Organocatalytic asymmetric Henry reaction of isatins: Highly enantioselective synthesis of 3-hydroxy-2-oxindoles†
Yong
Zhang
,
Zhi
Jun Li
,
Hai
Sen Xu
,
Yuan
Zhang
and
Wei
Wang
*
State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, Gansu 730000, P. R. China. E-mail: wang_wei@lzu.edu.cn; Fax: + 86 931 891 5557
First published on 18th August 2011
Abstract
Organocatalytic asymmetric Henry reaction of isatins with nitromethane has been achieved with the use of C6′-OH cinchona alkaloid catalyst, affording 3-substituted 3-hydroxy-oxindoles in excellent yields and high enantioselectivities, and this method was successfully applied to the total synthesis of (R)-(+)-dioxibrassinin.
3-Substituted 3-hydroxyoxindoles have been found to be crucial structural motifs in a variety of alkaloid natural products and pharmaceuticals (Fig. 1), the biological activities of which are greatly affected by the substituted group at the C3-position and the absolute configuration of the stereogenic centers.2 Therefore, the development of new methodologies for the stereoselective synthesis of chiral 3-hydroxyoxindoles with a certain configuration is of paramount importance. Over the past years, excellent synthetic approaches based on transition-metal catalysis or organocatalysis have been advanced to tackle this synthetic challenge.3 Among all the methods developed, the nucleophilic addition to isatins proves to be the most straightforward one, which has attracted much research interest recently.4 In this regard, significant progress has been made on asymmetric aldol reactions for the construction of a chiral 3-hydroxyoxindole scaffold. However, the synthesis of this subunit via a catalytic asymmetric Henry (or nitroaldol) reaction has not been reported until recently.1
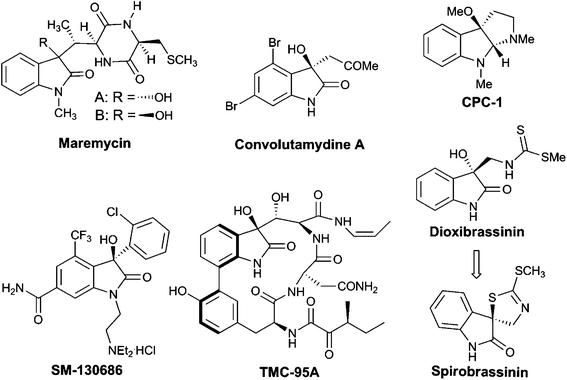 | ||
| Fig. 1 Selected examples of natural products and bioactive compounds containing 3-substituted 3-hydroxyoxindole motifs. | ||
The catalytic asymmetric Henry reaction provides an efficient strategy for constructing β-nitroalcohols, which are valuable building blocks in the synthesis of complex molecules.5 Although significant success has been made with aldehydes as the substrates in the asymmetric Henry reaction, the use of more challenging ketones6 as Henry acceptors to construct stereogenic quaternary carbon center7 has not been well explored.
Herein, we describe our research on the organocatalytic asymmetric Henry reaction of isatins for the construction of chiral 3-substituted 3-hydroxyoxindoles.1,8 This methodology was further applied as the key step for the total synthesis of (R)-(+)-dioxibrassinin and the formal synthesis of (S)-(−)-spirobrassinin.
