the hydration ability within polyester nanocomposites was also enhanced and resulted in accelerated degradation. Thus, the degradation data in PGS–CNT nanocomposites also indirectly demonstrated the esterification of carboxyl functionalized CNTs within the PGS network.
hMSC adhesion, proliferation, and differentiation on PGS–CNT nanocomposites
To evaluate the feasibility of the PGS–CNT nanocomposite network for tissue engineering applications, cell–matrix interactions were evaluated using hMSCs. PGS and PGS–CNT nanocomposites were seeded with hMSCs to determine the effect of CNTs on metabolic activity and ALP production. The metabolic activity of adhered hMSCs was determined by Alamar Blue assay for 10 days (Fig. 7a). Compared to the TCPS control, the metabolic activity of seeded hMSCs was almost half on PGS and PGS–CNT surfaces. However, no significant difference in the metabolic activity of seeded hMSCs was observed due to the addition of CNTs. The in vitro study was carried out for 10 days and all scaffolds were intact during this time frame. Under in vivo conditions, PGS might degrade and entrapped CNTs might emerge. It is expected that these CNTs might result in mild inflammation and fibrosis. However, additional in vivo studies are needed to determine the long-term efficacy of PGS–CNT nanocomposites.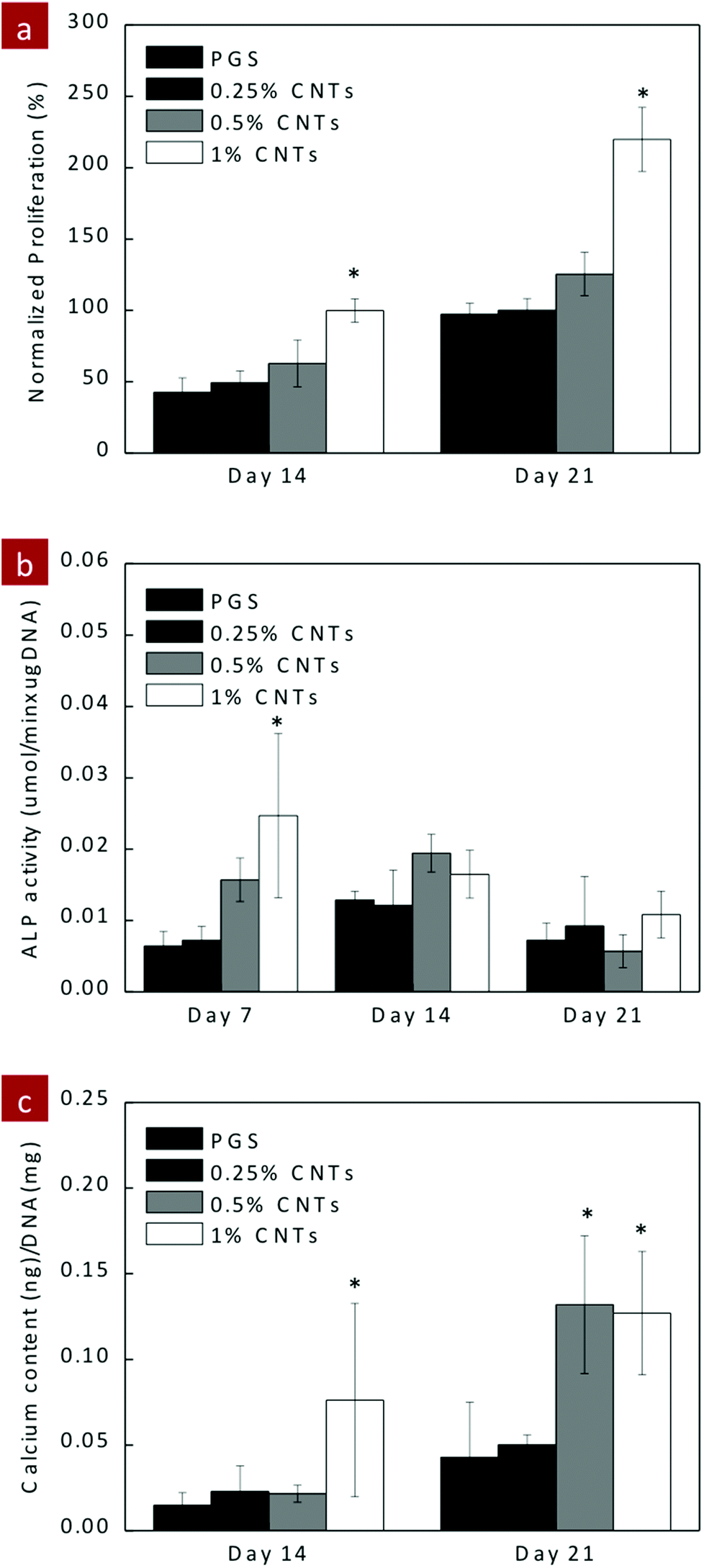 | ||
| Fig. 7 In vitro evaluation of PGS–CNT nanocomposites using hMSCs. (a) PGS and PGS-CNT nanocomposites support proliferation of hMSCs. The tissue culture polystyrene (TCPS) surface was used as positive control. (b) ALP activity of hMSCs seeded on PGS and PGS–CNTs was monitored on days 7, 14 and 21. The peak ALP activity (day 7) on PGS-1%CNT nanocomposites was significantly higher compared to PGS. No significant effect was observed on days 14 and 21. (c) The amount of calcium deposited by hMSCs was significantly higher on PGS-1%CNTs compared to PGS. (One-way ANOVA with Tukey post-hoc analysis, *p < 0.05.) | ||
Having established the ability to support hMSCs proliferation, we investigated the potential of PGS–CNT nanocomposite for orthopaedic tissue engineering by monitoring the osteogenic differentiation of hMSCs. We monitored ALP activity on days 7, 14 and 21, and calcium production on days 14 and 21. ALP is an early marker and hallmark of osteogenesis. The addition of CNTs resulted in significant (*p < 0.05) upregulation of ALP production on PGS-1%CNTs compared to pure PGS (Fig. 7b). This indicates that stiffness of the nanocomposite may play a significant role in osteogenesis. As expected on days 14 and 21, no significant difference in the ALP activity was observed. A similar trend was observed in calcium production