
MMP9-sensitive polymers mediate environmentally-responsive bivalirudin release and thrombin inhibition†
D. S.
Chu‡
a,
D. L.
Sellers‡
b,
M. J.
Bocek
a,
A. E.
Fischedick
b,
P. J.
Horner
*b and
S. H.
Pun
*a
aDepartment of Bioengineering and Molecular Engineering and Sciences Institute, University of Washington, Seattle, WA 98195, USA. E-mail: spun@u.washington.edu; Fax: (+)1-206-685-3488
bDepartment of Neurological Surgery and Institute for Stem Cell and Regenerative Medicine, University of Washington, Seattle, WA 98195, USA. E-mail: phorner@u.washington.edu
First published on 8th September 2014
Abstract
MMP9-responsive bivalirudin-HPMA copolymers were synthesized for direct, local administration in rat spinal cord contusion injury models. Polymer-conjugated bivalirudin peptides maintained activity while demonstrating enzyme-mediated release upon MMP9 exposure and prolonged release from hyaluronic acid/methylcellulose (HAMC) hydrogels compared to free bivalirudin peptide. Localized administration of bivalirudin copolymers in vivo at the site of rat spinal cord injury decreased cellular proliferation and astrogliosis, suggesting the bivalirudin copolymer and HAMC hydrogel system are a promising therapeutic intervention for reducing immediate inflammatory responses and long term scarring.
Spinal cord injuries (SCI) comprise a significant percentage of debilitating injuries, with over 250
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Americans suffering from SCI and 12000 new cases annually.1 Following acute SCI from blunt trauma, thrombin, a serine protease and key component of the coagulation cascade, rapidly increases from basal picomolar levels2 at injury sites and elevated levels are sustained for several days due to expression by endothelial and astrocytic cells.3,4 Thrombin administered to healthy rat brains induces histological changes resembling inflammation and glial scarring,5 indicating it may impede recovery of damaged neural networks.
000 Americans suffering from SCI and 12000 new cases annually.1 Following acute SCI from blunt trauma, thrombin, a serine protease and key component of the coagulation cascade, rapidly increases from basal picomolar levels2 at injury sites and elevated levels are sustained for several days due to expression by endothelial and astrocytic cells.3,4 Thrombin administered to healthy rat brains induces histological changes resembling inflammation and glial scarring,5 indicating it may impede recovery of damaged neural networks.
Modulation of thrombin activity post-SCI is therefore a potential method for improving outcome.3 Indeed, thrombin inhibition immediately following SCI has been shown to improve both histological and functional recovery.6 Systemic administration of recombinant thrombomodulin (rTM), a regulator of thrombin activity, reduced glial scarring and improved locomotor recovery in rats.7 However, systemic administration of thrombin inhibitors, particularly in a polytrauma patient, is associated with adverse outcomes. A sustained delivery of thrombin inhibitors localized to the injury site is likely to be more effective and safe.
Injectable hydrogels, typically formed from polymers that undergo a temperature transition near body temperature, have been investigated as delivery depots in the central nervous system (CNS). The Shoichet group has pioneered the use of natural biopolymer hydrogel formulations composed of hyaluronic acid and methylcellulose (HAMC) in the CNS. These materials exhibit low cellular adhesion, good biocompatibility, and tunable mechanical properties.8 However, peptide release from hydrogels typically occurs within hours due to their low molecular weight.9
Bivalirudin is a clinically approved, 20-amino acid direct thrombin inhibitor.10 N-terminal residues reversibly bind the catalytic pocket while C-terminal residues bind the fibrinogen-binding domain of thrombin. Bivalirudin is an attractive drug due to its low immunogenicity and large therapeutic window; however, poor proteolytic stability and small size results in rapid clearance. To improve peptide stability and local retention, peptides can be grafted to higher molecular weight polymers. Proteins loaded in HAMC hydrogels are released over two days,11 providing ideal release kinetics for protein-sized bivalirudin polymers.