We began our investigation with isatin (1a) and nitromethane (2) as the substrates, and with THF as the solvent for optimising the reaction conditions at 5 °C (Table 1). A series of bifunctional catalysts based on cinchona alkaloid scaffold were screened for this reaction. As shown in Table 1, the reaction catalysed by quinidine (QD) gave the desired product 3a in an almost quantitative yield, but with no enantioselectivity (entry 1); while quinidine-derived thiourea catalyst (QD-2) afforded the product 3a with 42% ee (entry 2). To our delight, the C6′-OH cinchona alkaloid catalyst9 (QD-1a) significantly improved the ee value to 76% (entry 3). This type of organocatalyst with different substituents at the C9-position (entries 3–8) had a great impact on the enantioselectivity of the product 3a. With –OBz as the substituent at C9-position, the catalyst QD-1d further improved the enantioselectivity to 88% ee (entry 6). When the catalyst Q-1d was used, another enantiomer of product 3a could be obtained with 87% ee (entry 7). Further improvement of ee value to 92% was found (entry 8), when the catalyst Q-1e6h with the more electron-withdrawing substituent, R = 3,5-(CF3)2-benzoyl, was employed.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time (h) | Yield (%)b | ee (%)c |
| a Reaction was performed with 0.1 mmol of 1a, 1.0 mmol of 2 and 10 mol% of catalyst in 1.0 mL of anhydrous solvent under argon. b Isolated yield after purification by column chromatography. c Determined by HPLC analysis using a Daicel chiral AD-H column. d 40 mg of 4 Å MS was added. e 25 μL of H2O was added. | |||||
| 1 | QD | THF | 5 | 99 | racemic |
| 2 | QD-2 | THF | 5 | 99 | 42 (S) |
| 3 | QD-1a | THF | 24 | 98 | 76 (S) |
| 4 | QD-1b | THF | 36 | 96 | 43 (S) |
| 5 | QD-1c | THF | 24 | 95 | 35 (S) |
| 6 | QD-1d | THF | 36 | 96 | 88 (S) |
| 7 | Q-1d | THF | 36 | 96 | 87 (R) |
| 8 | Q-1e | THF | 84 | 96 | 92 (R) |
| 9 | Q-1e | EtOAc | 60 | 96 | 79 (R) |
| 10 | Q-1e | Toluene | 36 | 93 | 57 (R) |
| 11 | Q-1e | CH2Cl2 | 36 | 93 | 4 (R) |
| 12 | Q-1e | EtOH | 20 | 98 | 30 (R) |
| 13 | Q-1e | Et2O | 96 | 94 | 82 (R) |
| 14d | Q-1e | THF | 84 | 96 | 92 (R) |
| 15e | Q-1e | THF | 84 | 96 | 70 (R) |
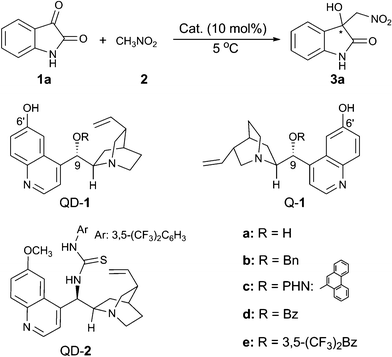
The influence of different solvents on the reaction was therefore investigated with Q-1e as the catalyst (Table 1, entries 8–14). All the solvents tested proved to be efficient, providing the product with excellent yield, however, the enantioselectivity varied significantly. With EtOAc (entry 9) or toluene (entry 10) as the solvent, the enantioselectivity is moderate. CH2Cl2 was not suitable for this reaction and gave an almost racemic product (entry 11). The reaction proceeded much faster in the polar protic solvent, EtOH, but the enantioselectivity is poor (entry 12). Amongst all the solvents screened, ethereal solvents proved to be more suitable for the reaction and THF emerged as the best. The addition of 4 Å molecular sieves (entry 14) did not have an obvious effect on the enantioselectivity. However, the ee value of 3a dramatically dropped in the presence of H2O (entry 15). Finally, the optimal reaction condition was determined to be: 10 mol% of Q-1e as the catalyst, THF as the solvent, and 5 °C as the reaction temperature.
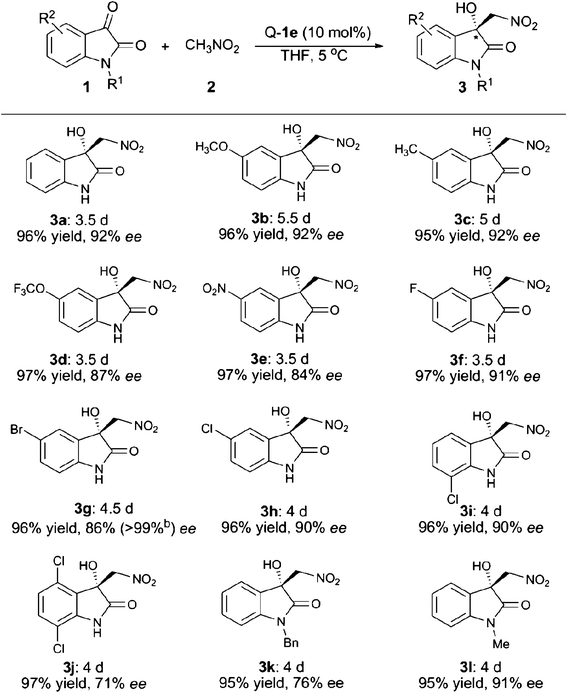