on days 14 and 21. The nanocomposite containing 1% CNTs shows a slightly higher amount of calcium on day 14 compared to PGS, although no significant difference was observed. But on day 21, nanocomposites containing 1% and 0.5% CNTs show a significantly (*p < 0.05) higher amount of calcium compared to PGS scaffolds (Fig. 7c). The increase in ALP expression and calcium production might be attributed to the increase in surface hydrophobicity, change in surface topography and increase in stiffness due to the addition of the CNTs to the PGS networks.
Conclusions
We have successfully designed and developed stiff, elastomeric nanocomposites from PGS and CNTs. To our knowledge, this is the first attempt to physically entrap and covalently conjugate the CNTs within the PGS structure. Imaging techniques revealed a uniform distribution of CNTs within the PGS network. The addition of 1% CNTs to PGS results in a five-fold increase in the tensile modulus and six-fold increase in the compressive modulus. The reported increase in mechanical properties is much superior compared to previously published literature for CNT-based nanocomposites. Despite a significant increase in mechanical stiffness, the elasticity of the network was not compromised and all the nanocomposites showed more than 94% recovery. The strong covalent interactions between PGS and CNTs restrict polymer chains movement during mechanical deformation and result in novel property combinations that are not observed in conventional nanocomposites. The addition of CNTs to PGS increases the surface hydrophobicity of nanocomposites and thus reduces the degradation rate up to 40% compared to PGS. Additionally, in vitro studies using hMSCs indicated that the addition of CNTs to PGS supports the differentiation potential of the seeded hMSCs as indicated by the increase in ALP activity and production of calcium. Overall, the unique property combinations of ultra-stiff, elastomeric CNT–PGS nanocomposites make them potentially suitable for biomedical applications such as scaffolding in musculoskeletal tissue engineering.Acknowledgements
A. K. G., A. P. and A. K. designed the study and wrote the manuscript. All the authors have contributed to experimental design and data analysis. All authors discussed the results and commented on the manuscript. The authors would like to thank Dr Daniel Siegwart for helping with PGS synthesis, and Shilpaa Mukundan and Silvia M. Mihaila for technical discussion. A. K. G. would like to thank Prof. Robert Langer for access to equipment and acknowledge financial support from MIT Portugal Program (MPP-09Call-Langer-47). A. P. would like to acknowledge postdoctoral fellowship (fellowship