In this work, we developed a material formulation for localized and prolonged bivalirudin delivery following SCI and demonstrated reduced proliferation and decreased gliosis in rats treated with these bivalirudin-release depots. Polymers displaying pendant bivalirudin were synthesized by reversible addition-fragmentation chain transfer (RAFT) polymerization of an HPMA-co-APMA polymer backbone followed by grafting of the BM9 peptide, comprised of bivalirudin fused to a protease substrate sequence for targeted proteolytic release from the polymer. Three peptide-HPMA copolymers (DP200, DP300, DP400) of varying molecular weight but comparable peptide incorporation were synthesized. The polymers were then loaded in HAMC hydrogels for localized delivery. We hypothesized that hydrogel-encapsulated, enzymatically-responsive bivalirudin polymers administered locally to the spinal cord would (1) improve bivalirudin stability and prolong residence time in the tissue through sustained hydrogel release and (2) allow for environmentally-responsive bivalirudin cleavage from polymer for enhanced tissue penetration at the site of injury after release from the hydrogel (Fig. 1).
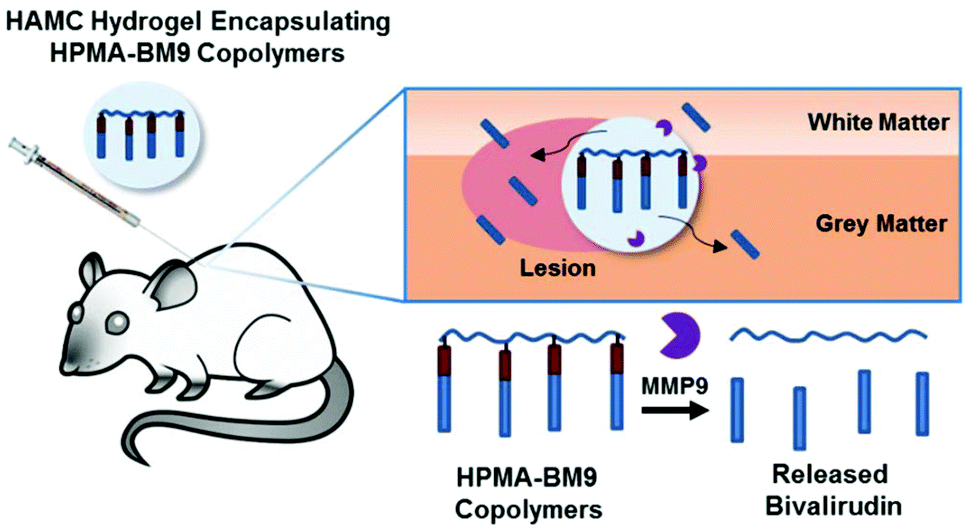 | ||
| Fig. 1 Localized depot delivery of bivalirudin through HPMA-BM9 thermoresponsive hydrogels. | ||
We first designed the BM9 peptide, which contains bivalirudin fused to an optimized matrix metalloproteinase-9 (MMP9) peptide substrate linker, PRQITAG.12 MMP9 was selected as the target protease for triggering bivalirudin release due to its maximal expression at 24 h following SCI, concurrent with upregulated thrombin expression.13 MMP9 activity on BM9 was confirmed via MALDI-TOF MS fragmentation analysis (Fig. S1a†), with >15% of peptide cleaved within 3 h (Fig. S1b†). Rapid recognition and specific cleavage of the bivalirudin-MMP9 peptide at the linker results in the emergence of a 2774 Da peak corresponding to fragmentation at the peptide linker (Table S1†). The peptide fragment is susceptible to further C-terminal exopeptidase degradation, leading to smaller, truncated peptide fragments over time. The N-terminus of the peptide shows resistance to exopeptidase activity, likely due to acetylation of the N-terminal amine and presence of a (D)-phenylalanine as the first residue.14,15 Cleavage at the linker sequence is expected to yield functional bivalirudin peptide.
A series of HPMA-BM9 copolymers was then synthesized as peptide carriers for localized thrombin inhibition (Scheme 1). The molecular weight and composition of synthesized copolymers are summarized in Table 1. The base HPMA-APMA polymer had a monomer feed ratio of 1:9 APMA to HPMA and were varied in size to investigate the effects of different polymer molecular weights. Base copolymers were synthesized with narrow polydispersities (<1.05) and molecular weights 28–51 kDa. The primary amines of the copolymers were converted to thiol-reactive maleimide groups and BM9 peptides conjugated via a C-terminal cysteine residue. Final BM9-conjugated copolymers contained 12–13% peptide, as determined by amino acid analysis, and ranged from 100–180 kDa in molecular weight with PDI < 1.5. Polymers contain ∼20–50 peptides per polymer chain depending on polymer molecular weight. Some bimodal polymer populations were observed by aqueous GPC (Fig. S2†), prominently for the DP400 copolymer, likely due to some inter-chain crosslinking during peptide bioconjugation. Alternative bioconjugation strategies with higher orthogonality, such as azide–alkyne “click” chemistries, could minimize crosslinking.