Next, we turned our focus to the substrate scope and the obtained results are summarised in Table 2. Whether electron-withdrawing or -donating groups at the C5- or the C7-position of the unprotected isatins (1a–1i) were employed, the reaction proceeded smoothly to give the product 3 in excellent yields (95–97%) along with high enantioselectivities (84–92% ee). However, for the substrate of 4,7-dichloroisatin (1j), only moderate enantioselectivity (71%) could be obtained. The protecting group on the N-atom also has an effect on the enantioselectivity. Utilization of N-benzyl isatin (1k) decreased the ee value to 76% while N-methyl isatin (1l) provided the corresponding product 3l with 91% ee. When nitroethane was used as the nucleophile, the reaction was rather slow and only 23% yield, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 5% ee were obtained after 5 days under the optimal reaction conditions. The enantiopure product of 3g (>99% ee) was obtained by slow recrystallisation from a mixture of acetone and hexane and the obtained single-crystals were suitable for X-ray analysis. As shown in Fig. 2, the absolute configuration of the quarternary carbon centre in the product 3g was determined to be R.10
:1 dr and 5% ee were obtained after 5 days under the optimal reaction conditions. The enantiopure product of 3g (>99% ee) was obtained by slow recrystallisation from a mixture of acetone and hexane and the obtained single-crystals were suitable for X-ray analysis. As shown in Fig. 2, the absolute configuration of the quarternary carbon centre in the product 3g was determined to be R.10
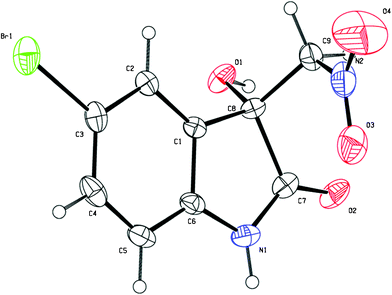 | ||
| Fig. 2 The X-ray single-crystal structure of the product 3g. | ||
| a The reaction was performed with 0.1 mmol of 1, 1.0 mmol of 2 and 10 mol% of catalyst Q-1e in 1 mL of anhydrous THF under argon. Yield refers to the isolated product. Enantiomeric excess was determined by HPLC analysis using a Daicel chiral AD-H or OJ-H column. b After recrystallisation. |
|---|
|
We have applied the above method to realise the catalytic asymmetric total synthesis of (R)-(+)-dioxibrassinin, which can be further transformed to (S)-(−)-spirobrassinin.1,11 As shown in Scheme 1, the use of isatin 1a as the substrate under optimised scale-up conditions gave the Henry product 3a with 95% yield and 91% ee. The product 3a was further reduced to the corresponding amino alcohol using Zn/HOAc conditions. We found that, after the treatment with HCl, the reaction residue could be directly used for the next transformation without isolation. In the presence of pyridine, the previous mixture was treated with CS2 and followed by MeI to give (R)-(+)-dioxibrassinin with 65% yield and 89% ee over two steps.
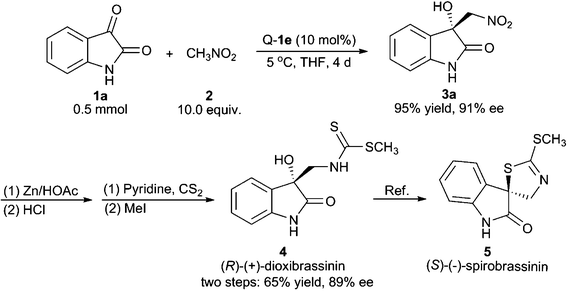 | ||
| Scheme 1 Asymmetric total synthesis of (R)-(+)-dioxibrassinin and formal synthesis of (S)-(−)-spirobrassinin. | ||
In summary, we have developed an organocatalytic asymmetric Henry reaction of isatins and nitromethane by using C6′-OH cinchona alkaloid catalyst. A variety of chiral 3-substituted 3-hydroxyoxindoles have been successfully synthesized in excellent yields and high enantioselectivities. This method was further applied in the total synthesis of (R)-(+)-dioxibrassinin.
Acknowledgements
The National Natural Foundation of China (No. 20972064) is gratefully acknowledged for financial support.References
- Presented as a poster at the 5th ChemComm International Symposium (May 18, 2011, Lanzhou). When we were about to submit the manuscript, independent but very similar work from the group of Prof. Wei Wang (University of New Mexico) appeared in advance: Chem.–Eur. J., 2011, 17, 7791. (published online on May 30, 2011; submitted on April 4, 2011). To avoid possible confusion, please note that the corresponding author of this manuscript, Prof. Wei Wang at Lanzhou University, is distinct from Prof. Wei Wang at University of New Mexico. Their native names are also different characters.
- S. Peddibhotla, Curr. Bioact. Compd., 2009, 5, 20 Search PubMed.
- (a) F. Zhou, Y.-L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 CAS; (b) J. J. Badillo, N. V.Hanhan and A. K. Franz, Curr. Opin. Drug Discovery Dev., 2010, 13, 758 CAS.