no. PDF-388346-2010) awarded by the Natural Science and Engineering Research Council, Canada. A. D. P. would like to thank the Danish Council for Independent Research grant (Technology and Production Sciences, 10-100118). This research was funded by the US Army Engineer Research and Development Center, the Institute for Soldier Nanotechnology (ISN), the NIH (EB009196; DE019024; EB007249; HL099073; AR057837), and the National Science Foundation CAREER award. Also, M. A. H. and A. K. would like to acknowledge the support of NSTIP strategic technologies program in the Kingdom of Saudi Arabia – award no. (11-NAN1544-03).Notes and references
- A. Oberlin, M. Endo and T. Koyama, J. Cryst. Growth, 1976, 32, 335–349 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Science, 2002, 297, 787–792 CrossRef CAS PubMed.
- A. Fennimore, T. Yuzvinsky, W.-Q. Han, M. Fuhrer, J. Cumings and A. Zettl, Nature, 2003, 424, 408–410 CrossRef CAS PubMed.
- R. H. Baughman, C. Cui, A. A. Zakhidov, Z. Iqbal, J. N. Barisci, G. M. Spinks, G. G. Wallace, A. Mazzoldi, D. De Rossi and A. G. Rinzler, Science, 1999, 284, 1340–1344 CrossRef CAS.
- K. Mylvaganam and L. Zhang, Nanotechnology, 2007, 18, 475701 CrossRef.
- E. Fortunati, F. D'Angelo, S. Martino, A. Orlacchio, J. M. Kenny and I. Armentano, Carbon, 2011, 49, 2370–2379 CrossRef CAS PubMed.
- T. Kashiwagi, F. Du, J. F. Douglas, K. I. Winey, R. H. Harris Jr. and J. R. Shields, Nat. Mater., 2005, 4, 928–933 CrossRef CAS PubMed.
- A. C. Dillon, K. Jones, T. Bekkedahl, C. Kiang, D. Bethune and M. Heben, Nature, 1997, 386, 377–379 CrossRef CAS.
- B. S. Harrison and A. Atala, Biomaterials, 2007, 28, 344–353 CrossRef CAS PubMed.
- L. P. Zanello, B. Zhao, H. Hu and R. C. Haddon, Nano Lett., 2006, 6, 562–567 CrossRef CAS PubMed.
- L. Lacerda, S. Raffa, M. Prato, A. Bianco and K. Kostarelos, Nano Today, 2007, 2, 38–43 CrossRef.
- A. Bianco, K. Kostarelos and M. Prato, Curr. Opin. Chem. Biol., 2005, 9, 674 CrossRef CAS PubMed.
- K. Besteman, J.-O. Lee, F. G. Wiertz, H. A. Heering and C. Dekker, Nano Lett., 2003, 3, 727–730 CrossRef CAS.
- J. Wang, Electroanalysis, 2005, 17, 7–14 CrossRef CAS.
- A. K. Hüttel, G. A. Steele, B. Witkamp, M. Poot, L. P. Kouwenhoven and H. S. J. van der Zant, Nano Lett., 2009, 9, 2547–2552 CrossRef PubMed.
- S. Sarojini, S. Rajasekar and K. Koumaravelou, Int. J. Pharm. Bio. Sci., 2010, 1, 644–649 CAS.
- P. A. Tran, L. Zhang and T. J. Webster, Adv. Drug Delivery Rev., 2009, 61, 1097–1114 CrossRef CAS PubMed.
- X. Chen, A. Kis, A. Zettl and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8218–8222 CrossRef CAS PubMed.
- A. K. Gaharwar, N. A. Peppas and A. Khademhosseini, Biotechnol. Bioeng., 2014, 111, 441–453 CrossRef CAS PubMed.