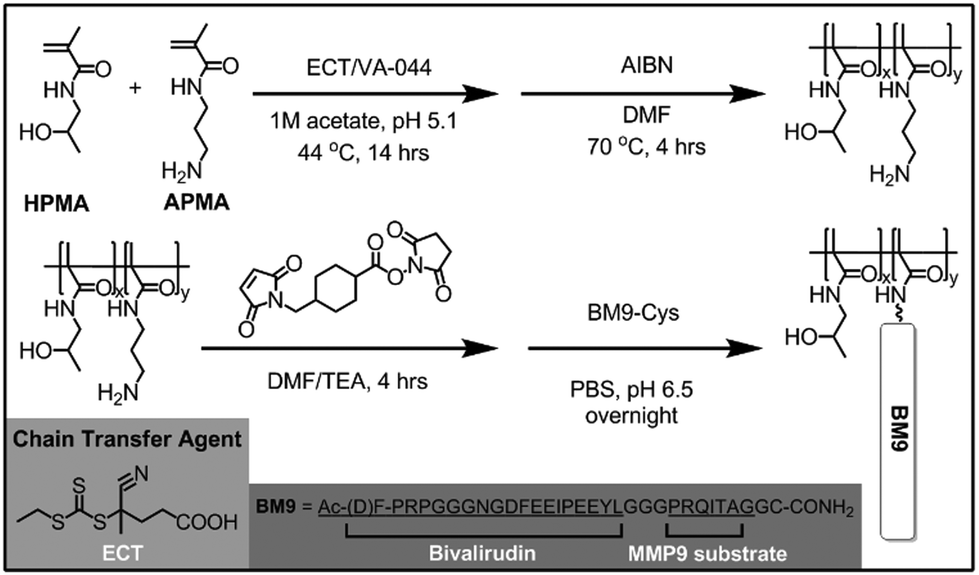 | ||
| Scheme 1 Synthetic scheme of HPMA-BM9 copolymers. | ||
Bivalirudin activity after peptide grafting onto polymers was confirmed using a colorimetric thrombin activity assay. All three HPMA-BM9 copolymers show similar thrombin inhibition kinetics and levels as free BM9 peptide regardless of polymer sizes (∼50% reduction in thrombin activity, Fig. S3†), confirming that polymer conjugation does not affect peptide activity (Fig. 2); pre-treatment of HPMA-BM9 polymers with MMP9 minimally affects thrombin inhibition (Fig. S4†). The retained bivalirudin activity when presented on a polymer backbone is likely due to adequate spacing between the bivalirudin sequences to minimize activity loss due to steric hindrance; spacing between grafted peptides was high (∼13 mol% peptide in polymer) as was spacing from the polymer backbone (12 amino acid linker). Peptides do not have to be cleaved from polymeric support for activity; therefore, MMP9-mediated bivalirudin release would be expected to improve the tissue penetration of bivalirudin peptides in vivo but is not required for activity.