- For recent examples of the nucleophilic addition to isatins: (a) G. Luppi, P. G. Cozzi, M. Monari, B. Kaptein, Q. B. Broxterman and C. Tomasini, J. Org. Chem., 2005, 70, 7418 CrossRef CAS; (b) R. Shintani, M. Inoue and T. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 3353 CrossRef CAS; (c) A. V. Malkov, M. A. Kabeshov, M. Bella, O. Kysilka, D. A. Malyshev, K. Pluháčková and P. Kočovský, Org. Lett., 2007, 9, 5473 CrossRef CAS; (d) S. Nakamura, N. Hara, H. Nakashima, K. Kubo, N. Shibata and T. Toru, Chem.–Eur. J., 2008, 14, 8079 CrossRef CAS; (e) R. Shintani, S. Hayashi, M. Murakami, M. Takeda and T. Hayashi, Org. Lett., 2009, 11, 3754 CrossRef CAS; (f) T. Itoh, H. Ishikawa and Y. Hayashi, Org. Lett., 2009, 11, 3854 CrossRef CAS; (g) N. Hara, S. Nakamura, N. Shibata and T. Toru, Chem.–Eur. J., 2009, 15, 6790 CrossRef CAS; (h) J. Itoh, S. B. Han and M. J. Krische, Angew. Chem., Int. Ed., 2009, 48, 6313 CrossRef CAS; (i) F. Xue, S. Zhang, L. Liu, W. Duan and W. Wang, Chem. –Asian. J., 2009, 4, 1664 CrossRef CAS; (j) N. V. Hanhan, A. H. Sahin, T. W. Chang, J. C. Fettinger and A. K. Franz, Angew. Chem., Int. Ed., 2010, 49, 744 CAS; (k) Y.-L. Liu, B.-L. Wang, J.-J. Cao, L. Chen, Y.-X. Zhang, C. Wang and J. Zhou, J. Am. Chem. Soc., 2010, 132, 15176 CrossRef CAS; (l) J. Deng, S. Zhang, P. Ding, H. Jiang, W. Wang and J. Li, Adv. Synth. Catal., 2010, 352, 833 CAS; (m) P. Chauhan and S. S. Chimni, Chem.–Eur. J., 2010, 16, 7709 CrossRef CAS; (n) X.-N. Wang, Y.-Y. Zhang and S. Ye, Adv. Synth. Catal., 2010, 352, 1892 CrossRef CAS; (o) Q. Guo, M. Bhanushali and C.-G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 9460 CrossRef CAS; (p) F. Zhong, G.-Y. Chen and Y. Lu, Org. Lett., 2011, 13, 82 CrossRef CAS; (q) X.-Y. Guan, Y. Wei and M. Shi, Chem. –Eur. J., 2010, 16, 13617 CrossRef; (r) K. Zheng, C. K. Yin, X. H. Liu, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2011, 50, 2573 CrossRef CAS. For examples via other methods, see: (s) T. Ishimaru, N. Shibata, J. Nagai, S. Nakamura, T. Toru and S. Kanemasa, J. Am. Chem. Soc., 2006, 128, 16488 CrossRef; (t) D. Sano, K. Nagata and T. Itoh, Org. Lett., 2008, 10, 1593 CrossRef CAS; (u) T. Bui, N. R. Candeias and C. F. Barbas III, J. Am. Chem. Soc., 2010, 132, 5574 CrossRef CAS; (v) Y.-X. Jia, J. M. Hillgren, E. L. Watson, S. P. Marsden and E. P. Kündig, Chem. Commun., 2008, 4040 RSC; (w) D. Tomita, K. Yamatsugu, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 6946 CrossRef CAS.
- For recent reviews on the catalytic asymmetric Henry reactions, see: (a) C. Palomo, M. Oiarbide and A. Laso, Eur. J. Org. Chem., 2007, 2561 CrossRef CAS; (b) J. Boruwa, N. Gogoi, P. P. Saikia and N. C. Barua, Tetrahedron: Asymmetry, 2006, 17, 3315 CrossRef CAS; (c) C. Palomo, M. Oiarbide and A. Mielgo, Angew. Chem., Int. Ed., 2004, 43, 5442 CrossRef CAS.