- A. K. Gaharwar, C. Rivera, C.-J. Wu, B. K. Chan and G. Schmidt, Mater. Sci. Eng., C, 2013, 33, 1800–1807 CrossRef CAS PubMed.
- S. Goenka, V. Sant and S. Sant, J. Controlled Release, 2013, 173, 75–88 CrossRef PubMed.
- M. Shim, N. W. Shi Kam, R. J. Chen, Y. Li and H. Dai, Nano Lett., 2002, 2, 285–288 CrossRef CAS.
- P.-C. Ma, N. A. Siddiqui, G. Marom and J.-K. Kim, Composites Part A, 2010, 41, 1345–1367 CrossRef PubMed.
- X.-L. Xie, Y.-W. Mai and X.-P. Zhou, Mater. Sci. Eng., R, 2005, 49, 89–112 CrossRef PubMed.
- A. Hirsch and O. Vostrowsky, in Functional molecular nanostructures, Springer, 2005, pp. 193–237 Search PubMed.
- H. Peng, L. B. Alemany, J. L. Margrave and V. N. Khabashesku, J. Am. Chem. Soc., 2003, 125, 15174–15182 CrossRef CAS PubMed.
- L. Zhang, V. U. Kiny, H. Peng, J. Zhu, R. F. Lobo, J. L. Margrave and V. N. Khabashesku, Chem. Mater., 2004, 16, 2055–2061 CrossRef CAS.
- Y. Wang, Z. Iqbal and S. V. Malhotra, Chem. Phys. Lett., 2005, 402, 96–101 CrossRef CAS PubMed.
- H. Wang, Curr. Opin. Colloid Interface Sci., 2009, 14, 364–371 CrossRef CAS PubMed.
- S. S. Karajanagi, H. Yang, P. Asuri, E. Sellitto, J. S. Dordick and R. S. Kane, Langmuir, 2006, 22, 1392–1395 CrossRef CAS PubMed.
- M. Zheng, A. Jagota, E. D. Semke, B. A. Diner, R. S. McLean, S. R. Lustig, R. E. Richardson and N. G. Tassi, Nat. Mater., 2003, 2, 338–342 CrossRef CAS PubMed.
- J. N. Coleman, U. Khan and Y. K. Gun'ko, Adv. Mater., 2006, 18, 689–706 CrossRef CAS.
- T. Liu and S. Guo, Properties of Polyurethane/Carbon Nanotube Nanocomposites, Scrivener Publishing, 2010 Search PubMed.
- K. Hayashida and H. Tanaka, Adv. Funct. Mater., 2012, 22, 2338–2344 CrossRef CAS.
- Y. Wang, Y. M. Kim and R. Langer, J. Biomed. Mater. Res., Part A, 2003, 66, 192–197 CrossRef PubMed.
- Y. D. Wang, G. A. Ameer, B. J. Sheppard and R. Langer, Nat. Biotechnol., 2002, 20, 602–606 CrossRef CAS PubMed.
- A. R. Webb, J. Yang and G. A. Ameer, Expert Opin. Biol. Ther., 2004, 4, 801–812 CrossRef CAS PubMed.
- R. Rai, M. Tallawi, A. Grigore and A. R. Boccaccini, Prog. Polym. Sci., 2012, 37, 1051–1078 CrossRef CAS PubMed.
- A. Patel, A. K. Gaharwar, G. Iviglia, H. Zhang, S. Mukundan, S. M. Mihaila, D. Demarchi and A. Khademhosseini, Biomaterials, 2013, 34, 3970–3983 CrossRef CAS PubMed.
- J. Y. Kim, Poly(butylene terephthalate) Nanocomposites Containing Carbon Nanotube, Advances in Nanocomposites - Synthesis, Characterization and Industrial Applications, InTech, 2011 Search PubMed.
- J. Y. Kim, S. I. Han and S. Hong, Polymer, 2008, 49, 3335–3345 CrossRef CAS PubMed.