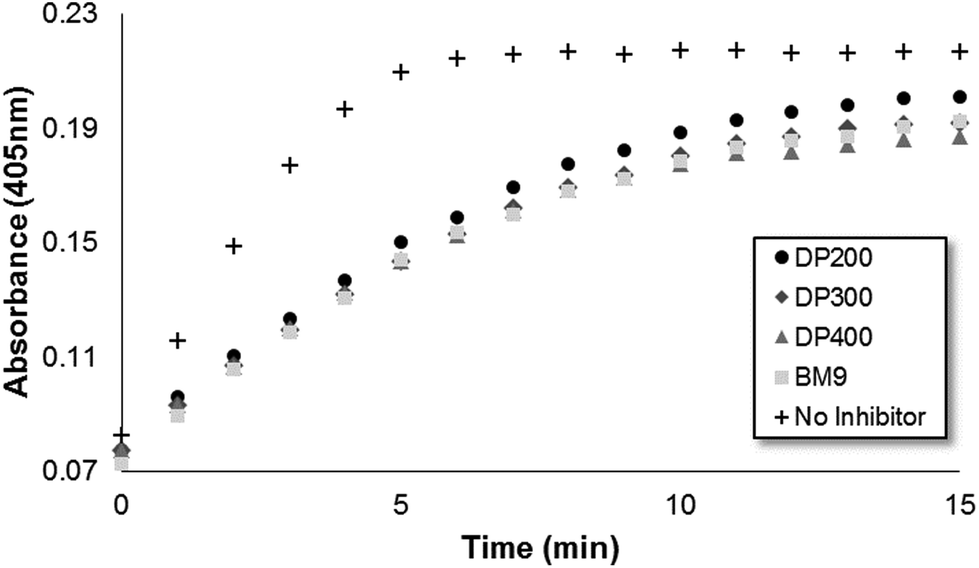 | ||
| Fig. 2 Thrombin activity measured by colorimetric substrate S-2238 as a function of time. | ||
MMP9-mediated degradation of HPMA-BM9 copolymers was evaluated by treating polymers with MMP9 and analyzing polymers at various time points by size exclusion chromatography (Fig. 3). Kinetics of BM9 peptide linker cleavage suggests >80% of peptide could be released within 24 h if similar enzyme activity is retained with polymers (Fig. S1b†). Degradation of DP200 begins within 4 h of incubation with MMP9 and continues for up to 24 h, as evidenced by increasing elution time as a function of MMP9 treatment time, indicating formation of lower molecular weight polymer populations. (Fig. 3a). Since BM9 peptide sequences are resistant to N-terminal peptidase activity, decreasing polymer molecular weight is attributed to the release of bivalirudin peptide following linker cleavage. DP300 and DP400 polymers degrade more slowly than DP200, showing noticeable shifts in molecular weight profiles within 4 h that last for at least 48 h (Fig. 3b and 3c). Slower peptide release kinetics could possibly be attributable to decreased enzymatic susceptibility due to more steric hindrance in larger polymers.
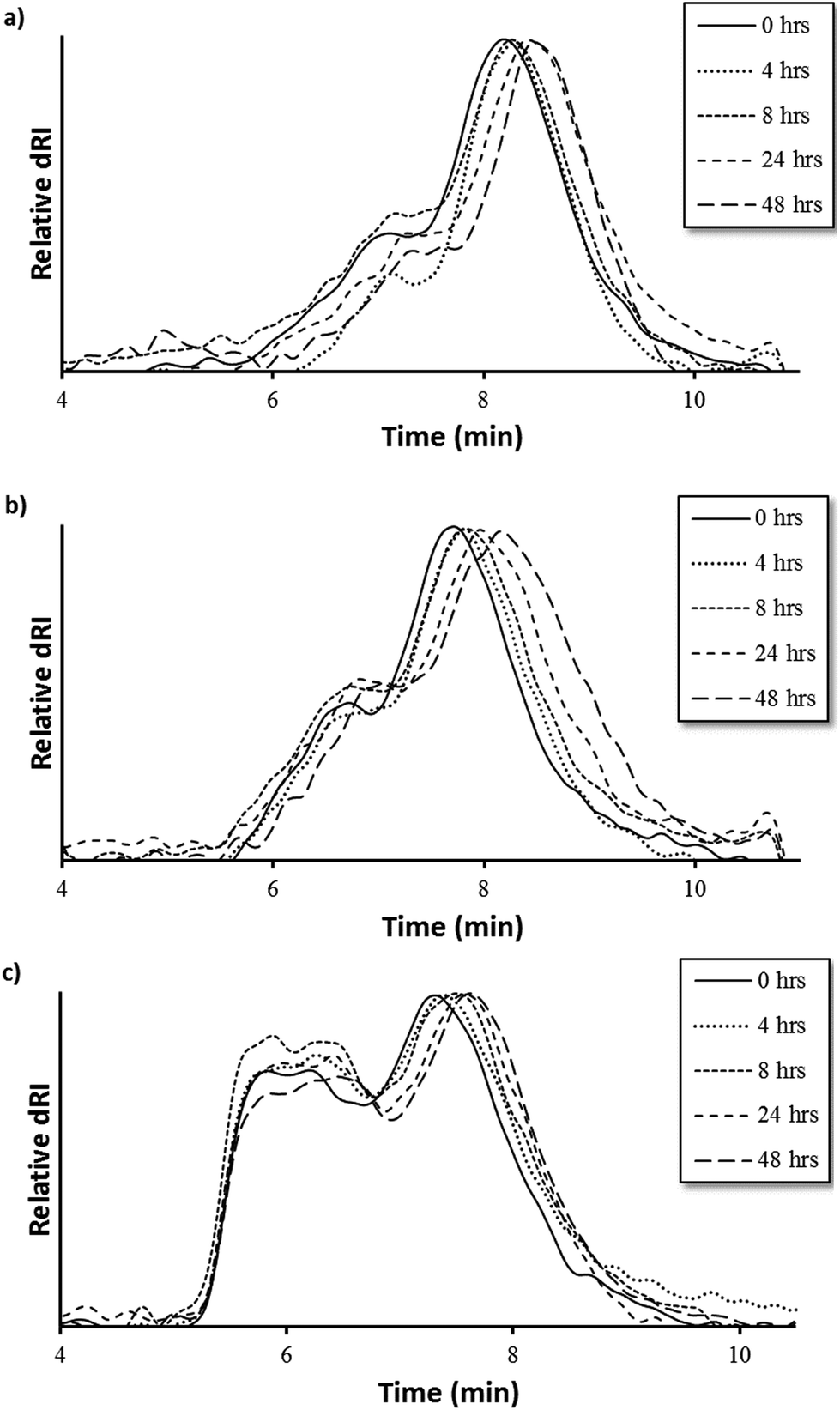 | ||
| Fig. 3 Polymer degradation as a function of elution time via size exclusion chromatography of (a) DP200, (b) DP300, and (c) DP400 HPMA-BM9 copolymers. | ||