- For selected examples, see: (a) C. Christensen, K. Juhl and K. A. Jørgensen, Chem. Commun., 2001, 2222 RSC; (b) C. Christensen, K. Juhl, R. G. Hazell and K. A. Jørgensen, J. Org. Chem., 2002, 67, 4875 CrossRef CAS; (c) Y. Misumi, R. A. Bulman and K. Matsumoto, Heterocycles, 2002, 56, 599 CrossRef CAS; (d) S. F. Lu, D. M. Du, S. W. Zhang and J. X. Xu, Tetrahedron: Asymmetry, 2004, 15, 119 CrossRef CAS; (e) D. M. Du, S. F. Lu, T. Fang and J. X. Xu, J. Org. Chem., 2005, 70, 3712 CrossRef CAS; (f) H. Li, B. Wang and L. Deng, J. Am. Chem. Soc., 2006, 128, 732 CrossRef CAS; (g) T. Mandal, S. Samanta and C.-G. Zhao, Org. Lett., 2007, 9, 943 CrossRef CAS; (h) M. Bandini, R. Sinisi and A. Umani-Ronchi, Chem. Commun., 2008, 4360 RSC.
- For recent reviews on the catalytic asymmetric synthesis of quaternary stereogenic carbon centers, see: (a) M. Bella and T. Gasperi, Synthesis, 2009, 1583 CrossRef CAS; (b) O. Riant and J. Hannedouche, Org. Biomol. Chem., 2007, 5, 873 RSC.
- For the racemic example, see: M. N. Elinson, A. I. llovaisky, V. M. Merkulova, F. Barba and B. Batanero, Tetrahedron, 2008, 64, 5915 Search PubMed.
- For examples of 6′-OH cinchona alkaloid catalysts in asymmetric reactions, see: (a) H. Li, Y. Wang, L. Tang and L. Deng, J. Am. Chem. Soc., 2004, 126, 9906 CrossRef CAS; (b) H. Li, Y. Wang, L. Tang, F. Wu, X. Liu, C. Guo, B. M. Foxman and L. Deng, Angew. Chem., Int. Ed., 2005, 44, 105 CrossRef CAS; (c) X. Liu, H. Li and L. Deng, Org. Lett., 2005, 7, 167 CrossRef CAS; (d) H. Li, J. Song, X. Liu and L. Deng, J. Am. Chem. Soc., 2005, 127, 8948 CrossRef CAS; (e) F. Wu, H. Li, R. Hong and L. Deng, Angew. Chem., Int. Ed., 2006, 45, 947 CrossRef CAS; (f) Y. Wang, X. Liu and L. Deng, J. Am. Chem. Soc., 2006, 128, 3928 CrossRef CAS; (g) F. Wu, R. Hong, J. Khan, X. Liu and L. Deng, Angew. Chem., Int. Ed., 2006, 45, 4301 CrossRef CAS; (h) X. Liu, B. Sun and L. Deng, Synlett, 2009, 1685 Search PubMed.
- Crystal data for 3g: C9H7BrN2O4, M = 287.08, monoclinic, a = 7.437(15) Å, b = 6.480(13) Å, c = 10.87(2) Å, α = 90.00°, β = 90.185(17)°, γ = 90.00°, V = 523.6(18) Å3, T = 296(2)K, space groupP21, Z = 2, 3567 reflections measured, 1895 independent reflections (Rint = 0.0669). The final R1 values were 0.0488 (I > 2σ(I)). The final wR(F2) values were 0.1074 (I > 2σ(I)). The final R1 values were 0.0776 (all data). The final wR(F2) values were 0.1191 (all data). Supplementary crystallographic data have been deposited at the Cambridge Crystallographic Data Centre, CCDC 828228.
- For the isolation of (R)-(+)-dioxibrassinin, see: (a) K. Monde, K. Sasaki, A. Shirata and M. Takasugi, Phytochemistry, 1991, 30, 2915 CrossRef CAS. For the isolation of (S)-(−)-spirobrassinin, see: (b) M. Takasugi, K. Monde, N. Katsui and A. Shirata, Chem. Lett., 1987, 1631 CAS. For the synthesis of (S)-(−)-spirobrassinin, see: (c) K. Monde, S. Osawa, N. Harada, M. Takasugi, M. Suchy, P. Kutschy, M. Dzurilla and E. Balentova, Chem. Lett., 2000, 886 CrossRef CAS; (d) M. Suchý, P. Kutschy, K. Monde, H. Goto, N. Harada, M. Takasugi, M. Dzurilla and E. Balentova, J. Org. Chem., 2001, 66, 3940 CrossRef CAS; (e) P. Kutschy, M. Suchý, K. Monde, N. Harada, R. Maruskova, Z. Curillova, M. Dzurilla, M. Miklosova, R. Mezencev and J. Mojzis, Tetrahedron Lett., 2002, 43, 9489 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: representative experimental procedures, full characterization of all novel compounds, crystallographic data of compound 3g. CCDC reference number 828228. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00477h |
| This journal is © The Royal Society of Chemistry 2011 |