- N. G. Sahoo, S. Rana, J. W. Cho, L. Li and S. H. Chan, Prog. Polym. Sci., 2010, 35, 837–867 CrossRef CAS PubMed.
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751–758 CrossRef CAS PubMed.
- I. Pomerantseva, N. Krebs, A. Hart, C. M. Neville, A. Y. Huang and C. A. Sundback, J. Biomed. Mater. Res., Part A, 2009, 91, 1038–1047 CrossRef PubMed.
- S. Huang, M. Wang, T. Liu, W.-D. Zhang, W. C. Tjiu, C. He and X. Lu, Polym. Eng. Sci., 2009, 49, 1063–1068 CAS.
- J. Y. Kim, S. Han, D. K. Kim and S. H. Kim, Composites Part A, 2009, 40, 45–53 CrossRef PubMed.
- W. Cai and C. Liu, Mater. Lett., 2008, 62, 2171–2173 CrossRef PubMed.
- S. L. Liang, W. D. Cook, G. A. Thouas and Q. Z. Chen, Biomaterials, 2010, 31, 8516–8529 CrossRef CAS PubMed.
- Y. Hou, J. Tang, H. Zhang, C. Qian, Y. Feng and J. Liu, ACS Nano, 2009, 3, 1057–1062 CrossRef CAS PubMed.
- I. Armentano, M. Dottori, D. Puglia and J. M. Kenny, J. Mater. Sci.: Mater. Med., 2007, 19, 2377–2387 CrossRef PubMed.
- A. A. Kovalchuk, V. G. Shevchenko, A. N. Shchegolikhin, P. M. Nedorezova, A. N. Klyamkina and A. M. Aladyshev, J. Mater. Sci., 2008, 43, 7132–7140 CrossRef CAS PubMed.
- D. Lahiri, F. Rouzaud, T. Richard, A. K. Keshri, S. R. Bakshi, L. Kos and A. Agarwal, Acta Biomater., 2010, 6, 3524–3533 CrossRef CAS PubMed.
- G. Jell, R. Verdejo, L. Safinia, M. S. P. Shaffer, M. M. Stevens and A. Bismarck, J. Mater. Chem., 2008, 18, 1865–1872 RSC.
- C.-L. Wu, H.-C. Lin, J.-S. Hsu, M.-C. Yip and W. Fang, Thin Solid Films, 2009, 517, 4895–4901 CrossRef CAS PubMed.
- L. Tan, Y. Chen, W. Zhou and S. Ye, Polym. Eng. Sci., 2012, 52, 2506–2517 CAS.
- S. Vidhate, L. Innocentini-Mei and N. A. D'Souza, Polym. Eng. Sci., 2012, 52, 1367–1374 CAS.
- H. H. So, J. W. Cho and N. G. Sahoo, Eur. Polym. J., 2007, 43, 3750–3756 CrossRef CAS PubMed.
- S. Ruan, P. Gao, X. Yang and T. Yu, Polymer, 2003, 44, 5643–5654 CrossRef CAS.
- H. Koerner, G. Price, N. A. Pearce, M. Alexander and R. A. Vaia, Nat. Mater., 2004, 3, 115–120 CrossRef CAS PubMed.
- S. I. Cha, K. T. Kim, S. N. Arshad, C. B. Mo and S. H. Hong, Adv. Mater., 2005, 17, 1377–1381 CrossRef CAS.
- I. Armentano, M. Dottori, D. Puglia and J. M. Kenny, J. Mater. Sci. Mater. Med., 2008, 19, 2377–2387 CrossRef CAS PubMed.
- Y. Zhao, Z. Qiu and W. Yang, Compos. Sci. Technol., 2009, 69, 627–632 CrossRef CAS PubMed.
Footnotes |
| † Currently at: Department of Biomedical Engineering and Department of Materials Science and Engineering, Texas A&M University, College Station, TX 77843, USA. |
| ‡ Dr A. K. Gaharwar and Dr A. Patel contributed equally. |
| § Currently at: Interdisciplinary Nanoscience Center (iNANO), Aarhus University, Aarhus (8000), Denmark. |
| This journal is © The Royal Society of Chemistry 2015 |