The release kinetics of HPMA-BM9 copolymers physically encapsulated in HAMC hydrogels was evaluated in vitro. Polymer release from the HAMC hydrogels was quantified by incubating loaded hydrogels with HBSS and then sampling at various time points to determine the amount of released polymer by hydrolysing the various samples and assaying for amine concentration. For all three polymers, complete hydrogel release is observed within 48 h (Fig. 4). Trends, though not statistically significant, of decreased release rate with increasing polymer size are observed, corresponding to expected decreased diffusivity with increased polymer size. Importantly, BM9 copolymers show significantly delayed release compared to free bivalirudin peptide, demonstrating that peptide grafting on polymeric support decreases hydrogel release rates. Direct conjugation of the peptides to the hydrogel is a useful alternative strategy for prolonged release in future studies. We therefore proceeded with the highest molecular weight (DP400) polymer for in vivo evaluation of the materials.
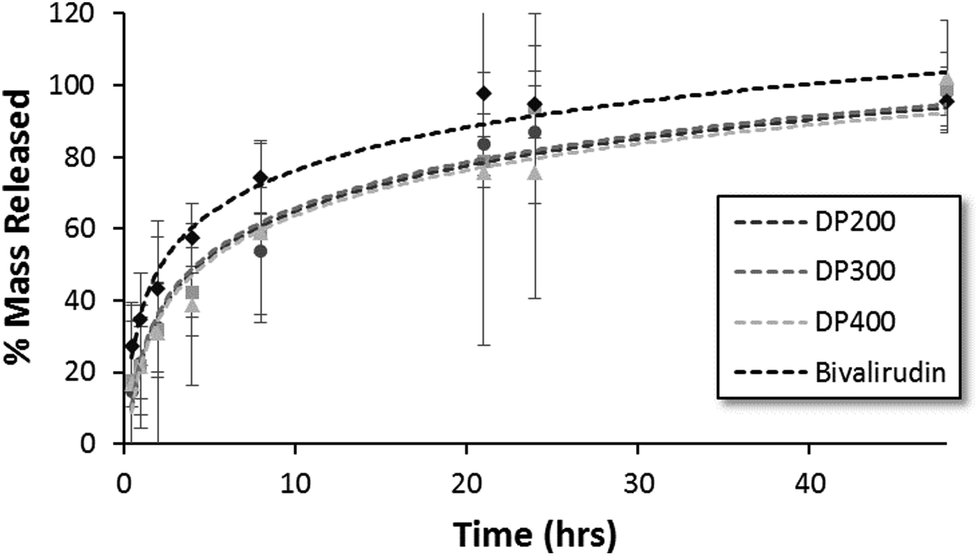 | ||
| Fig. 4 HPMA-BM9 copolymer and bivalirudin release from HAMC hydrogels as a function of time. | ||
To study the effects of localized thrombin inhibition on spinal cord recovery, we performed a laminectomy at cervical spinal level 4 (C4) and induced a lateral hemi-contusion injury.16 One hour following injury, rats received either no treatment, a localized intraspinal injection of DP400 base HPMA-APMA copolymer encapsulated in the HAMC hydrogel, or an injection of DP400 HPMA-BM9 polymer in a HAMC hydrogel within the lesion epicenter.
Previous studies have shown that proliferation of inflammatory cells, endogenous neural progenitors and glia is upregulated after spinal cord injury.16,17 To examine whether bivalirudin release affects cell proliferation after SCI, 5-bromo-2′-deoxyuridine (BrdU) was administered intraperitoneally 24 h post-injury (PI) to label mitotically-active cells (Fig. 5a and b). Spinal cord sections from the injury site show ∼45% decreased cellular proliferation for DP400 HPMA-BM9 treated animals compared to untreated control animals and pHPMA-APMA/HAMC treated animals (p < 0.05). These results confirm that the HAMC hydrogel and base HPMA-APMA polymer do not affect proliferative responses. Observed differences are therefore attributable to the thrombin-inhibition effects of the bivalirudin sequence.
 | ||
| Fig. 5 (a) Confocal images of BrdU and GFAP immunofluorescence in spinal cord sections at lesion epicenter. Bar, 100 μm. Inset shows BrdU at the lesion margin. Bar, 10 μm (b) Average BrdU+ cells as a measure of cellular proliferation in the C4 cervical vertebrae 24 h post-injury. (c) Average s100β+ cells as a measure of astrocyte density 30 days post-injury. *p < 0.05. Error bars = standard error. | ||
Since thrombin inhibition has been shown to reduce astrogliogenesis, we quantified astrocytes (s100β+ cells) proximal to the lesion epicenter (Fig. 5c). Rats treated with HPMA-BM9 polymer showed the lowest astrocyte density, a statistically-significant ∼15% decrease in astrocyte density (p < 0.05) compared to untreated injured animals. pHPMA-APMA/HAMC treated animals showed no significant difference. Analysis of the area around the lesion margin delimitated by GFAP-cells showed similar trends, but the differences did not show significance despite an apparent difference in density. Nonetheless, BM9 was able to reduce gliosis at the lesion without collateral effects on inflammation (Iba1-cells, Fig. S5†) or cell density (nuclei, Fig. S5 and S6†). BM9-treated rats tended to have increased oligodendrocyte populations (Fig. S6†), which could serve to promote functional regeneration in the long-term.
Following injury, many signals induce reactive gliosis.18 Astrogliosis is characterized by cellular hypertrophy, hyperplasia, increased glial fibrillary acidic protein (GFAP), proliferation, and ultimately scar formation;19 while playing important roles in restoration of extracellular ionic homeostasis and spatially limiting inflammation, astrogliosis can also significantly interfere with functional recovery through scar formation that impairs axon regeneration, leading to Wallarian degeneration (chronic demyelination), functional deficits and paralysis.20,21 Therefore, successful reduction in reactive gliosis would serve to reduce astrocyte proliferation and density to create a post-injury environment to augment recovery without an exacerbated paralytic response, which astrocyte ablation has been shown to affect.22 Our data show that polymer-loaded HAMC hydrogels do not have a negative impact on forelimb function or usage (Fig. S7†). Consequently, HPMA-BM9 reduced astrocyte numbers, suggesting lessened reactive gliosis during the post-injury response without collateral behavioral deficits. Since reactive gliosis is thought to impede recovery, BM9-loaded hydrogels might serve to remodel the post-injury response, inducing an environment more amenable to regenerative strategies after SCI.
Conclusions
In summary, HPMA-BM9 polymers are a promising platform for the grafting and display of functional peptides. Bivalirudin-grafted polymers maintain thrombin inhibition activity but demonstrate enzyme-mediated peptide release, making them a promising new formulation for active peptide delivery. Localized delivery of DP400 copolymer encapsulated in a HAMC hydrogel decreased cellular proliferation by 45% after 24 h and led to decreased astrocyte density after 30 days. These results suggest the DP400 copolymer and HAMC hydrogel system are effective therapeutics for reducing the immediate inflammatory response and long term scarring response, potentially leading to improved functional recovery following neural trauma.Acknowledgements
This work was funded by NIH 1R01NS064404 and the Craig H Neilsen Foundation (SAN 124679). D. Chu was supported by NIH T32 CA138312. We thank Drs Patrick Stayton and Anthony Convertine for generous donation of the ECT chain transfer agent.Notes and references
- K. A. Dunham and C. L. Floyd, in Animal Models of Movement Disorders: Volume II, ed. E. L. Lane and S. B. Dunnett, Humana Press, London, United Kingdom, 2011, vol. 62, pp. 345–362 Search PubMed.
- V. L. Turgeon and L. J. Houenou, Brain Res. Rev., 1997, 25, 85–95 CrossRef CAS.
- B. A. Citron, I. V. Smirnova, P. M. Arnold and B. W. Festoff, J. Neurotrauma, 2000, 17, 1191–1203 CrossRef CAS.
- S. Thuret, L. D. F. Moon and F. H. Gage, Nat. Rev. Neurosci., 2006, 7, 628–643 CrossRef CAS PubMed.
- A. Nishino, S. Michiyasu, H. Ohtani, O. Motohashi, K. Umezawa, H. Nagura and T. Toshimoto, J. Neurotrauma, 1993, 10, 167–179 CrossRef CAS.
- D. L. Sellers, T. H. Kim, C. W. Mount, S. H. Pun and P. J. Horner, Biomaterials, 2014, 35, 8895–8902 CrossRef CAS PubMed.
- B. W. Festoff, S. Ameenuddin, K. Santacruz, J. Morser, Z. Suo, P. M. Arnold, K. E. Stricker and B. A. Citron, J. Neurotrauma, 2004, 21, 907–922 CrossRef PubMed.
- M. J. Caicco, T. Zahir, A. J. Mothe, B. G. Ballios, A. J. Kihm, C. H. Tator and M. S. Shoichet, J. Biomed. Mater. Res., Part A, 2013, 101, 1472–1477 CrossRef PubMed.
- M. J. Caicco, M. J. Cooke, Y. Wang, A. Tuladhar, C. M. Morshead and M. S. Shoichet, J. Controlled Release, 2013, 166, 197–202 CrossRef CAS PubMed.
- T. E. Warkentin, A. Greinacher and A. Koster, Thromb. Haemostasis, 2008, 99, 830–839 CAS.
- K. Vulic and M. S. Shoichet, J. Am. Chem. Soc., 2012, 134, 882–885 CrossRef CAS PubMed.
- S. J. Kridel, E. Chen, L. P. Kotra, E. W. Howard, S. Mobashery and J. W. Smith, J. Biol. Chem., 2001, 276, 20572–20578 CrossRef CAS PubMed.
- L. J. Noble, F. Donovan, T. Igarashi, S. Goussev and Z. Werb, J. Neurosci., 2002, 22, 7526–7535 CAS.
- M. F. Powell, T. Stewart, L. Otvos Jr., L. Urge, F. C. Gaeta, A. Sette, T. Arrhenius, D. Thomson, K. Soda and S. M. Colon, Pharm. Res., 1993, 10, 1268–1273 CrossRef CAS.
- L. T. Nguyen, J. K. Chau, N. A. Perry, L. de Boer, S. A. Zaat and H. J. Vogel, PLoS One, 2010, 5, e12684 Search PubMed.
- S. E. Nutt, E.-A. Chang, S. T. Suhr, L. O. Schlosser, S. E. Mondello, C. T. Moritz, J. B. Cibelli and P. J. Horner, Exp. Neurol., 2013, 248, 491–503 CrossRef CAS PubMed.
- D. L. Sellers, D. O. Maris and P. J. Horner, J. Neurosci., 2009, 29, 6722–6733 CrossRef CAS PubMed.
- A. Pindon, M. Berry and D. Hantaï, J. Neurosci., 2000, 20, 2543–2550 CAS.
- M. Pekny and M. Nilsson, Glia, 2005, 50, 427–434 CrossRef PubMed.
- J. W. Fawcett and R. A. Asher, Brain Res. Bull., 1999, 49, 377–391 CrossRef CAS.
- J. M. Cregg, M. A. DePaul, A. R. Filous, B. T. Lang, A. Tran and J. Silver, Exp. Neurol., 2014, 253, 197–207 CrossRef PubMed.
- J. R. Faulkner, J. E. Herrmann, M. J. Woo, K. E. Tansey, N. B. Doan and M. V. Sofroniew, J. Neurosci., 2004, 24, 2143–2155 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4bm00259h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |